
Neurochemical Alterations in Social Anxiety Disorder (SAD): A Systematic Review of Proton Magnetic Resonance Spectroscopic Studies

 , and
, and
Abstract
:1. Introduction
2. Methods
2.1. Search Strategy
2.2. Search Selection
2.3. Data Extraction
3. Results and Discussion
3.1. Results
3.2. Discussion
3.2.1. What Are the Metabolite Differences Associated with SAD?
3.2.2. What Molecular Mechanisms Were Implicated for SAD?
3.2.3. Which Other SAD Molecular Mechanisms Should Be Investigated in Human 1H MRS Studies?
3.2.4. What Does the Evidence from 1H MRS Studies Imply about the ‘Fear Neurocircuitry’?
3.2.5. What Are the Study Limitations Discussed in this Review?
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kessler, R.C.; Berglund, P.; Demler, O.; Jin, R.; Merikangas, K.R.; Walters, E.E. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch. Gen. Psychiatry 2005, 62, 593–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, D.J.; Lim, C.C.W.; Roest, A.M.; de Jonge, P.; Aguilar-Gaxiola, S.; Al-Hamzawi, A.; Alonso, J.; Benjet, C.; Bromet, E.J.; Bruffaerts, R.; et al. Collaborators, WHOWMHS, The cross-national epidemiology of social anxiety disorder: Data from the World Mental Health Survey Initiative. BMC Med. 2017, 15, 143. [Google Scholar] [CrossRef] [PubMed]
- Leichsenring, F.; Leweke, F. Social Anxiety Disorder. N. Engl. J. Med. 2017, 376, 2255–2264. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, M.B.; Fowler, K.F. Social anxiety disorder in the Canadian population: Exploring gender differences in sociodemographic profile. J. Anxiety Disord. 2013, 27, 427–434. [Google Scholar] [CrossRef]
- Kim, Y.-K.; Yoon, H.-K. Common and distinct brain networks underlying panic and social anxiety disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 80, 115–122. [Google Scholar] [CrossRef]
- Wang, J.; Tian, Y.; Zeng, L.-H.; Xu, H. Prefrontal Disinhibition in Social Fear: A Vital Action of Somatostatin Interneurons. Front. Cell. Neurosci. 2020, 14, 611732. [Google Scholar] [CrossRef]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The Biology and Pathobiology of Glutamatergic, Cholinergic, and Dopaminergic Signaling in the Aging Brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef]
- Penzo, M.A.; Robert, V.; Tucciarone, J.; De Bundel, D.; Wang, M.; Van Aelst, L.; Darvas, M.; Parada, L.F.; Palmiter, R.D.; He, M.; et al. The paraventricular thalamus controls a central amygdala fear circuit. Nature 2015, 519, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Brehl, A.-K.; Kohn, N.; Fernandez, G.; Schene, A.H. A mechanistic model for individualised treatment of anxiety disorders based on predictive neural biomarkers. Psychol. Med. 2020, 50, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Pitts, M.W.; Todorovic, C.; Blank, T.; Takahashi, L.K. The central nucleus of the amygdala and corticotropin-releasing factor: Insights into contextual fear memory. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 7379–7388. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, J.-Z. From Structure to Behavior in Basolateral Amygdala-Hippocampus Circuits. Front. Neural. Circuits 2017, 11, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, S.G.; Klumpp, H.; Angstadt, M.; Nathan, P.J.; Phan, K.L. Amygdala and insula response to emotional images in patients with generalized social anxiety disorder. J. Psychiatry Neurosci. 2009, 34, 296–302. [Google Scholar] [PubMed]
- Klumpp, H.; Angstadt, M.; Phan, K.L. Insula reactivity and connectivity to anterior cingulate cortex when processing threat in generalized social anxiety disorder. Biol. Psychol. 2012, 89, 273–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uddin, L.Q.; Nomi, J.S.; Hébert-Seropian, B.; Ghaziri, J.; Boucher, O. Structure and Function of the Human Insula. J. Clin. Neurophysiol. Off. Publ. Am. Electroencephalogr. Soc. 2017, 34, 300–306. [Google Scholar] [CrossRef]
- Dorfman, J.; Benson, B.; Farber, M.; Pine, D.; Ernst, M. Altered striatal intrinsic functional connectivity in pediatric anxiety. Neuropsychologia 2016, 85, 159–168. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.P.I.; Simon, D.; Miltner, W.H.R.; Straube, T. Altered activation of the ventral striatum under performance-related observation in social anxiety disorder. Psychol. Med. 2017, 47, 2502–2512. [Google Scholar] [CrossRef]
- Bas-Hoogendam, J.M.; van Steenbergen, H.; Nienke Pannekoek, J.; Fouche, J.-P.; Lochner, C.; Hattingh, C.J.; Cremers, H.R.; Furmark, T.; Månsson, K.N.T.; Frick, A.; et al. Voxel-based morphometry multi-center mega-analysis of brain structure in social anxiety disorder. NeuroImage Clin. 2017, 16, 678–688. [Google Scholar] [CrossRef]
