
Multiomic Profiling Identified EGF Receptor Signaling as a Potential Inhibitor of Type I Interferon Response in Models of Oncolytic Therapy by Vesicular Stomatitis Virus

 , , , , ,
, , , , ,  ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
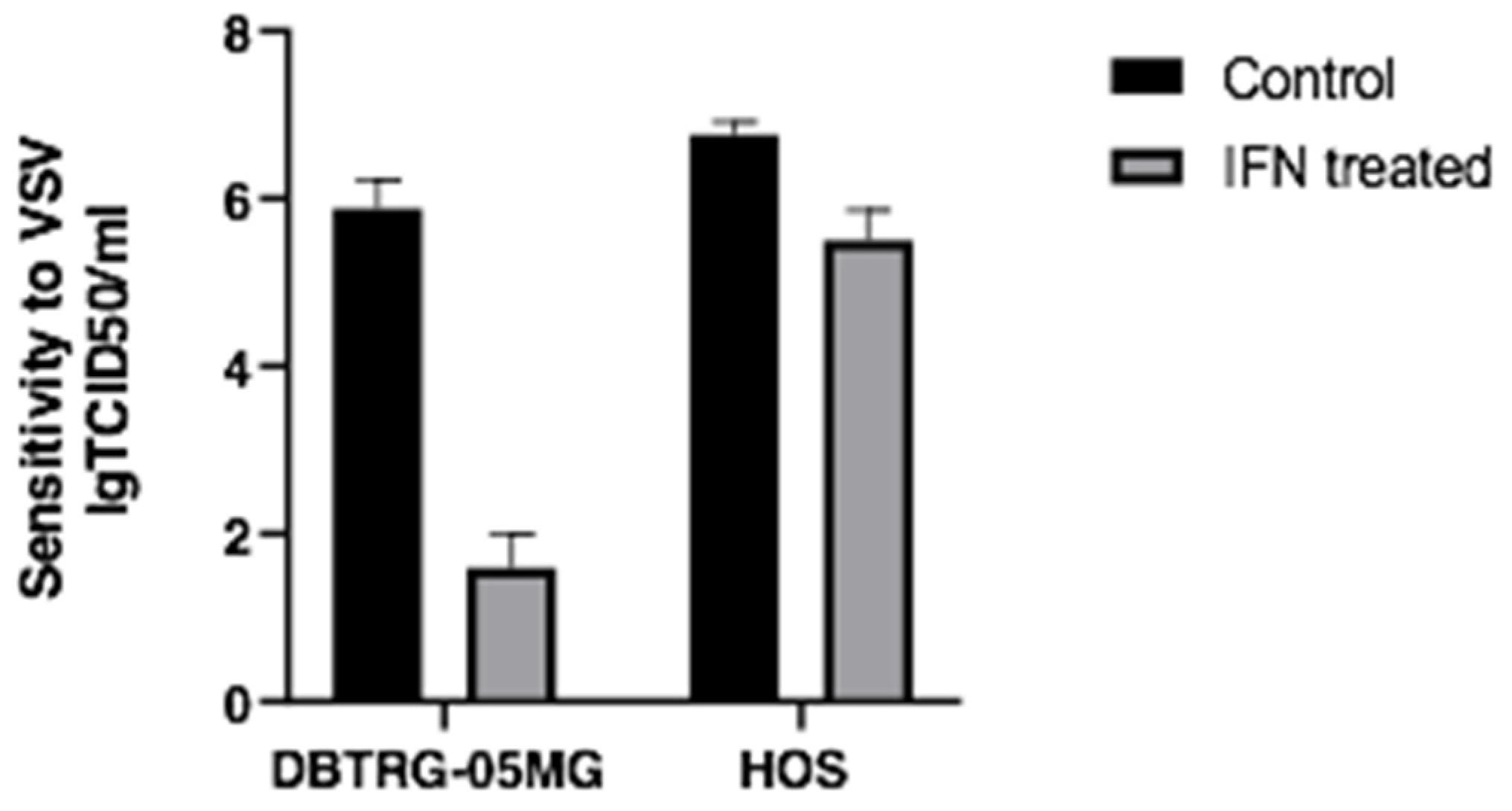
2.1. Type I Interferon Treatment Protects the DBTRG-05MG Cell Line But Does Not Prevent the HOS Cell Line from VSV Infection
2.2. Exome Analysis of DBTRG-05MG and HOS Cell Lines Confirmed Their Authenticity
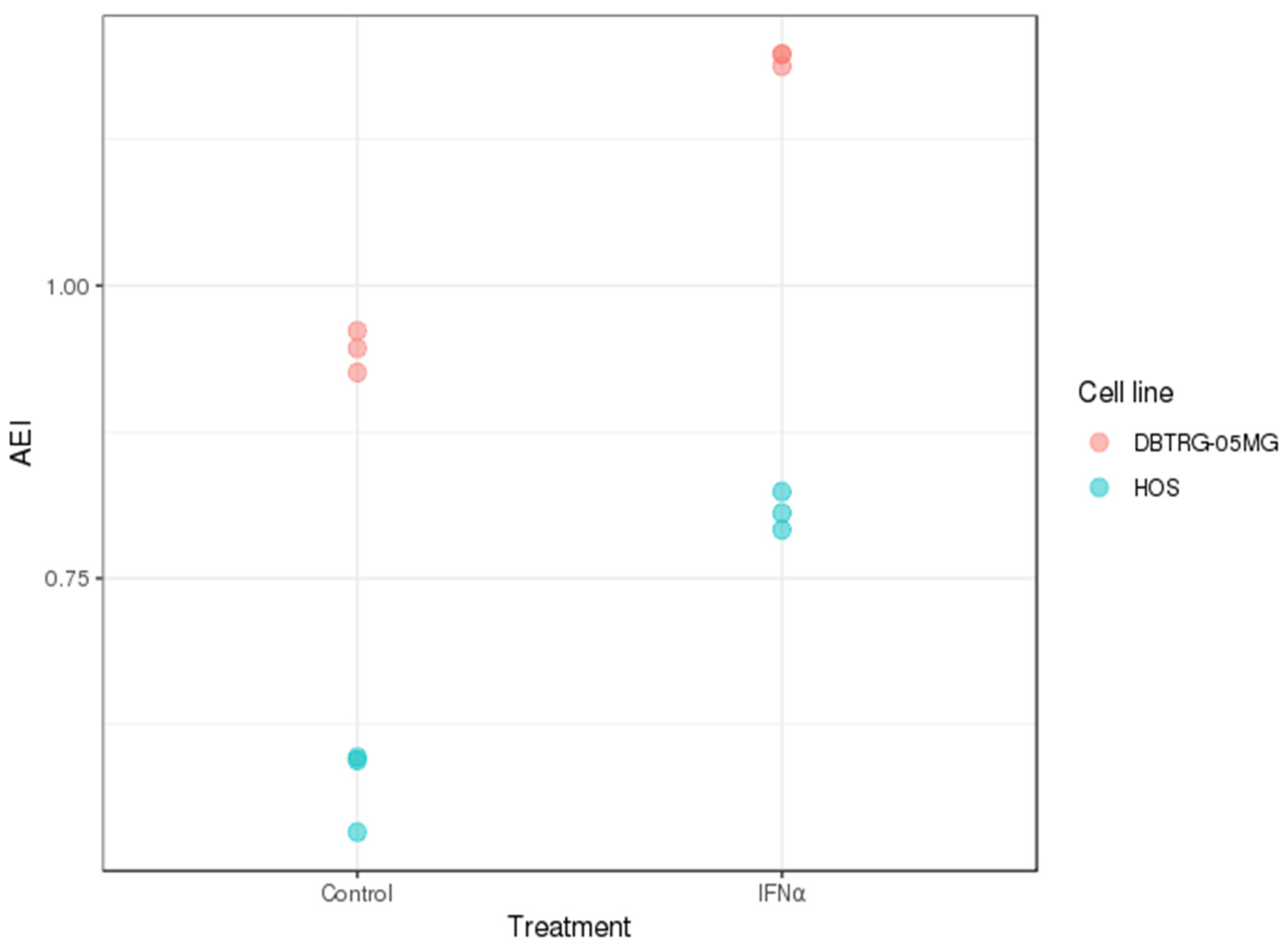
2.3. A-to-I RNA Editing in DBTRG-05MG and HOS Cell Lines before and after Type I Interferon Treatment
2.4. Combining Transcriptomic and Proteomic Data for Type I Interferon Response of DBTRG-05MG and HOS Cells Helps to Define Differentially Expressed Genes
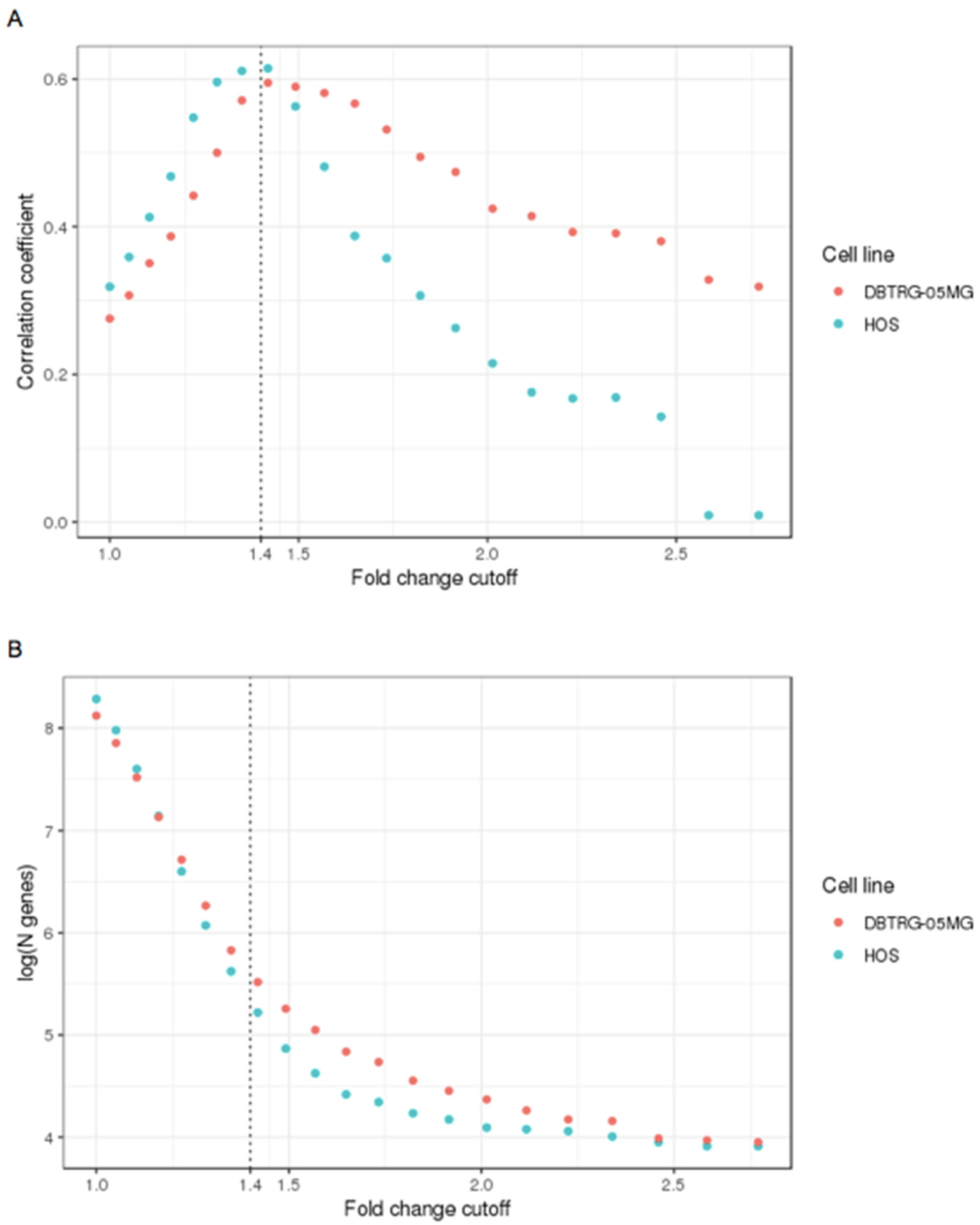
2.5. Differential Transcriptomics of Type I Interferon Responses in the VSV Protected DBTRG-05MG and VSV Sensitive HOS Cell Lines
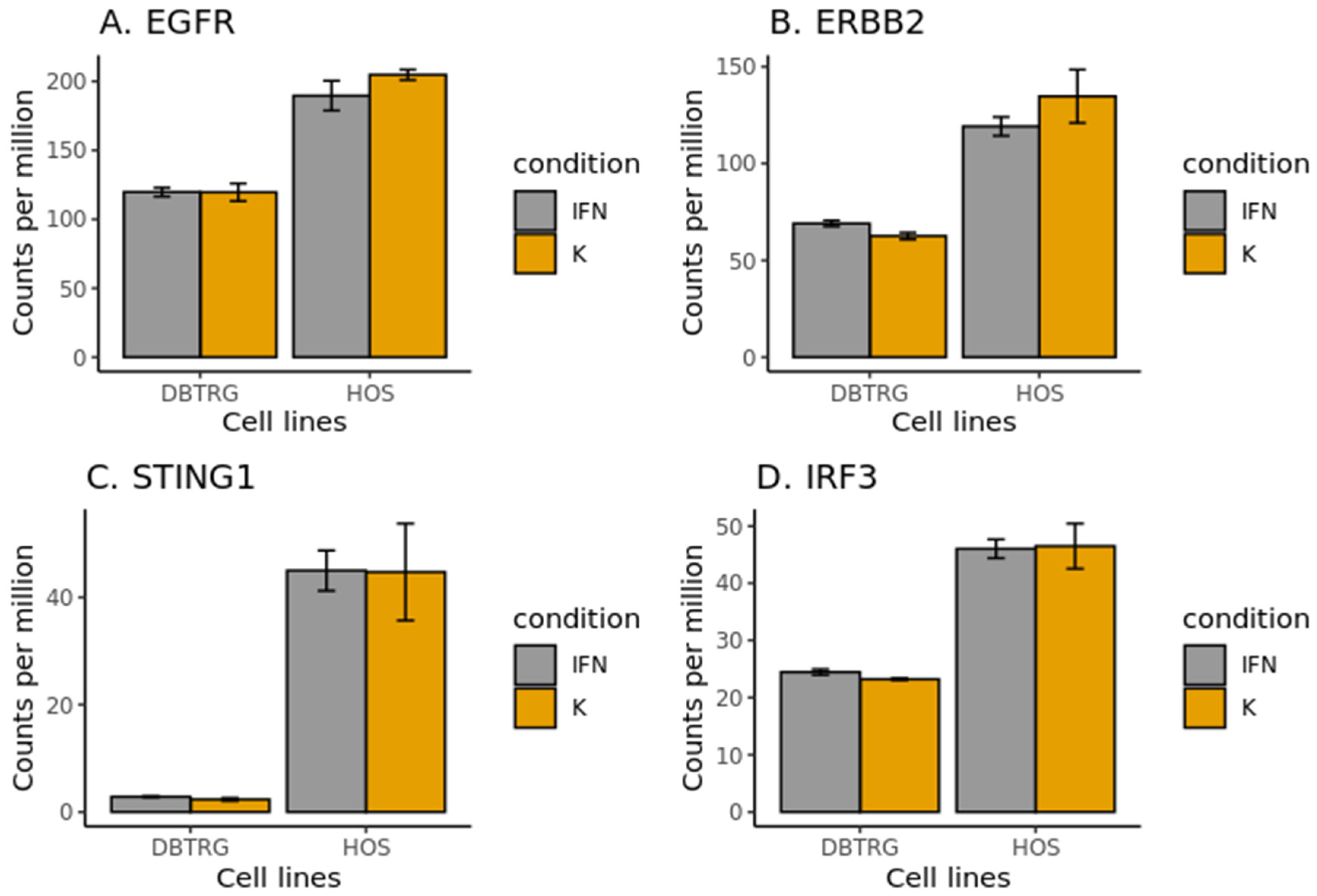
2.6. EGF Receptors in DBTRG-05MG and HOS Cell Lines

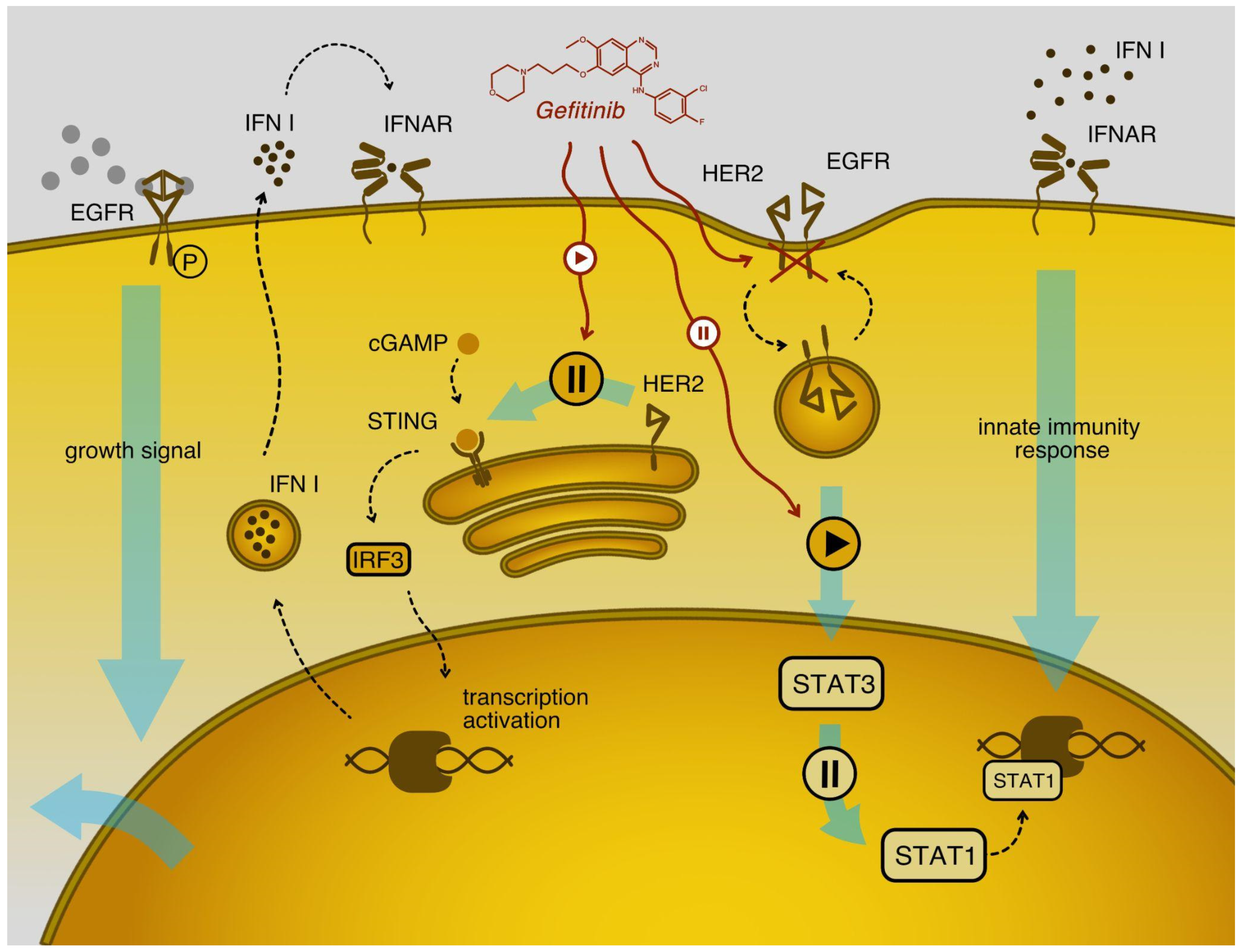
2.7. Gefitinib Stimulates Type I Interferon Signaling in Cancer Cell Models
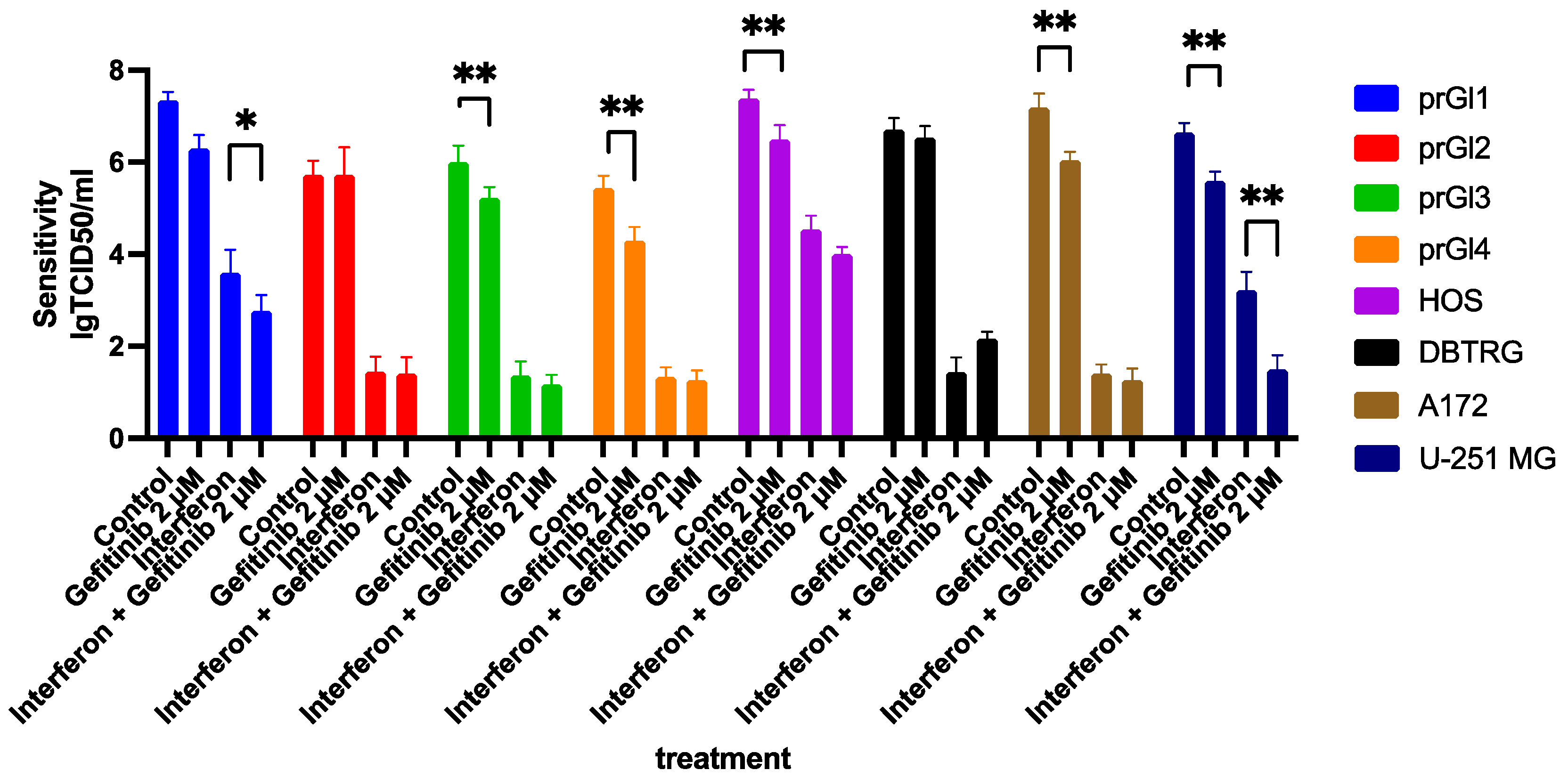
2.8. Gefitinib Influences the Type I Interferon Response in VSV Sensitive Cell Lines, Enhancing Their Protection from the Virus
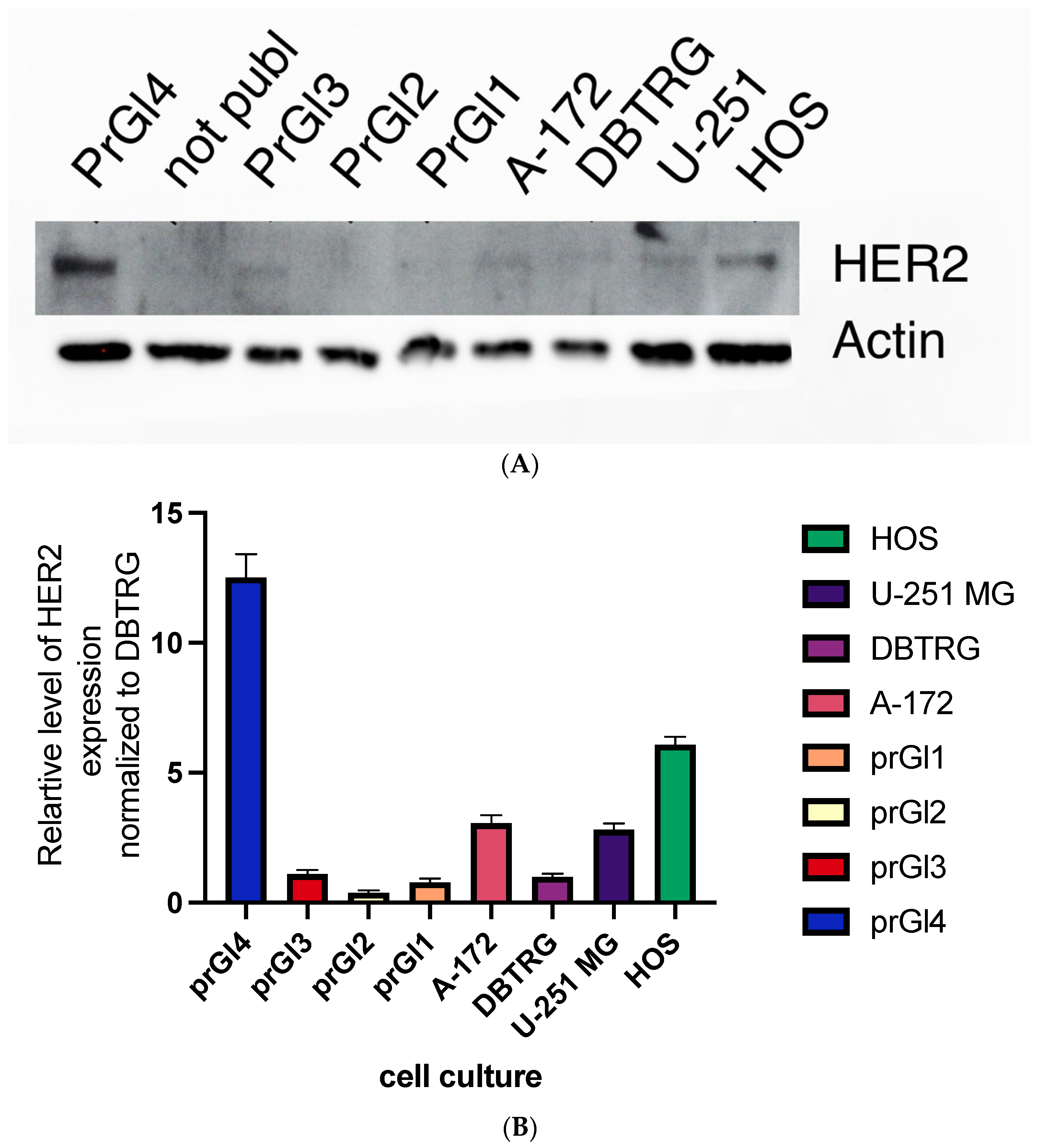
