
The Therapeutic Treatment with the GAG-Binding Chemokine Fragment CXCL9(74–103) Attenuates Neutrophilic Inflammation and Lung Dysfunction during Klebsiella pneumoniae Infection in Mice

 , , , , , , , and
, , , , , , , and 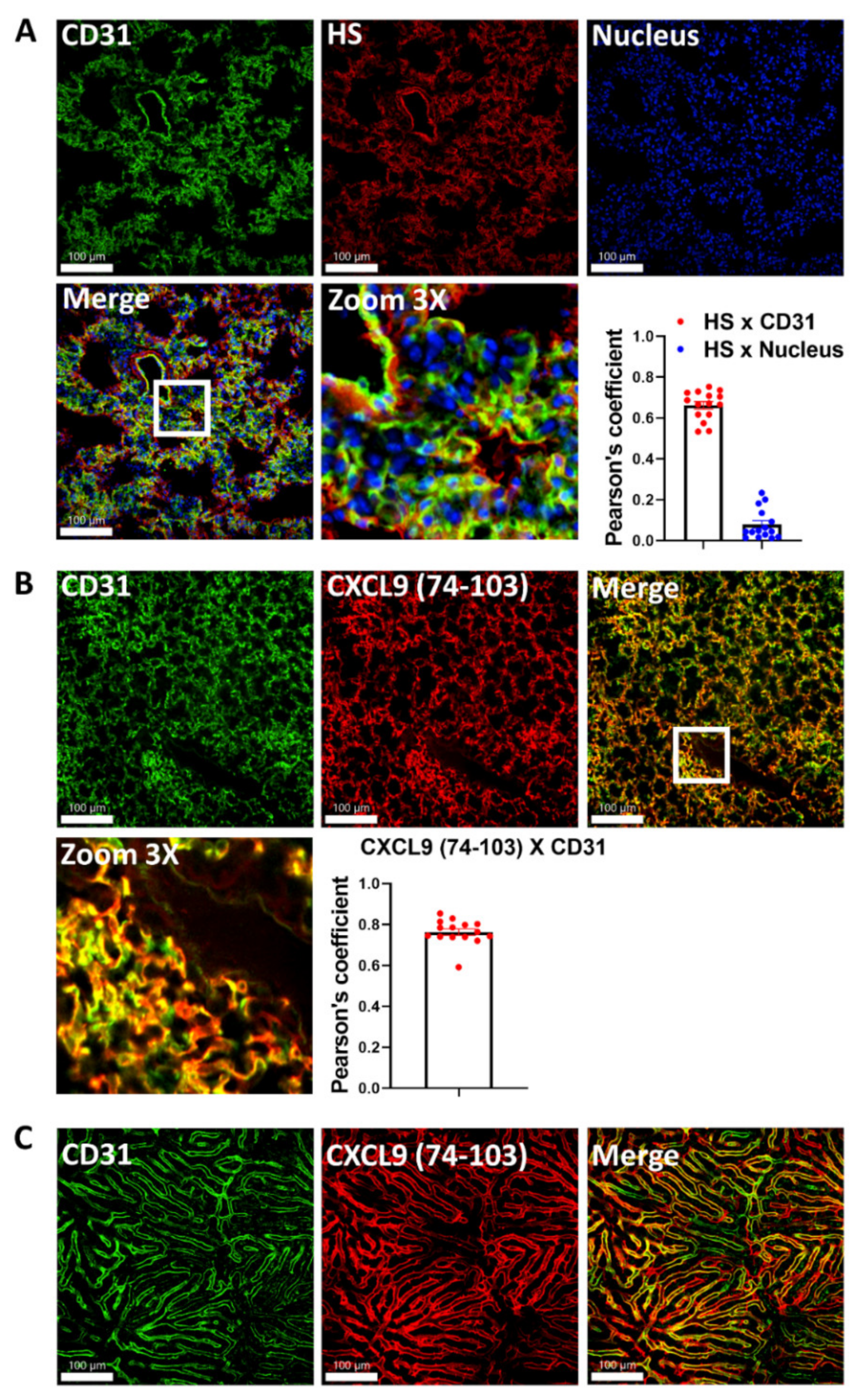{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
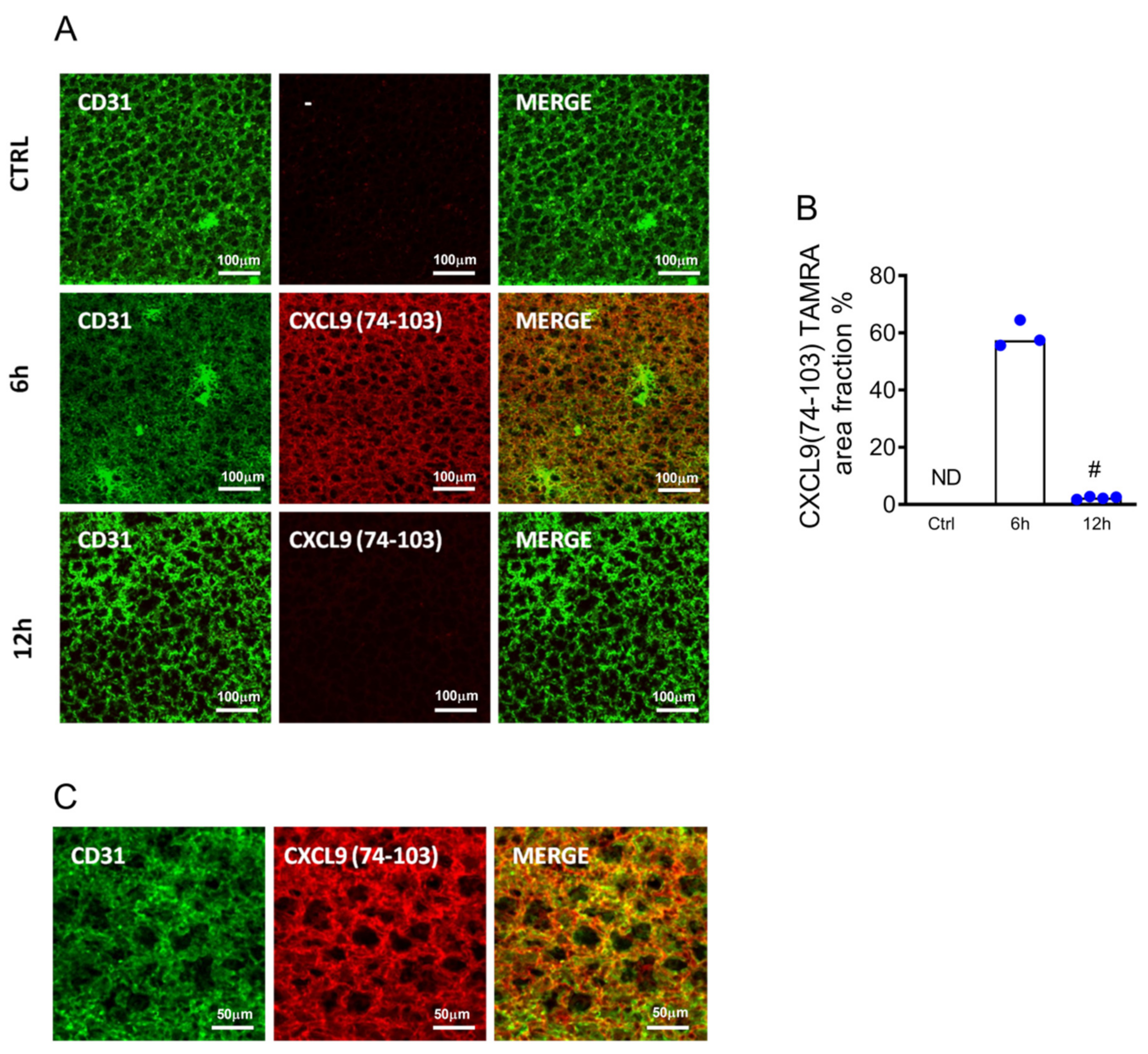
2.1. CXCL9(74–103) Binds to the Lung Endothelium In Vivo
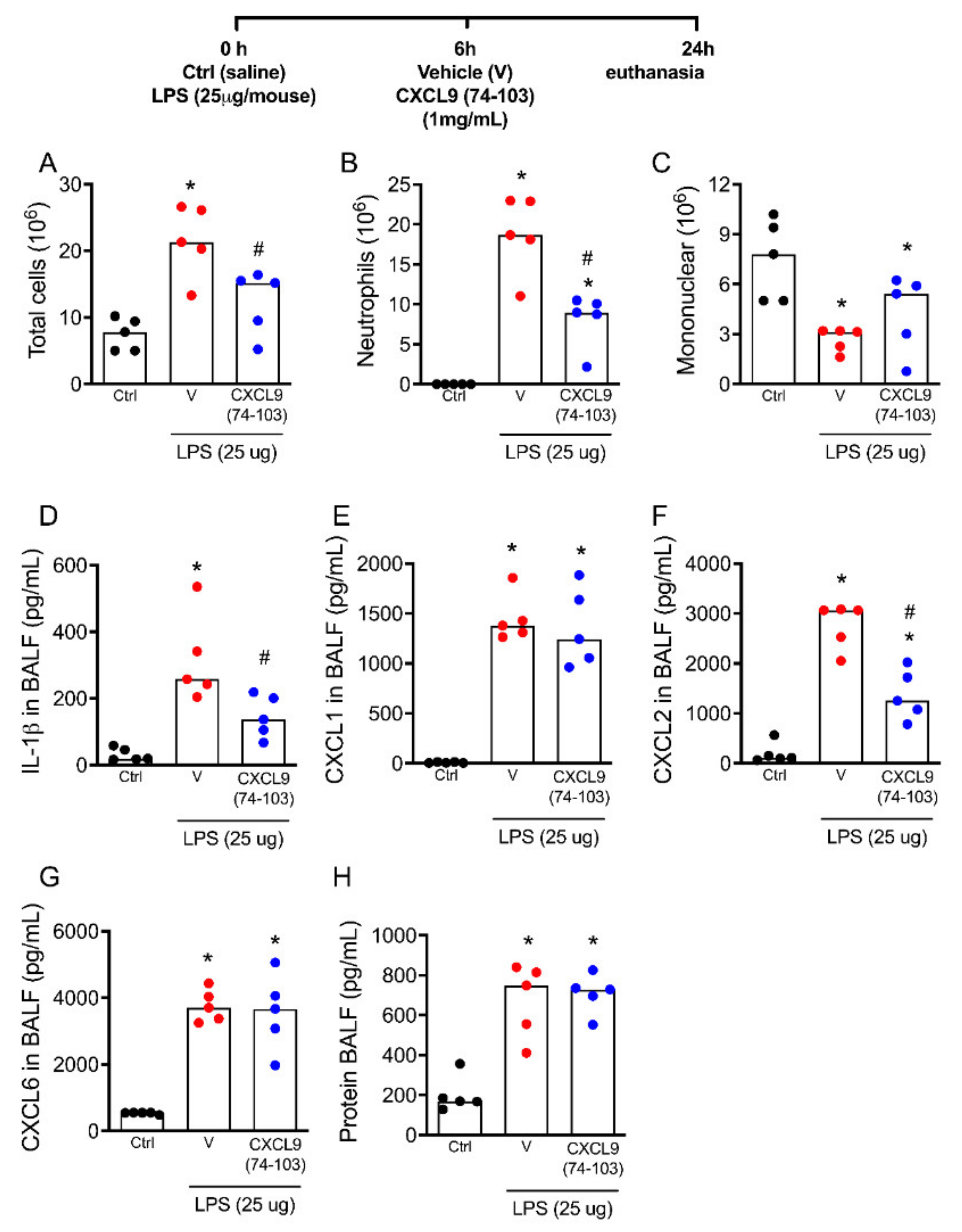
2.2. The Treatment with CXCL9(74–103) Decreases Lung Inflammation and Neutrophil Recruitment in Response to Lipopolysaccharide (LPS) In Vivo
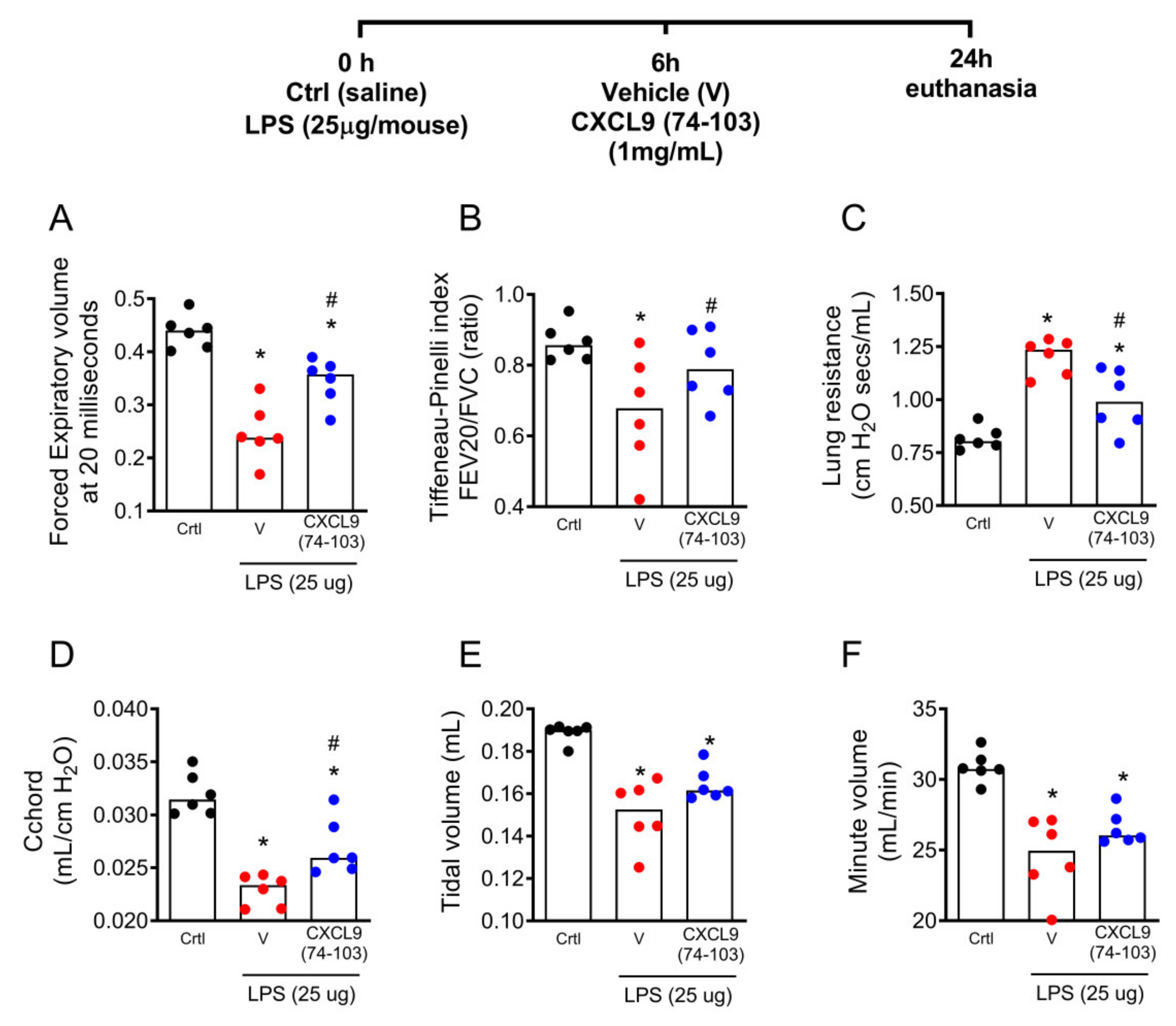
2.3. The Treatment with CXCL9(74–103) Improved Lung Function in LPS-Instilled Mice
2.4. The Treatment with CXCL9(74–103) Decreased the Inflammatory Response in Pneumonia Induced by Klebsiella pneumoniae Infection
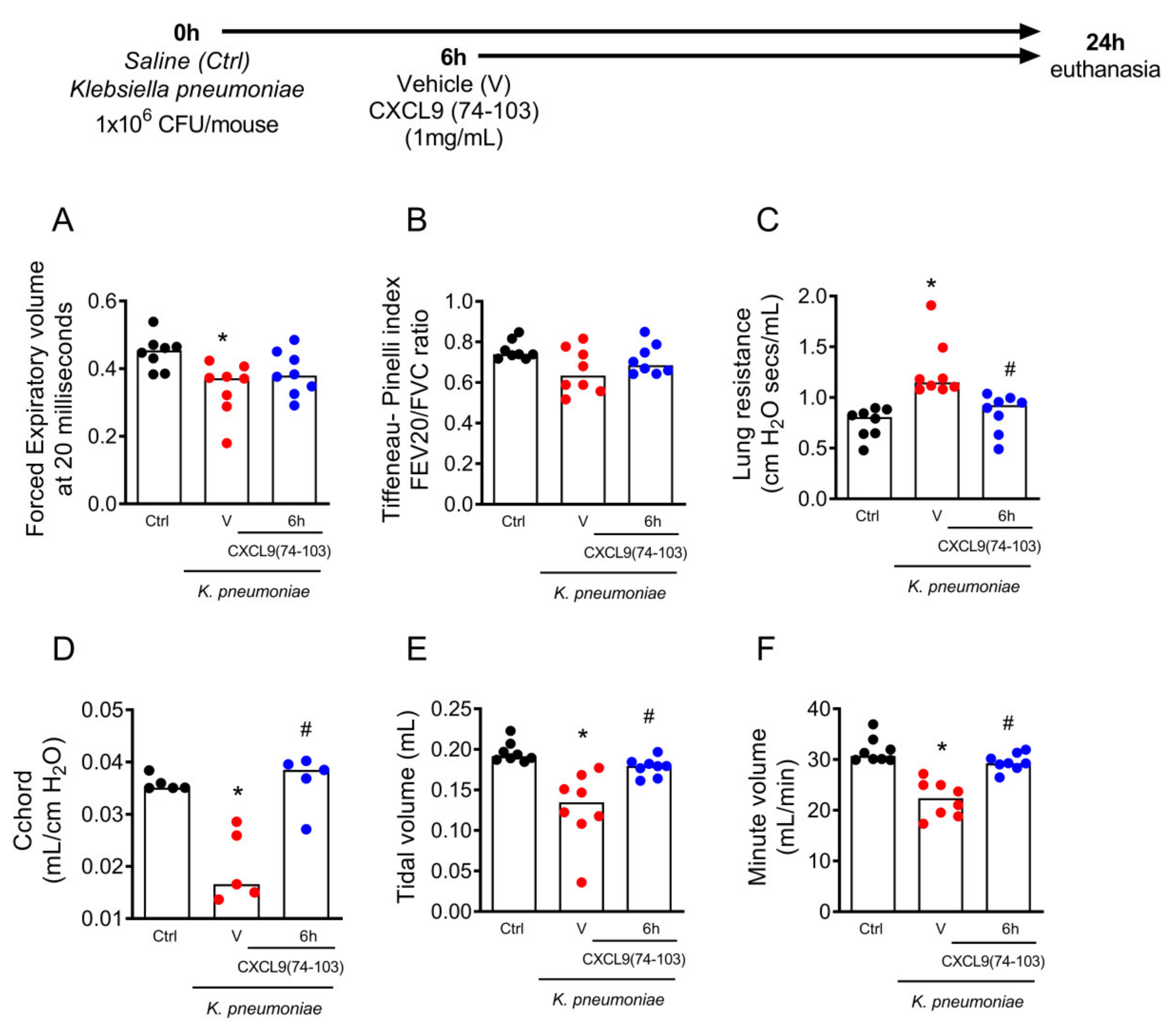
2.5. The Treatment with CXCL9(74–103) Improved Lung Function in Pneumonia Induced by Klebsiella pneumoniae
3. Discussion
4. Materials and Methods
4.1. Mice and Reagents
4.2. Solid-Phase Synthesis of the C-Terminal CXCL9-Derived Peptide
4.3. Intravital Microscopy
4.4. Bacterial Strain
4.5. LPS-Induced Acute Lung Inflammation
4.6. Klebsiella pneumoniae Lung Infection
4.7. Bronchoalveolar Lavage Fluid (BALF) and CFU Counts
4.8. Measurement of Chemokines and Cytokines
4.9. Assessment of Respiratory Mechanic Dysfunction
4.10. Cryosections and Immunostainings
4.11. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lanks, C.W.; Musani, A.I.; Hsia, D.W. Community-Acquired Pneumonia and Hospital-Acquired Pneumonia. Med. Clin. N. Am. 2019, 103, 487–501. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Revised WHO Classification and Treatment of Childhood Pneumonia at Health Facilities: Evidence Summaries; WHO: Geneva, Switzerland, 2014; ISBN 9789241507813. [Google Scholar]
- Torres, A.; Cilloniz, C.; Niederman, M.S.; Menéndez, R.; Chalmers, J.D.; Wunderink, R.G.; van der Poll, T. Pneumonia. Nat. Rev. Dis. Primers 2021, 7, 1–28. [Google Scholar] [CrossRef]
- Yin, Y.; Zhao, C.; Li, H.; Jin, L.; Wang, Q.; Wang, R.; Zhang, Y.; Zhang, J.; Wang, H.; Yang, C.; et al. Clinical and Microbiological Characteristics of Adults with Hospital-Acquired Pneumonia: A 10-Year Prospective Observational Study in China. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Ashurst, J.V. Klebsiella Pneumonia Pathophysiology; StatPearls: Treasure Island, FL, USA, 2020; pp. 1–5. [Google Scholar] [PubMed]