- de Graaf, R.A. In Vivo NMR Spectroscopy: Principles and Techniques, 2nd ed.; John Wiley & Sons Ltd.: London, UK, 2007. [Google Scholar]
- Xu, H.; Zhang, H.; Zhang, J.; Huang, Q.; Shen, Z.; Wu, R. Evaluation of neuron-glia integrity by in vivo proton magnetic resonance spectroscopy: Implications for psychiatric disorders. Neurosci. Biobehav. Rev. 2016, 71, 563–577. [Google Scholar] [CrossRef] [Green Version]
- Urenjak, J.; Williams, S.R.; Gadian, D.G.; Noble, M. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J. Neurochem. 1992, 59, 55–61. [Google Scholar] [CrossRef]
- Moffett, J.R.; Ross, B.; Arun, P.; Madhavarao, C.N.; Namboodiri, A.M. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Yan, G.; Xu, H.; Fang, Z.; Zhang, J.; Zhang, J.; Wu, R.; Kong, J.; Huang, Q. The recovery trajectory of adolescent social defeat stress-induced behavioral, (1)H-MRS metabolites and myelin changes in Balb/c mice. Sci. Rep. 2016, 6, 27906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warepam, M.; Ahmad, K.; Rahman, S.; Rahaman, H.; Kumari, K.; Singh, L.R. N-Acetylaspartate Is an Important Brain Osmolyte. Biomolecules 2020, 10, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaral, I.; Hadera, A.; Kotter, M.G.; Sonnewald, U. Oligodendrocytes Do Not Export NAA-Derived Aspartate In Vitro. Neurochem. Res. 2017, 42, 827–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordeen, K.; Heuser, C.; Rinholm, J.E.; Matalon, R.; Gundersen, V. Localisation of N-acetylaspartate in oligodendrocytes/myelin. Brain Struct. Funct. 2015, 220, 899–917. [Google Scholar] [CrossRef] [PubMed]
- Madhava Rao, C.N.; Arun, P.; Moffett, J.R.; Szucs, S.; Surendran, S.; Matalon, R.; Garber, J.; Hristova, D.; Johnson, A.; Jiang, W.; et al. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 5221. [Google Scholar] [CrossRef] [Green Version]
- Moffett, J.; Arun, P.; Ariyannur, P.; Namboodiri, A. N-Acetylaspartate reductions in brain injury: Impact on post-injury neuroenergetics, lipid synthesis, and protein acetylation. Front. Neuroenerg. 2013, 5, 11. [Google Scholar] [CrossRef] [Green Version]
- Narayana, P.A.; Johnston, D.; Flamig, D.P. In vivo proton magnetic resonance spectroscopy studies of human brain. Magn. Reason. Imaging 1991, 9, 303–308. [Google Scholar] [CrossRef]
- Mason, G.F.; Krystal, J.H. MR spectroscopy: Its potential role for drug development for the treatment of psychiatric diseases. NMR Biomed 2006, 19, 690–701. [Google Scholar] [CrossRef]
- de Kroon, A.I.P.M.; Rijken, P.J.; De Smet, C.H. Checks and balances in membrane phospholipid class and acyl chain homeostasis, the yeast perspective. Prog. Lipid Res. 2013, 52, 374–394. [Google Scholar] [CrossRef] [Green Version]
- Sestili, P.; Martinelli, C.; Bravi, G.; Piccoli, G.; Curci, R.; Battistelli, M.; Falcieri, E.; Agostini, D.; Gioacchini, A.M.; Stocchi, V. Creatine supplementation affords cytoprotection in oxidatively injured cultured mammalian cells via direct antioxidant activity. Free Radic. Biol. Med. 2006, 40, 837–849. [Google Scholar] [CrossRef]
- Sartorius, A.; Lugenbiel, P.; Mahlstedt, M.M.; Ende, G.; Schloss, P.; Vollmayr, B. Proton magnetic resonance spectroscopic creatine correlates with creatine transporter protein density in rat brain. J. Neurosci. Methods 2008, 172, 215–219. [Google Scholar] [CrossRef]
- Rackayova, V.; Cudalbu, C.; Pouwels, P.J.W.; Braissant, O. Creatine in the central nervous system: From magnetic resonance spectroscopy to creatine deficiencies. Anal. Biochem. 2017, 529, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Govindaraju, V.; Young, K.; Maudsley, A.A. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000, 13, 129–153. [Google Scholar] [CrossRef]
- Braissant, O.; Henry, H.; Loup, M.; Eilers, B.; Bachmann, C. Endogenous synthesis and transport of creatine in the rat brain: An in situ hybridization study. Brain Res. Mol. Brain Res. 2001, 86, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Braissant, O. Ammonia toxicity to the brain: Effects on creatine metabolism and transport and protective roles of creatine. Mol. Genet. Metab. 2010, 100 (Suppl. S1), S53–S58. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.; Richter-Landsberg, C.; Leibfritz, D. Multinuclear NMR studies on the energy metabolism of glial and neuronal cells. Dev. Neurosci. 1993, 15, 289–298. [Google Scholar] [CrossRef]
- Cecil, K.M. Proton magnetic resonance spectroscopy: Technique for the neuroradiologist. Neuroimaging Clin. N. Am. 2013, 23, 381–392. [Google Scholar] [CrossRef] [Green Version]
