
The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Aging and Aging-Related Disorders
1.2. Relaxin-Family Peptide Receptor 3
2. Intersection of RXFP3 Signaling with the Hallmarks of Aging
2.1. Metabolic and Mitochondrial Dysfunction
2.2. Oxidative Stress
2.3. DNA Damage
2.4. Epigenetic Alterations
2.5. Nutrient Sensing
2.6. Cell Senescence
2.7. Proteostasis/Fibrosis
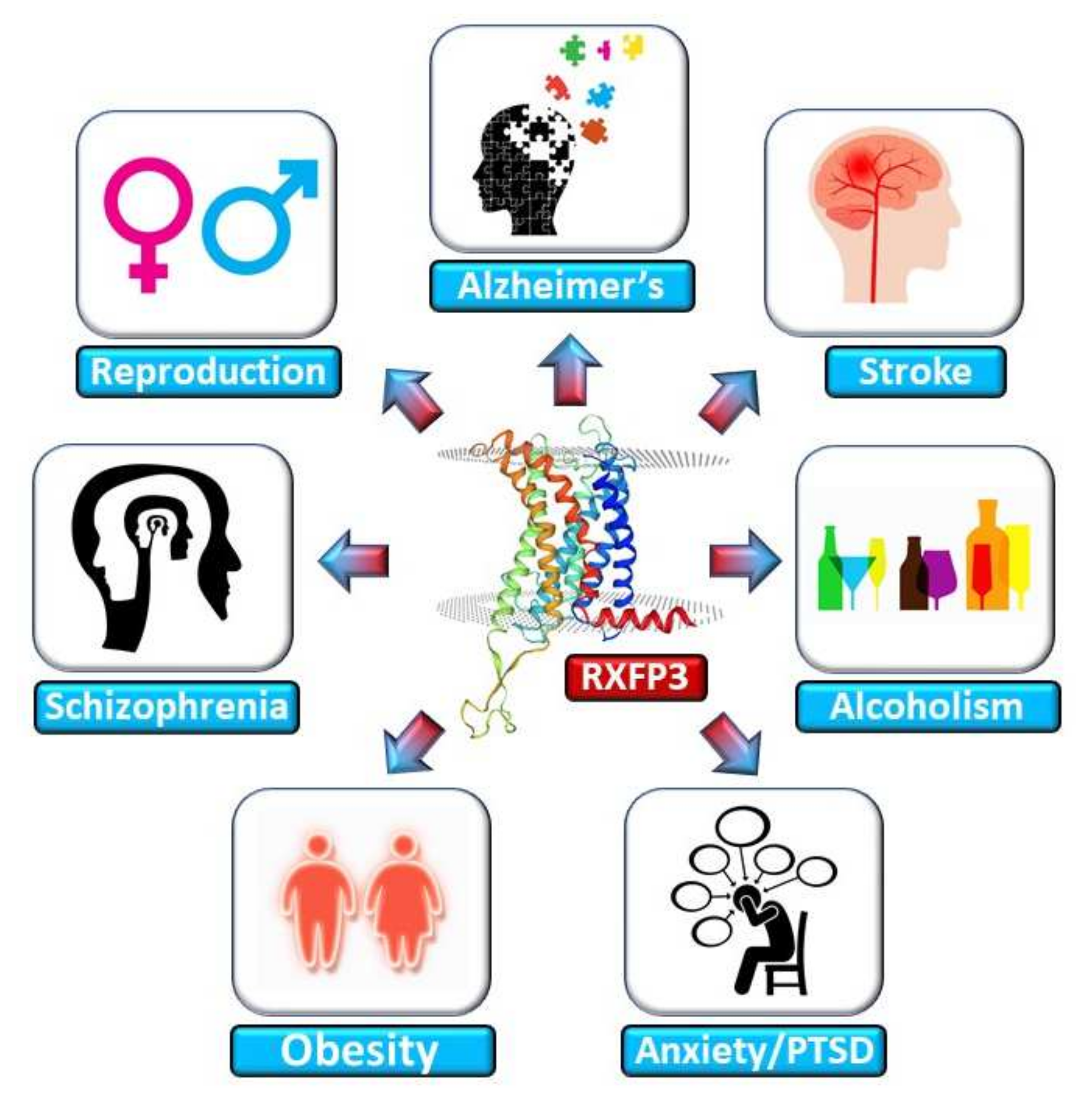
3. RXFP3 in Aging-Related Disorders
3.1. Alzheimer’s Disease
3.2. Anxiety and Post-Traumatic Stress Disorder
3.3. Schizophrenia
3.4. Obesity and Metabolic Dysfunction
3.5. Ischemic Stroke
3.6. Reproductive Aging
3.7. Alcohol Abuse
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging. 2018, 13, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadwick, W.; Martin, B.; Chapter, M.C.; Park, S.; Wang, L.; Daimon, C.M.; Brenneman, R.; Maudsley, S. GIT2 acts as a potential keystone protein in functional hypothalamic networks associated with age-related phenotypic changes in rats. PLoS ONE 2012, 7, e36975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Cong, W.N.; Ji, S.; Rothman, S.; Maudsley, S.; Martin, B. Metabolic dysfunction in Alzheimer’s disease and related neurodegenerative disorders. Curr. Alzheimer Res. 2012, 9, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mitchell, J.R.; Hasty, P. DNA double-strand breaks: A potential causative factor for mammalian aging? Mech. Ageing Dev. 2008, 129, 416–424. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Chadwick, W.; Janssens, J.; Premont, R.T.; Schmalzigaug, R.; Becker, K.G.; Lehrmann, E.; Wood, W.H.; Zhang, Y.; Siddiqui, S.; et al. GIT2 acts as a systems-level coordinator of neurometabolic activity and pathophysiological aging. Front. Endocrinol. 2016, 6, 191. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, W.; Zhou, Y.; Park, S.; Wang, L.; Mitchell, N.; Stone, M.D.; Becker, K.G.; Martin, B.; Maudsley, S. Minimal peroxide exposure of neuronal cells induces multifaceted adaptive responses. PLoS ONE 2010, 5, e14352. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.; Cai, H.; Park, S.S.; Siddiqui, S.; Premont, R.T.; Schmalzigaug, R.; Paramasivam, M.; Seidman, M.; Bodogai, I.; Biragyn, A.; et al. Nuclear GIT2 is an ATM substrate and promotes DNA repair. Mol. Cell. Biol. 2015, 35, 1081–1096. [Google Scholar] [CrossRef] [Green Version]
- Premont, R.T.; Claing, A.; Vitale, N.; Perry, S.J.; Lefkowitz, R.J. The GIT family of ADP-ribosylation factor GTPase-activating proteins. Functional diversity of GIT2 through alternative splicing. J. Biol. Chem. 2000, 275, 22373–22380. [Google Scholar] [CrossRef] [Green Version]
- Premont, R.T.; Perry, S.J.; Schmalzigaug, R.; Roseman, J.T.; Xing, Y.; Claing, A. The GIT/PIX complex: An oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell. Signal. 2004, 16, 1001–1011. [Google Scholar] [CrossRef]
- Van Gastel, J.; Etienne, H.; Azmi, A.; Maudsley, S. The synergistic GIT2-RXFP3 system in the brain and its importance in age-related disorders. Front. Aging Neurosci. 2016, 3, 8. [Google Scholar]
- Roux, B.T.; Cottrell, G.S. G protein-coupled receptors: What a difference a ‘partner’ makes. Int. J. Mol. Sci. 2014, 15, 1112–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bathgate, R.A.; Oh, M.H.; Ling, W.J.; Kaas, Q.; Hossain, M.A.; Gooley, P.R.; Rosengren, J. Elucidation of relaxin-3 binding interactions in the extracellular loops of RXFP3. Front. Endocrinol. 2013, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, M.; Kamohara, M.; Sugimoto, T.; Hidaka, K.; Takasaki, J.; Saito, T.; Okada, M.; Yamaguchi, T.; Furuichi, K. The novel G-protein coupled receptor SALPR shares sequence similarity with somatostatin and angiotensin receptors. Gene 2000, 248, 183–189. [Google Scholar] [CrossRef]
- Olucha-Bordonau, F.E.; Albert-Gascó, H.; Ros-Bernal, F.; Rytova, V.; Ong-Pålsson, E.K.E.; Ma, S.; Sánchez-Pérez, A.M.; Gundlach, A.L. Modulation of forebrain function by nucleus incertus and relaxin-3/RXFP3 signaling. CNS Neurosci. Ther. 2018, 24, 694–702. [Google Scholar] [CrossRef]
- Smith, C.M.; Shen, P.J.; Banerjee, A.; Bonaventure, P.; Ma, S.; Bathgate, R.A.D.; Sutton, S.W.; Gundlach, A.L. Distribution of relaxin-3 and RXFP3 within arousal, stress, affective, and cognitive circuits of mouse brain. J. Comp. Neurol. 2010, 518, 4016–4045. [Google Scholar] [CrossRef]
- Ganella, D.E.; Ma, S.; Gundlach, A.L. Relaxin-3/RXFP3 Signaling and Neuroendocrine Function—A Perspective on Extrinsic Hypothalamic Control. Front. Endocrinol. 2013, 4, 128. [Google Scholar] [CrossRef] [Green Version]
- Halls, M.L.; Bathgate, R.A.; Sutton, S.W.; Dschietzig, T.B.; Summers, R.J. International Union of Basic and Clinical Pharmacology. XCV. Recent advances in the understanding of the pharmacology and biological roles of relaxin family peptide receptors 1–4, the receptors for relaxin family peptides. Pharmacol. Rev. 2015, 67, 389–440. [Google Scholar] [CrossRef]
- Aaboud, M.; Aad, G.; Abbott, B.; Abdallah, J.; Abdinov, O.; Abeloos, B.; Aben, R.; AbouZeid, O.S.; Abraham, N.L.; Abramowicz, H.; et al. Measurement of the Inelastic Proton-Proton Cross Section at sqrt[s] = 13 TeV with the ATLAS Detector at the LHC. Phys. Rev. Lett. 2016, 117, 182002. [Google Scholar] [CrossRef] [Green Version]
- Van der Westhuizen, E.T.; Werry, T.D.; Sexton, P.M.; Summers, R.J. The relaxin family peptide receptor 3 activates extracellular signal-regulated kinase 1/2 through a protein kinase C-dependent mechanism. Mol. Pharmacol. 2007, 71, 1618–1629. [Google Scholar] [CrossRef]
- Wilkinson, T.N.; Speed, T.P.; Tregear, G.W.; Bathgate, R.A. Evolution of the relaxin-like peptide family: From neuropeptide to reproduction. Ann. N. Y. Acad. Sci. 2005, 1041, 530–533. [Google Scholar] [CrossRef]
- Yegorov, S.; Good-Avila, S.V.; Parry, L.; Wilson, B.C. Relaxin family genes in humans and teleosts. Ann. N. Y. Acad. Sci. 2009, 1160, 42–44. [Google Scholar] [CrossRef] [PubMed]
- Bathgate, R.A.; Samuel, C.S.; Burazin, T.C.; Layfield, S.; Claasz, A.A.; Grace, I.; Reytomas, T.; Dawson, N.F.; Zhao, C.; Bond, C.; et al. Human relaxin gene 3 (H3) and the equivalent mouse relaxin (M3) gene. Novel members of the relaxin peptide family. J. Biol. Chem. 2002, 277, 1148–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Good, S.; Yegorov, S.; Martijn, J.; Franck, J.; Bogerd, J. New insights into ligand-receptor pairing and coevolution of relaxin family peptides and their receptors in teleosts. Int. J. Evol. Biol. 2012, 2012, 310278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gastel, J.; Leysen, H.; Santos-Otte, P.; Hendrickx, J.O.; Azmi, A.; Martin, B.; Maudsley, S. The RXFP3 receptor is functionally associated with cellular responses to oxidative stress and DNA damage. Aging 2019, 11, 11268–11313. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Iijima, N.; Miyamoto, Y.; Fukusumi, S.; Itoh, Y.; Ozawa, H.; Ibata, Y. Neurons expressing relaxin 3/INSL 7 in the nucleus incertus respond to stress. Eur. J. Neurosci. 2005, 21, 1659–1670. [Google Scholar] [CrossRef]