2.9. Levels of HER2 Protein Are Higher in Cell Lines with Attenuated Type Interferon Response to VSV Infection
2.10. EGF Treatment of Cell Lines Confirmed That the Effect of Gefitinib Is Provided by EGF Signaling
3. Materials and Methods
3.1. Cells and Viruses
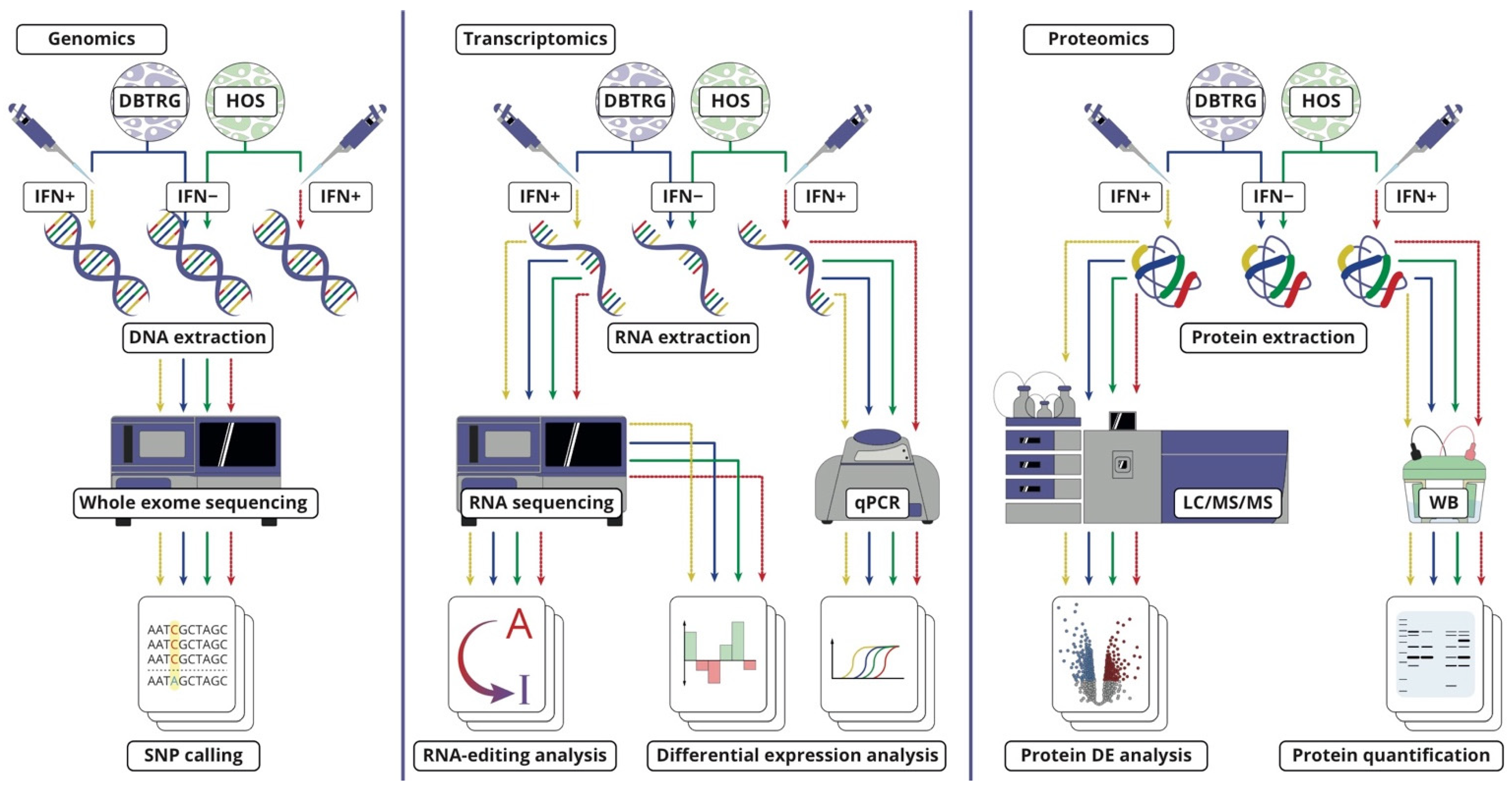
3.2. Cell Cultures for Multi-Omics Analysis
3.3. Interferon and Gefitinib Treatment and Infection
3.4. Western Blotting
3.5. Nucleic Acid Isolation
3.6. Real-Time PCR for ADARs and Functionally Related Genes
3.7. Library Construction and High-Throughput DNA and RNA Sequencing
3.8. Alu Editing Index Quantification
3.9. Differential Expression Analysis
3.10. Single Nucleotide Variation Calling
3.11. Proteomics Data Collecting
3.12. Proteomic Data Processing
3.13. Data Availability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spiesschaert, B.; Angerer, K.; Park, J.; Wollmann, G. Combining Oncolytic Viruses, and Small Molecule Therapeutics: Mutual Benefits. Cancers 2021, 13, 3386. [Google Scholar] [CrossRef] [PubMed]
- Leung, E.Y.L.; McNeish, I.A. Strategies to Optimise Oncolytic Viral Therapies: The Role of Natural Killer Cells. Viruses 2021, 13, 1450. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Song, X.; Wang, Y.; Liu, F.; Wei, J. Combining Oncolytic Viruses with Cancer Immunotherapy: Establishing a New Generation of Cancer Treatment. Front. Immunol. 2020, 11, 683. [Google Scholar] [CrossRef] [PubMed]