- Chhetri, D.R. Myo-Inositol and Its Derivatives: Their Emerging Role in the Treatment of Human Diseases. Front. Pharmacol. 2019, 10, 1172. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Uarquin, F.; Sommerfeld, V.; Rodehutscord, M.; Huber, K. Interrelationship of myo-inositol pathways with systemic metabolic conditions in two strains of high-performance laying hens during their productive life span. Sci. Rep. 2021, 11, 4641. [Google Scholar] [CrossRef]
- Holub, B.J. The nutritional significance, metabolism, and function of myo-inositol and phosphatidylinositol in health and disease. Adv. Nutr. Res. 1982, 4, 107–141. [Google Scholar]
- Ross, B.; Bluml, S. Magnetic resonance spectroscopy of the human brain. Anat. Rec. 2001, 265, 54–84. [Google Scholar] [CrossRef] [PubMed]
- Sonnewald, U.; Schousboe, A. Introduction to the Glutamate-Glutamine Cycle. Adv. Neurobiol. 2016, 13, 1–7. [Google Scholar] [PubMed]
- Xu, H.; Liu, L.; Tian, Y.; Wang, J.; Li, J.; Zheng, J.; Zhao, H.; He, M.; Xu, T.-L.; Duan, S.; et al. A Disinhibitory Microcircuit Mediates Conditioned Social Fear in the Prefrontal Cortex. Neuron 2019, 102, 668–682.e5. [Google Scholar] [CrossRef]
- Pham, T.H.; Gardier, A.M. Fast-acting antidepressant activity of ketamine: Highlights on brain serotonin, glutamate, and GABA neurotransmission in preclinical studies. Pharmacol. Ther. 2019, 199, 58–90. [Google Scholar] [CrossRef]
- Ramadan, S.; Lin, A.; Stanwell, P. Glutamate and glutamine: A review of in vivo MRS in the human brain. NMR Biomed. 2013, 26, 1630–1646. [Google Scholar] [CrossRef] [Green Version]
- Epperson, C.N.; Gueorguieva, R.; Czarkowski, K.A.; Stiklus, S.; Sellers, E.; Krystal, J.H.; Rothman, D.L.; Mason, G.F. Preliminary evidence of reduced occipital GABA concentrations in puerperal women: A 1H-MRS study. Psychopharmacology 2006, 186, 425–433. [Google Scholar] [CrossRef]
- Walls, A.B.; Waagepetersen, H.S.; Bak, L.K.; Schousboe, A.; Sonnewald, U. The glutamine-glutamate/GABA cycle: Function, regional differences in glutamate and GABA production and effects of interference with GABA metabolism. Neurochem. Res. 2015, 40, 402–409. [Google Scholar] [CrossRef]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.; Xu, A.; Cui, S.; Sun, M.R.; Xue, Y.C.; Wang, J.H. Impaired GABA synthesis, uptake, and release are associated with depression-like behaviors induced by chronic mild stress. Transl. Psychiatry 2016, 6, e910. [Google Scholar] [CrossRef]
- Puts, N.A.; Edden, R.A. In vivo magnetic resonance spectroscopy of GABA: A methodological review. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 60, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 statement: An updated guideline for reporting systematic reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Davidson, J.R.; Krishnan, K.R.; Charles, H.C.; Boyko, O.; Potts, N.L.; Ford, S.M.; Patterson, L. Magnetic resonance spectroscopy in social phobia: Preliminary findings. J. Clin. Psychiatry 1993, 54, 19–25. [Google Scholar]
- Tupler, L.A.; Davidson, J.R.T.; Smith, R.D.; Lazeyras, F.; Charles, H.C.; Krishnan, K.R.R. A Repeat Proton Magnetic Resonance Spectroscopy Study in Social Phobia. Biol. Psychiatry 1997, 42, 419–424. [Google Scholar] [CrossRef]
- Tukel, R.; Aydin, K.; Yuksel, C.; Ertekin, E.; Koyuncu, A. Proton Magnetic Resonance Spectroscopy in Social Anxiety Disorder. J. Neuropsychiatry Clin. Neurosci. 2016, 28, 138–142. [Google Scholar] [CrossRef] [Green Version]
- Phan, K.L.; Fitzgerald, D.A.; Cortese, B.M.; Seraji-Bozorgzad, N.; Tancer, M.E.; Moore, G.J. Anterior cingulate neurochemistry in social anxiety disorder: 1H-MRS at 4 Tesla. Neuroreport 2005, 16, 183–186. [Google Scholar] [CrossRef]
- Pollack, M.H.; Jensen, J.E.; Simon, N.M.; Kaufman, R.E.; Renshaw, P.F. High-field MRS study of GABA, glutamate, and glutamine in social anxiety disorder: Response to treatment with levetiracetam. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 739–743. [Google Scholar] [CrossRef]
- Grills, R. Brain Neurochemicals in Generalized Social Anxiety Disorder: A Proton Magnetic Resonance Spectroscopy Study. Master’s Thesis, University of Alberta, Edmonton, AB, Canada, 2011. [Google Scholar]
- Yue, Q.; Liu, M.; Nie, X.; Wu, Q.; Li, J.; Zhang, W.; Huang, X.; Gong, Q. Quantitative 3.0T MR spectroscopy reveals decreased creatine concentration in the dorsolateral prefrontal cortex of patients with social anxiety disorder. PLoS ONE 2012, 7, e48105. [Google Scholar] [CrossRef] [Green Version]
- Howells, F.M.; Hattingh, C.J.; Syal, S.; Breet, E.; Stein, D.J.; Lochner, C. (1)H-magnetic resonance spectroscopy in social anxiety disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2015, 58, 97–104. [Google Scholar] [CrossRef]
