
Blood–Brain Barrier Dysfunction and Astrocyte Senescence as Reciprocal Drivers of Neuropathology in Aging


1
Department of Integrative Biology, University of California, Berkeley, CA 94720, USA
2
Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA
3
Helen Wills Neuroscience Institute, University of California, Berkeley, CA 94720, USA
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(11), 6217; https://doi.org/10.3390/ijms23116217
Submission received: 29 April 2022
/
Revised: 26 May 2022
/
Accepted: 29 May 2022
/
Published: 1 June 2022
(This article belongs to the Special Issue Aging and Senescence 2.0)
{kind=link}
Abstract
:As the most abundant cell types in the brain, astrocytes form a tissue-wide signaling network that is responsible for maintaining brain homeostasis and regulating various brain activities. Here, we review some of the essential functions that astrocytes perform in supporting neurons, modulating the immune response, and regulating and maintaining the blood–brain barrier (BBB). Given their importance in brain health, it follows that astrocyte dysfunction has detrimental effects. Indeed, dysfunctional astrocytes are implicated in age-related neuropathology and participate in the onset and progression of neurodegenerative diseases. Here, we review two mechanisms by which astrocytes mediate neuropathology in the aging brain. First, age-associated blood–brain barrier dysfunction (BBBD) causes the hyperactivation of TGFβ signaling in astrocytes, which elicits a pro-inflammatory and epileptogenic phenotype. Over time, BBBD-associated astrocyte dysfunction results in hippocampal and cortical neural hyperexcitability and cognitive deficits. Second, senescent astrocytes accumulate in the brain with age and exhibit a decreased functional capacity and the secretion of senescent-associated secretory phenotype (SASP) factors, which contribute to neuroinflammation and neurotoxicity. Both BBBD and senescence progressively increase during aging and are associated with increased risk of neurodegenerative disease, but the relationship between the two has not yet been established. Thus, we discuss the potential relationship between BBBD, TGFβ hyperactivation, and senescence with respect to astrocytes in the context of aging and disease and identify future areas of investigation in the field.
1. Introduction
Age-onset neurodegenerative disorders such as Alzheimer’s disease (AD) and related dementias (ADRD) are debilitating conditions that progressively impair the memory, cognition, and daily functioning of afflicted individuals. As the average lifespan has increased in developed nations, so has the prevalence of symptomatic AD. An estimated 6.2 million adults aged 65 and older are living with Alzheimer’s dementia, presenting a substantial societal burden [1]. However, despite decades of research, negligible progress has been made in developing effective disease-modifying treatments capable of halting or reversing AD progression and associated cognitive decline. The unsatisfactory clinical trial outcomes of experimental AD therapies indicate the need for a greater understanding of the age-related processes that drive neurodegenerative diseases.
In recent years, increased focus has been placed on understanding the role of astrocytes in driving such neurological disorders. As the most abundant cell type in the adult brain, astrocytes form a sophisticated tissue-wide signaling network responsible for maintaining brain homeostasis and regulating various brain activities. Consequently, there are many potential mechanisms through which astrocytes may contribute to neural deficits in the context of advanced age and disease. Two such mechanisms are blood–brain barrier dysfunction (BBBD) and cellular senescence. Both progressively increase during aging and are associated with an increased risk of neurodegenerative disease, but the relationship between the two, if any, has not yet been established. Here, we discuss the causes and consequences of BBBD and senescence with respect to astrocytes and identify future areas of investigation in the field.
2. Astrocyte Functions
Astrocytes are stellated glial cells that interface with nearly every functional element of the brain. They are the most abundant glial cells in the brain and can comprise up to 50% of the tissue volume in some regions [2]. Their branch-like processes surround neurons, axons, synapses, and blood vessels, and perform numerous functions that are essential for brain homeostasis and neural functioning [3]. Therefore, astrocytes play a pivotal role in preserving brain health, as well as driving pathogenic processes [4,5]. Our understanding of astrocyte functions is continually growing as researchers study this heterogenous population of cells under different conditions, and it is increasingly evident that astrocytes perform unique functions, depending on their temporal and regional location within the brain [6,7,8,9]. Here, we briefly review the astrocyte functions related to neuronal support, immune modulation, and regulation of the blood–brain barrier (BBB), as three factors that go awry in aging and disease.
2.1. Neuronal Support
At the macrostructural level, astrocytes help form a functional glial network that extends from the ependyma to the pial surface via gap junctions [10,11]. The perivascular feet of astrocytes associate with the parenchymal basal lamina to form a plexus called the glia limitans. This thin, but dense, structure surrounds the pia mater, subpial space, and perivascular spaces, and plays an essential role in controlling the movement of substances from the blood or cerebrospinal fluid (CSF) into the brain parenchyma, where neurons are located [12].
In addition to their role in barrier function, astrocytes maintain the optimal conditions required for neurotransmission within the cerebral microenvironment. To transmit a signal, neurotransmitters are released from an axon terminal into the synaptic cleft, where they interact with post-synaptic receptors. Ending the transmission requires neurotransmitter uptake from the synaptic cleft by neurons and astrocytes. While neurons primarily uptake the inhibitory transmitter, gamma-amino butyric acid (GABA), astrocytes are responsible for the uptake and metabolism of the excitatory amino acid, glutamate [13,14,15]. Additionally, the propagation of nerve impulses involves cellular depolarization, which causes local extracellular changes in ion concentration. Astrocytes contain ion channels, enzymes, and receptors that enable them to modify extracellular ion concentrations and pH following depolarization to restore the surrounding milieu to its resting state [16,17,18,19]. Astrocytes are also critical in removing harmful metabolites and waste products from the brain. They can directly metabolize some soluble waste products, such as ammonia; alternatively, they collect and shuttle unwanted metabolites and soluble proteins, such as amyloid beta (Aβ) to the vasculature for elimination via the glymphatic system [15,20,21].
In addition to maintaining the cerebral microenvironment, astrocytes also participate in neurotransmission and are critical in shaping the complex circuitry of the brain [22]. Astrocytes form tripartite complexes with presynaptic and postsynaptic nerve terminals, through which they help define synaptic connections. Astrocytes are the major source of extracellular matrix proteins, cell adhesion molecules, and neurotrophic factors in the central nervous system (CNS), which are essential in promoting neurite growth and elongation [23,24,25,26,27]. Additionally, astrocytes physically associate with neuronal synapses via perisynaptic astrocytic processes (PAPs) that regulate many aspects of their formation, maturation, and function [22,28]. For example, astrocytes regulate synaptogenesis via the secretion of thrombospondins (TSP) [29] and TGFβ1 [30,31], and can specifically control the maturation and plasticity of certain circuits via the secretion of Hevin and SPARC [32,33].
2.2. Immune Modulation
Compared to other tissues, the brain is a relatively immune-privileged site because it lacks a significant resident lymphoid population, and the BBB substantially restricts the entry of circulating immune cells into the CNS. Astrocytes are not immune cells per se, but are capable of many immune functions, including phagocytosis, antigen presentation, and facilitating immune-cell trafficking [34,35]. Therefore, astrocytes may produce pro- or anti-inflammatory cytokines, such as IL-1β, IL-6, TNF, IL-10, IL-27, and TGF-β in response to disease, stress, or injury [34,36]. Within the CNS, they communicate bidirectionally with microglia to coordinate defense responses [37]. Along with microglia, astrocytes phagocytose neuronal material including synapses, apoptotic neurons, and degenerating axons, as well as toxic proteins, such as Aβ plaques in AD and α-synuclein in Parkinson’s disease [38,39]. Consistent with their ability to present antigens, astrocytes express major histocompatibility complex (MHC) antigens that are upregulated in response to disease [40]. For example, high levels of astrocytic MHC-II were found in the brains of patients with Parkinson’s disease (PD), which correlated with the load of pathological, phosphorylated alpha synuclein (αSYN) [41]. Notably, perivascular and infiltrated CD4+ T cells were surrounded by MHC-II expressing astrocytes, indicating astrocyte–T-cell cross-talk in the PD brain [41]. Astrocytic MHC-I is upregulated during aging, which appears to be protective, since it is associated with preserved cognitive function [42].
Due to their proximity to blood vessels, astrocytes play a critical role in mediating reciprocal communication between CNS-resident cells and the immune system. Depending on the subsets involved, astrocytes respond to T-cell signals to either boost or limit CNS inflammation. For instance, pathogenic Th17 cells signal to astrocytes via GM-CSF and IL-17 to boost neurotoxic astrogliosis [43,44,45]; however, FOXP3+ regulatory T cells secrete amphiregulin to suppress astrogliosis and promote recovery after ischemic stroke [46]. Conversely, they can also produce chemokines, such as CCL2, CXCL10, and CXCL12, which are involved in leukocyte recruitment into the CNS [47]. During aging, there is an increase in the production of astrocytic CXCL10, which serves as a chemoattractant for peripheral immune cells and aids in T-cell adhesion to endothelial cells [48]. In summary, astrocytes are critical regulators of immune responses in the CNS, as they may promote or dampen neuronal damage and inflammation, depending on the context and stimuli.
2.3. Blood–Brain Barrier Regulation and Maintenance
The BBB is a highly selective semipermeable barrier that restricts the entry of blood cells and plasma components into the brain parenchyma, facilitates the influx of essential nutrients, and mediates the efflux of neurotoxic products. Together, these tasks maintain an optimal environment for neuronal survival and function. The anatomical BBB consists of a continuous monolayer of endothelial cells (ECs) connected by tight junctions (TJ) and adherens junctions (containing claudin, occludin, and zonula occludens proteins). TJ proteins restrict paracellular permeability and also segregate the apical and basal domains of the cell membrane, which enables endothelial polarization [49]. The regulation of the BBB is accomplished by the neurovascular unit (NVU), a multicellular unit that functionally connects the brain and the cerebral vasculature. The NVU is composed of specialized ECs, pericytes, and perivascular astrocytes, whose end-feet sheath all cerebral blood vessels [50].
In addition to the paracellular pathway, several transcellular pathways across the BBB are carefully regulated by the NVU. Due to large surface areas of the lipophilic membranes in ECs, small gaseous molecules, such as O2 and CO2, and small lipid-soluble agents can diffuse freely though the endothelium. Specialized EC transporter proteins, such as glucose transporter 1 (GLUT1) and L-type amino acid transporter 1 (LAT1), supply the brain with glucose and amino acids, respectively. Additional transporters supply the brain with nucleosides, nucleobases, and other substances [51]. Some transporters, such as P-glycoprotein (Pgp), are energy-dependent and act as efflux transporters for neurotoxic molecules [52]. Other proteins, such as insulin and transferrin, are taken up by ECs via receptor-mediated endocytosis and then transported across the BBB in vesicles, in a process called receptor-mediated transcytosis (RMT) [53]. Native plasma proteins, such as albumin, are typically excluded from the healthy adult brain, but can cross the BBB via adsorptive-mediated transcytosis (AMT) under specific conditions [54]. AMT is also vesicle-mediated, but it involves nonspecific binding to the membrane surface charges before internalization and transport through EC cell bodies.