- Sattar, S.B.A.; Sharma, S. Bacterial Pneumonia; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Xiong, H.; Carter, R.A.; Leiner, I.M.; Tang, Y.W.; Chen, L.; Kreiswirth, B.N.; Pamer, E.G. Distinct Contributions of Neutrophils and CCR2+ Monocytes to Pulmonary Clearance of Different Klebsiella Pneumoniae Strains. Infect. Immun. 2015, 83, 3418–3427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Chen, Z.; Yuan, Y.; Jing, H.; Zou, J.; Zhang, X.; Zeng, X.; Zhang, W.; Zou, Q.; Zhang, J. Innate Immune Effectors Play Essential Roles in Acute Respiratory Infection Caused by Klebsiella Pneumoniae. J. Immunol. Res. 2020, 2020, 5291714. [Google Scholar] [CrossRef]
- Balamayooran, G.; Batra, S.; Fessler, M.B.; Happel, K.I.; Jeyaseelan, S. Mechanisms of Neutrophil Accumulation in the Lungs against Bacteria. Am. J. Respir. Cell Mol. Biol. 2010, 43, 5–16. [Google Scholar] [CrossRef]
- Hirche, T.O.; Gaut, J.P.; Heinecke, J.W.; Belaaouaj, A. Myeloperoxidase Plays Critical Roles in Killing Klebsiella Pneumoniae and Inactivating Neutrophil Elastase: Effects on Host Defense. J. Immunol. 2005, 174, 1557–1565. [Google Scholar] [CrossRef] [Green Version]
- Claushuis, T.A.M.; van der Donk, L.E.H.; Luitse, A.L.; van Veen, H.A.; van der Wel, N.N.; van Vught, L.A.; Roelofs, J.J.T.H.; de Boer, O.J.; Lankelma, J.M.; Boon, L.; et al. Role of Peptidylarginine Deiminase 4 in Neutrophil Extracellular Trap Formation and Host Defense during Klebsiella Pneumoniae—Induced Pneumonia-Derived Sepsis. J. Immunol. 2018, 201, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- Fleeman, R.M.; Macias, L.A.; Brodbelt, J.S.; Davies, B.W. Defining Principles That Influence Antimicrobial Peptide Activity against Capsulated Klebsiella Pneumoniae. Proc. Natl. Acad. Sci. USA 2020, 117, 27620–27626. [Google Scholar] [CrossRef]
- Zhao, Y.; Olonisakin, T.F.; Xiong, Z.; Hulver, M.; Sayeed, S.; Yu, M.T.; Gregory, A.D.; Kochman, E.J.; Chen, B.B.; Mallampalli, R.K.; et al. Thrombospondin-1 Restrains Neutrophil Granule Serine Protease Function and Regulates the Innate Immune Response during Klebsiella Pneumoniae Infection. Mucosal Immunol. 2015, 8, 896–905. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.D.; Porter, A.R.; Dorward, D.W.; Brinkworth, A.J.; Chen, L.; Kreiswirth, B.N.; Deleo, F.R. Phagocytosis and Killing of Carbapenem-Resistant ST258 Klebsiella Pneumoniae by Human Neutrophils. J. Infect. Dis. 2016, 213, 1615–1622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinton, L.J.; Mizgerd, J.P. Dynamics of Lung Defense in Pneumonia: Resistance, Resilience, and Remodeling. Annu. Rev. Physiol. 2015, 77, 407–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Happel, K.I.; Dubin, P.J.; Zheng, M.; Ghilardi, N.; Lockhart, C.; Quinton, L.J.; Odden, A.R.; Shellito, J.E.; Bagby, G.J.; Nelson, S.; et al. Divergent Roles of IL-23 and IL-12 in Host Defense against Klebsiella Pneumoniae. J. Exp. Med. 2005, 202, 761–769. [Google Scholar] [CrossRef]
