
The Effect of Activated FXIII, a Transglutaminase, on Vascular Smooth Muscle Cells


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
- Does the osteoblastic transformation of human aortic smooth muscle cells (HAoSMC) result in the expression of cFXIII?
- How does FXIIIa influence the proliferation, migration and collagen secretion of HAoSMCs?
- Does FXIIIa influence intracellular and cell-associated TSP-1 content of HAoSMCs and their TSP-1 synthesis?
2. Results
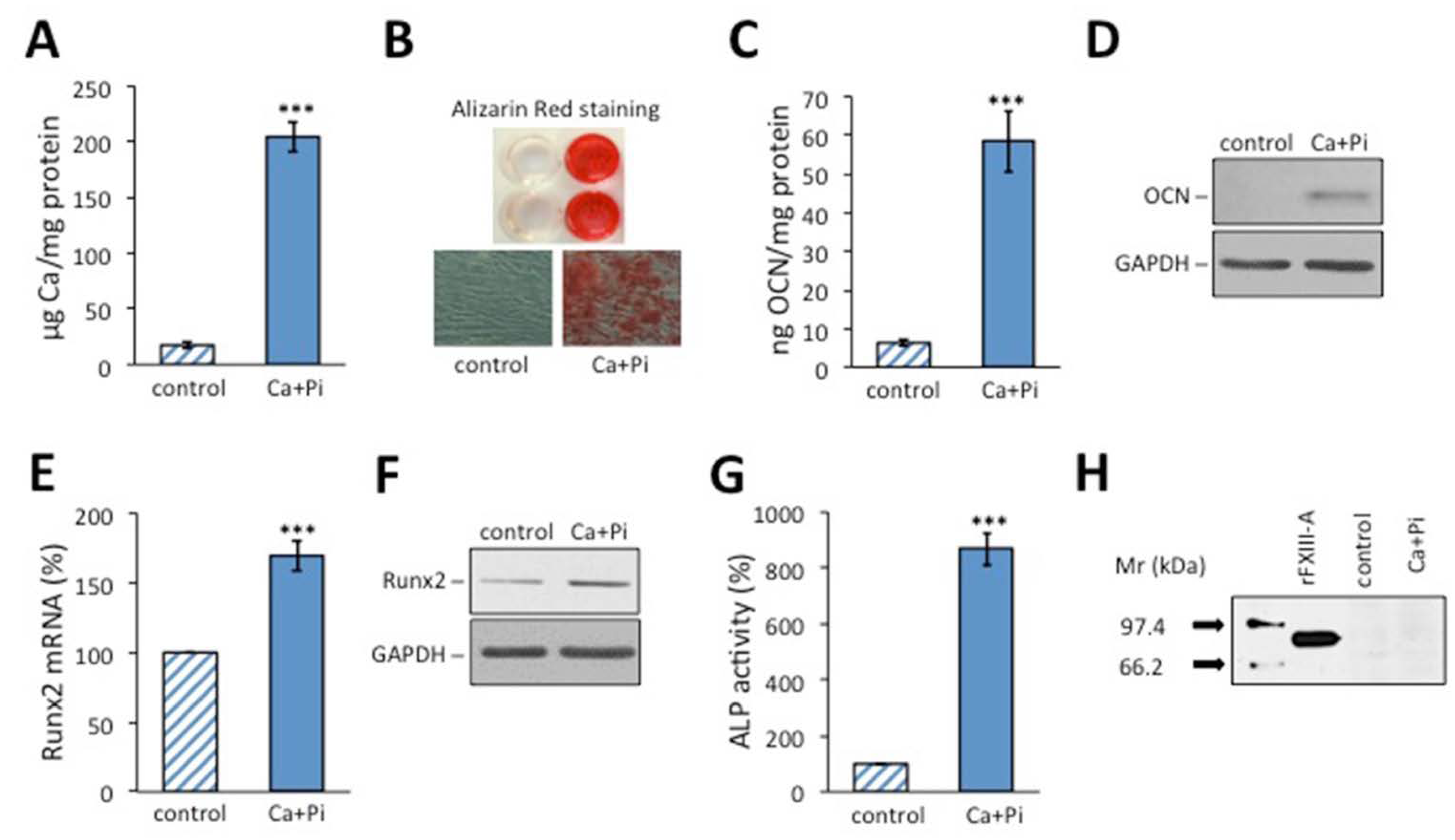
2.1. Osteoblastic Transformation Does Not Induce FXIII-A Expression in HAoSMCs
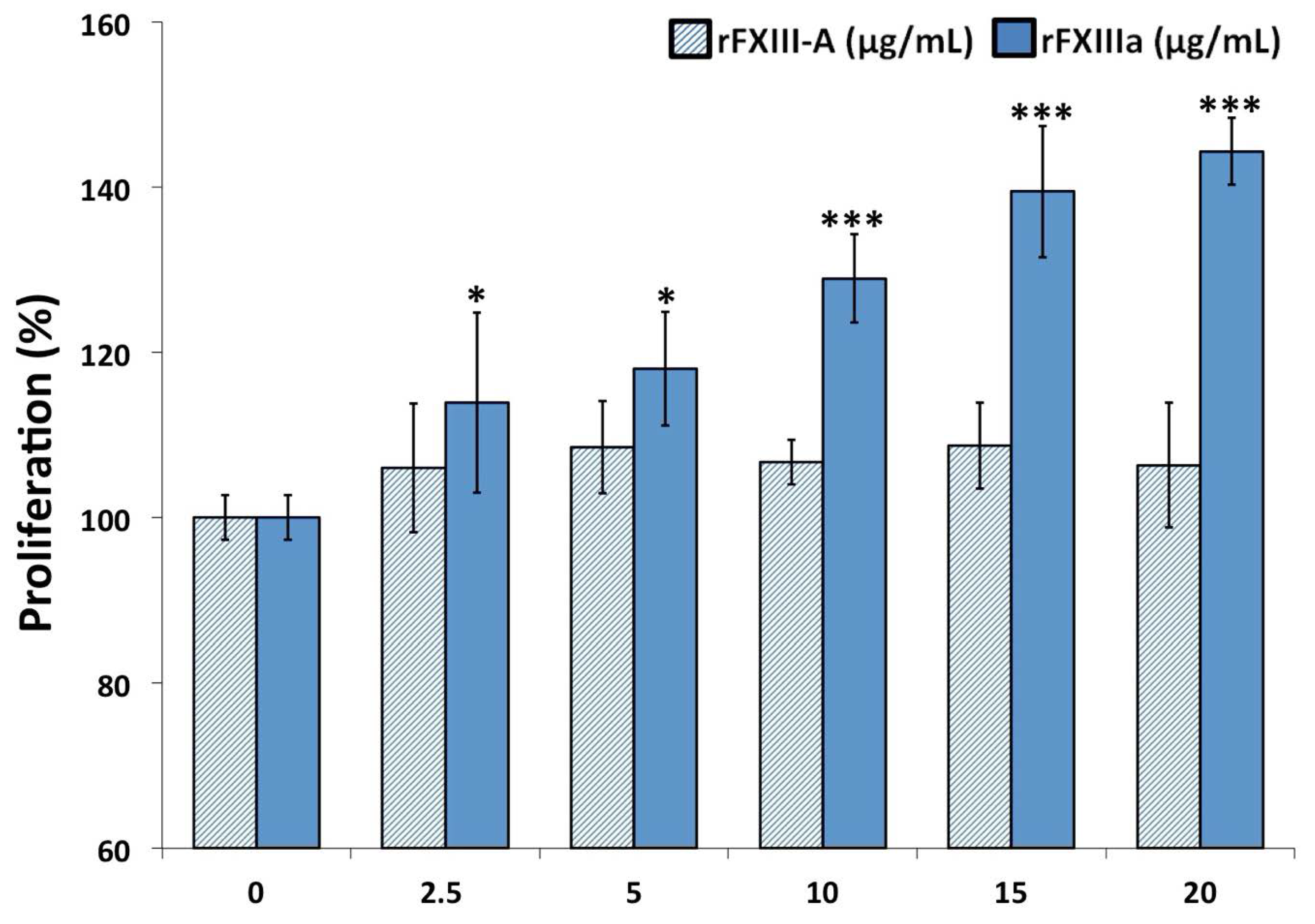
2.2. rFXIIIa Enhances the Proliferation of HAoSMCs
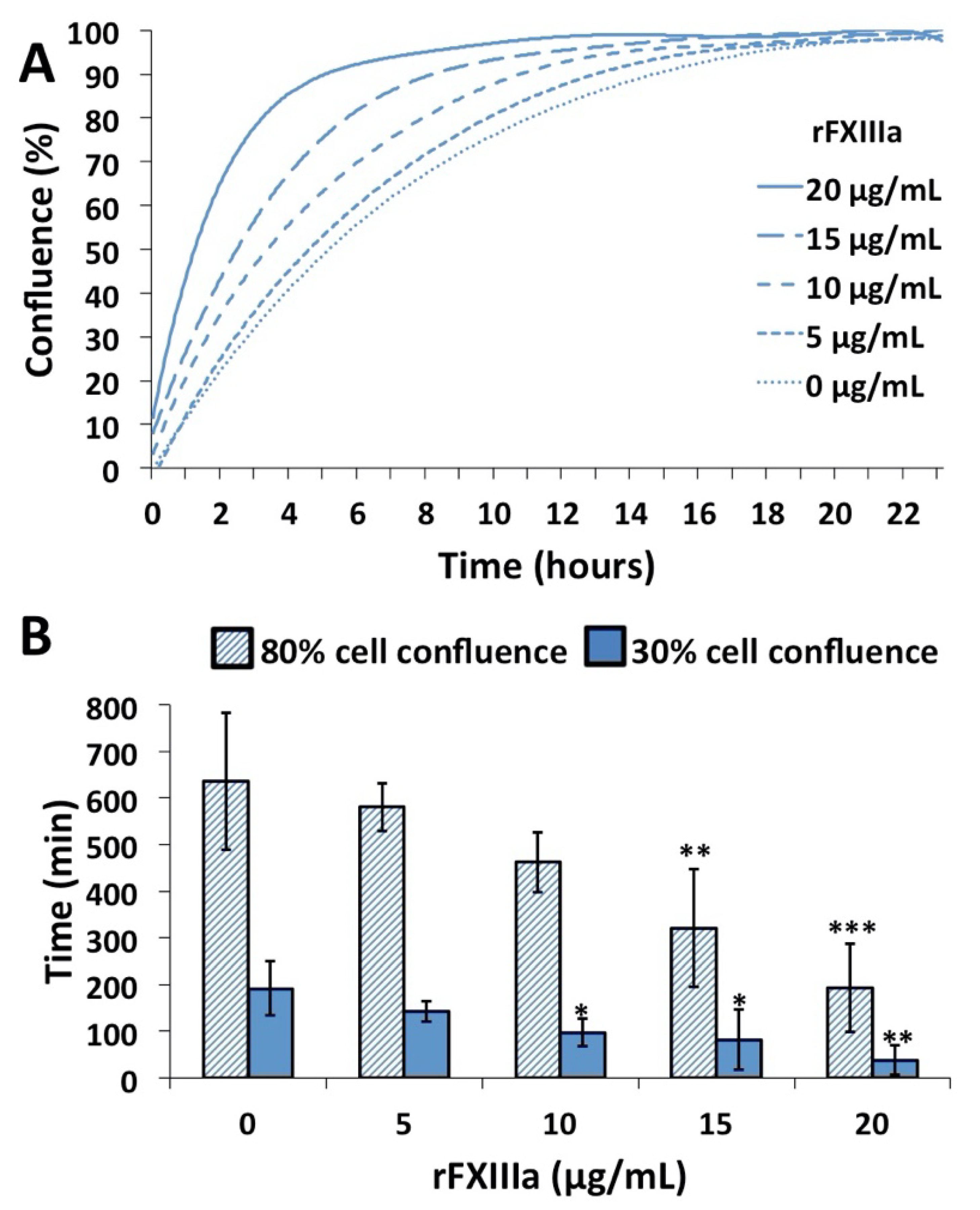
2.3. rFXIIIa Accelerates the Closure of In Vitro Gap Wound
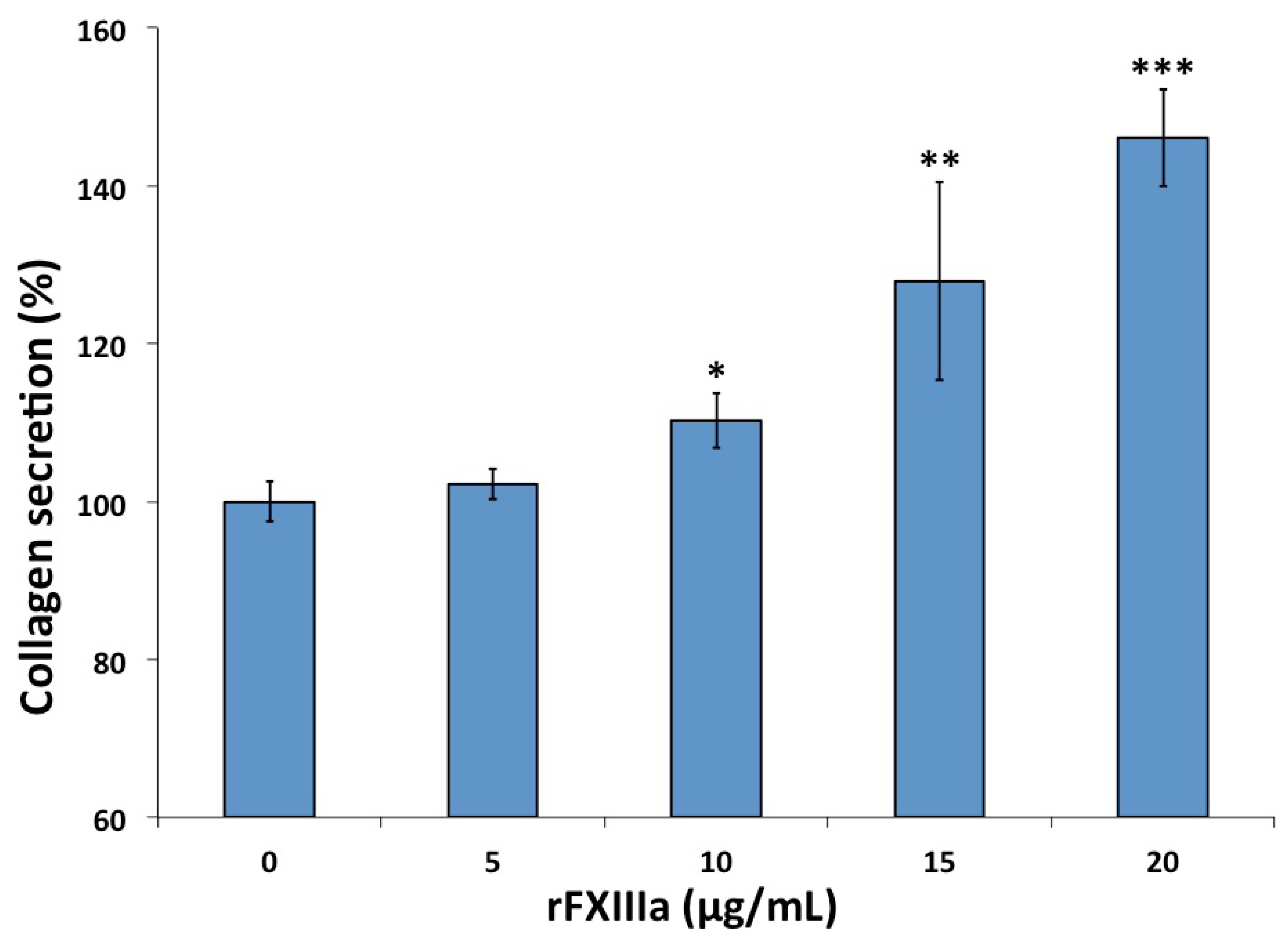
2.4. rFXIIIa Induced Elevated Collagen Secretion by HAoSMCs
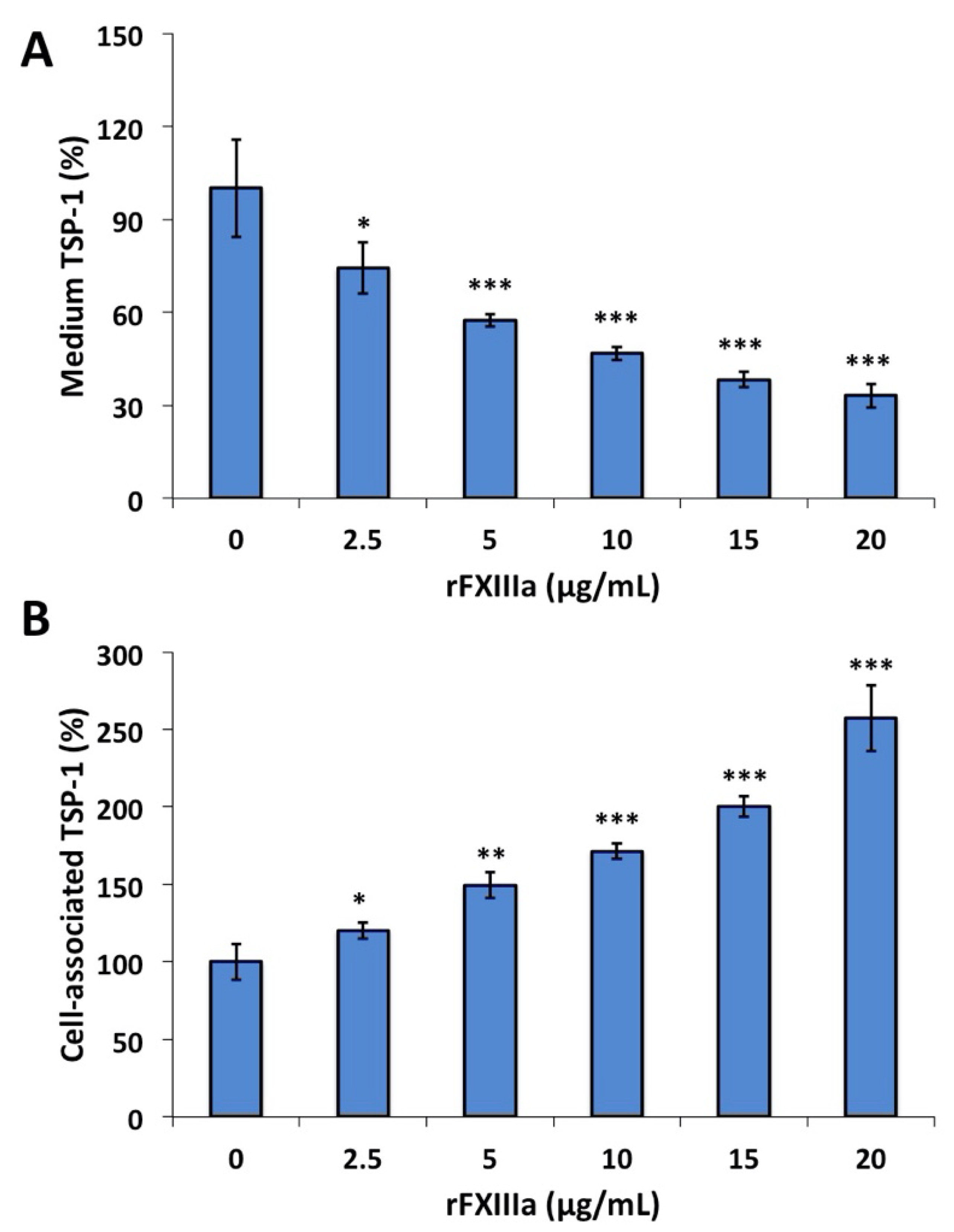
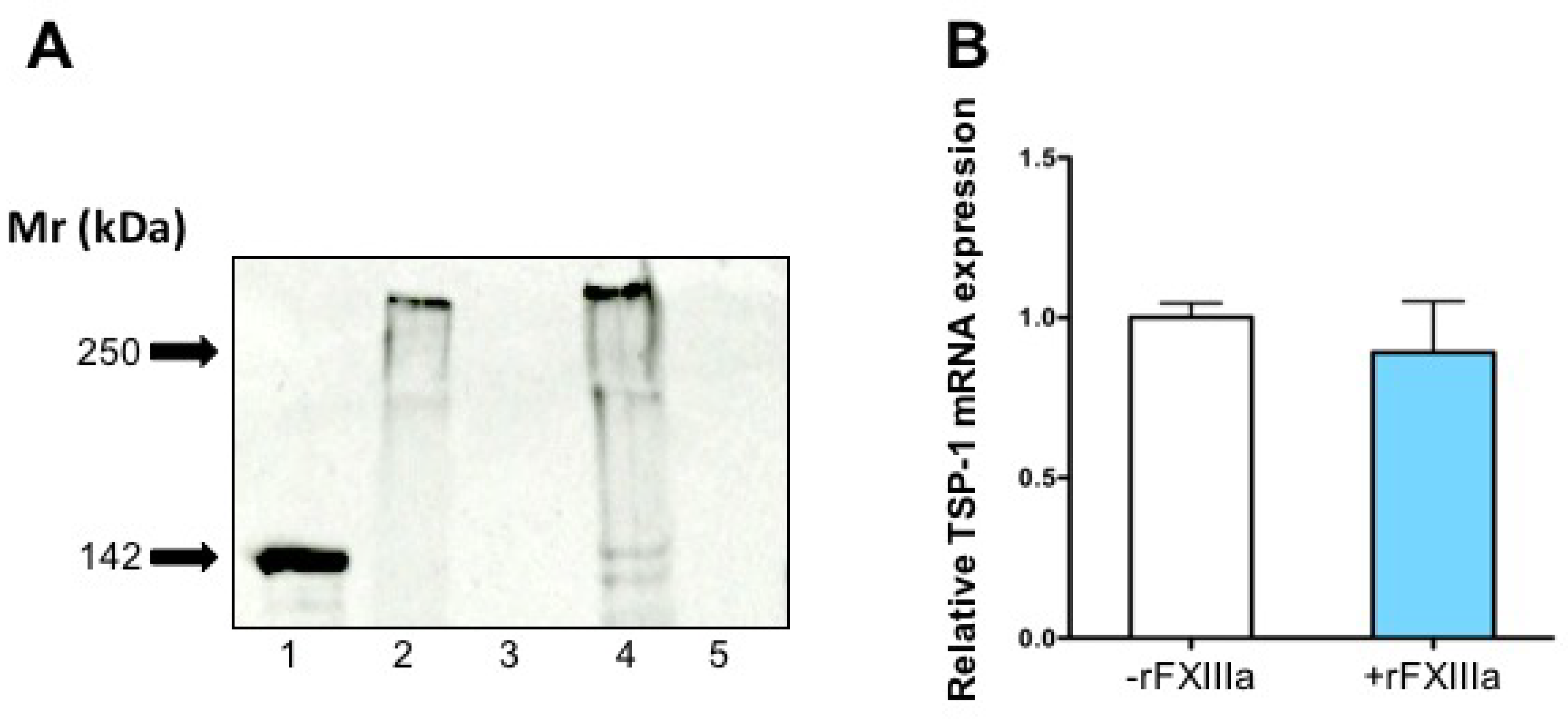
2.5. TSP-1 in the Culture Medium and Cell-Associated TSP-1 Following rFXIIIa Treatment of HAoSMCs
3. Discussion
4. Methods
4.1. Materials
4.2. Cell Culture
4.3. Assessment of Osteoblastic Differentiation
4.4. Treatment of Cells with Activated rFXIII
4.5. Cell Proliferation Assays
4.6. In Vitro Wound Healing Assay
4.7. Collagen Deposition in the Extracellular Matrix of HAoSMC
4.8. Measurement of Thrombospondin-1 in the Lysate and Culture Media of HAoSMCs by ELISA
4.9. Detection of Thrombospondin-1 in HAoSMCs and in Their Extracellular Matrix by Western Blotting
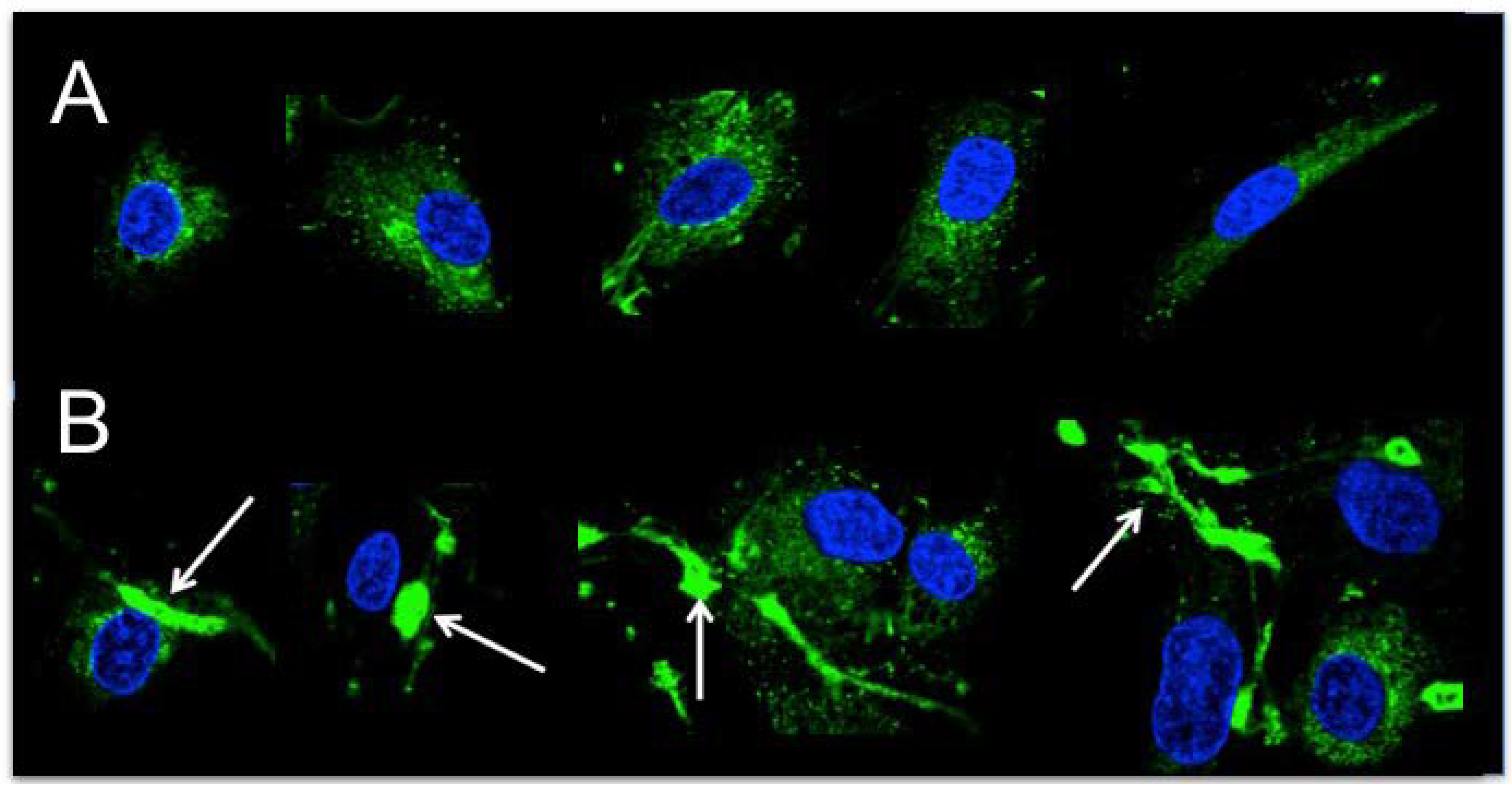
4.10. Immunofluorescent Staining of HAoSMCs for Thrombospondin-1
4.11. Real-Time PCR
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CM | Calcification medium |
| cFXIII | Cellular factor XIII |
| FXIIIa | Activated factor XIII |