- Ryan, P.J.; Buchler, E.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Lawrence, A.J.; Gundlach, A.L. Central relaxin-3 receptor (RXFP3) activation decreases anxiety- and depressive-like behaviours in the rat. Behav. Brain Res. 2013, 244, 142–151. [Google Scholar] [CrossRef]
- Lee, J.H.; Koh, S.Q.; Guadagna, S.; Francis, P.T.; Esiri, M.M.; Chen, C.P.; Wong, P.T.H.; Dawe, G.S.; Lai, M.K.P. Altered relaxin family receptors RXFP1 and RXFP3 in the neocortex of depressed Alzheimer’s disease patients. Psychopharmacology 2016, 233, 591–598. [Google Scholar] [CrossRef]
- Zhang, C.; Baimoukhametova, D.V.; Smith, C.M.; Bains, J.S.; Gundlach, A.L. Relaxin-3/RXFP3 signalling in mouse hypothalamus: No effect of RXFP3 activation on corticosterone, despite reduced presynaptic excitatory input onto paraventricular CRH neurons in vitro. Psychopharmacology 2017, 234, 1725–1739. [Google Scholar] [CrossRef]
- McGowan, B.M.; Stanley, S.A.; Smith, K.L.; Minnion, J.S.; Donovan, J.; Thompson, E.L.; Patterson, M.; Connolly, M.M.; Abbott, C.R.; Small, C.J.; et al. Effects of acute and chronic relaxin-3 on food intake and energy expenditure in rats. Regul. Pept. 2006, 136, 72–77. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Ryan, P.J.; Hosken, I.T.; Ma, S.; Gundlach, A.L. Relaxin-3 systems in the brain—The first 10 years. J. Chem. Neuroanat. 2011, 42, 262–275. [Google Scholar] [CrossRef]
- Ryan, P.J.; Kastman, H.E.; Krstew, E.V.; Rosengren, K.J.; Hossain, M.A.; Churilov, L.; Wade, J.D.; Gundlach, A.L.; Lawrence, A.J. Relaxin-3/RXFP3 system regulates alcohol-seeking. Proc. Natl. Acad. Sci. USA 2013, 110, 20789–20794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Rijt, S.; Molenaars, M.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front. Cell Dev. Biol. 2020, 8, 594416. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, N.; Huffman, D.M.; Muzumdar, R.H.; Bartke, A. The critical role of metabolic pathways in aging. Diabetes 2012, 61, 1315–1322. [Google Scholar] [CrossRef] [Green Version]
- Spinelli, R.; Parrillo, L.; Longo, M.; Florese, P.; Desiderio, A.; Zatterale, F.; Miele, C.; Raciti, G.A.; Beguint, F. Molecular basis of ageing in chronic metabolic diseases. J. Endocrinol. Investig. 2020, 43, 1373–1389. [Google Scholar] [CrossRef]
- Mendrick, D.L.; Diehl, A.M.; Topor, L.S.; Dietert, R.R.; Will, Y.; La Merrill, M.A.; Bouret, S.; Varma, V.; Hastings, K.L.; Schug, T.T.; et al. Metabolic Syndrome and Associated Diseases: From the Bench to the Clinic. Toxicol. Sci. 2018, 162, 36–42. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zhao, H.; Wang, J. Metabolism and Chronic Inflammation: The Links Between Chronic Heart Failure and Comorbidities. Front. Cardiovasc. Med. 2021, 8, 650278. [Google Scholar] [CrossRef]
- Schottker, B.; Brenner, H.; Jansen, E.H.; Gardiner, J.; Peasey, A.; Kubínová, R.; Pająk, A.; Topor-Madry, R.; Tamousiunas, A.; Saum, K.; et al. Evidence for the free radical/oxidative stress theory of ageing from the CHANCES consortium: A meta-analysis of individual participant data. BMC Med. 2015, 13, 300. [Google Scholar] [CrossRef] [Green Version]
- Smith, S. Telomerase can’t handle the stress. Genes Dev. 2018, 32, 597–599. [Google Scholar] [CrossRef]
- Maudsley, S.; Martin, B.; Luttrell, L.M. G protein-coupled receptor signaling complexity in neuronal tissue: Implications for novel therapeutics. Curr. Alzheimer Res. 2007, 4, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Golden, E.; Keselman, A.; Stone, M.; Mattson, M.P.; Egan, J.M.; Maudsley, S. Therapeutic perspectives for the treatment of Huntington’s disease: Treating the whole body. Histol. Histopathol. 2008, 23, 237–250. [Google Scholar] [PubMed]
- Martin, B.; Chadwick, W.; Yi, T.; Park, S.; Lu, D.; Ni, B.; Gadkaree, S.; Farhang, K.; Becker, K.G.; Maudsley, S. VENNTURE—A novel Venn diagram investigational tool for multiple pharmacological dataset analysis. PLoS ONE 2012, 7, e36911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, W.N.; Cai, H.; Wang, R.; Daimon, C.M.; Maudsley, S.; Raber, K.; Canneva, F.; von Hörsten, S.; Martin, B. Altered hypothalamic protein expression in a rat model of Huntington’s disease. PLoS ONE 2012, 7, e47240. [Google Scholar] [CrossRef]
- Liu, C.; Chen, J.; Kuei, C.; Sutton, S.; Nepomuceno, D.; Bonaventure, P.; Lovenbergl, T.W. Relaxin-3/insulin-like peptide 5 chimeric peptide, a selective ligand for G protein-coupled receptor (GPCR)135 and GPCR142 over leucine-rich repeat-containing G protein-coupled receptor 7. Mol. Pharmacol. 2005, 67, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Gundlach, A.L. Relaxin-family peptide and receptor systems in brain: Insights from recent anatomical and functional studies. Adv. Exp. Med. Biol. 2007, 612, 119–137. [Google Scholar] [PubMed]
- Sutton, S.W.; Bonaventure, P.; Kuei, C.; Rolan, B.; Chen, J.; Nepumuceno, D.; Lovenberg, T.W.; Liu, C. Distribution of G-protein-coupled receptor (GPCR)135 binding sites and receptor mRNA in the rat brain suggests a role for relaxin-3 in neuroendocrine and sensory processing. Neuroendocrinology 2004, 80, 298–307. [Google Scholar] [CrossRef] [PubMed]
- McGowan, B.M.; Minnion, J.S.; Murphy, K.G.; Roy, D.; Stanley, S.A.; Dhillo, W.S.; Gardiner, J.V.; Ghateri, M.A.; Bloom, S.R. Relaxin-3 stimulates the neuro-endocrine stress axis via corticotrophin-releasing hormone. J. Endocrinol. 2014, 221, 337–346. [Google Scholar] [CrossRef] [Green Version]
- DeAdder, N.P.; Gillam, H.J.; Wilson, B.C. Relaxin peptides reduce cellular damage in cultured brain slices exposed to transient oxygen–glucose deprivation: An effect mediated by nitric oxide. FACETS 2021, 6, 118. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, M.; Chen, W.; Liu, Z. The neuroprotective effects of intermittent fasting on brain aging and neurodegenerative diseases via regulating mitochondrial function. Free Radic. Biol. Med. 2022, 182, 206–218. [Google Scholar] [CrossRef]