- Craig, A.; Mai, J.; Cai, S.; Jeyaseelan, S. Neutrophil Recruitment to the Lungs during Bacterial Pneumonia. Infect. Immun. 2009, 77, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzemaekers, M.; Gouwy, M.; Proost, P. Neutrophil Chemoattractant Receptors in Health and Disease: Double-Edged Swords. Cell. Mol. Immunol. 2020, 17, 433–450. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Sperandio, M.; Mócsai, A. Neutrophils as Emerging Therapeutic Targets. Nat. Rev. Drug Discov. 2020, 19, 253–275. [Google Scholar] [CrossRef] [PubMed]
- Dyer, D.P.; Salanga, C.L.; Volkman, B.F.; Kawamura, T.; Handel, T.M. The Dependence of Chemokine-Glycosaminoglycan Interactions on Chemokine Oligomerization. Glycobiology 2015, 26, 312–326. [Google Scholar] [CrossRef]
- Russo, R.C.; Garcia, C.C.; Teixeira, M.M.; Amaral, F.A. The CXCL8/IL-8 Chemokine Family and Its Receptors in Inflammatory Diseases. Expert Rev. Clin. Immunol. 2014, 10, 593–619. [Google Scholar] [CrossRef] [Green Version]
- Hamel, D.J.; Sielaff, I.; Proudfoot, A.E.I.; Handel, T.M. Chapter 4. Interactions of Chemokines with Glycosaminoglycans. Methods Enzymol. 2009, 461, 71–102. [Google Scholar] [CrossRef]
- Crijns, H.; Vanheule, V.; Proost, P. Targeting Chemokine—Glycosaminoglycan Interactions to Inhibit Inflammation. Front. Immunol. 2020, 11, 483. [Google Scholar] [CrossRef]
- Vanheule, V.; Boff, D.; Mortier, A.; Janssens, R.; Petri, B.; Kolaczkowska, E.; Kubes, P.; Berghmans, N.; Struyf, S.; Kungl, A.J.; et al. CXCL9-Derived Peptides Differentially Inhibit Neutrophil Migration in Vivo through Interference with Glycosaminoglycan Interactions. Front. Immunol. 2017, 8, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crijns, H.; Adyns, L.; Ganseman, E.; Cambier, S.; Vandekerckhove, E.; Pörtner, N.; Vanbrabant, L.; Struyf, S.; Gerlza, T.; Kungl, A.; et al. Affinity and Specificity for Binding to Glycosaminoglycans Can Be Tuned by Adapting Peptide Length and Sequence. Int. J. Mol. Sci. 2022, 23, 447. [Google Scholar] [CrossRef] [PubMed]
- De Zutter, A.; Crijns, H.; Berghmans, N.; García-Caballero, M.; Vanbrabant, L.; Pörtner, N.; Vanheule, V.; Verscheure, P.; Siddiquei, M.M.; Abu El-Asrar, A.M.; et al. The Chemokine-Based Peptide, CXCL9(74–103), Inhibits Angiogenesis by Blocking Heparan Sulfate Proteoglycan-Mediated Signaling of Multiple Endothelial Growth Factors. Cancers 2021, 13, 5090. [Google Scholar] [CrossRef] [PubMed]
- Vanheule, V.; Janssens, R.; Boff, D.; Kitic, N.; Berghmans, N.; Ronsse, I.; Kungl, A.J.; Amaral, F.A.; Teixeira, M.M.; Van Damme, J.; et al. The Positively Charged COOH-Terminal Glycosaminoglycan-Binding CXCL9(74–103) Peptide Inhibits CXCL8-Induced Neutrophil Extravasation and Monosodium Urate Crystal-Induced Gout in Mice. J. Biol. Chem. 2015, 290, 21292–21304. [Google Scholar] [CrossRef] [Green Version]