| FXIII | Blood coagulation factor XIII |
| FXIII-A | Factor XIII A subunit |
| FXIII-B | Factor XIII B subunit |
| GAPDH | Glyceraldehyde 3-phosphate dehydrogenase |
| GM | Growth medium |
| HAoSMC | Human aortic smooth muscle cell |
| pFXIII | Plasma factor XIII |
| rFXIII | Recombinant factor XIII |
| TSP-1 | Thrombospondin-1 |
| VSMC | Vascular smooth muscle cell |
References
- Mitchell, J.L.; Mutch, N.J. Let′s cross-link: Diverse functions of the promiscuous cellular transglutaminase factor XIII-A. J. Thromb. Haemost. 2019, 17, 19–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muszbek, L.; Bereczky, Z.; Bagoly, Z.; Komáromi, I.; Katona, É. Factor XIII: A Coagulation Factor With Multiple Plasmatic and Cellular Functions. Physiol. Rev. 2011, 91, 931–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balogh, E.; Tóth, A.; Tolnai, E.; Bodó, T.; Bányai, E.; Szabó, D.J.; Petrovski, G.; Jeney, V. Osteogenic differentiation of human lens epithelial cells might contribute to lens calcification. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 1724–1731. [Google Scholar] [CrossRef]
- Schroeder, V.; Kohler, H.P. Factor XIII: Structure and Function. Semin. Thromb. Hemost. 2016, 42, 422–428. [Google Scholar]
- Komáromi, I.; Bagoly, Z.; Muszbek, L. Factor XIII: Novel structural and functional aspects. J. Thromb. Haemost. 2011, 9, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buluk, K. An unknown action of blood platelets; preliminary communication. Pol. Tyg. Lek. Wars 1955, 10, 191. [Google Scholar] [PubMed]
- Luscher, E.F. Fibrin-stabilizing factor from thrombocytes. Schweiz. Med. Wochenschr. 1957, 87, 1220–1221. [Google Scholar]
- Mitchell, J.L.; Mutch, N.J. Novel aspects of platelet factor XIII function. Thromb. Res. 2016, 141, S17–S21. [Google Scholar] [CrossRef] [Green Version]