- Wang, J.; Huo, K.; Ma, L.; Tang, L.; Li, D.; Huang, X.; Yuan, Y.; Li, C.; Wang, W.; Guan, W.; et al. Toward an understanding of the protein interaction network of the human liver. Mol. Syst. Biol. 2017, 13, 965. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Lien, J.; Chen, C.; Liu, Y.; Wang, C.; Ping, C.; Lin, Y.; Huang, A.; Lin, C. Antiviral Activity of a Novel Compound CW-33 against Japanese Encephalitis Virus through Inhibiting Intracellular Calcium Overload. Int. J. Mol. Sci. 2016, 17, 1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergeron, L.H.; Willcox, J.M.; Alibhai, F.J.; Connell, B.J.; Saleh, T.M.; Wilson, B.C.; Summerlee, A.J.S. Relaxin peptide hormones are protective during the early stages of ischemic stroke in male rats. Endocrinology 2015, 156, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Kato, K.; Oguri, M.; Horibe, H.; Fujimaki, T.; Yasukochi, Y.; Takeuchi, I.; Sakuma, J. Identification of 13 novel susceptibility loci for early-onset myocardial infarction, hypertension, or chronic kidney disease. Int. J. Mol. Med. 2018, 42, 2415–2436. [Google Scholar]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Buffenstein, R.; Edrey, Y.H.; Yang, T.; Mele, J. The oxidative stress theory of aging: Embattled or invincible? Insights from non-traditional model organisms. Age 2008, 30, 99–109. [Google Scholar]
- Doonan, R.; McElwee, J.J.; Matthijssens, F.; Walker, G.A.; Houthoofd, K.; Back, P.; Matscheski, A.; Vanfleteren, J.R.; Gems, D. Against the oxidative damage theory of aging: Superoxide dismutases protect against oxidative stress but have little or no effect on life span in Caenorhabditis elegans. Genes Dev. 2008, 22, 3236–3241. [Google Scholar]
- Del Valle, L.G. Oxidative stress in aging: Theoretical outcomes and clinical evidences in humans. Biomed. Aging Pathol. 2011, 1, 1–7. [Google Scholar]
- Chhunchha, B.; Singh, P.; Stamer, W.D.; Singh, D.P. Prdx6 retards senescence and restores trabecular meshwork cell health by regulating reactive oxygen species. Cell Death Discov. 2017, 3, 17060. [Google Scholar]
- Tsang, C.K.; Liu, Y.; Thomas, J.; Zhang, Y.; Zheng, X.F. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat. Commun. 2014, 5, 3446. [Google Scholar]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Crosstalk between Oxidative Stress and SIRT1: Impact on the Aging Process. Int. J. Mol. Sci. 2013, 14, 3834–3859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitsky, K.; Bar-Shira, A.; Gilad, S.; Rotman, G.; Ziv, Y.; Vanagaite, L.; Tagle, D.A.; Uziel, T.; Sfez, S.; Ashkenazi, M.; et al. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science 1995, 268, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Traven, A.; Heierhorst, J. SQ/TQ cluster domains: Concentrated ATM/ATR kinase phosphorylation site regions in DNA-damage-response proteins. Bioessays 2005, 27, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Deshpande, R.; Paull, T.T. ATM activation in the presence of oxidative stress. Cell Cycle 2010, 9, 4805–4811. [Google Scholar] [CrossRef] [Green Version]
- Irannejad, R.; von Zastrow, M. GPCR signaling along the endocytic pathway. Curr. Opin. Cell Biol. 2014, 27, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Estelles, A.; Sperinde, J.; Roulon, T.; Aguilar, B.; Bonner, C.; LePecq, J.-B.; Delcayre, A. Exosome nanovesicles displaying G protein-coupled receptors for drug discovery. Int. J. Nanomed. 2007, 2, 751–760. [Google Scholar]
- Staubert, C.; Schoneberg, T. GPCR Signaling from Intracellular Membranes—A Novel Concept. Bioessays 2017, 39, 12. [Google Scholar] [CrossRef]
- Jean-Alphonse, F.; Hanyaloglu, A.C. Regulation of GPCR signal networks via membrane trafficking. Mol. Cell Endocrinol. 2011, 331, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Wilbanks, A.M.; Laporte, S.A.; Bohn, L.M.; Barak, L.S.; Caron, M.G. Apparent loss-of-function mutant GPCRs revealed as constitutively desensitized receptors. Biochemistry 2002, 41, 11981–11989. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Bell, C.G.; Lowe, R.; Adams, P.D.; Baccarelli, A.A.; Beck, S.; Bell, J.T.; Christensen, B.C.; Gladyshev, V.N.; Heijmans, B.T.; Horvath, S.; et al. DNA methylation aging clocks: Challenges and recommendations. Genome Biol. 2019, 20, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, D.L.; Mousovich-Neto, F.; Tonon-da-Silva, G.; Salgueiro, W.G.; Mori, M.A. Epigenetic changes during ageing and their underlying mechanisms. Biogerontology 2020, 21, 423–443. [Google Scholar] [CrossRef] [PubMed]
- Hansel, A.; Steinbach, D.; Greinke, C.; Schmitz, M.; Eiselt, J.; Scheungraber, C.; Gajda, M.; Hoyer, H.; Runnebaum, I.B.; Dürst, M. A promising DNA methylation signature for the triage of high-risk human papillomavirus DNA-positive women. PLoS ONE 2014, 9, e91905. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.; Wunsch, K.; Hoyer, H.; Scheungraber, C.; Runnebaum, I.B.; Hansel, A.; Dürst, M. Performance of a methylation specific real-time PCR assay as a triage test for HPV-positive women. Clin. Epigenet. 2017, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.; Eichelkraut, K.; Schmidt, D.; Zeiser, I.; Hilal, Z.; Tettenborn, Z.; Hansel, A.; Ikenberg, H. Performance of a DNA methylation marker panel using liquid-based cervical scrapes to detect cervical cancer and its precancerous stages. BMC Cancer 2018, 18, 1197. [Google Scholar] [CrossRef]
- Dehan, P.; Canon, C.; Trooskens, G.; Rehli, M.; Munaut, C.; Van Criekinge, W.; Delvenne, P. Expression of type 2 orexin receptor in human endometrium and its epigenetic silencing in endometrial cancer. J. Clin. Endocrinol. Metab. 2013, 98, 1549–1557. [Google Scholar] [CrossRef] [Green Version]
- Kubarek, L.; Jagodzinski, P.P. Epigenetic up-regulation of CXCR4 and CXCL12 expression by 17 beta-estradiol and tamoxifen is associated with formation of DNA methyltransferase 3B4 splice variant in Ishikawa endometrial adenocarcinoma cells. FEBS Lett. 2007, 581, 1441–1448. [Google Scholar] [CrossRef] [Green Version]
- Yadav, S.S.; Prasad, S.B.; Das, M.; Kumari, S.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Epigenetic silencing of CXCR4 promotes loss of cell adhesion in cervical cancer. Biomed Res. Int. 2014, 2014, 581403. [Google Scholar] [CrossRef]
- Zhou, L.; Luo, L.; Qi, X.; Li, X.; Gorodeski, G.I. Regulation of P2X(7) gene transcription. Purinergic Signal. 2009, 5, 409–426. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Luo, J.; Weng, Y.; Mutch, D.G.; Goodfellow, P.J.; Miller, D.S.; Huang, T.M. Promoter hypermethylation of CIDEA, HAAO and RXFP3 associated with microsatellite instability in endometrial carcinomas. Gynecol. Oncol. 2010, 117, 239–247. [Google Scholar] [CrossRef] [Green Version]