- Marques, P.E.; Vandendriessche, S.; Oliveira, T.H.C.; Crijns, H.; Lopes, M.E.; Blanter, M.; Schuermans, S.; Yu, K.; Poosti, F.; Vanheule, V.; et al. Inhibition of Drug-Induced Liver Injury in Mice Using a Positively Charged Peptide That Binds DNA. Hepatol. Commun. 2021, 5, 1737–1754. [Google Scholar] [CrossRef]
- Eutamene, H.; Theodorou, V.; Schmidlin, F.; Tondereau, V.; Garcia-Villar, R.; Salvador-Cartier, C.; Chovet, M.; Bertrand, C.; Bueno, L. LPS-Induced Lung Inflammation Is Linked to Increased Epithelial Permeability: Role of MLCK. Eur. Respir. J. 2005, 25, 789–796. [Google Scholar] [CrossRef]
- Håkansson, H.F.; Smailagic, A.; Brunmark, C.; Miller-Larsson, A.; Lal, H. Altered Lung Function Relates to Inflammation in an Acute LPS Mouse Model. Pulm. Pharmacol. Ther. 2012, 25, 399–406. [Google Scholar] [CrossRef]
- Karaiskos, I.; Lagou, S.; Pontikis, K.; Rapti, V.; Poulakou, G. The “Old” and the “New” Antibiotics for MDR Gram-Negative Pathogens: For Whom, When, and How. Front. Public Health 2019, 7, 151. [Google Scholar] [CrossRef] [Green Version]
- Bassetti, M.; Righi, E.; Carnelutti, A.; Graziano, E.; Russo, A. Multidrug-Resistant Klebsiella Pneumoniae: Challenges for Treatment, Prevention and Infection Control. Expert Rev. Anti-Infect. Ther. 2018, 16, 749–761. [Google Scholar] [CrossRef]
- Navon-Venezia, S.; Kondratyeva, K.; Carattoli, A. Klebsiella Pneumoniae: A Major Worldwide Source and Shuttle for Antibiotic Resistance. FEMS Microbiol. Rev. 2017, 41, 252–275. [Google Scholar] [CrossRef]
- Boff, D.; Oliveira, V.L.S.; Queiroz Junior, C.M.; Silva, T.A.; Allegretti, M.; Verri, W.A.; Proost, P.; Teixeira, M.M.; Amaral, F.A. CXCR2 Is Critical for Bacterial Control and Development of Joint Damage and Pain in Staphylococcus Aureus-Induced Septic Arthritis in Mouse. Eur. J. Immunol. 2018, 48, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boff, D.; Crijns, H.; Teixeira, M.; Amaral, F.; Proost, P. Neutrophils: Beneficial and Harmful Cells in Septic Arthritis. Int. J. Mol. Sci. 2018, 19, 468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Cao, K.; Zhao, Y.; Du, J. Targeting Neutrophils in Sepsis: From Mechanism to Translation. Front. Pharmacol. 2021, 12, 644270. [Google Scholar] [CrossRef] [PubMed]
- Mattos, M.S.; Ferrero, M.R.; Kraemer, L.; Lopes, G.A.O.; Reis, D.C.; Cassali, G.D.; Oliveira, F.M.S.; Brandolini, L.; Allegretti, M.; Garcia, C.C.; et al. CXCR1 and CXCR2 Inhibition by Ladarixin Improves Neutrophil-Dependent Airway Inflammation in Mice. Front. Immunol. 2020, 11, 566953. [Google Scholar] [CrossRef]