- Lan, Q.; Xia, S.; Wang, Q.; Xu, W.; Huang, H.; Jiang, S.; Lu, L. Development of oncolytic virotherapy: From genetic modification to combination therapy. Front. Med. 2020, 14, 160–184. [Google Scholar] [CrossRef] [Green Version]
- Crupi, M.J.F.; Bell, J.C.; Singaravelu, R. Concise Review: Targeting Cancer Stem Cells and Their Supporting Niche Using Oncolytic Viruses. Stem Cells 2019, 37, 716–723. [Google Scholar] [CrossRef] [Green Version]
- Ferrucci, P.F.; Pala, L.; Conforti, F.; Cocorocchio, E. Talimogene Laherparepvec (T-VEC): An Intralesional Cancer Immunotherapy for Advanced Melanoma. Cancers 2021, 13, 1383. [Google Scholar] [CrossRef]
- Hastie, E.; Grdzelishvili, V.Z. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 2012, 93, 2529–2545. [Google Scholar] [CrossRef]
- Roy, D.G.; Geoffroy, K.; Marguerie, M.; Khan, S.T.; Martin, N.T.; Kmiecik, J.; Bobbala, D.; Aitken, A.S.; de Souza, C.T.; Stephenson, K.B.; et al. Adjuvant oncolytic virotherapy for personalized anti-cancer vaccination. Nat. Commun. 2021, 12, 2626. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.; Knowles, S.; Marius, R.; Atkins, H.; Sonenberg, N.; Bell, J.C. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat. Med. 2000, 6, 821–825. [Google Scholar] [CrossRef]
- Tarasova, I.A.; Tereshkova, A.V.; Lobas, A.A.; Solovyeva, E.M.; Sidorenko, A.S.; Gorshkov, V.; Kjeldsen, F.; Bubis, J.A.; Ivanov, M.V.; Ilina, I.Y.; et al. Comparative proteomics as a tool for identifying specific alterations within interferon response pathways in human glioblastoma multiforme cells. Oncotarget 2018, 9, 1785–1802. [Google Scholar] [CrossRef] [Green Version]