- Tang, P.; Low, H.B.; Png, C.W.; Torta, F.; Kumar, J.K.; Lim, H.Y.; Zhou, Y.; Yang, H.; Angeli, V.; Shabbir, A. Protective Function of Mitogen-Activated Protein Kinase Phosphatase 5 in Aging- and Diet-Induced Hepatic Steatosis and Steatohepatitis. Hepatol. Commun. 2019, 3, 748–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarente, L.; Kenyon, C. Genetic pathways that regulate ageing in model organisms. Nature 2000, 408, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Lund, J.; Tedesco, P.; Duke, K.; Wang, J.; Kim, S.K.; Johnson, T.E. Transcriptional profile of aging in C. elegans. Curr. Biol. 2002, 12, 1566–1573. [Google Scholar] [CrossRef] [Green Version]
- Corremans, R.; Neven, E.; Maudsley, S.; Leysen, H.; De Broe, M.E.; D’Haese, P.C.; Vervaet, B.A.; Verhulst, A. Progression of established non-diabetic chronic kidney disease is halted by metformin treatment in rats. Kidney Int. 2022, in press. [Google Scholar] [CrossRef]
- Van Gastel, J.; Cai, H.; Cong, W.N.; Chadwick, W.; Daimon, C.M.; Leysen, H.; Hendrickx, J.O.; De Schepper, R.; Vangenechten, L.; Van Turnhout, J.; et al. Multidimensional informatic deconvolution defines gender-specific roles of hypothalamic GIT2 in aging trajectories. Mech. Ageing Dev. 2019, 184, 111150. [Google Scholar] [CrossRef]
- Opdebeeck, B.; Maudsley, S.; Azmi, A.; De Maré, A.; De Leger, W.; Meijers, B.; Verhulst, A.; Evenepoel, P.; D’Haese, P.C.; Neven, E.J. Indoxyl Sulfate and p-Cresyl Sulfate Promote Vascular Calcification and Associate with Glucose Intolerance. Am. Soc. Nephrol. 2019, 30, 751–766. [Google Scholar] [CrossRef]
- Martin, B.; Wang, R.; Cong, W.N.; Daimon, C.M.; Wu, W.W.; Ni, B.; Becker, K.G.; Lehrmann, E.; Wood, W.H., 3rd; Zhang, Y.; et al. Altered learning, memory, and social behavior in type 1 taste receptor subunit 3 knock-out mice are associated with neuronal dysfunction. J. Biol. Chem. 2017, 292, 11508–11530. [Google Scholar] [CrossRef] [Green Version]
- Mou, Z.; Hyde, T.M.; Lipska, B.K.; Martinowich, K.; Wei, P.; Ong, C.J.; Hunter, L.A.; Palaguachi, G.I.; Morgun, E.; Teng, R.; et al. Human Obesity Associated with an Intronic SNP in the Brain-Derived Neurotrophic Factor Locus. Cell Rep. 2015, 13, 1073–1080. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ross, C.A.; Cai, H.; Cong, W.N.; Daimon, C.M.; Carlson, O.D.; Egan, J.M.; Siddiqui, S.; Maudsley, S.; Martin, B. Metabolic and hormonal signatures in pre-manifest and manifest Huntington’s disease patients. Front. Physiol. 2014, 5, 231. [Google Scholar] [CrossRef] [Green Version]
- Okun, E.; Griffioen, K.J.; Rothman, S.; Wan, R.; Cong, W.N.; De Cabo, R.; Martin-Montalvo, A.; Levette, A.; Maudsley, S.; Martin, B.; et al. Toll-like receptors 2 and 4 modulate autonomic control of heart rate and energy metabolism. Brain Behav. Immun. 2014, 36, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Cong, W.N.; Wang, R.; Cai, H.; Daimon, C.M.; Scheibye-Knudsen, M.; Bohr, V.A.; Turkin, R.; Wood, W.H., 3rd; Becker, K.G.; Moaddel, R.; et al. Long-term artificial sweetener acesulfame potassium treatment alters neurometabolic functions in C57BL/6J mice. PLoS ONE 2013, 8, e70257. [Google Scholar] [CrossRef] [PubMed]
- Stranahan, A.M.; Martin, B.; Chadwick, W.; Park, S.S.; Wang, L.; Becker, K.G.; Wood, W.H., 3rd; Zhang, Y.; Maudsley, S. Metabolic context regulates distinct hypothalamic transcriptional responses to antiaging interventions. Int. J. Endocrinol. 2012, 2012, 732975. [Google Scholar] [CrossRef] [PubMed]
- Maudsley, S.; Martin, B.; Egan, J.M. To be or not to be—Obese. Endocrinology 2011, 152, 3592–3596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCay, C.M.; Crowell, M.F.; Maynard, L.A. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar] [CrossRef]
- Lane, M.A.; Ingram, D.K.; Roth, G.S. Beyond the rodent model: Calorie restriction in rhesus monkeys. Age 1997, 20, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Colman, R.J.; Beasley, T.M.; Kemnitz, J.W.; Johnson, S.C.; Weindruch, R.; Anderson, R.M. Caloric restriction reduces age-related and all-cause mortality in rhesus monkeys. Nat. Commun. 2014, 5, 3557. [Google Scholar] [CrossRef]
- Martin, B.; Pearson, M.; Kebejian, L.; Golden, E.; Keselman, A.; Bender, M.; Carlson, O.; Egan, J.; Ladenheim, B.; Cadet, J.L.; et al. Sex-dependent metabolic, neuroendocrine, and cognitive responses to dietary energy restriction and excess. Endocrinology 2007, 148, 4318–4333. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Pearson, M.; Brenneman, R.; Golden, E.; Wood, W.H., 3rd; Prabhu, V.; Becker, K.G.; Mattson, M.P.; Maudsley, S. Gonadal transcriptome alterations in response to dietary energy intake: Sensing the reproductive environment. PLoS ONE 2009, 4, e4146. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Pearson, M.; Brenneman, R.; Golden, E.; Keselman, A.; Iyun, T.; Carlson, O.D.; Egan, J.M.; Becker, K.G.; Wood, W.H., 3rd; et al. Conserved and differential effects of dietary energy intake on the hippocampal transcriptomes of females and males. PLoS ONE 2008, 3, e2398. [Google Scholar] [CrossRef]
- Carlson, O.; Martin, B.; Stote, K.S.; Golden, E.; Maudsley, S.; Najjar, S.S.; Ferrucci, L.; Ingram, D.K.; Longo, D.L.; Rumpler, W.V. Impact of reduced meal frequency without caloric restriction on glucose regulation in healthy, normal-weight middle-aged men and women. Metabolism 2007, 56, 1729–1734. [Google Scholar] [CrossRef] [Green Version]
- Leysen, H.; Walter, D.; Christiaenssen, B.; Vandoren, R.; Harputluoğlu, İ.; Van Loon, N.; Maudsley, S. GPCRs Are Optimal Regulators of Complex Biological Systems and Orchestrate the Interface between Health and Disease. Int. J. Mol. Sci. 2021, 22, 13387. [Google Scholar] [CrossRef] [PubMed]
- Cokorinos, E.C.; Delmore, J.; Reyes, A.R.; Albuquerque, B.; Kjøbsted, R.; Jørgensen, N.O.; Tran, J.; Jatkar, A.; Cialdea, L.; Esquejo, R.M. Activation of skeletal muscle AMPK promotes glucose disposal and glucose lowering in non-human primates and mice. Cell Metab. 2017, 25, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Kume, S. Pathophysiological roles of nutrient-sensing mechanisms in diabetes and its complications. Diabetol. Int. 2019, 10, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Barone, E.; Di Domenico, F.; Perluigi, M.; Butterfield, D.A. The interplay among oxidative stress, brain insulin resistance and AMPK dysfunction contribute to neurodegeneration in type 2 diabetes and Alzheimer disease. Free Radic. Biol. Med. 2021, 176, 16–33. [Google Scholar] [CrossRef]
- Liu, Y.; Jurczak, M.J.; Lear, T.B.; Lin, B.; Larsen, M.B.; Kennerdell, J.R.; Chen, Y.; Huckestein, B.R.; Nguyen, M.K.; Tuncer, F. Fbxo48 inhibitor prevents pAMPKalpha degradation and ameliorates insulin resistance. Nat. Chem. Biol. 2021, 17, 298–306. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kennedy, B.K.; Liao, C.Y. Mechanistic target of rapamycin signaling in mouse models of accelerated aging. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 64–72. [Google Scholar] [CrossRef]