The perivascular end feet of astrocytes show several specialized features, including a high density of K+ transporters and aquaporin (AQP4) water channels, which are involved in ion recycling and brain-volume regulation, respectively [55]. Through a combination of cell–cell interactions and soluble factors, perivascular astrocytes regulate the expression of TJ proteins and directly modify the transport properties of the cerebral endothelium [55,56,57]. Compared with ECs cultured alone, ECs co-cultured with astrocytes or astrocyte-conditioned media were found to exhibit increased TJ formation, transporter expression, and overall improved barrier function [58]. Subsequent studies have identified the molecular mechanisms responsible for the astrocytic regulation of the BBB. For example, the astrocyte secretion of factors such as angiopoietin 1 (ANG1) and sonic hedgehog (SHH) cause ECs to upregulate TJ proteins, thus enhancing barrier tightness [59,60,61].
Beyond BBB regulation, a recent study showed that astrocytes perform a necessary and nonredundant function in adult BBB maintenance [62]. The study used a genetic DTA ablation system to conditionally and selectively ablate astrocytes from the adult mouse brain and observed significant BBBD, indicated by the leakage of cadaverine (~900 Da) and fibrinogen (340 kDa) into the parenchyma. The blood vessels within the regions of astrocyte loss had a lower expression of the TJ protein zonula occludens-1 (ZO-1), while the expression of the endothelial transporter GLUT1 remained undisturbed. BBBD persisted for several weeks following ablation, suggesting a lack of barrier repair [62].
Other roles of astrocytes in BBB function have been studied in the context of disease. For example, the E4 variant of apolipoprotein E (APOE), the main susceptibility gene for AD, leads to accelerated BBBD and cognitive decline in humans and animals [63,64]. APOE is primarily synthesized and secreted in the CNS by astrocytes and is required for BBB formation and maintenance [65]. A recent study used allele-specific knock-in mice with the human E4, E3, and E2 APOE variants and showed that the humanized APOE4, but not APOE2 or APOE3, mice exhibited BBBD, increased matrix metalloproteinases-9 (MMP9), impaired TJs, and reduced the astrocyte end-foot coverage of blood vessels [66]. This is a seminal example of how astrocyte dysfunction can directly lead to BBBD and subsequent neurological disease. Additional mechanisms through which astrocytes contribute to BBBD in aging and disease continue to be explored.
3. Mechanisms of Astrocyte-Mediated Neuropathology in the Aging Brain
Brain aging consists of a set of physiological and cellular changes that result in reduced neural function. The salient changes associated with aging include significant decreases in certain neuronal populations, dendritic spines, axonal arborization, post-synaptic density, and cortical volume [67]. Together, these physiological changes result in cognitive impairment and memory loss. Given the many diverse functions astrocytes perform in the brain, it is logical that astrocyte dysfunction would result in neural deficits and neuropathology. Thus, there is substantial interest in understanding the precise role of astrocytes in age-related neural dysfunction, especially in neurodegenerative diseases.
Several molecular changes have been observed in aging astrocytes that may negatively affect neurons, including the activation of complement system genes and increased oxidative stress. Aged astrocytes express C3 and C4B complement component genes [68], which may impair plasticity by causing structural changes in neurons [69]. Furthermore, oxidative stress increases during aging and leads to the intracellular accumulation of reactive oxygen species (ROS). In turn, this causes calcium overload in astrocytes, which is associated with hippocampal neuronal dysfunction [70]. While these pathways are relevant, this review focuses on two other critically important and potentially related mechanisms through which astrocytes contribute to neuropathology in aging: blood–brain barrier dysfunction (BBBD) and cellular senescence.
3.1. TGFβ Hyperactivation following Blood–Brain Barrier Dysfunction (BBBD)
One of the physiological consequences of advanced age is the progressive decline in NVU function, which results in increased BBB permeability and the leakage of blood-borne molecules into the neural microenvironment. Clinical studies have shown that BBBD is prevalent in aging individuals [71,72,73] and is correlated with neurodegenerative diseases, especially ADRD [74,75,76,77]. The BBBD phenotype is multifaceted and adversely affects nearly every element of the brain. The murine endothelium in BBBD is characterized by the decreased expression of TJ proteins [78,79], increased EC pinocytosis [80,81], and the decreased expression of the GLUT1 transporter [82]. In humans, BBBD is characterized by focal endothelium degeneration [83] and the decreased EC expression of the Pgp transporter [84,85]. The decreased GLUT1 expression in BBBD impairs the influx of glucose, and decreased Pgp expression impairs the efflux of neurotoxic molecules, such as Aβ, allowing them to accumulate in the brain tissue, where they contribute to inflammation and neural degeneration [84,86,87]. In humans and mice, pericytes and astrocytes have reduced vascular coverage in BBBD, and astrocytes exhibit the downregulation of AQP4, the protein primarily responsible for end-foot adhesion and polarization at the vascular wall [88,89,90,91,92]. The leakage of neurotoxic blood-derived proteins into the parenchyma triggers microglial activation and astrogliosis as an injury response, contributing to neuroinflammation [93,94,95]. Studies in mice have shown that in the short term, this inflammation triggers microglia to migrate to damaged vessels and maintain the BBB via the expression of the TJ protein, Claudin-5; however, during sustained inflammation, microglia phagocytose astrocytic end-feet, further impairing BBB integrity [96]. In addition to the leakage caused by the loss of BBB integrity, recent evidence suggests that there is a global age-related shift in BBB transcytosis mechanisms that further increases the influx of neurotoxic proteins such as albumin, fibrinogen, and autoantibodies into the aging brain [92]. Specifically, the aging BBB exhibits diminished RMT transport and an increased non-specific transport of macromolecules compared to young brains. Regardless of the mechanism of entry, the dysregulated presence of serum proteins causes astrogliosis and neuroinflammation, and the dysfunctional phenotype of the NVU components in BBBD creates a hostile environment for neurons. As a result, white matter, neuronal axons, and synapses are compromised in BBBD, leading to cognitive impairment [83].
The hippocampus, the brain region’s center for learning and memory, appears to be especially vulnerable to age-related BBBD. Hippocampal BBBD is associated with mild cognitive impairment (MCI) and occurs before tissue atrophy and dementia in AD patients [71,97]. Notably, hippocampal neural hyperexcitability is also an early biomarker of MCI in humans that precedes dementia [98,99] and is associated with disease progression in rodent AD models [100,101]. These findings suggest there is a relationship between BBBD and neural hyperexcitability in aging. Indeed, a recent study from our group identified astrocytic TGFβ signaling as a mechanistic link between BBBD and hyperexcitability in the aging hippocampus and established how these factors contribute to cognitive impairment in humans and mice [73,102]. This study was based on the observation that the context and symptomatology of age-related neural dysfunction resemble those observed in traumatic brain injury (TBI). Both aging and TBI involve a period of BBBD followed by secondary neural dysfunction and are associated with an increased risk of neurodegenerative disease and dementia [74,103]. In both cases, the neural dysfunction is characterized by an excitation–inhibition (E/I) imbalance, hippocampal and cortical circuit hyperexcitability, and cognitive deficits [73,102].
TBI causes sudden and pronounced BBBD in patients, which can persist for months to years after the initial injury [104,105,106]. Studies in mice and rats have shown that BBBD induced by deoxycholic acid sodium salt (DOC) or direct injury enables blood-derived serum albumin and fibrinogen molecules to extravasate into the brain, where they elicit a robust inflammatory response mediated by astrocytic TGFβ signaling [107,108,109]. Albumin binds to the type II TGFβ receptor (TGFβR) on astrocytes and activates the canonical ALK5 type I TGFβR after receptor dimerization. This results in the phosphorylation of the downstream effector protein SMAD2 (pSMAD2) and the subsequent activation of pro-inflammatory and epileptogenic transcriptional programs [110,111,112]. The molecular hallmarks of this altered astrocytic phenotype are the upregulation of the cytoskeletal intermediate filaments, GFAP and vimentin, as well as S100 calcium-binding proteins. In mouse and rat models of BBBD, the astrocytic inflammatory program involves the production of cytokines, such as interleukin 6 (IL-6), monocyte chemoattractant protein-1 (MCP-1; CCL2), and matrix metalloproteinase 9 (MMP-9), which degrades critical extracellular structures and destabilizes perineuronal nets (PNNs) around inhibitory interneurons [107,111]. In rodent models, the astrocytic epileptogenic program features the upregulation of excitatory glutamate receptors, the activation of complement components C1 and C2, and the downregulation of voltage-gated potassium channels (Kir4.1), plasticity-associated NMDA receptors, and genes involved in inhibitory GABAergic transmission [107,111]. Notably, albumin-induced TGFβ activation itself upregulates the astrocytic expression of TGFβ and its receptors, resulting in a positive feedback loop that can cause the altered phenotype to persist [73,110]. In this state, the normal homeostatic neurotransmitter recycling and ion buffering functions of astrocytes are impaired, leading to disrupted neural communication. The remodeling of neural circuits occurs over time, which causes network hyperexcitability, E/I imbalance, and cognitive deficits [108,112,113,114,115]. In mice, the astrocytic secretion of TGFβ was shown to increase the neuronal expression of c1q [116], a complement protein that mediates synapse elimination [117] and increases in the CNS with age [118].
In the case of aging, the onset of BBBD is more gradual than in TBI, but it is also temporally constrained. Depending on an individual’s lifestyle and risk factors, BBBD begins as early as middle age in the human hippocampus and progresses in scope and severity into advanced age [71,73,75,76]. Interestingly, our study found that albumin-induced astrocytic TGFβ activation is a mechanism underpinning key neural deficits in aging [73]. In mice, immunohistochemistry (IHC) revealed that albumin begins to accumulate in hippocampal astrocytes as early as 12 months (“middle age”) and is consistently elevated in aging up to the end of life at ~2 years, indicating BBBD. Concurrent with the time course of BBBD, aged mice showed a progressive increase in the amount of pSMAD2 colocalized with albumin-positive hippocampal astrocytes, indicating BBBD-induced TGFβ signaling activation. Additionally, a Western blot analysis showed an increased concentration of active TGFβ1 (a positive feedback output of the TGFβ pathway) in the hippocampi of aged mice. Compared to the young mice, the aged mice exhibited increased seizure vulnerability and paroxysmal slow-wave events (PSWEs), indicating increased neural hyperexcitability. The activation of TGFβ signaling via serum albumin infusion into the cerebral ventricles of young mice was sufficient to elicit increased hyperexcitability, aberrant neural activity, and learning impairment, as measured by the Morris Water Maze assay. The pharmacological inhibition of the ALK5 type I TGFβR with the small molecule drug IPW-5371 significantly reduced TGFβ activation and seizure vulnerability in young mice infused with albumin. Importantly, the conditional knockdown of the type II astrocytic TGFβR and systemic treatment with IPW were both able to reverse the BBBD-induced neuropathological changes observed in aged mice, providing further evidence for the mechanistic role of TGFβ activation in driving these changes [73,102].