- Muszbek, L.; Adány, R.; Szegedi, G.; Polgár, J.; Kávai, M. Factor XIII of blood coagulation in human monocytes. Thromb. Res. 1985, 37, 401–410. [Google Scholar] [CrossRef]
- Adány, R.; Belkin, A.; Vasilevskaya, T.; Muszbek, L. Identification of blood coagulation factor XIII in human peritoneal macrophages. Eur. J. Cell Biol. 1985, 38, 171–173. [Google Scholar]
- Nurminskaya, M.; Kaartinen, M.T. Transglutaminases in mineralized tissues. Front. Biosci. 2006, 11, 1591–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Jallad, H.F.; Nakano, Y.; Chen, J.L.; McMillan, E.; Lefebvre, C.; Kaartinen, M.T. Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol. 2006, 25, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Al-Jallad, H.F.; Myneni, V.D.; Piercy-Kotb, S.A.; Chabot, N.; Mulani, A.; Keillor, J.W.; Kaartinen, M.T. Plasma Membrane Factor XIIIA Transglutaminase Activity Regulates Osteoblast Matrix Secretion and Deposition by Affecting Microtubule Dynamics. PLoS ONE 2011, 6, e15893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nurminskaya, M.; Magee, C.; Nurminsky, D.; Linsenmayer, T.F. Plasma Transglutaminase in Hypertrophic Chondrocytes: Expression and Cell-specific Intracellular Activation Produce Cell Death and Externalization. J. Cell Biol. 1998, 142, 1135–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenthal, A.K.; Masuda, I.; Gohr, C.M.; Derfus, B.A.; Le, M. The transglutaminase, Factor XIIIA, is present in articular chondrocytes. Osteoarthr. Cartil. 2001, 9, 578–581. [Google Scholar] [CrossRef] [Green Version]
- Myneni, V.D.; Hitomi, K.; Kaartinen, M.T. Factor XIII-A transglutaminase acts as a switch between preadipocyte proliferation and differentiation. Blood 2014, 124, 1344–1353. [Google Scholar] [CrossRef]