- Carmona, J.J.; Michan, S. Biology of Healthy Aging and Longevity. Rev. Investig. Clin. 2016, 68, 7–16. [Google Scholar]
- Long, Y.C.; Tan, T.M.; Takao, I.; Tang, B.L. The biochemistry and cell biology of aging: Metabolic regulation through mitochondrial signaling. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E581–E591. [Google Scholar] [CrossRef] [Green Version]
- De Ávila, C.; Chometton, S.; Ma, S.; Pedersen, L.T.; Timofeeva, E.; Cifani, C.; Gundlach, A.L. Effects of chronic silencing of relaxin-3 production in nucleus incertus neurons on food intake, body weight, anxiety-like behaviour and limbic brain activity in female rats. Psychopharmacology 2020, 237, 1091–1106. [Google Scholar] [CrossRef]
- De Ávila, C.; Chometton, S.; Calvez, J.; Guèvremont, G.; Kania, A.; Torz, L.; Lenglos, C.; Blasiak, A.; Rosenkilde, M.M.; Holst, B. Estrous Cycle Modulation of Feeding and Relaxin-3/Rxfp3 mRNA Expression: Implications for Estradiol Action. Neuroendocrinology 2021, 111, 1201–1218. [Google Scholar] [CrossRef]
- Tanaka, M. Relaxin-3/insulin-like peptide 7, a neuropeptide involved in the stress response and food intake. FEBS J. 2010, 277, 4990–4997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Srikantan, S.; Tominaga, K.; Kang, M.; Yaniv, Y.; Martindale, J.L.; Yang, X.; Park, S.; Becker, K.G.; Subramanian, S.; et al. Growth inhibition by miR-519 via multiple p21-inducing pathways. Mol. Cell. Biol. 2012, 32, 2530–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Santos-Otte, P.; Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems and Their Role in Cellular Senescence. Comput. Struct. Biotechnol. J. 2019, 17, 1265–1277. [Google Scholar] [CrossRef]
- Donnelly, D.; Maudsley, S.; Gent, J.P.; Moser, R.N.; Hurrell, C.R.; Findlay, J.B.C. Conserved polar residues in the transmembrane domain of the human tachykinin NK2 receptor: Functional roles and structural implications. Biochem. J. 1999, 339, 55–61. [Google Scholar] [CrossRef]
- Wang, L.; Chadwick, W.; Park, S.; Zhou, Y.; Silver, N.; Martin, B.; Maudsley, S. Gonadotropin-releasing hormone receptor system: Modulatory role in aging and neurodegeneration. CNS Neurol. Disord. Drug Targets 2010, 9, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Gesty-Palmer, D.; Yuan, L.; Martin, B.; Wood, W.H., 3rd; Lee, M.; Janech, M.G.; Tsoi, L.C.; Zheng, J.; Luttrell, L.M.; Maudsley, S. β-Arrestin-Selective G Protein-Coupled Receptor Agonists Engender Unique Biological Efficacy in Vivo. Mol. Endo. 2013, 2, 296–314. [Google Scholar] [CrossRef] [Green Version]
- Maudsley, S.; Patel, S.A.; Park, S.; Luttrell, L.M.; Martin, B. Functional signaling biases in G protein-coupled receptors: Game Theory and receptor dynamics. Mini Rev. Med. Chem. 2012, 12, 831–840. [Google Scholar] [CrossRef]
- Van Gastel, J.; Leysen, H.; Boddaert, J.; Vangenechten, L.; Luttrell, L.M.; Martin, B.; Maudsley, S. Aging-related modifications to G protein-coupled receptor signaling diversity. Pharmacol. Ther. 2021, 223, 107793. [Google Scholar] [CrossRef]
- Gusach, A.; Maslov, I.; Luginina, A.; Borshchevskiy, V.; Mishin, A.; Cherezov, V. Beyond structure: Emerging approaches to study GPCR dynamics. Curr. Opin. Struct. Biol. 2020, 63, 18–25. [Google Scholar] [CrossRef]
- Leysen, H.; van Gastel, J.; Hendrickx, J.O.; Santos-Otte, P.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. Int. J. Mol. Sci. 2018, 19, 2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Lei, W.I.; Lee, L.T.O. The Role of Neuropeptide-Stimulated cAMP-EPACs Signalling in Cancer Cells. Molecules 2022, 27, 311. [Google Scholar] [CrossRef] [PubMed]
- Anckaerts, C.; van Gastel, J.; Leysen, V.; Hinz, R.; Azmi, A.; Simoens, P.; Shah, D.; Kara, F.; Langbeen, A.; Bols, P.; et al. Image-guided phenotyping of ovariectomized mice: Altered functional connectivity, cognition, myelination, and dopaminergic functionality. Neurobiol. Aging 2019, 74, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Meadows, K.L.; Byrnes, E.M. Sex- and age-specific differences in relaxin family peptide receptor expression within the hippocampus and amygdala in rats. Neuroscience 2015, 284, 337–348. [Google Scholar] [CrossRef]
- Salovska, B.; Kondelova, A.; Pimkova, K.; Liblova, Z.; Pribyl, M.; Fabrik, I.; Bartek, J.; Vajrychova, M.; Hodny, Z. Peroxiredoxin 6 protects irradiated cells from oxidative stress and shapes their senescence-associated cytokine landscape. Redox Biol. 2022, 49, 102212. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef]
- Maudsley, S.; Devanarayan, V.; Martin, B.; Geerts, H. Brain Health Modeling Initiative (BHMI). Intelligent and effective informatic deconvolution of “Big Data” and its future impact on the quantitative nature of neurodegenerative disease therapy. Alzheimers Dement. 2018, 14, 961–975. [Google Scholar] [CrossRef]
- Moons, R.; Konijnenberg, A.; Mensch, C.; Van Elzen, R.; Johannessen, C.; Maudsley, S.; Lambeir, A.M.; Sobott, F. Metal ions shape α-synuclein. Sci. Rep. 2020, 10, 16293. [Google Scholar] [CrossRef]
- Janssens, J.; Etienne, H.; Idriss, S.; Azmi, A.; Martin, B.; Maudsley, S. Systems-Level G Protein-Coupled Receptor Therapy Across a Neurodegenerative Continuum by the GLP-1 Receptor System. Front. Endocrinol. 2014, 5, 142. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Lopez de Maturana, R.; Brenneman, R.; Walent, T.; Mattson, M.P.; Maudsley, S. Class II G protein-coupled receptors and their ligands in neuronal function and protection. Neuromol. Med. 2005, 7, 3–36. [Google Scholar] [CrossRef] [Green Version]