- Grünvogel, O.; Esser-Nobis, K.; Windisch, M.P.; Frese, M.; Trippler, M.; Bartenschlager, R.; Lohmann, V.; Binder, M. Type I and type II interferon responses in two human liver cell lines (Huh-7 and HuH6). Genom. Data 2016, 7, 166–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Péré, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020, 369, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Kruse, C.A.; Mitchell, D.H.; Kleinschmidt-DeMasters, B.K.; Franklin, W.A.; Morse, H.G.; Spector, E.B.; Lillehei, K.O. Characterization of a continuous human glioma cell line DBTRG-05MG: Growth kinetics, karyotype, receptor expression, and tumor suppressor gene analyses. In Vitro Cell. Dev. Biol. 1992, 28A, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, T.; Futaesaku, Y.; Tsuchida, N. In vitro differentiation of the human osteosarcoma cell lines, HOS and KHOS. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol. 1992, 62, 199–206. [Google Scholar] [CrossRef]
- Mannion, N.M.; Greenwood, S.M.; Young, R.; Cox, S.; Brindle, J.; Read, D.; Nellåker, C.; Vesely, C.; Ponting, C.P.; McLaughlin, P.J.; et al. The RNA-Editing Enzyme ADAR1 Controls Innate Immune Responses to RNA. Cell Rep. 2014, 9, 1482–1494. [Google Scholar] [CrossRef] [Green Version]