3.2. Cellular Senescence
Another consequence of advanced age is the accumulation of senescent cells [119,120,121,122,123,124]. Cellular senescence is an important anti-cancer mechanism that occurs in replication-competent cell types. The senescence response permanently arrests cell growth in response to stresses that can promote malignant transformation [125]. There are two main types of senescence: replicative senescence is triggered by extreme telomere erosion caused by repeated cell division [126], while stress-induced premature senescence (SIPS) is triggered by sublethal levels of cellular stress in the absence of telomere erosion. A common trigger of SIPS is DNA damage, which can be caused by factors such as environmental toxins, viral infection, and ionizing radiation [127,128,129]. Other stressors can trigger SIPS by activating molecular pathways that are associated with cellular transformation. For example, epigenetic factors, mitochondrial dysfunction, disrupted nutrient signaling, chronic inflammation, oxidative stress, and proteotoxic stress can also cause SIPS [130]. The senescence response ultimately activates the p53/p21WAF1 and p16INK4A/pRB tumor suppressor pathways, which establish and maintain growth arrest [127,131,132,133]. The senescent phenotype is not precisely defined and differs among cell types and conditions but is generally characterized by permanent cell-cycle arrest, increased lysosomal mass, the loss of nuclear integrity, decreased functional capacity, and the expression of the senescence-associated secretory phenotype (SASP) [119,134,135,136,137].
Senescent cells accumulate with age in a variety of tissues and contribute to age-associated tissue dysfunction and disease. A seminal study in the field showed that senescent cells accumulate in a progeroid mouse model of accelerated aging, and that the elimination of senescent cells via a drug-inducible transgene extends lifespan and prevents the onset of aging phenotypes, including cataracts, sarcopenia, and subcutaneous fat loss [138]. There are two known mechanisms through which senescent cells drive tissue aging and dysfunction. The first is through loss of function, in which senescent cells have a reduced ability to perform their vital activities within t tissues. The second is through the SASP, in which senescent cells secrete inflammatory cytokines, chemokines, growth factors, and proteases to communicate cellular damage to surrounding cells [130,139,140]. Many SASP factors are considered beneficial in limited quantities because they stimulate tissue repair and increase immune surveillance; however, their abundance in aging tissue has been shown to impair tissue structure and function by disrupting intercellular signaling and maintaining a pro-inflammatory environment [139,141,142,143,144].
While normally quiescent in the adult brain, astrocytes maintain their ability to replicate, and are thus susceptible to SIPS [145]. Astrocytic SIPS has been observed in response to oxidative stress [146], proteasome inhibition [146], HIV infection [147], methamphetamine [147], Aβ oligomers [148], and the environmental toxin, paraquat [149]. Human and animal studies have demonstrated that senescent cells accumulate in the aging CNS and are associated with neuroinflammation and neurodegeneration [150,151,152,153,154]. In human brain samples, the prevalence of senescent astrocytes in the brain was shown to increase with age, with a significantly higher burden observed in the cortices of AD patients compared to age-matched controls [148]. Recent studies in animal models have demonstrated a causal relationship between glial senescence and neurodegeneration. The accumulation of senescent astrocytes and microglia was found in the cortices and hippocampi of a mouse model of tau-dependent pathology, and the genetic ablation of these cells reduced tau deposition and prevented neuronal degeneration [155]. Similarly, senescent astrocytes were found in abundance in the substantia nigra of a mouse model of paraquat-induced Parkinson’s disease, and their selective genetic ablation was sufficient to reduce nigral dopaminergic cell loss and improve motor function [149]. Notably, novel pharmacological agents have been developed that can selectively eliminate senescent cells, and they are being explored in clinical trials for the treatment of age-related diseases [156,157,158]. These findings indicate that senescent astrocytes actively participate in driving age-associated neural dysfunction and pathology, including neurodegeneration, and can be leveraged as therapeutic targets.
As with other senescent cell types, senescent astrocytes appear to drive neural dysfunction via both loss of function and their SASP. Given the important role of astrocytes in supporting brain homeostasis and maintaining the neural microenvironment, the senescence-associated loss of astrocyte function is especially detrimental to neuronal survival and performance [159]. Indeed, substantial impairments have been reported in rat astrocyte functions cultured in vitro, including impaired wound healing ability, phagocytic uptake, metabolic function, and neuroprotective capacity [160,161]. Furthermore, the downregulation of potassium (Kir4.1) and glutamate (EAAT1/2) transporters in human senescent astrocytes promotes neurotoxicity in cortical neurons [162]. As a heterogenous population, it is likely that other specialized astrocyte functions are also impaired that have not yet been identified. Additionally, the astrocytic SASP participates in driving pathological changes in the aging brain by generating a chronic inflammatory environment. Senescent astrocytes from humans and mice secrete inflammatory and proteolytic SASP factors, such as IL-6, IL-1β, CCL2, MMP-3, and MMP-9 [148,149,155,163,164]. Interestingly, these factors are elevated in the cerebrospinal fluid (CSF) and sera of AD patients, suggesting that senescence-associated inflammation accompanies and may contribute to the progression of AD [165,166]. In particular, IL-6 is a salient SASP component and a biomarker of AD [167], whose overexpression drives neurodegeneration in AD models [168,169,170].
4. Perspectives on BBBD, TGFβ Signaling, and Astrocyte Senescence in Aging
As detailed above, a growing body of evidence suggests that both BBBD and astrocyte senescence progressively increase in the aging brain and are independently associated with an increased risk of neurodegenerative disease, but the relationship between the two has not been established. Given the salience of TGFβ signaling activation in aging phenotypes and neural dysfunction, BBBD-induced TGFβ hyperactivation should be explored as a potential mechanistic link between BBBD and senescence in the aging brain.
Studies show that TGFβ activation mediates senescence in various cell types in vitro, including fibroblasts [171,172,173], keratinocytes [174], mesenchymal stem cells (MSCs) [175], glioblastoma cells [176], cardiomyocytes [177], and lung epithelial cells [178]. Furthermore, TGFβ1, as a SASP component, was found to be an important mediator of paracrine senescence in vivo [179]. Furthermore, the pharmacological attenuation of age-associated TGFβ activation has rejuvenating effects in the aged brain [73,180], including the suppression of cellular senescence [181]. In astrocytes, previous reports have identified triggers of astrocyte senescence, such as oxidative stress [146], Aβ oligomers [148,182], HIV infection, and drug abuse [147]. Notably, TGFβ signaling is also implicated in these senescence mechanisms. In the context of oxidative stress, TGFβ activation increases the production of reactive oxygen species (ROS) by impairing mitochondrial function and inducing NADPH oxidases [183,184]. Indeed, TGFβ1 was found to induce the senescence of bone marrow MSCs via increased mitochondrial ROS [175]. TGFβ activation also suppresses the synthesis of antioxidant enzymes, such as glutathione, causing redox imbalance and promoting further oxidative stress [185]. In the case of Aβ pathology, astrocytic TGFβ overexpression increases Aβ generation in transgenic mice [186,187,188], and TGFβ1 expression is elevated in cortical astrocytes surrounding Aβ plaques in AD patients [189,190]. HIV infection and drug abuse were found to mediate astrocyte senescence in a β-catenin-dependent manner [147]. Notably, TGFβ1 operates through the canonical WNT/β-catenin pathway, and these two pathways stimulate each other via SMAD effector proteins [191]. The precise molecular mechanisms through which TGFβ signaling mediates senescence in astrocytes are unknown, but some evidence suggests that epigenetic alterations may be involved. A recent study found that canonical TGF-β/SMAD signaling promotes SIPS in cardiomyocytes via the miR-29-induced loss of H4K20me3, and that disrupting TGFβ signaling improves cardiac function in aged mice [177].
Together, these findings indicate that TGFβ activation is a salient factor in age-related pathological changes and can directly mediate the senescence response under certain conditions. Indeed, the results from a recent study suggest that BBBD-induced TGFβ activation via albumin extravasation into the brain parenchyma is a potential physiological trigger of astrocyte senescence [192]. This study utilized an animal model of BBBD in which albumin was continuously infused into the lateral ventricles of adult mice. One week of albumin infusion significantly increased TGFβ signaling activation and the burden of senescent astrocytes in hippocampal tissue. The pharmacological inhibition of TGFβR ALK5 or the conditional genetic knockdown of astrocytic TGFβR prior to albumin infusion were sufficient to prevent albumin-induced astrocyte senescence. In addition to activating canonical TGFβ signaling, albumin uptake activates the p38MAPK pathway in astrocytes [94], and it is known to play a central role in the regulation of astrocyte senescence and SASP expression [148,193]. Thus, it follows that the simultaneous activation of canonical TGFβ, p38MAPK, and, potentially, other unknown pathways by albumin can mediate astrocyte senescence.
Reciprocally, the molecular and functional effects of senescent astrocytes on BBB integrity have not been established. Certainly, astrocytes produce SASP factors, such as MMPs and IL-6, which are known to increase endothelial barrier permeability. MMPs induce BBBD via the disruption of TJ proteins [194,195], and IL-6 upregulates cell adhesion molecules (CAMs) on ECs, which facilitates leukocyte transmigration across the endothelium. Interestingly, one study found that the accumulation of senescent ECs and pericytes is associated with compromised BBB integrity due to reduced TJs; however, the blood vessel coverage by astrocytic end-feet was not altered [196]. This suggests that EC and pericyte senescence is not the proximal cause of the reduced astrocyte end-foot coverage observed in age-associated BBBD. These findings raise another possibility—that each cellular component of the NVU is differentially susceptible to physiological triggers of senescence. Indeed, TGFβ signaling is complex and context-dependent [197,198]; therefore, TGFβ hyperactivation may have variable effects in different cell types and conditions. Future studies should seek to elucidate the context in which various triggers promote senescence during aging and the extent of their respective contributions.
5. Concluding Remarks and Future Directions
Astrocytes are a diverse population of cells that perform essential functions in the brain related to neuronal support, immune modulation, and blood–brain barrier regulation and maintenance. Collectively, these functions ensure that the brain environment remains optimal for neuronal function. In aging, astrocytes adopt a neurotoxic phenotype following age-related BBBD that is mediated by TGFβ hyperactivation. Furthermore, aging is associated with an accumulation of senescent astrocytes, which contribute to neuroinflammation and neurodegeneration. Given that TGFβ is a salient factor in aging pathologies and is known to mediate senescence in various cell types, BBBD-induced TGFβ activation via blood protein extravasation should be further explored as a potential trigger of astrocyte senescence in the aging brain. Identifying the physiological triggers of astrocyte senescence is important for understanding the complex series of events that facilitates the development of neurodegenerative disease.