- Orosz, Z.Z.; Bárdos, H.; Shemirani, A.H.; Debreceni, I.B.; Lassila, R.; Riikonen, A.S.; Hovinga, J.A.K.; Seiler, T.G.; van Dorland, H.A.; Schroeder, V.; et al. Cellular Factor XIII, a Transglutaminase in Human Corneal Keratocytes. Int. J. Mol. Sci. 2019, 20, 5963. [Google Scholar] [CrossRef] [Green Version]
- Polgar, J.; Hidasi, V.; Muszbek, L. Non-proteolytic activation of cellular protransglutaminase (placenta macrophage factor XIII). Biochem. J. 1990, 267, 557–560. [Google Scholar] [CrossRef] [Green Version]
- Muszbek, L.; Polgar, J.; Boda, Z. Platelet factor XIII becomes active without the release of activation peptide during platelet activation. Thromb. Haemost. 1993, 69, 282–285. [Google Scholar] [CrossRef]
- Muszbek, L.; Haramura, G.; Polgár, J. Transformation of cellular factor XIII into an active zymogen transglutaminase in thrombin-stimulated platelets. Thromb. Haemost. 1995, 73, 702–705. [Google Scholar] [CrossRef]
- Alshehri, F.S.M.; Whyte, C.S.; Mutch, N.J. Factor XIII-A: An Indispensable “Factor” in Haemostasis and Wound Healing. Int. J. Mol. Sci. 2021, 22, 3055. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Bereczky, Z.; Cohan, N.; Muszbek, L. Factor XIII Deficiency. Semin. Thromb. Hemost. 2009, 35, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Muszbek, L.; Katona, E. Diagnosis and Management of Congenital and Acquired FXIII Deficiencies. Semin. Thromb. Hemost. 2016, 42, 429–439. [Google Scholar] [PubMed]
- Duckert, F. The fibrin stabilizing factor, factor XIII. Blut 1973, 26, 177–179. [Google Scholar] [CrossRef] [PubMed]