- Moonen, C.T.; von Kienlin, M.; van Zijl, P.C.; Cohen, J.; Gillen, J.; Daly, P.; Wolf, G. Comparison of single-shot localization methods (STEAM and PRESS) for in vivo proton NMR spectroscopy. NMR Biomed. 1989, 2, 201–208. [Google Scholar] [CrossRef]
- Cichocka, M.; Bereś, A. From fetus to older age: A review of brain metabolic changes across the lifespan. Ageing Res. Rev. 2018, 46, 60–73. [Google Scholar] [CrossRef]
- Kreis, R.; Boer, V.; Choi, I.-Y.; Cudalbu, C.; de Graaf, R.A.; Gasparovic, C.; Heerschap, A.; Krššák, M.; Lanz, B.; Maudsley, A.A.; et al. Terminology and concepts for the characterization of in vivo MR spectroscopy methods and MR spectra: Background and experts’ consensus recommendations. NMR Biomed. 2021, 34, e4347. [Google Scholar] [CrossRef] [PubMed]
- Alger, J.R. Quantitative proton magnetic resonance spectroscopy and spectroscopic imaging of the brain: A didactic review. Top. Magn. Reason. Imaging 2010, 21, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Near, J.; Harris, A.D.; Juchem, C.; Kreis, R.; Marjańska, M.; Öz, G.; Slotboom, J.; Wilson, M.; Gasparovic, C. Preprocessing, analysis, and quantification in single-voxel magnetic resonance spectroscopy: Experts’ consensus recommendations. NMR Biomed. 2021, 34, e4257. [Google Scholar] [CrossRef]
- Hollis, F.; van der Kooij Michael, A.; Zanoletti, O.; Lozano, L.; Cantó, C.; Sandi, C. Mitochondrial function in the brain links anxiety with social subordination. Proc. Natl. Acad. Sci. USA 2015, 112, 15486–15491. [Google Scholar] [CrossRef] [Green Version]
- Allen, P.J. Creatine metabolism, and psychiatric disorders: Does creatine supplementation have therapeutic value? Neurosci. Biobehav. Rev. 2012, 36, 1442–1462. [Google Scholar] [CrossRef] [Green Version]
- Marenco, S.; Bertolino, A.; Weinberger, D.R. In vivo NMR measures of NAA and the neurobiology of schizophrenia. Adv. Exp. Med. Biol. 2006, 576, 227–240. [Google Scholar]
- Yan, G.; Xuan, Y.; Dai, Z.; Shen, Z.; Zhang, G.; Xu, H.; Wu, R. Brain metabolite changes in subcortical regions after exposure to cuprizone for 6 weeks: Potential implications for schizophrenia. Neurochem. Res. 2015, 40, 49–58. [Google Scholar] [CrossRef]
- Amiri, S.; Amini-Khoei, H.; Haj-Mirzaian, A.; Rahimi-Balaei, M.; Naserzadeh, P.; Dehpour, A.; Mehr, S.E.; Hosseini, M.-J. Tropisetron attenuated the anxiogenic effects of social isolation by modulating nitrergic system and mitochondrial function. Biochim. Biophys. Acta (BBA)—Gen. Particip. 2015, 1850, 2464–2475. [Google Scholar] [CrossRef]
- Okwuofu, E.O.; Ogundepo, G.E.; Akhigbemen, A.M.; Abiola, A.L.; Ozolua, R.I.; Igbe, I.; Chinazamoku, O. Creatine attenuates seizure severity, anxiety and depressive-like behaviors in pentylenetetrazole kindled mice. Metab. Brain Dis. 2021, 36, 571–579. [Google Scholar] [CrossRef]
- Stork, C.; Renshaw, P.F. Mitochondrial dysfunction in bipolar disorder: Evidence from magnetic resonance spectroscopy research. Mol. Psychiatry 2005, 10, 900–919. [Google Scholar] [CrossRef] [Green Version]
- Raparia, E.; Coplan, J.D.; Abdallah, C.G.; Hof, P.R.; Mao, X.; Mathew, S.J.; Shungu, D.C. Impact of childhood emotional abuse on neocortical neurometabolites and complex emotional processing in patients with generalized anxiety disorder. J. Affect. Disord. 2016, 190, 414–423. [Google Scholar] [CrossRef] [PubMed]
- Moon, C.-M.; Jeong, G.-W. Brain morphological alterations and cellular metabolic changes in patients with generalized anxiety disorder: A combined DARTEL-based VBM and (1)H-MRS study. Magn. Reson. Imaging 2016, 34, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Kaur, G. Withania somnifera as a Potential Anxiolytic and Anti-inflammatory Candidate Against Systemic Lipopolysaccharide-Induced Neuroinflammation. Neuromol. Med. 2018, 20, 343–362. [Google Scholar] [CrossRef]
- Shaldubina, A.; Buccafusca, R.; Johanson, R.A.; Agam, G.; Belmaker, R.H.; Berry, G.T.; Bersudsky, Y. Behavioural phenotyping of sodium-myo-inositol cotransporter heterozygous knockout mice with reduced brain inositol. Genes Brain Behav. 2007, 6, 253–259. [Google Scholar] [CrossRef]
- Filipović, D.; Todorović, N.; Bernardi, R.E.; Gass, P. Oxidative and nitrosative stress pathways in the brain of socially isolated adult male rats demonstrating depressive- and anxiety-like symptoms. Brain Struct. Funct. 2017, 222, 1–20. [Google Scholar] [CrossRef]
- Fedoce, A.d.G.; Ferreira, F.; Bota, R.G.; Bonet-Costa, V.; Sun, P.Y.; Davies, K.J.A. The role of oxidative stress in anxiety disorder: Cause or consequence? Free Radic. Res. 2018, 52, 737–750. [Google Scholar] [CrossRef]