Astrocytes play an important role in BBB regulation and maintenance through their involvement in the NVU. Since the NVU is highly interdependent, the impacts of perivascular astrocyte senescence on each component of the NVU merit investigation. Genetic tools that enable the selective ablation of senescent astrocytes [149] should be leveraged to investigate the role of senescent astrocytes in the context of BBBD. Another unanswered question in the context of aging is how the global shift in transcytosis mechanisms manifests in each NVU cell type. Specifically, how do perivascular astrocytes experience this shift, and does cellular senescence play a role in facilitating this shift? In other words, do senescent cells experience more nonspecific transcytosis than their youthful counterparts?
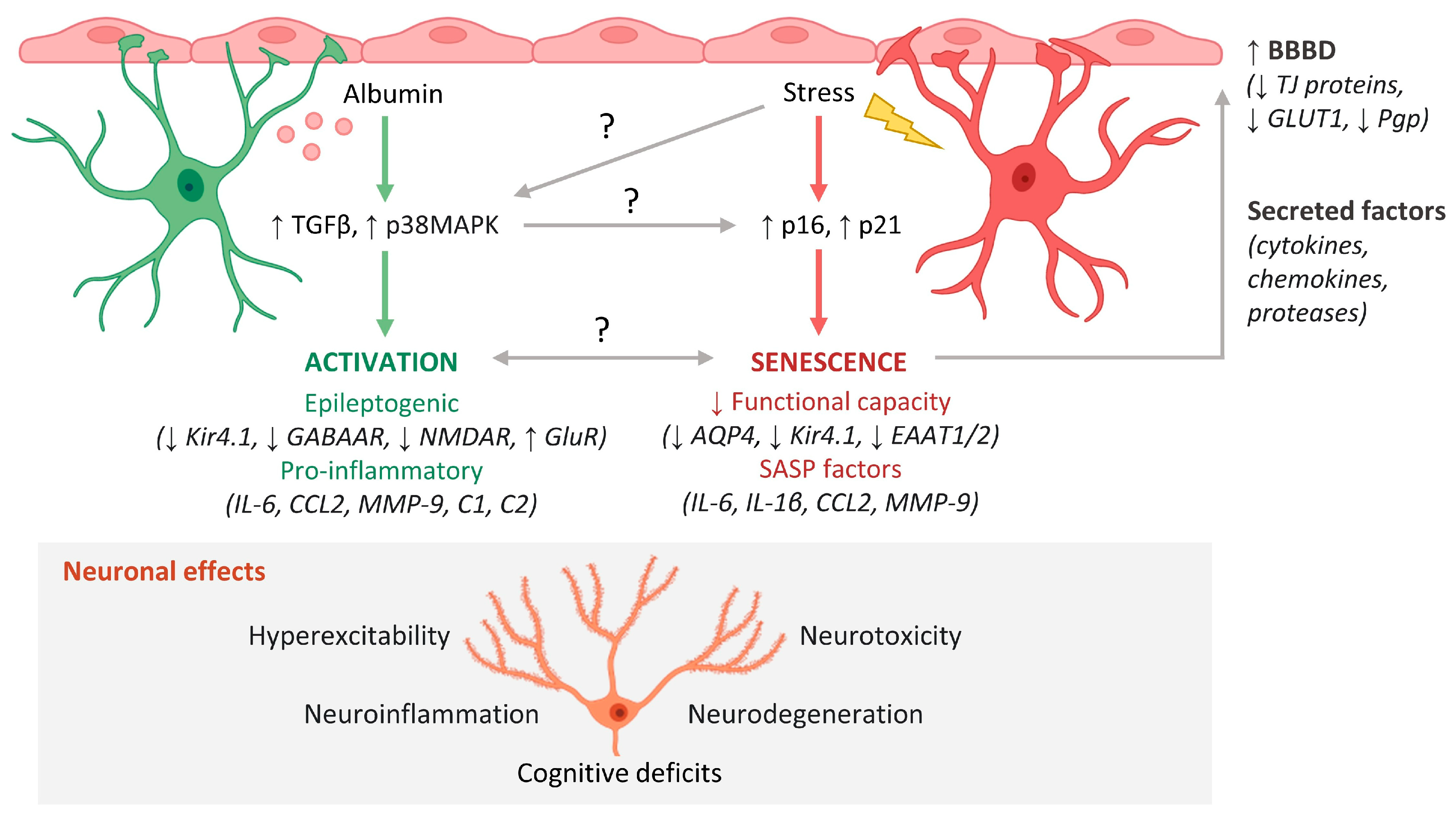
Given the importance of astrocytes in preserving the BBB, it is likely that initial BBBD-induced astrocyte senescence establishes a positive feedback loop that results in progressive neurological decline. In this scenario, the accumulation of functionally impaired senescent astrocytes causes more BBBD and, thus, more BBBD-induced astrocyte senescence—perpetuating a vicious cycle of BBBD and associated functional decline (Figure 1). Further studies should investigate how this process is regulated and explore potential strategies to prevent its onset.
Author Contributions
M.K.P. conceived and wrote the manuscript with input from D.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by a Bakar Foundation Fellowship (D.K.), the Archer Foundation Award (D.K.), the Borstein Family Foundation award (D.K.), a NSF GRFP fellowship (M.K.P.), and a NIH T32 fellowship (GM 098218; M.K.P.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| AD | Alzheimer’s disease |
| BBB | blood-brain barrier |
| BBBD | blood-brain barrier dysfunction |
| CNS | central nervous system |
| EC | endothelial cells |
| SASP | senescence-associated secretory phenotype |
| MCI | mild cognitive impairment |
| NVU | neurovascular unit |
| SIPS | stress-induced premature senescence |
| TBI | traumatic brain injury |
| TJ | tight junctions |
References
- Alzheimer’s Association. Disease Facts and Figures. Alzheimers. Dement. 2021, 17, 327–406. [Google Scholar] [CrossRef]
- Herculano-Houzel, S. The Human Brain in Numbers: A Linearly Scaled-up Primate Brain. Front. Hum. Neurosci. 2009, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Vasile, F.; Dossi, E.; Rouach, N. Human Astrocytes: Structure and Functions in the Healthy Brain. Brain Struct. Funct. 2017, 222, 2017–2029. [Google Scholar] [CrossRef] [Green Version]
- Sidoryk-Wegrzynowicz, M.; Wegrzynowicz, M.; Lee, E.; Bowman, A.B.; Aschner, M. Role of Astrocytes in Brain Function and Disease. Toxicol. Pathol. 2011, 39, 115–123. [Google Scholar] [CrossRef]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef] [Green Version]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of Region-Specific Astrocyte Subtypes at Single Cell Resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef] [Green Version]
- Pestana, F.; Edwards-Faret, G.; Belgard, T.G.; Martirosyan, A.; Holt, M.G. No Longer Underappreciated: The Emerging Concept of Astrocyte Heterogeneity in Neuroscience. Brain Sci. 2020, 10, 168. [Google Scholar] [CrossRef] [Green Version]
- Huang, A.Y.S.; Woo, J.; Sardar, D.; Lozzi, B.; Bosquez Huerta, N.A.; Lin, C.C.J.; Felice, D.; Jain, A.; Paulucci-Holthauzen, A.; Deneen, B. Region-Specific Transcriptional Control of Astrocyte Function Oversees Local Circuit Activities. Neuron 2020, 106, 992–1008.e9. [Google Scholar] [CrossRef]
- Chaboub, L.S.; Deneen, B. Developmental Origins of Astrocyte Heterogeneity: The Final Frontier of CNS Development. Dev. Neurosci. 2013, 34, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Rash, J.E.; Duffy, H.S.; Dudek, F.E.; Bilhartz, B.L.; Whalen, L.R.; Yasumura, T. Grid-Mapped Freeze-Fracture Analysis of Gap Junctions in Gray and White Matter of Adult Rat Central Nervous System, with Evidence for a “panglial Syncytium” That Is Not Coupled to Neurons. J. Comp. Neurol. 1997, 388, 265–292. [Google Scholar] [CrossRef]
- Nagy, J.I.; Dudek, F.E.; Rash, J.E. Update on Connexins and Gap Junctions in Neurons and Glia in the Mammalian Nervous System. Brain Res. Rev. 2004, 47, 191–215. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, Maintenance and Disruption of the Blood-Brain Barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Amundson, R.H.; Goderie, S.K.; Kimelberg, H.K. Uptake of [3H]Serotonin and [3H]Glutamate by Primary Astrocyte Cultures. II. Differences in Cultures Prepared from Different Brain Regions. Glia 1992, 6, 9–18. [Google Scholar] [CrossRef]
- Sonnewald, U.; Westergaard, N.; Schousboe, A. Glutamate Transport and Metabolism in Astrocytes. Glia 1997, 21, 56–63. [Google Scholar] [CrossRef]
- Kaneko, T.; Shigemoto, R.; Mizuno, N. Metabolism of glutamate and ammonia in astrocyte: An immunocytochemical study. Brain Res. 1988, 457, 160–164. [Google Scholar] [CrossRef]
- Hertz, L. Autonomic control of neuronal-astrocytic interactions, regulating metabolic activities, and ion fluxes in the CNS. Brain Res. Bull. 1992, 29, 303–313. [Google Scholar] [CrossRef]
- Bellot-Saez, A.; Kékesi, O.; Morley, J.W.; Buskila, Y. Astrocytic modulation of neuronal excitability through K + spatial buffering. Neurosci. Biobehav. Rev. 2017, 77, 87–97. [Google Scholar] [CrossRef]
- Walz, W. Role of astrocytes in the clearance of excess extracellular potassium. Neurochem. Int. 2000, 36, 291–300. [Google Scholar] [CrossRef]
- Wuttke, W.A.; Walz, W. Sodium- and bicarbonate-independent regulation of intracellular pH in cultured mouse astrocytes. Neurosci. Lett. 1990, 117, 105–110. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A Paravascular Pathway Facilitates CSF Flow through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef] [Green Version]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of Amyloid-β Peptide Across the Blood-Brain Barrier: Implication for Therapies in Alzheimers Disease. CNS Neurol. Disord. Drug. Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, A.Y.; Espinosa De Los Monteros, A.; Cole, R.A.; Loera, S.; De Vellis, J. Laminin and s-laminin are produced and released by astrocytes, schwann cells, and schwannomas in culture. Glia 1991, 4, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Shea, T.B.; Beermann, M.L.; Nixon, R.A. Sequential effects of astroglial-derived factors on neurite outgrowth: Initiation by protease inhibitors and potentiation by extracellular matrix components. J. Neurosci. Res. 1992, 31, 309–317. [Google Scholar] [CrossRef]
- Rudge, J.S.; Alderson, R.F.; Pasnikowski, E.; McClain, J.; Ip, N.Y.; Lindsay, R.M. Expression of Ciliary Neurotrophic Factor and the Neurotrophins—Nerve Growth Factor, Brain-Derived Neurotrophic Factor and Neurotrophin 3-in Cultured Rat Hippocampal Astrocytes. Eur. J. Neurosci. 1992, 4, 459–471. [Google Scholar] [CrossRef]
- Seil, F.J.; Eckenstein, F.P.; Reier, P.J. Induction of dendritic spine proliferation by an astrocyte secreted factor. Exp. Neurol. 1992, 117, 85–89. [Google Scholar] [CrossRef]
- Bukalo, O.; Dityatev, A. Synaptic Cell Adhesion Molecules. Adv. Exp. Med. Biol. 2012, 970, 97–128. [Google Scholar] [CrossRef]
- Baldwin, K.T.; Eroglu, C. Molecular mechanisms of astrocyte-induced synaptogenesis. Curr. Opin. Neurobiol. 2017, 45, 113–120. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.A.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins Are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Diniz, L.P.; Almeida, J.C.; Tortelli, V.; Lopes, C.V.; Setti-Perdigao, P.; Stipursky, J.; Kahn, S.A.; Romão, L.F.; de Miranda, J.; Alves-Leon, S.V.; et al. Astrocyte-induced Synaptogenesis Is Mediated by Transforming Growth Factor β Signaling through Modulation of d-Serine Levels in Cerebral Cortex Neurons. J. Biol. Chem. 2012, 287, 41432–41445. [Google Scholar] [CrossRef]
- Diniz, L.P.; Tortelli, V.; Garcia, M.N.; Araújo, A.P.B.; Melo, H.M.; da Silva, G.S.S.; De Felice, F.G.; Alves-Leon, S.V.; de Souza, J.M.; Romão, L.F.; et al. Astrocyte transforming growth factor beta 1 promotes inhibitory synapse formation via CaM kinase II signaling. Glia 2014, 62, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Kucukdereli, H.; Allen, N.J.; Lee, A.T.; Feng, A.; Ozlu, M.I.; Conatser, L.M.; Chakraborty, C.; Workman, G.; Weaver, M.; Sage, E.H.; et al. Control of excitatory CNS synaptogenesis by astrocyte-secreted proteins Hevin and SPARC. Proc. Natl. Acad. Sci. USA 2011, 108, E440–E449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Risher, W.C.; Patel, S.; Kim, I.H.; Uezu, A.; Bhagat, S.; Wilton, D.K.; Pilaz, L.-J.; Alvarado, J.S.; Calhan, O.Y.; Silver, D.L.; et al. Astrocytes refine cortical connectivity at dendritic spines. eLife 2014, 3, e04047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.L. Astrocytes: Form, Functions, and Roles in Disease. Vet. Pathol. 1994, 31, 145–167. [Google Scholar] [CrossRef]