- Korfei, M.; MacKenzie, B.; Meiners, S. The ageing lung under stress. Eur. Respir. Rev. 2020, 29, 200126. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.A.; Man, B.C.S.; Zhao, C.; Xu, Q.; Du, X.-J.; Wade, J.D.; Samuel, C.S. H3 Relaxin Demonstrates Antifibrotic Properties via the RXFP1 Receptor. Biochemistry 2011, 50, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fu, Y.; Li, H.; Shen, L.; Chang, Q.; Pan, L.; Hong, S.; Yin, X. H3 relaxin inhibits the collagen synthesis via ROS- and P2X7R-mediated NLRP3 inflammasome activation in cardiac fibroblasts under high glucose. J. Cell. Mol. Med. 2018, 22, 1816–1825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Pan, L.; Yang, K.; Fu, Y.; Liu, Y.; Chen, W.; Ma, X.; Yin, X. Alterations of relaxin and its receptor system components in experimental diabetic cardiomyopathy rats. Cell Tissue Res. 2017, 370, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Doyle, M.E.; Liu, Z.; Lao, Q.; Shin, Y.K.; Carlson, O.D.; Kim, H.S.; Thomas, S.; Napora, J.K.; Lee, E.K.; et al. Cannabinoids inhibit insulin receptor signaling in pancreatic beta-cells. Diabetes 2011, 60, 1198–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, B.; Shin, Y.K.; White, C.M.; Ji, S.; Kim, W.; Carlson, O.D.; Napora, J.K.; Chadwick, W.; Chapter, M.; Waschek, J.A.; et al. Vasoactive intestinal peptide-null mice demonstrate enhanced sweet taste preference, dysglycemia, and reduced taste bud leptin receptor expression. Diabetes 2010, 59, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapter, M.C.; White, C.M.; DeRidder, A.; Chadwick, W.; Martin, B.; Maudsley, S. Chemical modification of class II G protein-coupled receptor ligands: Frontiers in the development of peptide analogs as neuroendocrine pharmacological therapies. Pharmacol. Ther. 2010, 125, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Livak, M.F.A.; Bernier, M.; Muller, D.C.; Carlson, O.D.; Elahi, D.; Maudsley, S.; Egan, J.M. Ubiquitination is involved in glucose-mediated downregulation of GIP receptors in islets. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E538–E547. [Google Scholar] [CrossRef] [Green Version]
- Camell, C.D. Adipose tissue microenvironments during aging: Effects on stimulated lipolysis. Biochim. Biophys. Acta. Mol. Cell Biol. Lipids 2022, 1867, 159118. [Google Scholar] [CrossRef]
- Lu, B.; Huang, L.; Cao, J.; Li, L.; Wu, W.; Chen, X.; Ding, C.J. Adipose tissue macrophages in aging-associated adipose tissue function. Physiol. Sci. 2021, 71, 38. [Google Scholar] [CrossRef]
- Fonseca, G.W.P.D.; von Haehling, S. The fatter, the better in old age: The current understanding of a difficult relationship. Curr. Opin. Clin. Nutr. Metab. Care 2022, 25, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Shimokawa, H.; Haga, T.; Fukui, Y.; Iguchi, K.; Unno, K.; Hoshino, M.; Takeda, A. The expression of relaxin-3 in adipose tissue and its effects on adipogenesis. Protein Pept. Lett. 2014, 21, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Gallander, G.E.; Bathgate, R.A.D. Relaxin family peptide systems and the central nervous system. Cell. Mol. Life Sci. 2010, 67, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.; Skrobot, O.; Sanyoura, M.; Kay, V.; Susce, M.T.; Glaser, P.E.; de Leon, J.; Blakemore, A.I.; Arranz, M.J. Relaxin polymorphisms associated with metabolic disturbance in patients treated with antipsychotics. J. Psychopharmacol. 2012, 26, 374–379. [Google Scholar] [CrossRef] [PubMed]
- De Avila, C.; Chometton, S.; Lenglos, C.; Calvez, J.; Gundlach, A.L.; Timofeeva, E. Differential effects of relaxin-3 and a selective relaxin-3 receptor agonist on food and water intake and hypothalamic neuronal activity in rats. Behav. Brain Res. 2018, 336, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miquel, S.; Champ, C.; Day, J.; Aarts, E.; Bahr, B.A.; Bakker, M.; Bánáti, D.; Calabrese, V.; Cederholm, T.; Cryan, J.; et al. Poor cognitive ageing: Vulnerabilities, mechanisms and the impact of nutritional interventions. Ageing Res. Rev. 2018, 42, 40–55. [Google Scholar] [CrossRef] [Green Version]
- Maudsley, S.; Chadwick, W. Progressive and unconventional pharmacotherapeutic approaches to Alzheimer’s disease therapy. Curr. Alzheimer Res. 2012, 9, 1–4. [Google Scholar] [CrossRef]
- Haidar, M.; Tin, K.; Zhang, C.; Nategh, M.; Covita, J.; Wykes, A.D.; Rogers, J.; Gundlach, A.L. Septal GABA and Glutamate Neurons Express RXFP3 mRNA and Depletion of Septal RXFP3 Impaired Spatial Search Strategy and Long-Term Reference Memory in Adult Mice. Front. Neuroanat. 2019, 13, 30. [Google Scholar] [CrossRef] [Green Version]
- Haidar, M.; Guèvremont, G.; Zhang, C.; Bathgate, R.A.D.; Timofeeva, E.; Smith, C.M.; Gundlach, A.L. Relaxin-3 inputs target hippocampal interneurons and deletion of hilar relaxin-3 receptors in "floxed-RXFP3" mice impairs spatial memory. Hippocampus 2017, 27, 529–546. [Google Scholar] [CrossRef]
- García-Díaz, C.; Gil-Miravet, I.; Albert-Gasco, H.; Mañas-Ojeda, A.; Ros-Bernal, F.; Castillo-Gómez, E.; Gundlach, A.L.; Olucha-Bordonau, F.E. Relaxin-3 Innervation from the Nucleus Incertus to the Parahippocampal Cortex of the Rat. Front. Neuroanat. 2021, 15, 674649. [Google Scholar] [CrossRef]
- Vida, C.; González, E.M.; De la Fuente, M. Increase of oxidation and inflammation in nervous and immune systems with aging and anxiety. Curr. Pharm. Des. 2014, 20, 4656–4678. [Google Scholar] [CrossRef] [PubMed]
- Green, E.; Fairchild, J.K.; Kinoshita, L.M.; Noda, A.; Yesavage, J. Effects of Posttraumatic Stress Disorder and Metabolic Syndrome on Cognitive Aging in Veterans. Gerontologist 2016, 56, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutter, L.A.; Vahia, I.V.; Passell, E.; Forester, B.P.; Germine, L. The role of intraindividual cognitive variability in posttraumatic stress syndromes and cognitive aging: A literature search and proposed research agenda. Int. Psychogeriatr. 2021, 33, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Mañas-Ojeda, A.; Ros-Bernal, F.; Olucha-Bordonau, F.E.; Castillo-Gómez, E. Becoming Stressed: Does the Age Matter? Reviewing the Neurobiological and Socio-Affective Effects of Stress throughout the Lifespan. Int. J. Mol. Sci. 2020, 21, 5819. [Google Scholar] [CrossRef]
- Rytova, V.; Ganella, D.E.; Hawkes, D.; Bathgate, R.A.D.; Ma, S.; Gundlach, A.L. Chronic activation of the relaxin-3 receptor on GABA neurons in rat ventral hippocampus promotes anxiety and social avoidance. Hippocampus 2019, 29, 905–920. [Google Scholar] [CrossRef]
- Zhang, C.; Chua, B.E.; Yang, A.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Rosengren, K.J.; Smith, C.M.; Gundlach, A.L. Central relaxin-3 receptor (RXFP3) activation reduces elevated, but not basal, anxiety-like behaviour in C57BL/6J mice. Behav. Brain Res. 2015, 292, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Palmer, B.W.; Shir, C.; Chang, H.; Mulvaney, M.; Hall, J.M.H.; Shu, I.W.; Jin, H.; Lohr, J.B. Increased Prevalence of Metabolic Syndrome in Veterans with PTSD Untreated with Antipsychotic Medications. Int. J. Ment. Health 2021, 5, 10. [Google Scholar] [CrossRef]
- Mayer, A.; Mizdrak, M.; Babić, M.; Mastelić, T.; Glavina, T.; Božić, J.; Kurir, T.T. Knowledge, Attitudes, and Screening for Obstructive Sleep Apnea and Diabetes Mellitus among War Veterans Seeking Treatment of Posttraumatic Stress Disorder. Healthcare 2021, 9, 1698. [Google Scholar] [CrossRef]