- Inbal, A.; Lubetsky, A.; Krapp, T.; Castel, D.; Shaish, A.; Dickneitte, G.; Modis, L.; Muszbek, L.; Inbal, A. Impaired wound healing in factor XIII deficient mice. Thromb. Haemost. 2005, 94, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Dardik, R.; Solomon, A.; Loscalzo, J.; Eskaraev, R.; Bialik, A.; Goldberg, I.; Schiby, G.; Inbal, A. Novel Proangiogenic Effect of Factor XIII Associated With Suppression of Thrombospondin 1 Expression. Arter. Thromb. Vasc. Biol. 2003, 23, 1472–1477. [Google Scholar] [CrossRef] [Green Version]
- Dardik, R.; Loscalzo, J.; Eskaraev, R.; Inbal, A. Molecular Mechanisms Underlying the Proangiogenic Effect of Factor XIII. Arter. Thromb. Vasc. Biol. 2005, 25, 526–532. [Google Scholar] [CrossRef] [Green Version]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Melnichenko, A.A.; Myasoedova, V.A.; Grechko, A.; Orekhov, A.N. Thrombospondins: A Role in Cardiovascular Disease. Int. J. Mol. Sci. 2017, 18, 1540. [Google Scholar] [CrossRef] [Green Version]
- Streit, M.; Riccardi, L.; Velasco, P.; Brown, L.F.; Hawighorst, T.; Bornstein, P.; Detmar, M. Thrombospondin-2: A potent endogenous inhibitor of tumor growth and angiogenesis. Proc. Natl. Acad. Sci. USA 1999, 96, 14888–14893. [Google Scholar] [CrossRef] [Green Version]
- Lawler, J. Thrombospondin-1 as an endogenous inhibitor of angiogenesis and tumor growth. J. Cell. Mol. Med. 2002, 6, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.W.; Slayter, H.S.; Miller, B.E.; McDonagh, J. Characterization of thrombospondin as a substrate for factor XIII transglutaminase. J. Biol. Chem. 1987, 262, 1772–1778. [Google Scholar] [CrossRef]
- Dardik, R.; Krapp, T.; Rosenthal, E.; Loscalzo, J.; Inbal, A. Effect of FXIII on Monocyte and Fibroblast Function. Cell. Physiol. Biochem. 2007, 19, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Basatemur, G.L.; Jørgensen, H.F.; Clarke, M.; Bennett, M.; Mallat, Z. Vascular smooth muscle cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 727–744. [Google Scholar] [CrossRef]