- Cao, Q.; Li, B.; Wang, X.; Sun, K.; Guo, Y. Therapeutic Inhibition of CXC Chemokine Receptor 2 by SB225002 Attenuates LPS-Induced Acute Lung Injury in Mice. Arch. Med. Sci. 2018, 14, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Russo, R.C.; Guabiraba, R.; Garcia, C.C.; Barcelos, L.S.; Roffê, E.; Souza, A.L.S.; Amaral, F.A.; Cisalpino, D.; Cassali, G.D.; Doni, A.; et al. Role of the Chemokine Receptor CXCR2 in Bleomycin-Induced Pulmonary Inflammation and Fibrosis. Am. J. Respir. Cell Mol. Biol. 2009, 40, 410–421. [Google Scholar] [CrossRef]
- Capucetti, A.; Albano, F.; Bonecchi, R. Multiple Roles for Chemokines in Neutrophil Biology. Front. Immunol. 2020, 11, 1259. [Google Scholar] [CrossRef]
- Gschwandtner, M.; Strutzmann, E.; Teixeira, M.M.; Anders, H.J.; Diedrichs-Möhring, M.; Gerlza, T.; Wildner, G.; Russo, R.C.; Adage, T.; Kungl, A.J. Glycosaminoglycans Are Important Mediators of Neutrophilic Inflammation in Vivo. Cytokine 2017, 91, 65–73. [Google Scholar] [CrossRef]
- Wei, J.; Peng, J.; Wang, B.; Qu, H.; Wang, S.; Faisal, A.; Cheng, J.W.; Gordon, J.R.; Li, F. CXCR1/CXCR2 Antagonism Is Effective in Pulmonary Defense against Klebsiella Pneumoniae Infection. BioMed Res. Int. 2013, 2013, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Schilter, H.C.; Collison, A.; Russo, R.C.; Foot, J.S.; Yow, T.T.; Vieira, A.T.; Tavares, L.D.; Mattes, J.; Teixeira, M.M.; Jarolimek, W. Effects of an Anti-Inflammatory VAP-1/SSAO Inhibitor, PXS-4728A, on Pulmonary Neutrophil Migration. Respir. Res. 2015, 16, 42. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Aloe, C.; Wilson, N.; Bozinovski, S. G-CSFR Antagonism Reduces Neutrophilic Inflammation during Pneumococcal and Influenza Respiratory Infections without Compromising Clearance. Sci. Rep. 2019, 9, 17732. [Google Scholar] [CrossRef] [PubMed]
- Toews, G.B. Cytokines and the Lung. Eur. Respir. J. Suppl. 2001, 18, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzemaekers, M.; Cambier, S.; Blanter, M.; Vandooren, J.; de Carvalho, A.C.; Malengier-Devlies, B.; Vanderbeke, L.; Jacobs, C.; Coenen, S.; Martens, E.; et al. Kinetics of Peripheral Blood Neutrophils in Severe Coronavirus Disease 2019. Clin. Transl. Immunol. 2021, 10, e1271. [Google Scholar] [CrossRef] [PubMed]
- Vanderbeke, L.; Van Mol, P.; Van Herck, Y.; De Smet, F.; Humblet-Baron, S.; Martinod, K.; Antoranz, A.; Arijs, I.; Boeckx, B.; Bosisio, F.M.; et al. Monocyte-Driven Atypical Cytokine Storm and Aberrant Neutrophil Activation as Key Mediators of COVID-19 Disease Severity. Nat. Commun. 2021, 12, 4117. [Google Scholar] [CrossRef]