- Schmalzigaug, R.; Rodriguiz, R.M.; Phillips, L.E.; Davidson, C.E.; Wetsel, W.C.; Premont, R.T. Anxiety-like behaviors in mice lacking GIT2. Neurosci. Lett. 2009, 451, 156–161. [Google Scholar] [CrossRef] [Green Version]
- Albert-Gasco, H.; Sanchez-Sarasua, S.; Ma, S.; García-Díaz, C.; Gundlach, A.L.; Sanchez-Perez, A.M.; Olucha-Bordonau, F.E. Central relaxin-3 receptor (RXFP3) activation impairs social recognition and modulates ERK-phosphorylation in specific GABAergic amygdala neurons. Brain Struct. Funct. 2019, 224, 453–469. [Google Scholar] [CrossRef]
- Ganella, D.E.; Ryan, P.J.; Bathgate, R.A.D.; Gundlach, A.L. Increased feeding and body weight gain in rats after acute and chronic activation of RXFP3 by relaxin-3 and receptor-selective peptides: Functional and therapeutic implications. Behav. Pharmacol. 2012, 23, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Kania, A.; Szlaga, A.; Sambak, P.; Gugula, A.; Blasiak, E.; Di Bonaventura, M.V.M.; Hossain, M.A.; Cifani, C.; Hess, G.; Gundlach, A.L.; et al. RLN3/RXFP3 Signaling in the PVN Inhibits Magnocellular Neurons via M-like Current Activation and Contributes to Binge Eating Behavior. J. Neurosci. 2020, 40, 5362–5375. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, B.; Park, S.; Vong, L.; Bamford, D.; Cho, H.; Duvadie, R.; Fedolak, A.; Hogan, C.; Honda, T.; Pandey, P.; et al. Discovery of a small molecule RXFP3/4 agonist that increases food intake in rats upon acute central administration. Bioorg. Med. Chem. Lett. 2019, 29, 991–994. [Google Scholar] [CrossRef] [PubMed]
- Walker, A.W.; Smith, C.M.; Chua, B.E.; Krstew, E.V.; Zhang, C.; Gundlach, A.L.; Lawrence, A.J. Relaxin-3 receptor (RXFP3) signalling mediates stress-related alcohol preference in mice. PLoS ONE 2015, 10, e0122504. [Google Scholar] [CrossRef]
- Van Gastel, J.; Hendrickx, J.O.; Leysen, H.; Martin, B.; Veenker, L.; Beuning, S.; Coppens, V.; Morrens, M.; Maudsley, S. Enhanced Molecular Appreciation of Psychiatric Disorders Through High-Dimensionality Data Acquisition and Analytics. Methods Mol. Biol. 2019, 2011, 671–723. [Google Scholar]
- Solomon, H.V.; Sinopoli, M.; DeLisi, L.E. Ageing with schizophrenia: An update. Curr. Opin. Psychiatry 2021, 34, 266–274. [Google Scholar] [CrossRef]
- Jones, R.; MacCabe, J.H.; Price, M.J.; Liu, X.; Upthegrove, R. Effect of age on the relative efficacy of clozapine in schizophrenia. Acta Psychiatr. Scand. 2020, 142, 109–120. [Google Scholar] [CrossRef]
- Kumar, J.R.; Rajkumar, R.; Jayakody, T.; Marwari, S.; Mei Hong, J.; Ma, S. Relaxin’ the brain: A case for targeting the nucleus incertus network and relaxin-3/RXFP3 system in neuropsychiatric disorders. Br. J. Pharmacol. 2017, 174, 1061–1076. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.M.; Walker, A.W.; Hosken, I.T.; Chua, B.E.; Zhang, C.; Haidar, M.; Gundlach, A.L. Relaxin-3/RXFP3 networks: An emerging target for the treatment of depression and other neuropsychiatric diseases? Front. Pharmacol. 2014, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Hosken, I.T.; Sutton, S.W.; Smith, C.M.; Gundlach, A.L. Relaxin-3 receptor (Rxfp3) gene knockout mice display reduced running wheel activity: Implications for role of relaxin-3/RXFP3 signalling in sustained arousal. Behav. Brain Res. 2015, 278, 167–175. [Google Scholar] [CrossRef]
- Lee, S.; Huang, K. Epigenetic profiling of human brain differential DNA methylation networks in schizophrenia. BMC Med. Genomics 2016, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattison, J.A.; Wang, M.; Bernier, M.; Zhang, J.; Park, S.; Maudsley, S.; An, S.S.; Santhanam, L.; Martin, B.; Faulkner, S.; et al. Resveratrol prevents high fat/sucrose diet-induced central arterial wall inflammation and stiffening in nonhuman primates. Cell Metab. 2014, 20, 183–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colleluori, G.; Villareal, D.T. Aging, obesity, sarcopenia and the effect of diet and exercise intervention. Exp. Gerontol. 2021, 155, 111561. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Cuevas, J.; Galicia-Moreno, M.; Monroy-Ramírez, H.C.; Sandoval-Rodriguez, A.; García-Bañuelos, J.; Santos, A.; Armendariz-Borunda, J. The Role of NRF2 in Obesity-Associated Cardiovascular Risk Factors. Antioxidants 2022, 11, 235. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef]
- Mezhnina, V.; Ebeigbe, O.P.; Poe, A.; Kondratov, R.V. Circadian Control of Mitochondria in Reactive Oxygen Species Homeostasis. Antioxid. Redox Signal. 2022, in press. [Google Scholar] [CrossRef]
- Smith, C.M.; Chua, B.E.; Zhang, C.; Walker, A.W.; Haidar, M.; Hawkes, D.; Shabanpoor, F.; Hossain, M.A.; Wade, J.D.; Rosengren, K.J.; et al. Central injection of relaxin-3 receptor (RXFP3) antagonist peptides reduces motivated food seeking and consumption in C57BL/6J mice. Behav. Brain Res. 2014, 268, 117–126. [Google Scholar] [CrossRef]
- Calvez, J.; de Ávila, C.; Timofeeva, E. Sex-specific effects of relaxin-3 on food intake and body weight gain. Br. J. Pharmacol. 2017, 174, 1049–1060. [Google Scholar] [CrossRef] [Green Version]
- Lenglos, C.; Mitra, A.; Guèvremont, G.; Timofeeva, E. Regulation of expression of relaxin-3 and its receptor RXFP3 in the brain of diet-induced obese rats. Neuropeptides 2014, 48, 119–132. [Google Scholar] [CrossRef]
- Hida, T.; Takahashi, E.; Shikata, K.; Hirohashi, T.; Sawai, T.; Seiki, T.; Tanaka, H.; Kawai, T.; Ito, O.; Arai, T.; et al. Chronic intracerebroventricular administration of relaxin-3 increases body weight in rats. J. Recept. Signal Transduct. Res. 2006, 26, 147–158. [Google Scholar] [CrossRef]
- Hossain, M.A.; Smith, C.M.; Ryan, P.J.; Büchler, E.; Bathgate, R.A.; Gundlach, A.L.; Wade, J.D. Chemical synthesis and orexigenic activity of rat/mouse relaxin-3. Amino Acids. 2013, 44, 1529–1536. [Google Scholar] [CrossRef] [PubMed]
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef] [PubMed]
- Yousufuddin, M.; Young, N. Aging and ischemic stroke. Aging 2019, 11, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Podolak, A.; Woclawek-Potocka, I.; Lukaszuk, K. The Role of Mitochondria in Human Fertility and Early Embryo Development: What Can We Learn for Clinical Application of Assessing and Improving Mitochondrial DNA? Cells 2022, 11, 797. [Google Scholar] [CrossRef]
- DePina, A.S.; Iser, W.B.; Park, S.S.; Maudsley, S.; Wilson, M.A.; Wolkow, C.A. Regulation of Caenorhabditis elegans vitellogenesis by DAF-2/IIS through separable transcriptional and posttranscriptional mechanisms. BMC Physiol. 2011, 11, 11. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Golden, E.; Carlson, O.D.; Egan, J.M.; Mattson, M.P.; Maudsley, S. Caloric restriction: Impact upon pituitary function and reproduction. Ageing Res. Rev. 2008, 7, 209–224. [Google Scholar] [CrossRef] [Green Version]