- Robin, T.; Capes-Davis, A.; Bairoch, A. CLASTR: The Cellosaurus STR similarity search tool—A precious help for cell line authentication. Int. J. Cancer 2020, 146, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Dankner, M.; Rose, A.A.N.; Rajkumar, S.; Siegel, P.M.; Watson, I.R. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene 2018, 37, 3183–3199. [Google Scholar] [CrossRef]
- Goncharov, A.O.; Kliuchnikova, A.A.; Nasaev, S.S.; Moshkovskii, S.A. RNA Editing by ADAR Adenosine Deaminases: From Molecular Plasticity of Neural Proteins to the Mechanisms of Human Cancer. Biochemistry 2019, 84, 896–904. [Google Scholar] [CrossRef]
- Rice, G.I.; Kasher, P.R.; Forte, G.M.A.; Mannion, N.M.; Greenwood, S.M.; Szynkiewicz, M.; Dickerson, J.E.; Bhaskar, S.S.; Zampini, M.; Briggs, T.A.; et al. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet. 2012, 44, 1243–1248. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K.; et al. Dynamic landscape and regulation of RNA editing in mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, R.; Zhang, F.; Srinivasan, H.; Charles Richard, J.L.; Liu, K.I.; Zhang, X.; Woo, C.W.A.; Chua, Z.H.M.; Buschdorf, J.P.; Meaney, M.J.; et al. SRSF9 selectively represses ADAR2-mediated editing of brain-specific sites in primates. Nucleic Acids Res. 2018, 46, 7379–7395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, S.H.; Levanon, E.Y.; Eisenberg, E. Genome-wide quantification of ADAR adenosine-to-inosine