- Cai, S.; Batra, S.; Wakamatsu, N.; Pacher, P.; Jeyaseelan, S. NLRC4 Inflammasome-Mediated Production of IL-1β Modulates Mucosal Immunity in the Lung against Gram-Negative Bacterial Infection. J. Immunol. 2012, 188, 5623–5635. [Google Scholar] [CrossRef] [Green Version]
- He, X.; Qian, Y.; Li, Z.; Fan, E.K.; Li, Y.; Wu, L.; Billiar, T.R.; Wilson, M.A.; Shi, X.; Fan, J. TLR4-Upregulated IL-1β and IL-1RI Promote Alveolar Macrophage Pyroptosis and Lung Inflammation through an Autocrine Mechanism. Sci. Rep. 2016, 6, 31663. [Google Scholar] [CrossRef] [Green Version]
- Cai, S.; Batra, S.; Lira, S.A.; Kolls, J.K.; Jeyaseelan, S. CXCL1 Regulates Pulmonary Host Defense to Klebsiella Infection via CXCL2, CXCL5, NF-ΚB, and MAPKs. J. Immunol. 2010, 185, 6214–6225. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, M.; Zemans, R.L.; Jeyaseelan, S. Role of Chemokines in the Pathogenesis of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2012, 46, 566–572. [Google Scholar] [CrossRef] [Green Version]
- Kolb, M.; Margetts, P.J.; Anthony, D.C.; Pitossi, F.; Gauldie, J. Transient Expression of IL-1beta Induces Acute Lung Injury and Chronic Repair Leading to Pulmonary Fibrosis. J. Clin. Investig. 2001, 107, 1529–1536. [Google Scholar] [CrossRef] [Green Version]
- Ganter, M.T.; Roux, J.; Miyazawa, B.; Howard, M.; Frank, J.A.; Su, G.; Sheppard, D.; Violette, S.M.; Weinreb, P.H.; Horan, G.S.; et al. Interleukin-1β Causes Acute Lung Injury via Avβ5 and Avβ6 Integrin-Dependent Mechanisms. Circ. Res. 2008, 102, 804–812. [Google Scholar] [CrossRef] [Green Version]
- Schultz, M.J.; Rijneveld, A.W.; Florquin, S.; Edwards, C.K.; Dinarello, C.A.; Van Der Poll, T. Role of Interleukin-1 in the Pulmonary Immune Response during Pseudomonas Aeruginosa Pneumonia. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2002, 282, 285–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labrousse, D.; Perret, M.; Hayez, D.; Da Silva, S.; Badiou, C.; Couzon, F.; Bes, M.; Chavanet, P.; Lina, G.; Vandenesch, F.; et al. Kineret®/IL-1Ra Blocks the IL-1/IL-8 Inflammatory Cascade during Recombinant Panton Valentine Leukocidin-Triggered Pneumonia but Not during S. Aureus Infection. PLoS ONE 2014, 9, e97546. [Google Scholar] [CrossRef] [PubMed]
- Peiró, T.; Patel, D.F.; Akthar, S.; Gregory, L.G.; Pyle, C.J.; Harker, J.A.; Birrell, M.A.; Lloyd, C.M.; Snelgrove, R.J. Neutrophils Drive Alveolar Macrophage IL-1β Release during Respiratory Viral Infection. Thorax 2018, 73, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadatomo, A.; Inoue, Y.; Ito, H.; Karasawa, T.; Kimura, H.; Watanabe, S.; Mizushina, Y.; Nakamura, J.; Kamata, R.; Kasahara, T.; et al. Interaction of Neutrophils with Macrophages Promotes IL-1β Maturation and Contributes to Hepatic Ischemia-Reperfusion Injury. J. Immunol. 