- Kim, H.; McGrath, B.M.; Silverstone, P.H. A review of the possible relevance of inositol and the phosphatidylinositol second messenger system (PI-cycle) to psychiatric disorders—Focus on magnetic resonance spectroscopy (MRS) studies. Hum. Psychopharmacol. 2005, 20, 309–326. [Google Scholar] [CrossRef]
- Nutt, D.J.; Bell, C.J.; Malizia, A.L. Brain mechanisms of social anxiety disorder. J. Clin. Psychiatry 1998, 59, 4–11. [Google Scholar]
- Montoya, A.; Bruins, R.; Katzman, M.A.; Blier, P. The noradrenergic paradox: Implications in the management of depression and anxiety. Neuropsychiatr. Dis. Treat. 2016, 12, 541–557. [Google Scholar] [CrossRef] [Green Version]
- Yizhar, O.; Fenno, L.E.; Prigge, M.; Schneider, F.; Davidson, T.J.; O’Shea, D.J.; Sohal, V.S.; Goshen, I.; Finkelstein, J.; Paz, J.T.; et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 2011, 477, 171. [Google Scholar] [CrossRef]
- Toth, I.; Neumann, I.D.; Slattery, D.A. Social fear conditioning: A novel and specific animal model to study social anxiety disorder. Neuropsychopharmacology 2012, 37, 1433–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, I.; Neumann, I.D. Animal models of social avoidance and social fear. Cell Tissue Res. 2013, 354, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Zhou, L.; Bai, Y.; Zhou, R.; Chen, L. Hypothalamic-pituitary-adrenal axis hyperactivity accounts for anxiety-and depression-like behaviors in rats perinatally exposed to bisphenol A. J. Biomed. Res. 2015, 29, 250. [Google Scholar]
- Atmaca, M.; Tezcan, E.; Kuloglu, M.; Ustundag, B.; Tunckol, H. Antioxidant enzyme and malondialdehyde values in social phobia before and after citalopram treatment. Eur. Arch. Psychiatry Clin. Neurosci. 2004, 254, 231–235. [Google Scholar] [CrossRef]
- Atmaca, M.; Kuloglu, M.; Tezcan, E.; Ustundag, B. Antioxidant enzyme and malondialdehyde levels in patients with social phobia. Psychiatry Res. 2008, 159, 95–100. [Google Scholar] [CrossRef]
- Plitman, E.; de la Fuente-Sandoval, C.; Reyes-Madrigal, F.; Chavez, S.; Gómez-Cruz, G.; León-Ortiz, P.; Graff-Guerrero, A. Elevated Myo-Inositol, Choline, and Glutamate Levels in the Associative Striatum of Antipsychotic-Naive Patients With First-Episode Psychosis: A Proton Magnetic Resonance Spectroscopy Study With Implications for Glial Dysfunction. Schizophr. Bull. 2016, 42, 415–424. [Google Scholar] [CrossRef] [Green Version]
- Colla, M.; Schubert, F.; Bubner, M.; Heidenreich, J.O.; Bajbouj, M.; Seifert, F.; Luborzewski, A.; Heuser, I.; Kronenberg, G. Glutamate as a spectroscopic marker of hippocampal structural plasticity is elevated in long-term euthymic bipolar patients on chronic lithium therapy and correlates inversely with diurnal cortisol. Mol. Psychiatry 2009, 14, 696–704. [Google Scholar] [CrossRef]
- Neu, A.; Neuhoff, H.; Trube, G.; Fehr, S.; Ullrich, K.; Roeper, J.; Isbrandt, D. Activation of GABA(A) receptors by guanidinoacetate: A novel pathophysiological mechanism. Neurobiol. Dis. 2002, 11, 298–307. [Google Scholar] [CrossRef] [Green Version]
- Koga, Y.; Takahashi, H.; Oikawa, D.; Tachibana, T.; Denbow, D.M.; Furuse, M. Brain creatine functions to attenuate acute stress responses through GABAnergic system in chicks. Neuroscience 2005, 132, 65–71. [Google Scholar] [CrossRef]
- Ryu, H.; Ferrante, R.J. Emerging chemotherapeutic strategies for Huntington’s disease. Expert Opin. Emerg. Drugs 2005, 10, 345–363. [Google Scholar] [CrossRef]
- Bertholdo, D.; Watcharakorn, A.; Castillo, M. Brain proton magnetic resonance spectroscopy: Introduction and overview. Neuroimaging Clin. N. Am. 2013, 23, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Oz, G.; Alger, J.R.; Barker, P.B.; Bartha, R.; Bizzi, A.; Boesch, C.; Bolan, P.J.; Brindle, K.M.; Cudalbu, C.; Dincer, A.; et al. Clinical proton MR spectroscopy in central nervous system disorders. Radiology 2014, 270, 658–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.P.; Dai, H.Y.; Dai, Z.Z.; Xu, C.T.; Wu, R.H. Anterior cingulate cortex and cerebellar hemisphere neurometabolite changes in depression treatment: A 1H magnetic resonance spectroscopy study. Psychiatry Clin. Neurosci. 2014, 68, 357–364. [Google Scholar] [CrossRef]
- Chiappelli, J.; Rowland, L.M.; Wijtenburg, S.A.; Muellerklein, F.; Tagamets, M.; McMahon, R.P.; Gaston, F.; Kochunov, P.; Hong, L.E. Evaluation of Myo-Inositol as a Potential Biomarker for Depression in Schizophrenia. Neuropsychopharmacology 2015, 40, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]




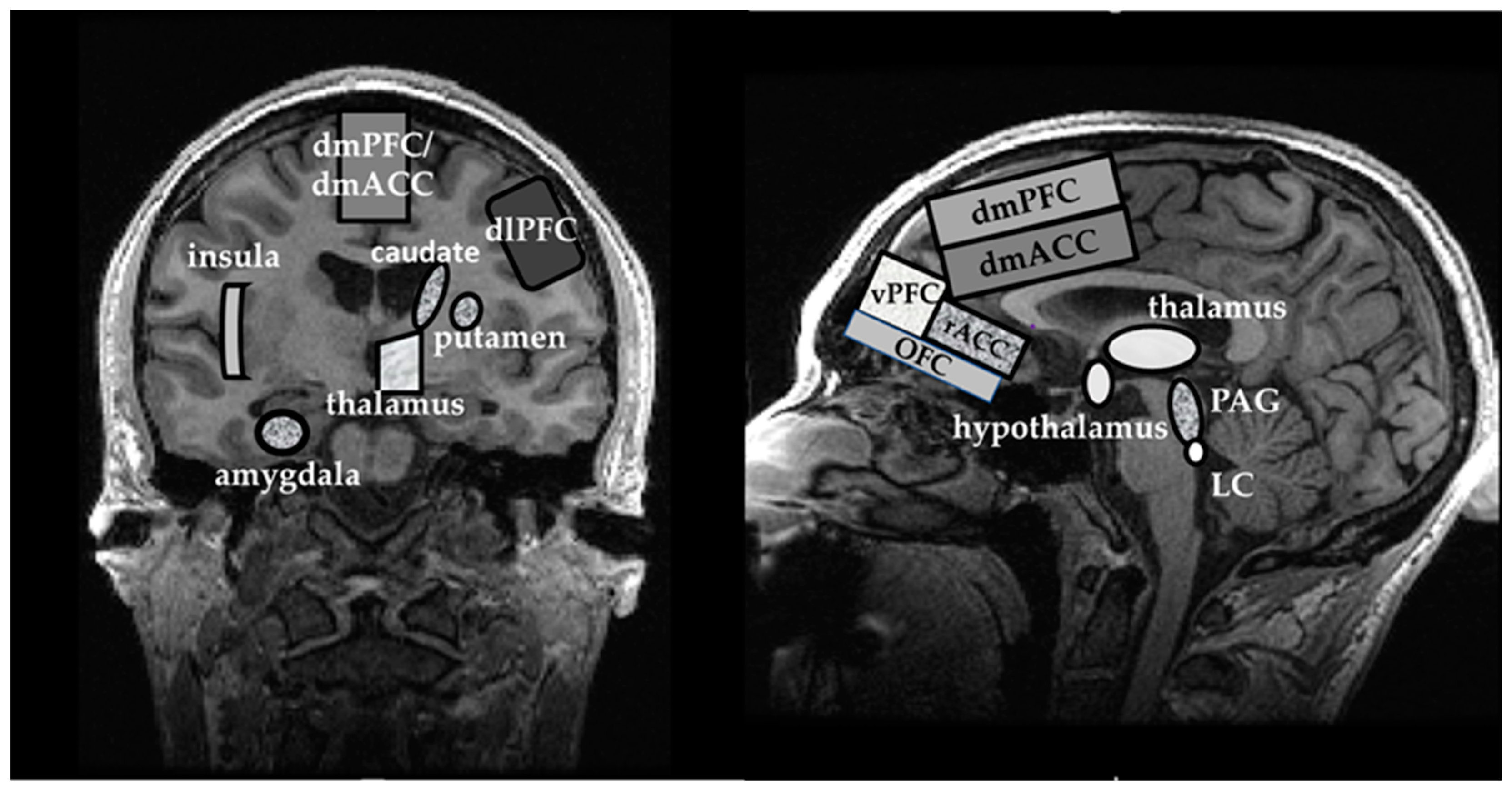{kind=link}
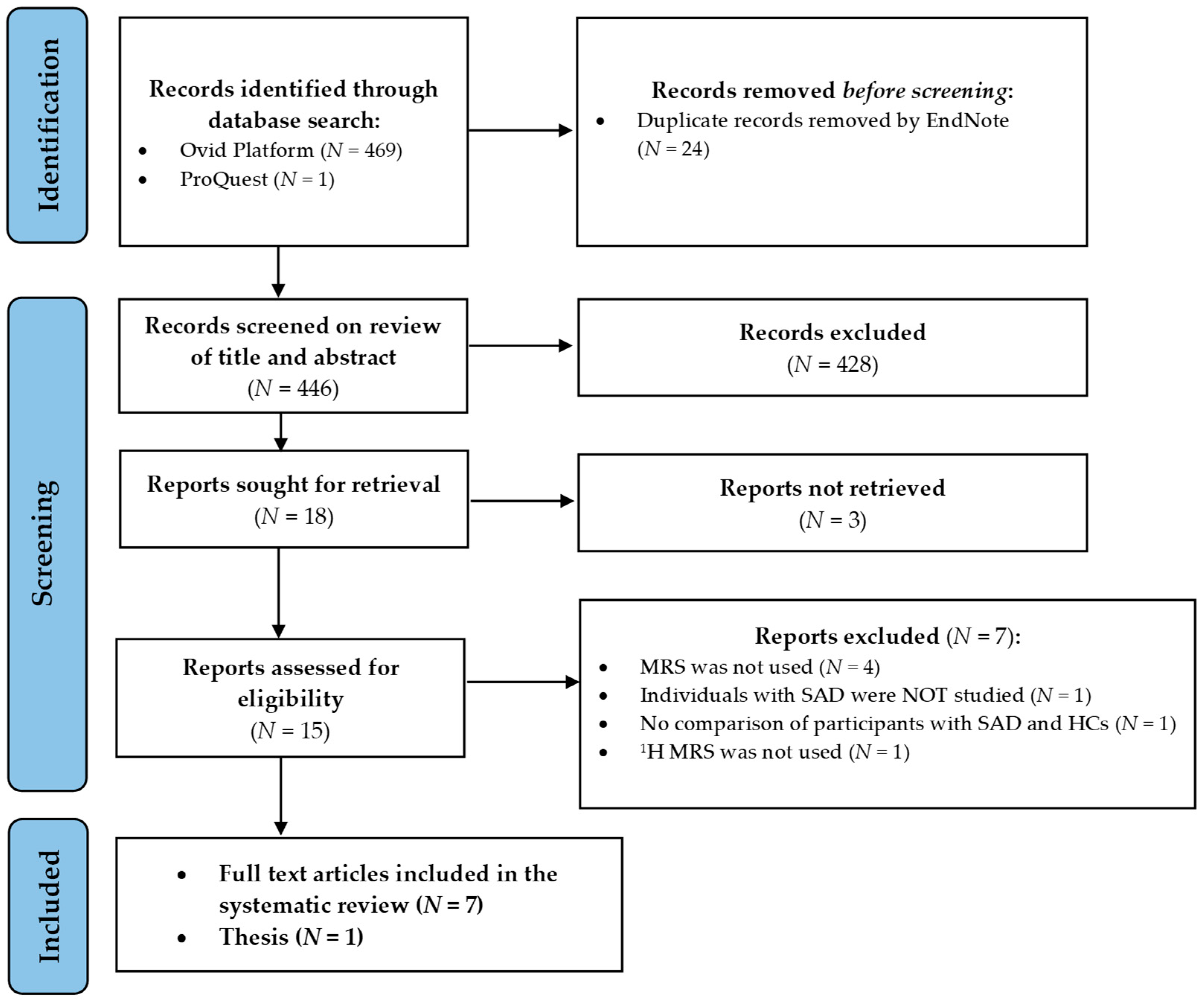{kind=link}
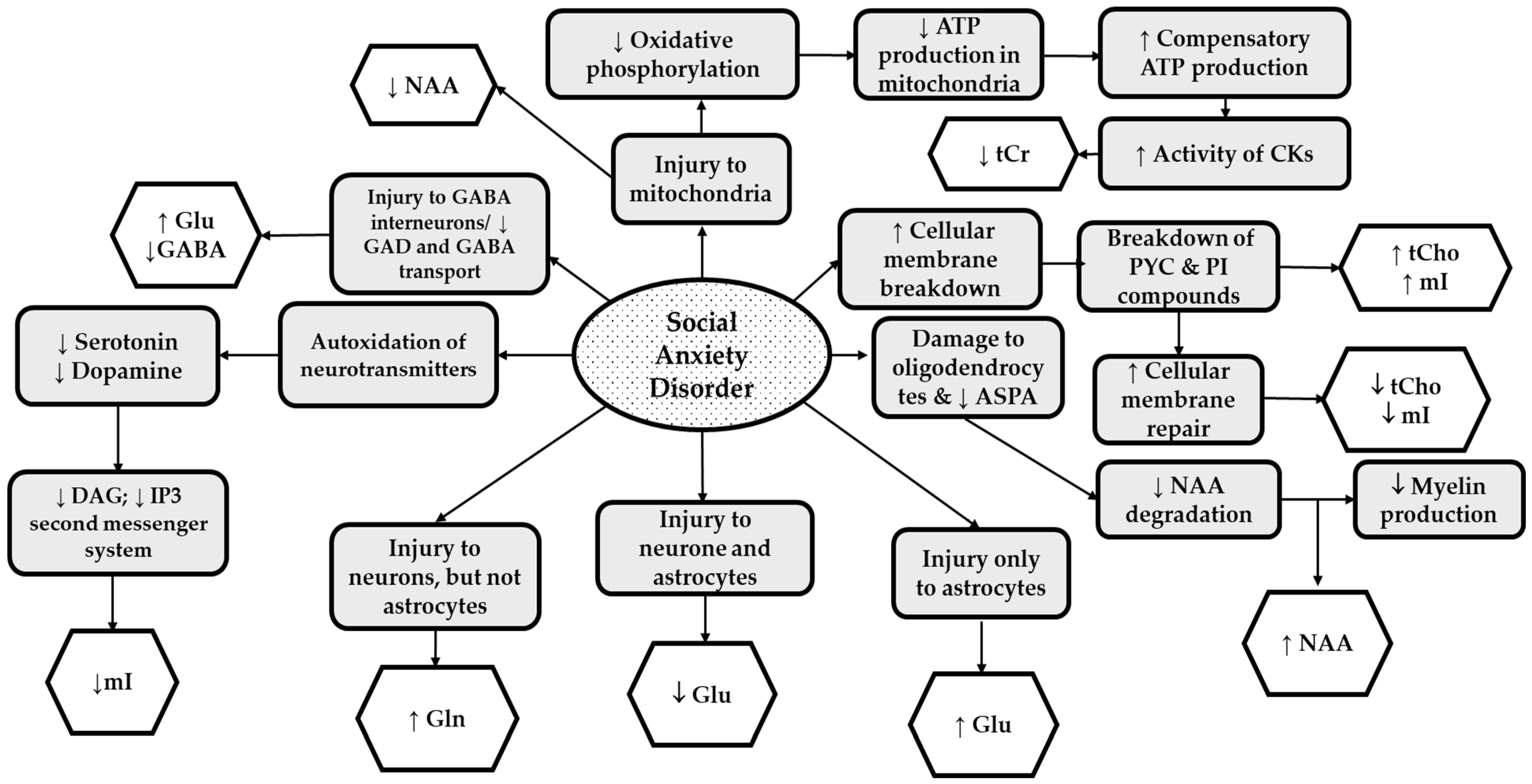{kind=link}