RNA editing activity. Nat. Methods 2019, 16, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beyer, A.; Aebersold, R. On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 2016, 165, 535–550. [Google Scholar] [CrossRef] [Green Version]
- Bathke, J.; Konzer, A.; Remes, B.; McIntosh, M.; Klug, G. Comparative analyses of the variation of the transcriptome and proteome of Rhodobacter sphaeroides throughout growth. BMC Genom. 2019, 20, 358. [Google Scholar] [CrossRef] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [Green Version]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Lupberger, J.; Duong, F.H.T.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef]
- Wu, S.; Zhang, Q.; Zhang, F.; Meng, F.; Liu, S.; Zhou, R.; Wu, Q.; Li, X.; Shen, L.; Huang, J.; et al. HER2 recruits AKT1 to disrupt STING signalling and suppress antiviral defence and antitumour immunity. Nat. Cell Biol. 2019, 21, 1027–1040. [Google Scholar] [CrossRef] [PubMed]
- Gong, K.; Guo, G.; Panchani, N.; Bender, M.E.; Gerber, D.E.; Minna, J.D.; Fattah, F.; Gao, B.; Peyton, M.; Kernstine, K.; et al. EGFR inhibition triggers an adaptive response by co-opting antiviral signaling pathways in lung cancer. Nat. Cancer 2020, 1, 394–409. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Flavell, R.A. HER2 joins AKT to inhibit STING immunity. Nat. Cell Biol. 2019, 21, 917–918. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, R.; Barbie, D.A. EGFR blockade activates interferon. Nat. Cancer 2020, 1, 376–378. [Google Scholar] [CrossRef] [PubMed]