2017, 199, 3306–3315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grailer, J.J.; Canning, B.A.; Kalbitz, M.; Haggadone, M.D.; Dhond, R.M.; Andjelkovic, A.V.; Zetoune, F.S.; Ward, P.A. Critical Role for the NLRP3 Inflammasome during Acute Lung Injury. J. Immunol. 2014, 192, 5974–5983. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, A.E.I.; Johnson, Z.; Bonvin, P.; Handel, T.M. Glycosaminoglycan Interactions with Chemokines Add Complexity to a Complex System. Pharmaceuticals 2017, 10, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boff, D.; Crijns, H.; Janssens, R.; Vanheule, V.; Menezes, G.B.; Macari, S.; Silva, T.A.; Amaral, F.A.; Proost, P. The Chemokine Fragment CXCL9(74–103) Diminishes Neutrophil Recruitment and Joint Inflammation in Antigen-Induced Arthritis. J. Leukoc. Biol. 2018, 104, 413–422. [Google Scholar] [CrossRef]
- Jin, L.; Batra, S.; Douda, D.N.; Palaniyar, N.; Jeyaseelan, S. CXCL1 Contributes to Host Defense in Polymicrobial Sepsis via Modulating T Cell and Neutrophil Functions. J. Immunol. 2014, 193, 3549–3558. [Google Scholar] [CrossRef]
- Baines, K.J.; Backer, V.; Gibson, P.G.; Powel, H.; Porsbjerg, C.M. Impaired Lung Function Is Associated with Systemic Inflammation and Macrophage Activation. Eur. Respir. J. 2015, 45, 557–559. [Google Scholar] [CrossRef] [Green Version]
- Laitinen, L.A.; Miettinen, A.K.; Kuosma, E.; Huhtala, L.; Lehtomaki, K. Lung Function Impairment Following Mycoplasmal and Other Acute Pneumonias. Eur. Respir. J. 1992, 5, 670–674. [Google Scholar]
- Lappalainen, U.; Whitsett, J.A.; Wert, S.E.; Tichelaar, J.W.; Bry, K. Interleukin-1β Causes Pulmonary Inflammation, Emphysema, and Airway Remodeling in the Adult Murine Lung. Am. J. Respir. Cell Mol. Biol. 2005, 32, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Mizgerd, J.P. Inflammation and Pneumonia: Why Are Some More Susceptible than Others? Clin. Chest Med. 2018, 39, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Lazaar, A.L.; Miller, B.E.; Donald, A.C.; Keeley, T.; Ambery, C.; Russell, J.; Watz, H.; Tal-Singer, R. CXCR2 Antagonist for Patients with Chronic Obstructive Pulmonary Disease with Chronic Mucus Hypersecretion: A Phase 2b Trial. Respir. Res. 2020, 21, 149. [Google Scholar] [CrossRef] [PubMed]
- Sokulsky, L.A.; Garcia-Netto, K.; Nguyen, T.H.; Girkin, J.L.N.; Collison, A.; Mattes, J.; Kaiko, G.; Liu, C.; Bartlett, N.W.; Yang, M.; et al. A Critical Role for the CXCL3/CXCL5/CXCR2 Neutrophilic Chemotactic Axis in the Regulation of Type 2 Responses in a Model of Rhinoviral-Induced Asthma Exacerbation. J. Immunol. 2020, 205, 2468–2478. [Google Scholar] [CrossRef]