| Study, Magnetic Field, and Scanner Brand | Participants (N; # females; Mean Age ± SD; Age–Sex Matching) | LSAS/BSPS Scores (Mean ± SD) | Psychiatric Comorbidities | Medication Status | 1H MRS Method; Sequence and Sequence Parameters | Studied, Voxel Size (mm3) and Reported Neuro-Metabolites |
|---|---|---|---|---|---|---|
| [55] 1.5 T Philips Medical Systems | SAD: 24; 12F, 28.5 ± 6.63 HC: 24; 12F; 28.38 ± 5.84 Age–sex matched Right-handed only | SAD: 74.04 ± 27.39 HC: Not reported | None reported | Medication-free 6 weeks prior to study enrollment | SVS; STEAM TR = 2500 ms TE = 30 ms TM not reported | Left ACC (2169); Left caudate (1000), Left putamen (1400), Left insula (1920) All voxels: NAA/tCr; tCho/tCr; mI/tCr |
| [60] 3 T Allergra Siemens | SAD: 18; 11F; 31 ± 9.89 HC: 19; 8F; 29.2 ± 8.15 Age–sex matched | SAD: 88.6 ± 24.82 HC: 21.55 ± 21.19 | None reported | Medication-free during the study | SVS for ACC; PRESS TR = 1500 ms TE = 30 ms 2D CSI rest; PRESS TR = 2000 ms TE = 30 ms | ACC (4000) NAA met/Cr; NAA/tCr; Glx/tCr; Glu/tCr, Cho/tCr; Cho/tCr; mI/tCr Bilateral caudate putamen and thalamus (800) NAA met./tCr; Cho/tCr |
| [59] 3T Philips Achieva | SAD: 9; 4F; 21.6 ± 2.5 HC: 9′ 4F; 21.2 ± 2.0 Age–sex matched | SAD: 57.3 ± 11.5 HC: 26.7 ± 6.0 | None reported | Medication-free during the study | SVS; STEAM TR = 2000 ms TE = 20 ms | Left dlPFC (3500), Left ACC (3400), Left putamen (2800), Right putamen (2900), Left thalamus (3200) All regions NAA/H2O; Cho/H2O; Cr/H2O; NAA/tCr; Cho/tCr |
| [58] 3T Magnex Scientific | Two group comparison All SAD: 36; 19F; 29.86 ± 8.80 HC: 75; 55F; 31.49 ± 9.68 Age–sex not matched Three group comparison SAD: 15; 7F; 28.87 ± 10.13 SAD + MDD: 21; 13F30.57 ± 7.89 HC: 75; 55F; 31.49 ± 9.68 Age–sex not matched | Two-group comparison Median for SAD 75.50 Median for HC 12 Three-group comparison Means ± SD or medians not reported | Two-group comparison None specifically reported Three-group comparison None specifically reported | Two-group comparison Medication-free 4 weeks before enrollment Three-group comparison Medication-free 4 weeks prior to enrollment | All SVS; PRESS For Glu TR = 2400 ms TE = 130 ms For NAA and mI TR = 2400 ms TE = 132 ms PRESS with DQF-S For GABA TR = 2400 ms TE = 130 ms | dmPFC/ACC (2250) GABA/H2O Glu/H2O NAA/H2O mI/H2O |
| [57] 4T Varian, Unity-INOVA | SAD: 10; 2F; 37.2 ± 11.8 HC: 9; 2F; 33.2 ± 11.6 Age–sex matched | SAD: 81.4 ± 19.4 HC: Not reported | GAD: 3 Past MDD: 5 Past alcohol abuse: 5 | Medication-free at least 2 weeks prior to enrollment | 2D-CSI; MEGA-PRESS with J-editing TR = 1400 ms TE = 30–490 ms | Thalamus (1600) Whole brain All regions GABA/tCr; Glu/tCr; Gln/tCr |
| [56] 4T Siemens Medical Systems | SAD: 10; 5F; 26.7 ± 6.8 HC: 10; 5F; 26.6 ± 6.8 Age–sex matched Right-handed only | SAD: 72.1 ± 20.6 HC: 9.8 ± 8.9 | AD with depressed mood: 1 Past MDE: 1 | Medication-free and naive | SVS; STEAM TR = 2000 ms TE = 10 ms TM = 10 ms | ACC (800); OC (800) All voxels tCr; Glu/tCr; NAA/tCr; Cho/tCr; mI/tCr |
| [54] 1.5T General Electric | SAD: 19; 14F; 42.0 ± 11.6 HC: 10; 4F; 37.8 ± 10.5 Age-sex not matched | LSAS scores not reported | DD: 4 SP: 4 DD and SP: 1 | Medication-free at least 2 weeks prior enrollment | 2D-CSI; STEAM TR = 1500 ms TE = 20 ms TM = 26 ms | CGM; SCGM; WM All voxels Cho/H2O; NAA/H2O; Cho/tCr; NAA/tCr; mI/tCr; NAA/tCho; mI/Cho; mI/NAA |
| [53] 1.5T General Electric | SAD: 20; 9F; 35.7 ± 6.7 HC: 20; 10F; 34.6 ± 9.1 Age–sex matched | LSAS not administered Means ± SDs not reported for BSPS | APD: 8 SP: 4 DD: 4 MDD: 1 | Medication-free at least 2 weeks prior to the study enrollment | 2D-CSI; STEAM TR = 2000 ms TE = 270 ms TM = 10.6 ms | Voxel 63 (3000): 75% WM, 10% NCGM; 15% V (mostly WM) Voxel 64 (3000): 70% NCGM, 30% WM (WM + NCGM; mostly thalamus) Voxel 65 (3000): 60% NCGM, 30% WM, 10% CGM (mostly GM, including caudate) All regions: tCr SNR; Cho SNR, NAA SNR; NAA/tCr, NAA/Cho |
| ROI | Study | Metabolite | SAD Group | HC Group | Stat. | p-Value | Results | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | N | Mean | SD | N | ||||||
| NAA | |||||||||||
| ACC | [55] | NAA/tCr | 1.86 | 0.05 | 24 | 1.80 | 0.04 | 24 | t = 4.48 | <0.001 * | ↑ in SAD group |
| [60] | NAA met/tCr | 1.20 | 0.07 | 18 | 1.23 | 0.10 | 18 | n/a | n/a | ↔ | |
| NAA/tCr | 1.09 | 0.07 | 18 | 1.13 | 0.09 | 18 | n/a | n/a | ↔ | ||
| [59] | NAA/tCr | 1.034 | 0.184 | 9 | 1.019 | 0.159 | 9 | t = 0.176 | 0.863 | ↔ | |
| NAA/H2O | 14.84 | 2.081 | 9 | 15.173 | 3.564 | 9 | t = −0.23 | 0.817 | ↔ | ||
| [56] | NAA/tCr | 1.62 | 0.22 | 10 | 1.44 | 0.15 | 10 | t = 2.19 | 0.04 * | ↑ in SAD group | |
| dmPFC/ACC | [58] | NAA/H2O M | 9.08 | 1.11 | 17 | 8.5 | 0.89 | 20 | F = 1.15 | 0.3 | ↔ |