- Ivell, R.; Kotula-Balak, M.; Glynn, D.; Heng, K.; Anand-Ivell, R. Relaxin family peptides in the male reproductive system--a critical appraisal. Mol. Hum. Reprod. 2011, 17, 71–84. [Google Scholar] [CrossRef]
- Wilson, B.C.; Burnett, D.; Rappaport, R.; Parry, L.J.; Fletcher, E.K. Relaxin-3 and RXFP3 expression, and steroidogenic actions in the ovary of teleost fish. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2009, 153, 69–74. [Google Scholar] [CrossRef]
- McGowan, B.M.; Stanley, S.A.; Donovan, J.; Thompson, E.L.; Patterson, M.; Semjonous, N.M.; Gardiner, J.V.; Murphy, K.G.; Ghatei, M.A.; Bloom, S.R. Relaxin-3 stimulates the hypothalamic-pituitary-gonadal axis. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E278–E286. [Google Scholar] [CrossRef]
- Pomrenze, M.B.; Millan, E.Z.; Hopf, F.W.; Keiflin, R.; Maiya, R.; Blasio, A.; Dadgar, J.; Khazaria, V.; De Guglielmo, G.; Crawford, E.; et al. A transgenic rat for investigating the anatomy and function of corticotrophin releasing factor circuits. Front. Neurosci. 2015, 9, 487. [Google Scholar] [CrossRef]
- Kilpatrick, D.G.; McAlhany, D.A.; McCurdy, R.L.; Shaw, D.L.; Roitzsch, J.C. Aging, alcoholism, anxiety, and sensation seeking: An exploratory investigation. Addict. Behav. 1982, 7, 97–100. [Google Scholar] [CrossRef]
- Matthews, D.B.; Imhoff, B.M. Age modifies the effect of ethanol on behavior: Investigations in adolescent, adult and aged rats. Int. Rev. Neurobiol. 2022, 161, 251–275. [Google Scholar] [PubMed]
- Leber, W.R.; Parsons, O.A. Premature aging and alcoholism. Int. J. Addict. 1982, 17, 61–88. [Google Scholar] [CrossRef] [PubMed]
- Topiwala, A.; Ebmeier, K.P. Effects of drinking on late-life brain and cognition. Evid. Based Ment. Health 2018, 21, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.V.; Pfefferbaum, A. Brain-behavior relations and effects of aging and common comorbidities in alcohol use disorder: A review. Neuropsychology 2019, 33, 760–780. [Google Scholar] [CrossRef] [PubMed]
- León, B.E.; Kang, S.; Franca-Solomon, G.; Shang, P.; Choi, D.S. Alcohol-Induced Neuroinflammatory Response and Mitochondrial Dysfunction on Aging and Alzheimer’s Disease. Front. Behav. Neurosci. 2022, 15, 778456. [Google Scholar] [CrossRef]
- Nunes, P.T.; Kipp, B.T.; Reitz, N.L.; Savage, L.M. Aging with alcohol-related brain damage: Critical brain circuits associated with cognitive dysfunction. Int. Rev. Neurobiol. 2019, 148, 101–168. [Google Scholar]
- Shirahase, T.; Aoki, M.; Watanabe, R.; Watanabe, Y.; Tanaka, M. Increased alcohol consumption in relaxin-3 deficient male mice. Neurosci. Lett. 2016, 612, 155–160. [Google Scholar] [CrossRef]
- Zhang, B.; Trapp, A.; Kerepesi, C.; Gladyshev, V.N. Emerging rejuvenation strategies—Reducing the biological age. Aging Cell 2022, 21, e13538. [Google Scholar] [CrossRef]
- Shetty, A.K.; Kodali, M.; Upadhya, R.; Madhu, L.N. Emerging Anti-Aging Strategies—Scientific Basis and Efficacy. Aging Dis. 2018, 9, 1165–1184. [Google Scholar] [CrossRef] [Green Version]
- Mehdi, M.M.; Solanki, P.; Singh, P. Oxidative stress, antioxidants, hormesis and calorie restriction: The current perspective in the biology of aging. Arch. Gerontol. Geriatr. 2021, 95, 104413. [Google Scholar] [CrossRef] [PubMed]
- Davidsohn, N.; Pezone, M.; Vernet, A.; Graveline, A.; Oliver, D.; Slomovic, S.; Punthambaker, S.; Sun, X.; Liao, R.; Bonventre, J.V.; et al. A single combination gene therapy treats multiple age-related diseases. Proc. Natl. Acad. Sci. USA 2019, 116, 23505–23511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Brommer, B.; Tian, X.; Krishnan, A.; Meer, M.; Wang, C.; Vera, D.L.; Zeng, Q.; Yu, D.; Bonkowski, M.S.; et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature 2020, 588, 124–129. [Google Scholar] [CrossRef]
- Stranahan, A.M.; Zhou, Y.; Martin, B.; Maudsley, S. Pharmacomimetics of exercise: Novel approaches for hippocampally-targeted neuroprotective agents. Curr. Med. Chem. 2009, 16, 4668–4678. [Google Scholar] [CrossRef] [Green Version]
- Stranahan, A.M.; Martin, B.; Maudsley, S. Anti-inflammatory effects of physical activity in relationship to improved cognitive status in humans and mouse models of Alzheimer’s disease. Curr. Alzheimer Res. 2012, 9, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Alhassan, S.; Kim, S.; Bersamin, A.; King, A.C.; Gardner, C.D. Dietary adherence and weight loss success among overweight women: Results from the A to Z weight loss study. Int. J. Obes. 2008, 32, 985–991. [Google Scholar] [CrossRef] [Green Version]
- Cypser, J.R.; Tedesco, P.; Johnson, T.E. Hormesis and aging in Caenorhabditis elegans. Exp. Gerontol. 2006, 41, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Gems, D.; Partridge, L. Stress-response hormesis and aging: “That which does not kill us makes us stronger”. Cell Metab. 2008, 7, 200–203. [Google Scholar] [CrossRef] [Green Version]
- Chadwick, W.; Maudsley, S. The devil is in the dose: Complexity of receptor systems and responses. In Hormesis; Mattson, M., Calabrese, E., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 95–108. [Google Scholar] [CrossRef]
- Ravichandran, S.; Singh, N.; Donnelly, D.; Migliore, M.; Johnson, P.; Fishwick, C.; Luke, B.T.; Martin, B.; Maudsley, S.; Fugmann, S.D.; et al. Pharmacophore model of the quercetin binding site of the SIRT6 protein. J. Mol. Graph. Model. 2014, 49, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Liao, D.; Dong, Y.; Pu, R. Clinical effectiveness of quercetin supplementation in the management of weight loss: A pooled analysis of randomized controlled trials. Diabetes Metab. Syndr. Obes. 2019, 12, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leysen, H.; Walter, D.; Clauwaert, L.; Hellemans, L.; van Gastel, J.; Vasudevan, L.; Martin, B.; Maudsley, S. The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease. Int. J. Mol. Sci. 2022, 23, 4387. https://doi.org/10.3390/ijms23084387
Leysen H, Walter D, Clauwaert L, Hellemans L, van Gastel J, Vasudevan L, Martin B, Maudsley S. The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease. International Journal of Molecular Sciences. 2022; 23(8):4387. https://doi.org/10.3390/ijms23084387
Chicago/Turabian StyleLeysen, Hanne, Deborah Walter, Lore Clauwaert, Lieselot Hellemans, Jaana van Gastel, Lakshmi Vasudevan, Bronwen Martin, and Stuart Maudsley. 2022. "The Relaxin-3 Receptor, RXFP3, Is a Modulator of Aging-Related Disease" International Journal of Molecular Sciences 23, no. 8: 4387. https://doi.org/10.3390/ijms23084387