- Athar, A.; Füllgrabe, A.; George, N.; Iqbal, H.; Huerta, L.; Ali, A.; Snow, C.; Fonseca, N.A.; Petryszak, R.; Papatheodorou, I.; et al. ArrayExpress update—From bulk to single-cell expression data. Nucleic Acids Res. 2019, 47, D711–D715. [Google Scholar] [CrossRef] [PubMed]
- Branca, R.M.M.; Orre, L.M.; Johansson, H.J.; Granholm, V.; Huss, M.; Pérez-Bercoff, Å.; Forshed, J.; Käll, L.; Lehtiö, J. HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 2014, 11, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Barrett, T.; Wilhite, S.E.; Ledoux, P.; Evangelista, C.; Kim, I.F.; Tomashevsky, M.; Marshall, K.A.; Phillippy, K.H.; Sherman, P.M.; Holko, M.; et al. NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Res. 2012, 41, D991–D995. [Google Scholar] [CrossRef] [Green Version]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D481–D487. [Google Scholar] [CrossRef]
- Gene Ontology Consortium. The Gene Ontology resource: Enriching a GOld mine. Nucleic Acids Res. 2021, 49, D325–D334. [Google Scholar] [CrossRef]
- Pontén, J.; Macintyre, E.H. Long term culture of normal and neoplastic human glia. Acta Pathol. Microbiol. Scand. 1968, 74, 465–486. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Broad Institute. Picard. Available online: http://broadinstitute.github.io/picard/ (accessed on 1 October 2021).