- Falsig, J.; Pörzgen, P.; Lund, S.; Schrattenholz, A.; Leist, M. The inflammatory transcriptome of reactive murine astrocytes and implications for their innate immune function. J. Neurochem. 2006, 96, 893–907. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Valiente, M. The Potential of Astrocytes as Immune Modulators in Brain Tumors. Front. Immunol. 2019, 10, 1314. [Google Scholar] [CrossRef]
- Lee, S.Y.; Chung, W.S. The roles of astrocytic phagocytosis in maintaining homeostasis of brains. J. Pharmacol. Sci. 2020, 145, 223–227. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, J.Y.; Noh, S.; Lee, H.; Lee, S.Y.; Mun, J.Y.; Park, H.; Chung, W.S. Astrocytes phagocytose adult hippocampal synapses for circuit homeostasis. Nature 2020, 590, 612–617. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Estes, M.L. Astrocyte Expression of Major Histocompatibility Complex Gene Products in Multiple Sclerosis Brain Tissue Obtained by Stereotactic Biopsy. Arch. Neurol. 1991, 48, 1244–1246. [Google Scholar] [CrossRef]
- Rostami, J.; Fotaki, G.; Sirois, J.; Mzezewa, R.; Bergström, J.; Essand, M.; Healy, L.; Erlandsson, A. Astrocytes have the capacity to act as antigen-presenting cells in the Parkinson’s disease brain. J. Neuroinflamm. 2020, 17, 119. [Google Scholar] [CrossRef] [PubMed]
- Lazarczyk, M.J.; Kemmler, J.E.; Eyford, B.A.; Short, J.A.; Varghese, M.; Sowa, A.; Dickstein, D.R.; Yuk, F.J.; Puri, R.; Biron, K.E.; et al. Major Histocompatibility Complex class I proteins are critical for maintaining neuronal structural complexity in the aging brain. Sci. Rep. 2016, 6, 26199. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Altuntas, C.Z.; Gulen, M.F.; Liu, C.; Giltiay, N.; Qin, H.; Liu, L.; Qian, W.; Ransohoff, R.M.; Bergmann, C.; et al. Astrocyte-Restricted Ablation of Interleukin-17-Induced Act1-Mediated Signaling Ameliorates Autoimmune Encephalomyelitis. Immunity 2010, 32, 414–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- You, T.; Bi, Y.; Li, J.; Zhang, M.; Chen, X.; Zhang, K.; Li, J. IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci. Rep. 2017, 7, 41779. [Google Scholar] [CrossRef]
- Ito, M.; Komai, K.; Mise-Omata, S.; Iizuka-Koga, M.; Noguchi, Y.; Kondo, T.; Sakai, R.; Matsuo, K.; Nakayama, T.; Yoshie, O.; et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 2019, 565, 246–250. [Google Scholar] [CrossRef]
- Ubogu, E.E.; Cossoy, M.B.; Ransohoff, R.M. The expression and function of chemokines involved in CNS inflammation. Trends Pharmacol. Sci. 2006, 27, 48–55. [Google Scholar] [CrossRef]
- Sorensen, E.W.; Lian, J.; Ozga, A.J.; Miyabe, Y.; Ji, S.W.; Bromley, S.K.; Mempel, T.R.; Luster, A.D. CXCL10 stabilizes T cell–brain endothelial cell adhesion leading to the induction of cerebral malaria. JCI Insight 2018, 3, e98911. [Google Scholar] [CrossRef]
- Wolburg, H.; Lippoldt, A. Tight junctions of the blood–brain barrier: Development, composition and regulation. Vasc. Pharmacol. 2002, 38, 323–337. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [Green Version]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug Res. 2003, 61, 39–78. [Google Scholar] [PubMed]
- Schinkel, A.H. P-Glycoprotein, a Gatekeeper in the Blood-Brain Barrier. Adv. Drug Deliv. Rev. 1999, 36, 179–194. [Google Scholar] [CrossRef]
- Pulgar, V.M. Transcytosis to Cross the Blood Brain Barrier, New Advancements and Challenges. Front. Neurosci. 2019, 12, 1019. [Google Scholar] [CrossRef] [PubMed]
- Hervé, F.; Ghinea, N.; Scherrmann, J.-M. CNS Delivery Via Adsorptive Transcytosis. AAPS J. 2008, 10, 455–472. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte-Endothelial Interactions at the Blood-Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [Google Scholar] [CrossRef]
- Michinaga, S.; Koyama, Y. Dual Roles of Astrocyte-Derived Factors in Regulation of Blood-Brain Barrier Function after Brain Damage. Int. J. Mol. Sci. 2019, 20, 571. [Google Scholar] [CrossRef] [Green Version]
- Abbott, N.J. Astrocyte-Endothelial Interactions and Blood-Brain Barrier Permeability. J. Anat. 2002, 200, 523–534. [Google Scholar] [CrossRef]
- Hayashi, Y.; Nomura, M.; Yamagishi, S.I.; Harada, S.I.; Yamashita, J.; Yamamoto, H. Induction of Various Blood-Brain Barrier Properties in Non-Neural Endothelial Cells by Close Apposition to Co-Cultured Astrocytes. Glia 1997, 19, 13–26. [Google Scholar] [CrossRef]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Blood-Brain Barrier Integrity and CNS Immune Quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Fabre, P.; Ifergan, I.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonnière, L.; Bernard, M.; et al. The Hedgehog Pathway Promotes Optimal Blood Brain Barrier Functioning. Glia 2013, 61, S190. [Google Scholar]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood–brain barrier maintenance in the adult mouse brain. Glia 2020, 69, 436–472. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.D.; Winkler, E.A.; Singh, I.; Sagare, A.P.; Deane, R.; Wu, Z.; Holtzman, D.M.; Betsholtz, C.; Armulik, A.; Sallstrom, J.; et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature 2012, 485, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Montagne, A.; Nation, D.A.; Sagare, A.P.; Barisano, G.; Sweeney, M.D.; Chakhoyan, A.; Pachicano, M.; Joe, E.; Nelson, A.R.; D’Orazio, L.M.; et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature 2020, 581, 71–76. [Google Scholar] [CrossRef]
- Soto, I.; Graham, L.C.; Richter, H.J.; Simeone, S.N.; Radell, J.E.; Grabowska, W.; Funkhouser, W.K.; Howell, M.C.; Howell, G.R. APOE Stabilization by Exercise Prevents Aging Neurovascular Dysfunction and Complement Induction. PLoS Biol. 2015, 13, e1002279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, R.J.; Meltzer, J.C.; Nguyen, H.; Commins, C.; Bennett, R.E.; Hudry, E.; Hyman, B.T. APOE4 derived from astrocytes leads to blood–brain barrier impairment. Brain 2021, awab478. [Google Scholar] [CrossRef]
- Yankner, B.A.; Lu, T.; Loerch, P. The Aging Brain. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 41–66. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [Green Version]
- Pekny, M.; Wilhelmsson, U.; Bogestål, Y.R.; Pekna, M. The Role of Astrocytes and Complement System in Neural Plasticity. Int. Rev. Neurobiol. 2007, 82, 95–111. [Google Scholar] [CrossRef]
- Ishii, T.; Takanashi, Y.; Sugita, K.; Miyazawa, M.; Yanagihara, R.; Yasuda, K.; Onouchi, H.; Kawabe, N.; Nakata, M.; Yamamoto, Y.; et al. Endogenous reactive oxygen species cause astrocyte defects and neuronal dysfunctions in the hippocampus: A new model for aging brain. Aging Cell 2016, 16, 39–51. [Google Scholar] [CrossRef]
- Montagne, A.; Barnes, S.R.; Sweeney, M.D.; Halliday, M.R.; Sagare, A.P.; Zhao, Z.; Toga, A.W.; Jacobs, R.E.; Liu, C.Y.; Amezcua, L.; et al. Blood-Brain Barrier Breakdown in the Aging Human Hippocampus. Neuron 2015, 85, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, B.; Fang, C.; Chang, J. Blood–Brain Barrier Breakdown: An Emerging Biomarker of Cognitive Impairment in Normal Aging and Dementia. Front. Neurosci. 2021, 15, 688090. [Google Scholar] [CrossRef] [PubMed]
- Senatorov, V.V.; Friedman, A.R.; Milikovsky, D.Z.; Ofer, J.; Saar-Ashkenazy, R.; Charbash, A.; Jahan, N.; Chin, G.; Mihaly, E.; Lin, J.M.; et al. Blood-Brain Barrier Dysfunction in Aging Induces Hyper-Activation of TGF-Beta Signaling and Chronic yet Reversible Neural Dysfunction. bioRxiv 2019. bioRxiv:537431. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef]
- Farrall, A.J.; Wardlaw, J.M. Blood–brain barrier: Ageing and microvascular disease–systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef]
- Skillbäck, T.; Delsing, L.; Synnergren, J.; Mattsson-Carlgren, N.; Janelidze, S.; Nägga, K.; Kilander, L.; Hicks, R.; Wimo, A.; Winblad, B.; et al. CSF/serum albumin ratio in dementias: A cross-sectional study on 1861 patients. Neurobiol. Aging 2017, 59, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van De Haar, H.J.; Burgmans, S.; Jansen, J.F.A.; Van Osch, M.J.P.; Van Buchem, M.A.; Muller, M.; Hofman, P.A.M.; Verhey, F.R.J.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Bake, S.; Friedman, J.A.; Sohrabji, F. Reproductive age-related changes in the blood brain barrier: Expression of IgG and tight junction proteins. Microvasc. Res. 2009, 78, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Elahy, M.; Jackaman, C.; Mamo, J.C.; Lam, V.; Dhaliwal, S.S.; Giles, C.; Nelson, D.; Takechi, R. Blood–brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun. Ageing 2015, 12, 2. [Google Scholar] [CrossRef] [Green Version]
- Cipolla, M.J.; Crete, R.; Vitullo, L.; Rix, R.D. Transcellular transport as a mechanism of blood-brain barrier disruption during stroke. Front. Biosci. 2004, 9, 777–785. [Google Scholar] [CrossRef] [Green Version]