| NAA/H2O F | 8.77 | 1.38 | 19 | 9.97 | 1.4 | 55 | F = 4.81 | 0.03 * | ↓ in SAD group | ||
| OC | [56] | NAA/tCr | 1.06 | 0.09 | 10 | 1.15 | 0.14 | 10 | t = −2.17 | 0.04 * | ↓ in SAD group |
| Left insula | [55] | NAA/tCr | 1.84 | 0.05 | 24 | 1.8 | 0.03 | 24 | t = 3.07 | 0.004 * | ↑ in SAD group |
| dlPFC | [59] | NAA/tCr | 1.398 | 0.16 | 9 | 1.23 | 0.167 | 9 | t = 2.186 | 0.044 * | ↑ in SAD group |
| NAA/H2O | 15.573 | 1.571 | 9 | 16.196 | 1.56 | 9 | t = −0.842 | 0.412 | ↔. | ||
| Cortical GM | [54] | NAA/tCr | 1.331 | 0.081 | 19 | 1.373 | 0.097 | 10 | n/a | n/a | ↔ |
| Left caudate | [55] | NAA/tCr | 1.86 | 0.03 | 24 | 1.85 | 0.04 | 24 | t = 1.34 | 0.19 | ↔ |
| [59] | NAA/tCr | 0.97 | 0.33 | 14 | 1.01 | 0.2 | 16 | n/a | n/a | ↔ | |
| Right caudate | [60] | NAA/tCr | 1.1 | 0.52 | 14 | 0.98 | 0.17 | 16 | n/a | n/a | ↔ |
| NCGM + WM | [53] | NAA SNR | 9.69 | 6.19 | 12 | 23.29 | 7.2 | 13 | z = 3.67 | 0.0002 * | ↓ in SAD group |
| NAA/tCr | 1.78 | 0.45 | 12 | 2.12 | 0.49 | 13 | t = 1.95 | 0.06 | ↔ | ||
| Subcortical GM | [54] | NAA/tCr | 1.228 | 0.094 | 19 | 1.276 | 0.082 | 10 | n/a | n/a | ↔. |
| Left putamen | [55] | NAA/tCr | 1.86 | 0.03 | 24 | 1.85 | 0.04 | 24 | t = 1.34 | 0.19 | ↔ |
| [60] | NAA/tCr | 0.97 | 0.33 | 14 | 1.01 | 0.2 | 16 | n/a | n/a | ↔ | |
| [59] | NAA/tCr | 0.987 | 0.158 | 9 | 0.95 | 0.207 | 9 | t = 0.438 | 0.667 | ↔ | |
| NAA/H2O | 13.398 | 1.552 | 9 | 12.871 | 1.612 | 9 | t = 0.719 | 0.482 | ↔ | ||
| Right putamen | [60] | NAA/tCr | 1.02 | 0.21 | 17 | 1.06 | 0.16 | 18 | n/a | n/a | ↔ |
| [59] | NAA/tCr | 1.02 | 0.21 | 17 | 1.06 | 0.16 | 18 | t = 0.191 | 0.851 | ↔ | |
| NAA/H2O | 12.31 | 1.786 | 9 | 12.809 | 1.692 | 9 | t = −0.608 | 0.551 | ↔ | ||
| Left thalamus | [60] | NAA/tCr | 1.39 | 0.39 | 17 | 1.17 | 0.22 | 17 | z = 1.92 | 0.054 | ↔ |
| [59] | NAA/tCr | 1.475 | 0.296 | 9 | 1.362 | 0.399 | 9 | t = 0.715 | 0.484 | ↔ | |
| NAA/H2O | 14.96 | 2.404 | 9 | 14.209 | 4.071 | 9 | t = 0.506 | 0.62 | ↔ | ||
| Right thalamus | [60] | NAA/tCr | 1.49 | 0.39 | 17 | 1.21 | 0.25 | 17 | z = 2.14 | 0.031 * | ↑ in SAD group |
| Mostly NCGM | [53] | NAA SNR | 15.68 | 7.04 | 20 | 26.39 | 10.78 | 19 | z = 3.16 | 0.001 * | ↓ in SAD group |
| Mostly WM | [53] | NAA SNR | 11.14 | 4.83 | 20 | 15.9 | 5.4 | 17 | z = 2.66 | 0.0007 * | ↓ in SAD group |
| NAA/tCr | 1.78 | 0.41 | 20 | 1.99 | 0.32 | 17 | t = 1.72 | 0.09 | ↔ | ||
| NAA/Cho | 1.93 | 0.53 | 20 | 2.26 | 0.40 | 17 | t = 2.14 | 0.03 * | ↓ in SAD group | ||
| WM | [54] | NAA/tCr | 1.312 | 0.119 | 19 | 1.368 | 0.125 | 10 | n/a | n/a | ↔ |
| tCho | |||||||||||
| ACC | [55] | tCho/tCr | 0.83 | 0.05 | 24 | 0.84 | 0.05 | 24 | t = −0.2 | 0.84 | ↔ |
| [60] | tCho met/tCr | 0.28 | 0.03 | 18 | 0.29 | 0.03 | 18 | n/a | n/a | ↔ | |
| [59] | tCho/tCr | 0.292 | 0.08 | 9 | 0.253 | 0.063 | 9 | t = 1.06 | 0.307 | ↔ | |
| tCho/H2O | 4.186 | 1.057 | 9 | 3.71 | 0.929 | 9 | t = 0.942 | 0.362 | ↔ | ||
| [56] | tCho/tCr | 0.49 | 0.07 | 10 | 0.57 | 0.09 | 10 | t = −2.19 | 0.04 * | ↓ in SAD group | |
| Left insula | [55] | tCho/tCr | 0.77 | 0.03 | 24 | 0.77 | 0.19 | 24 | t = 0.02 | 0.99 | ↔ |
| dlPFC | [59] | tCho/tCr | 0.209 | 0.034 | 9 | 0.207 | 0.034 | 9 | t = 0.149 | 0.883 | ↔ |
| tCho/H2O | 2.373 | 0.597 | 9 | 2.757 | 0.657 | 9 | t = −1.296 | 0.213 | ↔ | ||
| Cortical GM | [54] | tCho/tCr | 0.876 | 0.071 | 19 | 0.806 | 0.046 | 10 | n/a | <0.01 * | ↑ in SAD group |
| Left caudate | [55] | tCho/tCr | 0.78 | 0.05 | 24 | 0.77 | 0.04 | 24 | t = 0.5 | 0.62 | ↔ |
| [60] | tCho met/tCr | 0.23 | 0.06 | 14 | 0.24 | 0.04 | 16 | n/a | n/a | ↔ | |
| Right caudate | [60] | tCho met/tCr | 0.22 | 0.04 | 14 | 0.25 | 0.07 | 16 | n/a | n/a | ↔ |
| NCGM + WM | [53] | tCho SNR | 6.24 | 4.48 | 12 | 12.5 | 3.64 | 13 | z = 3.18 | 0.001 * | ↓ in SAD group |
| Subcortical GM | [54] | tCho/tCr | 0.882 | 0.055 | 19 | 0.845 | 0.058 | 10 | n/a | <0.1 | ↔ |
| Left putamen | [55] | tCho/tCr | 0.87 | 0.03 | 24 | 0.86 | 0.06 | 24 | t = 0.44 | 0.66 | ↔ |
| [60] | tCho met/tCr | 0.25 | 0.06 | 17 | 0.26 | 0.03 | 18 | n/a | n/a | ↔ | |
| [59] | tCho/tCr | 0.191 | 0.038 | 9 | 0.216 | 0.027 | 9 | t = −1.595 | 0.129 | ↔ | |
| tCho/H2O | 2.596 | 0.483 | 9 | 3.006 | 0.664 | 9 | t = −1.565 | 0.136 | ↔ | ||
| Right putamen | [60] | tCho met/tCr | 0.23 | 0.06 | 17 | 0.28 | 0.03 | 18 | t = −2.86 | 0.0042 * | ↓ in SAD group |
| [59] | tCho/tCr | 0.154 | 0.078 | 9 | 0.184 | 0.047 | 9 | t = −0.977 | 0.343 | ↔ | |
| tCho/H2O | 2.164 | 1.32 | 9 | 2.557 | 0.735 | 9 | t = −0.779 | 0.447 | ↔ | ||
| Left thalamus | [60] | tCho met/tCr | 0.29 | 0.04 | 17 | 0.29 | 0.04 | 17 | n/a | n/a | ↔ |
| [59] | tCho/tCr | 0.269 | 0.08 | 9 | 0.215 | 0.073 | 9 | t = 1.489 | 0.155 | ↔ | |