- Koenig, L.M.; Boehmer, D.F.R.; Metzger, P.; Schnurr, M.; Endres, S.; Rothenfusser, S. Blocking Inflammation on the Way: Rationale for CXCR2 Antagonists for the Treatment of COVID-19. J. Exp. Med. 2020, 217, 9–11. [Google Scholar] [CrossRef]
- Loos, T.; Mortier, A.; Proost, P. Chapter 1 Isolation, Identification, and Production of Posttranslationally Modified Chemokines. Methods Enzymol. 2009, 461, 3–29. [Google Scholar] [CrossRef]
- Asti, C.; Ruggieri, V.; Porzio, S.; Chiusaroli, R.; Melillo, G.; Caselli, G.F. Lipopolysaccharide-Induced Lung Injury in Mice. I. Concomitant Evaluation of Inflammatory Cells and Haemorrhagic Lung Damage. Pulm. Pharmacol. Ther. 2000, 13, 61–69. [Google Scholar] [CrossRef]
- Vieira, A.T.; Rocha, V.M.; Tavares, L.; Garcia, C.C.; Teixeira, M.M.; Oliveira, S.C.; Cassali, G.D.; Gamba, C.; Martins, F.S.; Nicoli, J.R. Control of Klebsiella Pneumoniae Pulmonary Infection and Immunomodulation by Oral Treatment with the Commensal Probiotic Bifidobacterium Longum 51A. Microbes Infect. 2016, 18, 180–189. [Google Scholar] [CrossRef]
- Russo, R.C.; Savino, B.; Mirolo, M.; Buracchi, C.; Germano, G.; Anselmo, A.; Zammataro, L.; Pasqualini, F.; Mantovani, A.; Locati, M.; et al. The Atypical Chemokine Receptor ACKR2 Drives Pulmonary Fibrosis by Tuning Influx of CCR2+ and CCR5+ IFNγ-Producing ΓδT Cells in Mice. Am. J. Physiol.—Lung Cell. Mol. Physiol. 2018, 314, L1010–L1025. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boff, D.; Russo, R.C.; Crijns, H.; de Oliveira, V.L.S.; Mattos, M.S.; Marques, P.E.; Menezes, G.B.; Vieira, A.T.; Teixeira, M.M.; Proost, P.; et al. The Therapeutic Treatment with the GAG-Binding Chemokine Fragment CXCL9(74–103) Attenuates Neutrophilic Inflammation and Lung Dysfunction during Klebsiella pneumoniae Infection in Mice. Int. J. Mol. Sci. 2022, 23, 6246. https://doi.org/10.3390/ijms23116246
Boff D, Russo RC, Crijns H, de Oliveira VLS, Mattos MS, Marques PE, Menezes GB, Vieira AT, Teixeira MM, Proost P, et al. The Therapeutic Treatment with the GAG-Binding Chemokine Fragment CXCL9(74–103) Attenuates Neutrophilic Inflammation and Lung Dysfunction during Klebsiella pneumoniae Infection in Mice. International Journal of Molecular Sciences. 2022; 23(11):6246. https://doi.org/10.3390/ijms23116246
Chicago/Turabian StyleBoff, Daiane, Remo Castro Russo, Helena Crijns, Vivian Louise Soares de Oliveira, Matheus Silvério Mattos, Pedro Elias Marques, Gustavo Batista Menezes, Angélica Thomaz Vieira, Mauro Martins Teixeira, Paul Proost, and et al. 2022. "The Therapeutic Treatment with the GAG-Binding Chemokine Fragment CXCL9(74–103) Attenuates Neutrophilic Inflammation and Lung Dysfunction during Klebsiella pneumoniae Infection in Mice" International Journal of Molecular Sciences 23, no. 11: 6246. https://doi.org/10.3390/ijms23116246