- Hashizume, K.; Black, K.L. Increased Endothelial Vesicular Transport Correlates with Increased Blood-Tumor Barrier Permeability Induced by Bradykinin and Leukotriene C4. J. Neuropathol. Exp. Neurol. 2002, 61, 725–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, F.; Yao, J.; Rettberg, J.R.; Chen, S.; Brinton, R.D. Early Decline in Glucose Transport and Metabolism Precedes Shift to Ketogenic System in Female Aging and Alzheimer’s Mouse Brain: Implication for Bioenergetic Intervention. PLoS ONE 2013, 8, e79977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.; Miller, M.C.; Monahan, R.; Osgood, D.P.; Stopa, E.G.; Silverberg, G.D. P-glycoprotein expression and amyloid accumulation in human aging and Alzheimer’s disease: Preliminary observations. Neurobiol. Aging 2015, 36, 2475–2482. [Google Scholar] [CrossRef] [PubMed]
- Langford, D.; Grigorian, A.; Hurford, R.; Adame, A.; Ellis, R.J.; Hansen, L.; Masliah, E. Altered P-Glycoprotein Expression in AIDS Patients with HIV Encephalitis. J. Neuropathol. Exp. Neurol. 2004, 63, 1038–1047. [Google Scholar] [CrossRef] [Green Version]
- Van Assema, D.M.E.; Lubberink, M.; Boellaard, R.; Schuit, R.C.; Windhorst, A.D.; Scheltens, P.; Lammertsma, A.A.; Van Berckel, B.N.M. P-Glycoprotein Function at the Blood–Brain Barrier: Effects of Age and Gender. Mol. Imaging Biol. 2012, 14, 771–776. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Bodles-Brakhop, A.M.; Barger, S.W. A Role for P-Glycoprotein in Clearance of Alzheimer Amyloid?—Peptide from the Brain. Curr. Alzheimer Res. 2016, 13, 615–620. [Google Scholar] [CrossRef]
- Duncombe, J.; Lennen, R.J.; Jansen, M.A.; Marshall, I.; Wardlaw, J.M.; Horsburgh, K. Ageing causes prominent neurovascular dysfunction associated with loss of astrocytic contacts and gliosis. Neuropathol. Appl. Neurobiol. 2017, 43, 477–491. [Google Scholar] [CrossRef] [Green Version]
- Mills, W.A.; Jiang, S.; Martin, J.; Woo, A.M.; Bergstresser, M.; Kimbrough, I.F.; Sontheimer, H. Astrocyte Plasticity Ensures Continued Endfoot Coverage of Cerebral Blood Vessels and Integrity of the Blood Brain Barrier, with Plasticity Declining with Normal Aging. bioRxiv 2021. bioRxiv:2021.05.08.443259. [Google Scholar] [CrossRef]
- Szu, J.I.; Binder, D.K. The Role of Astrocytic Aquaporin-4 in Synaptic Plasticity and Learning and Memory. Front. Integr. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Goodall, E.F.; Wang, C.; Simpson, J.E.; Baker, D.J.; Drew, D.R.; Heath, P.R.; Saffrey, M.J.; Romero, I.A.; Wharton, S.B. Age-associated changes in the blood-brain barrier: Comparative studies in human and mouse. Neuropathol. Appl. Neurobiol. 2017, 44, 328–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nature 2020, 583, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Hooper, C.; Pinteaux-Jones, F.; Fry, V.A.H.; Sevastou, I.G.; Baker, D.; Heales, S.J.; Pocock, J.M. Differential effects of albumin on microglia and macrophages; implications for neurodegeneration following blood-brain barrier damage. J. Neurochem. 2009, 109, 694–705. [Google Scholar] [CrossRef] [PubMed]
- Ranaivo, H.R.; Wainwright, M.S. Albumin activates astrocytes and microglia through mitogen-activated protein kinase pathways. Brain Res. 2010, 1313, 222–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Haberman, R.P.; Branch, A.; Gallagher, M. Targeting Neural Hyperactivity as a Treatment to Stem Progression of Late-Onset Alzheimer’s Disease. Neurotherapeutics 2017, 14, 662–676. [Google Scholar] [CrossRef] [Green Version]
- Yassa, M.A.; Stark, S.M.; Bakker, A.; Albert, M.S.; Gallagher, M.; Stark, C.E.L. High-resolution structural and functional MRI of hippocampal CA3 and dentate gyrus in patients with amnestic Mild Cognitive Impairment. NeuroImage 2010, 51, 1242–1252. [Google Scholar] [CrossRef] [Green Version]
- Fontana, R.; Agostini, M.; Murana, E.; Mahmud, M.; Scremin, E.; Rubega, M.; Sparacino, G.; Vassanelli, S.; Fasolato, C. Early hippocampal hyperexcitability in PS2APP mice: Role of mutant PS2 and APP. Neurobiol. Aging 2017, 50, 64–76. [Google Scholar] [CrossRef]
- Palop, J.J.; Chin, J.; Roberson, E.D.; Wang, J.; Thwin, M.T.; Bien-Ly, N.; Yoo, J.; Ho, K.O.; Yu, G.-Q.; Kreitzer, A.; et al. Aberrant Excitatory Neuronal Activity and Compensatory Remodeling of Inhibitory Hippocampal Circuits in Mouse Models of Alzheimer’s Disease. Neuron 2007, 55, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Milikovsky, D.Z.; Ofer, J.; Senatorov, V.V.; Friedman, A.R.; Prager, O.; Sheintuch, L.; Elazari, N.; Veksler, R.; Zelig, D.; Weissberg, I.; et al. Paroxysmal slow cortical activity in Alzheimer’s disease and epilepsy is associated with blood-brain barrier dysfunction. Sci. Transl. Med. 2019, 11, eaaw8954. [Google Scholar] [CrossRef] [PubMed]
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.A.; Zhang, X.L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 2018, 141, 422–458. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.R.; Johnson, V.E.; Young, A.M.H.; Smith, D.H.; Stewart, W. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years after Traumatic Brain Injury in Humans. J. Neuropathol. Exp. Neurol. 2015, 74, 1147–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomkins, O.; Feintuch, A.; Benifla, M.; Cohen, A.; Friedman, A.; Shelef, I. Blood-Brain Barrier Breakdown Following Traumatic Brain Injury: A Possible Role in Posttraumatic Epilepsy. Cardiovasc. Psychiatry Neurol. 2011, 2011, 765923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, E.; Aboghazleh, R.; Mumby, G.; Veksler, R.; Ofer, J.; Newton, J.; Smith, R.; Kamintsky, L.; Jones, C.M.A.; O’Keeffe, E.; et al. Concussion Susceptibility Is Mediated by Spreading Depolarization-Induced Neurovascular Dysfunction. Brain 2021. [Google Scholar] [CrossRef]
- Cacheaux, L.P.; Ivens, S.; David, Y.; Lakhter, A.J.; Bar-Klein, G.; Shapira, M.Y.; Heinemann, U.; Friedman, A.; Kaufer, D. Transcriptome Profiling Reveals TGF-Beta Signaling Involvement in Epileptogenesis. J. Neurosci. 2009, 29, 8927–8935. [Google Scholar] [CrossRef]
- Ivens, S.; Kaufer, D.; Flores, L.P.; Bechmann, I.; Zumsteg, D.; Tomkins-Netzer, O.; Seiffert, E.; Heinemann, U.; Friedman, A. TGF-Beta receptor-mediated albumin uptake into astrocytes is involved in neocortical epileptogenesis. Brain 2006, 130, 535–547. [Google Scholar] [CrossRef] [Green Version]
- Schachtrup, C.; Ryu, J.K.; Helmrick, M.J.; Vagena, E.; Galanakis, D.K.; Degen, J.L.; Margolis, R.U.; Akassoglou, K. Fibrinogen Triggers Astrocyte Scar Formation by Promoting the Availability of Active TGF-Beta after Vascular Damage. J. Neurosci. 2010, 30, 5843–5854. [Google Scholar] [CrossRef]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng-Hathaway, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-β signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Kim, S.Y.; Senatorov, V.V., Jr.; Morrissey, C.S.; Lippmann, K.; Vazquez, O.; Milikovsky, D.Z.; Gu, F.; Parada, I.; Prince, D.A.; Becker, A.J.; et al. TGFβ signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Sci. Rep. 2017, 7, 7711. [Google Scholar] [CrossRef] [PubMed]
- Weissberg, I.; Wood, L.; Kamintsky, L.; Vazquez, O.; Milikovsky, D.Z.; Alexander, A.; Oppenheim, H.; Ardizzone, C.; Becker, A.; Frigerio, F.; et al. Albumin induces excitatory synaptogenesis through astrocytic TGF-β/ALK5 signaling in a model of acquired epilepsy following blood–brain barrier dysfunction. Neurobiol. Dis. 2015, 78, 115–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, N.; Milikovsky, D.Z.; Baranauskas, G.; Vinogradov, E.; David, Y.; Ketzef, M.; Abutbul, S.; Weissberg, I.; Kamintsky, L.; Fleidervish, I.; et al. Differential TGF-β Signaling in Glial Subsets Underlies IL-6–Mediated Epileptogenesis in Mice. J. Immunol. 2015, 195, 1713–1722. [Google Scholar] [CrossRef] [Green Version]
- Seiffert, E.; Dreier, J.P.; Ivens, S.; Bechmann, I.; Tomkins-Netzer, O.; Heinemann, U.; Friedman, A. Lasting Blood-Brain Barrier Disruption Induces Epileptic Focus in the Rat Somatosensory Cortex. J. Neurosci. 2004, 24, 7829–7836. [Google Scholar] [CrossRef] [Green Version]
- Holmes, G.L. Cognitive impairment in epilepsy: The role of network abnormalities. Epileptic Disord. 2015, 17, 101–116. [Google Scholar] [CrossRef]
- Bialas, A.R.; Stevens, B. TGF-β signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat. Neurosci. 2013, 16, 1773–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Stephan, A.H.; Madison, D.V.; Mateos, J.M.; Fraser, D.A.; Lovelett, E.A.; Coutellier, L.; Kim, L.; Tsai, H.-H.; Huang, E.J.; Rowitch, D.H.; et al. A Dramatic Increase of C1q Protein in the CNS during Normal Aging. J. Neurosci. 2013, 33, 13460–13474. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O.; et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Erusalimsky, J.D.; Kurz, D.J. Cellular senescence in vivo: Its relevance in ageing and cardiovascular disease. Exp. Gerontol. 2005, 40, 634–642. [Google Scholar] [CrossRef]