| tCho/H2O | 2.761 | 0.84 | 9 | 2.293 | 0.792 | 9 | t = 1.228 | 0.236 | ↔ | ||
| Right thalamus | [60] | tCho met/tCr | 0.3 | 0.06 | 17 | 0.28 | 0.05 | 17 | n/a | n/a | ↔ |
| Mostly NCGM | [53] | tCho SNR | 8.53 | 3.18 | 20 | 13.29 | 5.15 | 19 | z = 3.02 | 0.002 * | ↓ in SAD group |
| WM | [54] | tCho/tCr | 0.928 | 0.065 | 19 | 0.924 | 0.089 | 10 | n/a | n/a | ↔ |
| Mostly WM | [53] | tCho SNR | 5.79 | 2.51 | 20 | 6.79 | 2.94 | 17 | n/a | n/a | ↔ |
| mI | |||||||||||
| ACC | [55] | mI/tCr | 0.31 | 0.04 | 24 | 0.33 | 0.05 | 24 | t = −1.31 | 0.2 | ↔ |
| [60] | mI/tCr | 0.98 | 0.1 | 18 | 0.98 | 0.09 | 18 | n/a | n/a | ↔ | |
| dmPFC/ACC | [58] | mI/H2O | 4.54 | 0.62 | 36 | 5.25 | 0.97 | 75 | t = 3.64 | 0.001 * | ↓ in SAD group |
| Left insula | [55] | mI/tC | 0.26 | 0.03 | 24 | 0.28 | 0.04 | 24 | t = −1.65 | 0.11 | ↔ |
| Cortical GM | [54] | mI/tC | 0.994 | 0.089 | 19 | 0.887 | 0.093 | 10 | n/a | <0.01 * | ↑ in SAD group |
| Left caudate | [55] | mI/tC | 0.35 | 0.03 | 24 | 0.36 | 0.05 | 24 | t = −1.14 | 0.26 | ↔ |
| Left putamen | [55] | mI/tC | 0.35 | 0.03 | 24 | 0.33 | 0.04 | 24 | t = 2.51 | 0.16 | ↔ |
| Subcortical GM | [54] | mI/tC | 0.982 | 0.113 | 19 | 0.89 | 0.101 | 10 | n/a | <0.05 * | ↑ in SAD group |
| WM | [54] | mI/tC | 1.031 | 0.097 | 19 | 0.989 | 0.073 | 10 | n/a | n/a | ↔ |
| tCr | |||||||||||
| ACC | [59] | tCr | 14.737 | 3.223 | 9 | 14.836 | 2.276 | 9 | t = −0.069 | 0.946 | ↔ |
| [56] | tCr | 4.18 | 0.5 | 10 | 4.38 | 0.61 | 10 | t = −0.8 | 0.43 | ↔ | |
| dlPFC | [59] | tCr | 11.217 | 1.297 | 9 | 13.392 | 2.22 | 9 | t = −2.539 | 0.022 * | ↓ in SAD group |
| OC | [56] | tCr | 4.39 | 0.21 | 10 | 4.43 | 0.42 | 10 | t = −0.31 | 0.76 | ↔ |
| Left putamen | [59] | tCr | 13.73 | 1.63 | 9 | 13.834 | 1.769 | 9 | t = −0.132 | 0.896 | ↔ |
| Right putamen | [59] | tCr | 13.303 | 2.234 | 9 | 13.913 | 2.063 | 9 | t = −0.602 | 0.556 | ↔ |
| NCGM + WM | [53] | tCr SNR | 5.59 | 3.97 | 12 | 11.23 | 4.08 | 13 | z = 3.07 | 0.001 * | ↓ in SAD group |
| Left thalamus | [59] | tCr | 10.402 | 2.136 | 9 | 11.053 | 3.687 | 9 | t = −0.487 | 0.633 | ↔ |
| NCGM + WM | [53] | tCr SNR | 7.64 | 2.98 | 20 | 12.78 | 3.94 | 19 | z = 3.75 | 0.0002 * | ↓ in SAD group |
| Mostly WM | [53] | tCr SNR | 6.37 | 2.8 | 20 | 7.93 | 3.4 | 17 | n/a | n/a | ↔ |
| Glu | |||||||||||
| ACC | [60] | Glu/tCr | 1.62 | 0.193 | 18 | 1.76 | 0.18 | 18 | F = 5.07 | 0.031 * | ↓ in SAD group |
| [56] | Glu/tCr | 1.37 | 0.18 | 10 | 1.21 | 0.11 | 10 | t = 2.39 | 0.03 * | ↑ in SAD group | |
| dmPFC/ACC | [58] M | Glu/H2O | 11.98 | 1.42 | 17 | 10.48 | 1.3 | 20 | t = −2.61 | 0.02 * | ↑ in SAD group |
| [58] F | Glu/H2O | 11.17 | 1.03 | 19 | 11.33 | 1.4 | 55 | t = 0.44 | 0.66 | ↔ | |
| OC | [56] | Glu/tCr | 1.07 | 0.09 | 10 | 1.16 | 0.14 | 10 | t = −1.69 | 0.11 | ↔ |
| Thalamus | [57] | Glu/tCr | 1.07 | 0.22 | 10 | 0.91 | 0.22 | 10 | t = −1.41 | 0.18 | ↔ |
| The Whole Brain | [57] | Glu/tCr | 1.37 | 0.43 | 10 | 0.99 | 0.21 | 10 | t = −2.22 | 0.04 * | ↑ in SAD group |
| tGln | |||||||||||
| Thalamus | [57] | Gln/tCr | 0.43 | 0.18 | 10 | 0.19 | 0.06 | 10 | t = −3.24 | 0.008 * | ↑ in SAD group |
| The Whole Brain | [57] | Gln/tCr | 0.57 | 0.33 | 10 | 0.23 | 0.06 | 10 | t = −2.88 | 0.01 * | ↑ in SAD group |
| tGlx | |||||||||||
| ACC | [60] | Glx/tCr | 2.01 | 0.27 | 18 | 2.17 | 0.33 | 18 | n/a | n/a | ↔ |
| GABA | |||||||||||
| dmPFC/ACC | [58] | GABA/H2O | 0.97 | 0.26 | 36 | 1.1 | 0.24 | 75 | t = 2.58 | 0.01 * | ↓ in SAD group |
| Thalamus | [57] | GABA/tCr | 0.05 | 0.02 | 10 | 0.12 | 0.07 | 10 | t = 2.17 | 0.05 * | ↓ in SAD group |
| The Whole Brain | [57] | GABA/tCr | 0.09 | 0.09 | 10 | 0.11 | 0.07 | 10 | t = 0.42 | 0.69 | ↔ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsaid, S.; Rubin-Kahana, D.S.; Kloiber, S.; Kennedy, S.H.; Chavez, S.; Le Foll, B. Neurochemical Alterations in Social Anxiety Disorder (SAD): A Systematic Review of Proton Magnetic Resonance Spectroscopic Studies. Int. J. Mol. Sci. 2022, 23, 4754. https://doi.org/10.3390/ijms23094754
Elsaid S, Rubin-Kahana DS, Kloiber S, Kennedy SH, Chavez S, Le Foll B. Neurochemical Alterations in Social Anxiety Disorder (SAD): A Systematic Review of Proton Magnetic Resonance Spectroscopic Studies. International Journal of Molecular Sciences. 2022; 23(9):4754. https://doi.org/10.3390/ijms23094754
Chicago/Turabian StyleElsaid, Sonja, Dafna S. Rubin-Kahana, Stefan Kloiber, Sidney H. Kennedy, Sofia Chavez, and Bernard Le Foll. 2022. "Neurochemical Alterations in Social Anxiety Disorder (SAD): A Systematic Review of Proton Magnetic Resonance Spectroscopic Studies" International Journal of Molecular Sciences 23, no. 9: 4754. https://doi.org/10.3390/ijms23094754