- Miano, J.M.; Fisher, E.A.; Majesky, M.W. Fate and State of Vascular Smooth Muscle Cells in Atherosclerosis. Circulation 2021, 143, 2110–2116. [Google Scholar] [CrossRef]
- Durham, A.L.; Speer, M.Y.; Scatena, M.; Giachelli, C.M.; Shanahan, C.M. Role of smooth muscle cells in vascular calcification: Implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018, 114, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef]
- Nakano, Y.; Al-Jallad, H.F.; Mousa, A.; Kaartinen, M.T. Expression and Localization of Plasma Transglutaminase Factor XIIIA in Bone. J. Histochem. Cytochem. 2007, 55, 675–685. [Google Scholar] [CrossRef]
- Wirka, R.C.; Wagh, D.; Paik, D.T.; Pjanic, M.; Nguyen, T.; Miller, C.L.; Kundu, R.; Nagao, M.; Coller, J.; Koyano, T.K.; et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat. Med. 2019, 25, 1280–1289. [Google Scholar] [CrossRef]
- Coppinger, J.A.; Cagney, G.; Toomey, S.; Kislinger, T.; Belton, O.; McRedmond, J.P.; Cahill, D.J.; Emili, A.; Fitzgerald, D.J.; Maguire, P.B. Characterization of the proteins released from activated platelets leads to localization of novel platelet proteins in human atherosclerotic lesions. Blood 2004, 103, 2096–2104. [Google Scholar] [CrossRef] [Green Version]
- Kapusta, P.; Wypasek, E.; Natorska, J.; Grudzien, G.; Sobczyk, D.; Sadowski, J.; Undas, A. Factor XIII expression within aortic valves and its plasma activity in patients with aortic stenosis: Association with severity of disease. Thromb. Haemost. 2012, 108, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.L.; Lionikiene, A.S.; Fraser, S.R.; Whyte, C.S.; Booth, N.A.; Mutch, N.J. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood 2014, 124, 3982–3990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somodi, L.; Debreceni, I.B.; Kis, G.; Cozzolino, M.; Kappelmayer, J.; Antal, M.; Panyi, G.; Bárdos, H.; Mutch, N.J.; Muszbek, L. Activation mechanism dependent surface exposure of cellular factor XIII on activated platelets and platelet microparticles. J. Thromb. Haemost. 2022, 20, 1223–1235. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, F.S.M.; Whyte, C.S.; Tuncay, A.; Williams, M.L.; Wilson, H.M.; Mutch, N.J. Monocytes Expose Factor XIII-A and Stabilize Thrombi against Fibrinolytic Degradation. Int. J. Mol. Sci 2021, 22, 6591. [Google Scholar] [CrossRef] [PubMed]
- Jaberi, N.; Soleimani, A.; Pashirzad, M.; Abdeahad, H.; Mohammadi, F.; Khoshakhlagh, M.; Khazaei, M.; Ferns, G.A.; Avan, A.; Hassanian, S.M. Role of thrombin in the pathogenesis of atherosclerosis. J. Cell. Biochem. 2019, 120, 4757–4765. [Google Scholar] [CrossRef]
- Stouffer, G.A.; Smyth, S.S. Effects of thrombin on interactions between beta3-integrins and extracellular matrix in platelets and vascular cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1971–1978. [Google Scholar] [CrossRef] [Green Version]
- Lacolley, P.; Regnault, V.; Segers, P.; Laurent, S. Vascular Smooth Muscle Cells and Arterial Stiffening: Relevance in Development, Aging, and Disease. Physiol. Rev. 2017, 97, 1555–1617. [Google Scholar] [CrossRef]
- Dardik, R.; Leor, J.; Skutelsky, E.; Castel, D.; Holbova, R.; Schiby, G.; Shaish, A.; Dickneite, G.; Loscalzo, J.; Inbal, A. Evaluation of the pro-angiogenic effect of factor XIII in heterotopic mouse heart allografts and FXIII-deficient mice. Thromb. Haemost. 2006, 95, 546–550. [Google Scholar] [CrossRef]
- Dardik, R.; Loscalzo, J.; Inbal, A. Factor XIII (FXIII) and angiogenesis. J. Thromb. Haemost. 2006, 4, 19–25. [Google Scholar] [CrossRef]