- Jeyapalan, J.C.; Ferreira, M.; Sedivy, J.M.; Herbig, U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech. Ageing Dev. 2007, 128, 36–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishi, S. Functional Aging and Gradual Senescence in Zebrafish. Ann. N. Y. Acad. Sci. 2004, 1019, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Melk, A.; Kittikowit, W.; Sandhu, I.; Halloran, K.M.; Grimm, P.; Schmidt, B.M.W.; Halloran, P.F. Cell senescence in rat kidneys in vivo increases with growth and age despite lack of telomere shortening. Kidney Int. 2003, 63, 2134–2143. [Google Scholar] [CrossRef] [Green Version]
- Paradis, V.; Youssef, N.; Dargère, D.; Bâ, N.; Bonvoust, F.; Deschatrette, J.; Bedossa, P. Replicative senescence in normal liver, chronic hepatitis C, and hepatocellular carcinomas. Hum. Pathol. 2001, 32, 327–332. [Google Scholar] [CrossRef]
- Campisi, J. From Cells to Organisms: Can We Learn about Aging from Cells in Culture? Exp. Gerontol. 2001, 34, 607–618. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E. Senescence and Immortalization: Role of Telomeres and Telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2017, 217, 65–77. [Google Scholar] [CrossRef]
- Adams, P.D. Healing and Hurting: Molecular Mechanisms, Functions, and Pathologies of Cellular Senescence. Mol. Cell 2009, 36, 2–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtani, N.; Yamakoshi, K.; Takahashi, A.; Hara, E. The p16INK4a-RB pathway: Molecular link between cellular senescence and tumor suppression. J. Med. Investig. 2004, 51, 146–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and aging: The critical roles of p53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef] [PubMed]
- Dodig, S.; Čepelak, I.; Pavić, I. Hallmarks of senescence and aging. Biochem. Med. 2019, 29, 483–497. [Google Scholar] [CrossRef]
- Freund, A.; Laberge, R.-M.; Demaria, M.; Campisi, J. Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 2012, 23, 2066–2075. [Google Scholar] [CrossRef]
- Wang, A.S.; Dreesen, O. Biomarkers of Cellular Senescence and Skin Aging. Front. Genet. 2018, 9, 247. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Riessland, M. Cellular Senescence in Health, Disease and Aging: Blessing or Curse? Life 2021, 11, 541. [Google Scholar] [CrossRef]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of senescence-associated secretory phenotype and its potential as a therapeutic target for senescence-associated diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Pulido, T.; Velarde, M.C.; Alimirah, F. The senescence-associated secretory phenotype: Fueling a wound that never heals. Mech. Ageing Dev. 2021, 199, 111561. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, M.; Kavanagh, T.J.; Costa, L.G. Measurements of Astrocyte Proliferation. Methods Mol. Biol. 2011, 758, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Sell, C.; Crowe, E.; Lorenzini, A.; Malaguti, M.; Hrelia, S.; Torres, C. Stress-induced senescence in human and rodent astrocytes. Exp. Cell Res. 2010, 316, 2961–2968. [Google Scholar] [CrossRef]
- Yu, C.; Narasipura, S.D.; Richards, M.H.; Hu, X.-T.; Yamamoto, B.; Al-Harthi, L. HIV and drug abuse mediate astrocyte senescence in a β-catenin-dependent manner leading to neuronal toxicity. Aging Cell 2017, 16, 956–965. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte Senescence as a Component of Alzheimer’s Disease. PLoS ONE 2012, 7, e45069. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Cué, C.; Rueda, N. Cellular Senescence in Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 16. [Google Scholar] [CrossRef]
- Kritsilis, M.; Rizou, S.V.; Koutsoudaki, P.N.; Evangelou, K.; Gorgoulis, V.G.; Papadopoulos, D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int. J. Mol. Sci. 2018, 19, 2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, D.J.; Petersen, R.C. Cellular senescence in brain aging and neurodegenerative diseases: Evidence and perspectives. J. Clin. Investig. 2018, 128, 1208–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, X.; Liu, X.; Wang, F.; Wang, F.; Geng, X. Role of Senescence and Neuroprotective Effects of Telomerase in Neurodegenerative Diseases. Rejuvenation Res. 2020, 23, 150–158. [Google Scholar] [CrossRef]
- Zhang, P.; Sung, M.-H. Cellular senescence in neurodegenerative diseases. Cell. Senescence Dis. 2021, 14, 363–381. [Google Scholar] [CrossRef]
- Bussian, T.J.; Aziz, A.; Meyer, C.F.; Swenson, B.L.; Van Deursen, J.M.; Baker, D.J. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018, 562, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; Van Deursen, J.M. Senescent cells: An emerging target for diseases of ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Lam, E.W.-F.; Tchkonia, T.; Kirkland, J.L.; Sun, Y. Senescent Cells: Emerging Targets for Human Aging and Age-Related Diseases. Trends Biochem. Sci. 2020, 45, 578–592. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; di Fagagna, F.D. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2020, 22, 75–95. [Google Scholar] [CrossRef]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef] [Green Version]
- Bang, M.; Gonzales, E.L.; Shin, C.Y.; Kwon, K.J. Late Passage Cultivation Induces Aged Astrocyte Phenotypes in Rat Primary Cultured Cells. Biomol. Ther. 2021, 29, 144–153. [Google Scholar] [CrossRef]
- Pertusa, M.; García-Matas, S.; Rodríguez-Farré, E.; Sanfeliu, C.; Cristòfol, R. Astrocytes aged in vitro show a decreased neuroprotective capacity. J. Neurochem. 2007, 101, 794–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Lye, J.J.; Latorre, E.; Lee, B.P.; Bandinelli, S.; Holley, J.E.; Gutowski, N.J.; Ferrucci, L.; Harries, L.W. Astrocyte senescence may drive alterations in GFAPα, CDKN2A p14 (ARF), and TAU3 transcript expression and contribute to cognitive decline. GeroScience 2019, 41, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmnacher, K.; Krach, F.; Schneider, Y.; Alecu, J.E.; Mautner, L.; Klein, P.; Roybon, L.; Prots, I.; Xiang, W.; Winner, B. Unique signatures of stress-induced senescent human astrocytes. Exp. Neurol. 2020, 334, 113466. [Google Scholar] [CrossRef] [PubMed]
- Blum-Degena, D.; Müller, T.; Kuhn, W.; Gerlach, M.; Przuntek, H.; Riederer, P. Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci. Lett. 1995, 202, 17–20. [Google Scholar] [CrossRef]
- Horstmann, S.; Budig, L.; Gardner, H.; Koziol, J.; Deuschle, M.; Schilling, C.; Wagner, S. Matrix metalloproteinases in peripheral blood and cerebrospinal fluid in patients with Alzheimer’s disease. Int. Psychogeriatr. 2010, 22, 966–972. [Google Scholar] [CrossRef] [PubMed]
- Bradburn, S.; Sarginson, J.; Murgatroyd, C.A. Association of Peripheral Interleukin-6 with Global Cognitive Decline in Non-demented Adults: A Meta-Analysis of Prospective Studies. Front. Aging Neurosci. 2018, 9, 438. [Google Scholar] [CrossRef]
- Campbell, I.L.; Abraham, C.R.; Masliah, E.; Kemper, P.; Inglis, J.D.; Oldstone, M.B.; Mucke, L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc. Natl. Acad. Sci. USA 1993, 90, 10061–10065. [Google Scholar] [CrossRef] [Green Version]
- Campbell, I.L. Structural and Functional Impact of the Transgenic Expression of Cytokines in the CNS. Ann. N. Y. Acad. Sci. 1998, 840, 83–96. [Google Scholar] [CrossRef]
- Campbell, I.L.; Hofer, M.J.; Pagenstecher, A. Transgenic models for cytokine-induced neurological disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2010, 1802, 903–917. [Google Scholar] [CrossRef]
- Frippiat, C.; Chen, Q.M.; Zdanov, S.; de Magalhaes, J.P.; Remacle, J.; Toussaint, O. Subcytotoxic H2O2 Stress Triggers a Release of Transforming Growth Factor-β1, Which Induces Biomarkers of Cellular Senescence of Human Diploid Fibroblasts. J. Biol. Chem. 2001, 276, 2531–2537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rapisarda, V.; Borghesan, M.; Miguela, V.; Encheva, V.; Snijders, A.P.; Lujambio, A.; O’Loghlen, A. Integrin Beta 3 Regulates Cellular Senescence by Activating the TGF-β Pathway. Cell Rep. 2017, 18, 2480–2493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated exposure of human skin fibroblasts to UVB at subcytotoxic level triggers premature senescence through the TGF-β1 signaling pathway. J. Cell Sci. 2005, 118, 743–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayachandra, K.; Higgins, W.; Lee, J.; Glick, A. Induction of p16ink4a and p19ARF by TGFβ1 contributes to growth arrest and senescence response in mouse keratinocytes. Mol. Carcinog. 2008, 48, 181–186. [Google Scholar] [CrossRef]
- Wu, J.; Niu, J.; Li, X.; Wang, X.; Guo, Z.; Zhang, F. TGF-β1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production. BMC Dev. Biol. 2014, 14, 21. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Gont, A.; Perkins, T.J.; Hanson, J.E.L.; Lorimer, I.A.J. Induction of senescence in primary glioblastoma cells by serum and TGFβ. Sci. Rep. 2017, 7, 2156. [Google Scholar] [CrossRef] [Green Version]