- Levitsky, L.I.; Ivanov, M.V.; Lobas, A.A.; Bubis, J.A.; Tarasova, I.A.; Solovyeva, E.M.; Pridatchenko, M.L.; Gorshkov, M.V. IdentiPy: An Extensible Search Engine for Protein Identification in Shotgun Proteomics. J. Proteome Res. 2018, 17, 2249–2255. [Google Scholar] [CrossRef]
- Teleman, J.; Chawade, A.; Sandin, M.; Levander, F.; Malmström, J. Dinosaur: A Refined Open-Source Peptide MS Feature Detector. J. Proteome Res. 2016, 15, 2143–2151. [Google Scholar] [CrossRef]
- Zhang, B.; Pirmoradian, M.; Zubarev, R.; Käll, L. Covariation of Peptide Abundances Accurately Reflects Protein Concentration Differences. Mol. Cell. Proteom. 2017, 16, 936–948. [Google Scholar] [CrossRef] [Green Version]
- Vizcaíno, J.A.; Deutsch, E.W.; Wang, R.; Csordas, A.; Reisinger, F.; Ríos, D.; Dianes, J.A.; Sun, Z.; Farrah, T.; Bandeira, N.; et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 2014, 32, 223–226. [Google Scholar] [CrossRef]
- Li, Y.M.; Pan, Y.; Wei, Y.; Cheng, X.; Zhou, B.P.; Tan, M.; Zhou, X.; Xia, W.; Hortobagyi, G.N.; Yu, D.; et al. Upregulation of CXCR4 is essential for HER2-mediated tumor metastasis. Cancer Cell 2004, 6, 459–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, M.; Firestein, B.L. DNA transfection: Calcium phosphate method. Methods Mol. Biol. 2013, 1018, 107–108. [Google Scholar] [CrossRef] [PubMed]
- Fukumoto, Y.; Obata, Y.; Ishibashi, K.; Tamura, N.; Kikuchi, I.; Aoyama, K.; Hattori, Y.; Tsuda, K.; Nakayama, Y.; Yamaguchi, N. Cost-effective gene transfection by DNA compaction at pH 4.0 using acidified, long shelf-life polyethylenimine. Cytotechnology 2010, 62, 73–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hallmark Pathway | DBTRG-05MG Cell Line, Enrichment p-Value for All Differentially Expressed Genes, n = 963 * | DBTRG-05MG Cell Line, Enrichment p-Value for Genes Differentially Explicitly Expressed in This Cell Line, n = 727 | HOS Cell Line, Enrichment p-Value for All Differentially Expressed Genes, n = 442 |
|---|---|---|---|
| Interferon-alpha response | 10−84 | Not enriched specifically | 10−99 |
| Interferon-gamma response | 10−75 | 0.008 | 10−87 |
| Inflammatory response | 10−11 | 10−4 | 10−5 |
| Allograft rejection | 10−8 | 0.01 | 10−6 |
| IL6-Jak-Stat3 signaling | 10−6 | 0.007 | 10−4 |
| Complement | 10−5 | Not enriched specifically | 10−6 |
| TNFα signaling via NFkB | 10−4 | 0.01 | Not enriched |
| Apoptosis | 0.003 | Not enriched specifically | 10−4 |
| Estrogen response, early | 0.003 | 0.007 | Not enriched |
| Estrogen response, late | 0.01 | 0.007 | Not enriched |
| Gene Names | Functional Group |
|---|---|
| CXCL6, CXCL10 *, CXCL11 *, TNFSF10, TNFSF15, TNFAIP6, EREG, SPHK1 | Secreted regulators of inflammatory response |
| APLNR, IL1R1 *, TNFRSF9,TNFRSF1B * | Hormone and cytokine receptors |
| ICAM1, ITGA5, SELL | Immune cell adhesion molecules |
| NLRP3, CYBB, TAPBP | Pathogen response |
| BTG2, RNF144B | Antiproliferative and/or proapoptotic |
| RTP4 | Chaperone for G-protein coupled receptors |
| Human Cell Lines Treated by Gefitinib | Interferon Alpha/Beta Signaling, Reactome ID R-HSA-909733; an Enrichment Rank/p-Value Adjusted | Type I Interferon Signaling Pathway, GO: 0060337; an Enrichment Rank/p-Value |
|---|---|---|
| A-431 epidermoid carcinoma, 24 h treatment | 1/5 × 104 | 8/0.005 |
| PC-9 lung adenocarcinoma, 2 w treatment | 1/10−8 | 12/0.0002 |
| MGH119 lung non-small cell carcinoma, 2 w treatment | 1/0.00003 | 2/0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikitina, A.S.; Lipatova, A.V.; Goncharov, A.O.; Kliuchnikova, A.A.; Pyatnitskiy, M.A.; Kuznetsova, K.G.; Hamad, A.; Vorobyev, P.O.; Alekseeva, O.N.; Mahmoud, M.; et al. Multiomic Profiling Identified EGF Receptor Signaling as a Potential Inhibitor of Type I Interferon Response in Models of Oncolytic Therapy by Vesicular Stomatitis Virus. Int. J. Mol. Sci. 2022, 23, 5244. https://doi.org/10.3390/ijms23095244
Nikitina AS, Lipatova AV, Goncharov AO, Kliuchnikova AA, Pyatnitskiy MA, Kuznetsova KG, Hamad A, Vorobyev PO, Alekseeva ON, Mahmoud M, et al. Multiomic Profiling Identified EGF Receptor Signaling as a Potential Inhibitor of Type I Interferon Response in Models of Oncolytic Therapy by Vesicular Stomatitis Virus. International Journal of Molecular Sciences. 2022; 23(9):5244. https://doi.org/10.3390/ijms23095244
Chicago/Turabian StyleNikitina, Anastasia S., Anastasia V. Lipatova, Anton O. Goncharov, Anna A. Kliuchnikova, Mikhail A. Pyatnitskiy, Ksenia G. Kuznetsova, Azzam Hamad, Pavel O. Vorobyev, Olga N. Alekseeva, Marah Mahmoud, and et al. 2022. "Multiomic Profiling Identified EGF Receptor Signaling as a Potential Inhibitor of Type I Interferon Response in Models of Oncolytic Therapy by Vesicular Stomatitis Virus" International Journal of Molecular Sciences 23, no. 9: 5244. https://doi.org/10.3390/ijms23095244