- Katona, E.E.; Ajzner, E.; Toth, K.; Karpati, L.; Muszbek, L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates. J. Immunol. Methods 2001, 258, 127–135. [Google Scholar] [CrossRef]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Zavaczki, E.; Balla, G.; Balla, J. Ferritin ferroxidase activity: A potent inhibitor of osteogenesis. Off. J. Am. Soc. Bone Miner. Res. 2010, 25, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Oros, M.; Zavaczki, E.; Vadasz, C.; Jeney, V.; Tosaki, A.; Lekli, I.; Balla, G.; Nagy, L.; Balla, J. Ethanol increases phosphate-mediated mineralization and osteoblastic transformation of vascular smooth muscle cells. J. Cell. Mol. Med. 2012, 16, 2219–2226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becs, G.; Zarjou, A.; Agarwal, A.; Kovács, K.; Becs, A.; Nyitrai, M.; Balogh, E.; Bányai, E.; Eaton, J.W.; Arosio, P.; et al. Pharmacological induction of ferritin prevents osteoblastic transformation of smooth muscle cells. J. Cell. Mol. Med. 2016, 20, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bogáti, R.; Katona, É.; Shemirani, A.H.; Balogh, E.; Bárdos, H.; Jeney, V.; Muszbek, L. The Effect of Activated FXIII, a Transglutaminase, on Vascular Smooth Muscle Cells. Int. J. Mol. Sci. 2022, 23, 5845. https://doi.org/10.3390/ijms23105845
Bogáti R, Katona É, Shemirani AH, Balogh E, Bárdos H, Jeney V, Muszbek L. The Effect of Activated FXIII, a Transglutaminase, on Vascular Smooth Muscle Cells. International Journal of Molecular Sciences. 2022; 23(10):5845. https://doi.org/10.3390/ijms23105845
Chicago/Turabian StyleBogáti, Réka, Éva Katona, Amir H. Shemirani, Enikő Balogh, Helga Bárdos, Viktória Jeney, and László Muszbek. 2022. "The Effect of Activated FXIII, a Transglutaminase, on Vascular Smooth Muscle Cells" International Journal of Molecular Sciences 23, no. 10: 5845. https://doi.org/10.3390/ijms23105845