- Lyu, G.; Guan, Y.; Zhang, C.; Zong, L.; Sun, L.; Huang, X.; Huang, L.; Zhang, L.; Tian, X.-L.; Zhou, Z.; et al. TGF-β signaling alters H4K20me3 status via miR-29 and contributes to cellular senescence and cardiac aging. Nat. Commun. 2018, 9, 2560. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Lee, J.-H.; Hwang, S.-C.; Choi, K.S.; Yoon, G. TGF β1 induces prolonged mitochondrial ROS generation through decreased complex IV activity with senescent arrest in Mv1Lu cells. Oncogene 2005, 24, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Yousef, H.; Conboy, M.J.; Morgenthaler, A.; Schlesinger, C.; Bugaj, L.; Paliwal, P.; Greer, C.; Conboy, I.M.; Schaffer, D. Systemic attenuation of the TGF-β pathway by a single drug simultaneously rejuvenates hippocampal neurogenesis and myogenesis in the same old mammal. Oncotarget 2015, 6, 11959–11978. [Google Scholar] [CrossRef] [Green Version]
- Mehdipour, M.; Etienne, J.; Chen, C.-C.; Gathwala, R.; Rehman, M.; Kato, C.; Liu, C.; Liu, Y.; Zuo, Y.; Conboy, M.J.; et al. Rejuvenation of brain, liver and muscle by simultaneous pharmacological modulation of two signaling determinants, that change in opposite directions with age. Aging 2019, 11, 5628–5645. [Google Scholar] [CrossRef]
- Amram, S.; Iram, T.; Lazdon, E.; Vassar, R.; Ben-Porath, I.; Frenkel, D. Astrocyte Senescence in an Alzheimer’s Disease Mouse Model Is Mediated by TGF-Β1 and Results in Neurotoxicity. bioRxiv 2019. bioRxiv:700013. [Google Scholar] [CrossRef]
- Liu, R.-M.; Desai, L.P. Reciprocal regulation of TGF-β and reactive oxygen species: A perverse cycle for fibrosis. Redox Biol. 2015, 6, 565–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krstić, J.; Trivanović, D.; Mojsilović, S.; Santibanez, J.F. Transforming Growth Factor-Beta and Oxidative Stress Interplay: Implications in Tumorigenesis and Cancer Progression. Oxid. Med. Cell. Longev. 2015, 2015, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.-M.; Pravia, K.G. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesne, S.; Docagne, F.; Gabriel, C.; Liot, G.; Lahiri, D.K.; Buee, L.; Plawinski, L.; Delacourte, A.; MacKenzie, E.T.; Buisson, A.; et al. Transforming Growth Factor-β1 Potentiates Amyloid-β Generation in Astrocytes and in Transgenic Mice. J. Biol. Chem. 2003, 278, 18408–18418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyss-Coray, T.; Masliah, E.; Mallory, M.; McConlogue, L.; Johnson-Wood, K.; Lin, C.; Mucke, L. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer’s disease. Nature 1997, 389, 603–606. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Feng, L.; Masliah, E.; Ruppe, M.D.; Lee, H.S.; Toggas, S.M.; Rockenstein, E.M.; Mucke, L. Increased central nervous system production of extracellular matrix components and development of hydrocephalus in transgenic mice overexpressing transforming growth factor-beta 1. Am. J. Pathol. 1995, 147, 53–67. [Google Scholar]
- Apelt, J.; Schliebs, R. β-Amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001, 894, 21–30. [Google Scholar] [CrossRef]
- Van der Wal, E.A.; Gómez-Pinilla, F.; Cotman, C.W. Transforming growth factor-β1 is in plaques in Alzheimer and Down pathologies. NeuroReport 1993, 4, 69–72. [Google Scholar] [CrossRef]
- Vallée, A.; LeCarpentier, Y.; Guillevin, R.; Vallée, J.N. Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis. Oncotarget 2017, 8, 90579–90604. [Google Scholar] [CrossRef] [Green Version]
- Preininger, M.; Zaytseva, D.; Lin, J.M.; Kaufer, D. Blood-Brain Barrier Dysfunction Promotes Astrocyte Senescence through Albumin-Induced TGFβ Signaling Activation. bioRxiv 2022. bioRxiv:2022.04.25.489438. [Google Scholar] [CrossRef]
- Mombach, J.C.M.; Vendrusculo, B.; Bugs, C.A. A Model for p38MAPK-Induced Astrocyte Senescence. PLoS ONE 2015, 10, e0125217. [Google Scholar] [CrossRef] [PubMed]
- Spampinato, S.F.; Merlo, S.; Sano, Y.; Kanda, T.; Sortino, M.A. Astrocytes contribute to Aβ-induced blood-brain barrier damage through activation of endothelial MMP9. J. Neurochem. 2017, 142, 464–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brkic, M.; Balusu, S.; Van Wonterghem, E.; Gorlé, N.; Benilova, I.; Kremer, A.; Van Hove, I.; Moons, L.; De Strooper, B.; Kanazir, S.; et al. Amyloid β Oligomers Disrupt Blood-CSF Barrier Integrity by Activating Matrix Metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Baker, D.J.; Tachibana, M.; Liu, C.-C.; Van Deursen, J.M.; Brott, T.G.; Bu, G.; Kanekiyo, T. Vascular Cell Senescence Contributes to Blood–Brain Barrier Breakdown. Stroke 2016, 47, 1068–1077. [Google Scholar] [CrossRef]
- Derynck, R.; Budi, E.H. Specificity, Versatility, and Control of TGF-b Family Signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [Green Version]
- Budi, E.H.; Duan, D.; Derynck, R. Transforming Growth Factor-β Receptors and Smads: Regulatory Complexity and Functional Versatility. Trends Cell Biol. 2017, 27, 658–672. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of astrocyte-mediated neuropathology in the aging brain. Blood–brain barrier dysfunction (BBBD) and cellular senescence progressively increase during aging and are associated with an increased risk of neurodegenerative disease. Two processes that result in dysfunctional astrocyte phenotypes and subsequent neuropathology are BBBD-associated astrocyte activation and astrocyte senescence. BBBD causes albumin to leak into the brain, where it activates TGFβ and p38MAPK signaling in astrocytes, which elicits an epileptogenic and pro-inflammatory astrocyte phenotype. The epileptogenic phenotype is characterized by downregulation of K+ transporters (Kir4.1), inhibitory GABAA receptors (GABAAR), and NMDA receptors (NMDAR) and upregulation of excitatory glutamate receptors (GluR). Second, certain types of cellular stress cause expression of tumor suppressors p16 and p21, which elicit a senescent phenotype characterized by decreased functional capacity and SASP expression. This decreased capacity involves downregulation of aquaporin 4 (AQP4), K+ transporters (Kir4.1), and glutamate transporters (EAAT1/2). Factors such as cytokines, chemokines, and proteases secreted by activated and senescent astrocytes may act on the endothelium to further exacerbate BBBD via downregulation of tight junction (TJ) proteins, glucose transporters (GLUT1), and waste efflux transporters (Pgp). Dysfunctional astrocytes contribute to neuronal hyperexcitability, neuroinflammation, neurotoxicity, and neurodegeneration, which manifest as cognitive deficits in the aging brain. Questions remain regarding the mechanistic links and relationships between BBBD-associated astrocyte activation and senescence. This figure was created with cell images from BioRender.
Figure 1.
Mechanisms of astrocyte-mediated neuropathology in the aging brain. Blood–brain barrier dysfunction (BBBD) and cellular senescence progressively increase during aging and are associated with an increased risk of neurodegenerative disease. Two processes that result in dysfunctional astrocyte phenotypes and subsequent neuropathology are BBBD-associated astrocyte activation and astrocyte senescence. BBBD causes albumin to leak into the brain, where it activates TGFβ and p38MAPK signaling in astrocytes, which elicits an epileptogenic and pro-inflammatory astrocyte phenotype. The epileptogenic phenotype is characterized by downregulation of K+ transporters (Kir4.1), inhibitory GABAA receptors (GABAAR), and NMDA receptors (NMDAR) and upregulation of excitatory glutamate receptors (GluR). Second, certain types of cellular stress cause expression of tumor suppressors p16 and p21, which elicit a senescent phenotype characterized by decreased functional capacity and SASP expression. This decreased capacity involves downregulation of aquaporin 4 (AQP4), K+ transporters (Kir4.1), and glutamate transporters (EAAT1/2). Factors such as cytokines, chemokines, and proteases secreted by activated and senescent astrocytes may act on the endothelium to further exacerbate BBBD via downregulation of tight junction (TJ) proteins, glucose transporters (GLUT1), and waste efflux transporters (Pgp). Dysfunctional astrocytes contribute to neuronal hyperexcitability, neuroinflammation, neurotoxicity, and neurodegeneration, which manifest as cognitive deficits in the aging brain. Questions remain regarding the mechanistic links and relationships between BBBD-associated astrocyte activation and senescence. This figure was created with cell images from BioRender.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Preininger, M.K.; Kaufer, D. Blood–Brain Barrier Dysfunction and Astrocyte Senescence as Reciprocal Drivers of Neuropathology in Aging. Int. J. Mol. Sci. 2022, 23, 6217. https://doi.org/10.3390/ijms23116217
AMA Style
Preininger MK, Kaufer D. Blood–Brain Barrier Dysfunction and Astrocyte Senescence as Reciprocal Drivers of Neuropathology in Aging. International Journal of Molecular Sciences. 2022; 23(11):6217. https://doi.org/10.3390/ijms23116217
Chicago/Turabian StylePreininger, Marcela K., and Daniela Kaufer. 2022. "Blood–Brain Barrier Dysfunction and Astrocyte Senescence as Reciprocal Drivers of Neuropathology in Aging" International Journal of Molecular Sciences 23, no. 11: 6217. https://doi.org/10.3390/ijms23116217
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.