
Role of Bevacizumab on Vascular Endothelial Growth Factor in Apolipoprotein E Deficient Mice after Traumatic Brain Injury

 , , , , , , , , , and
, , , , , , , , , and 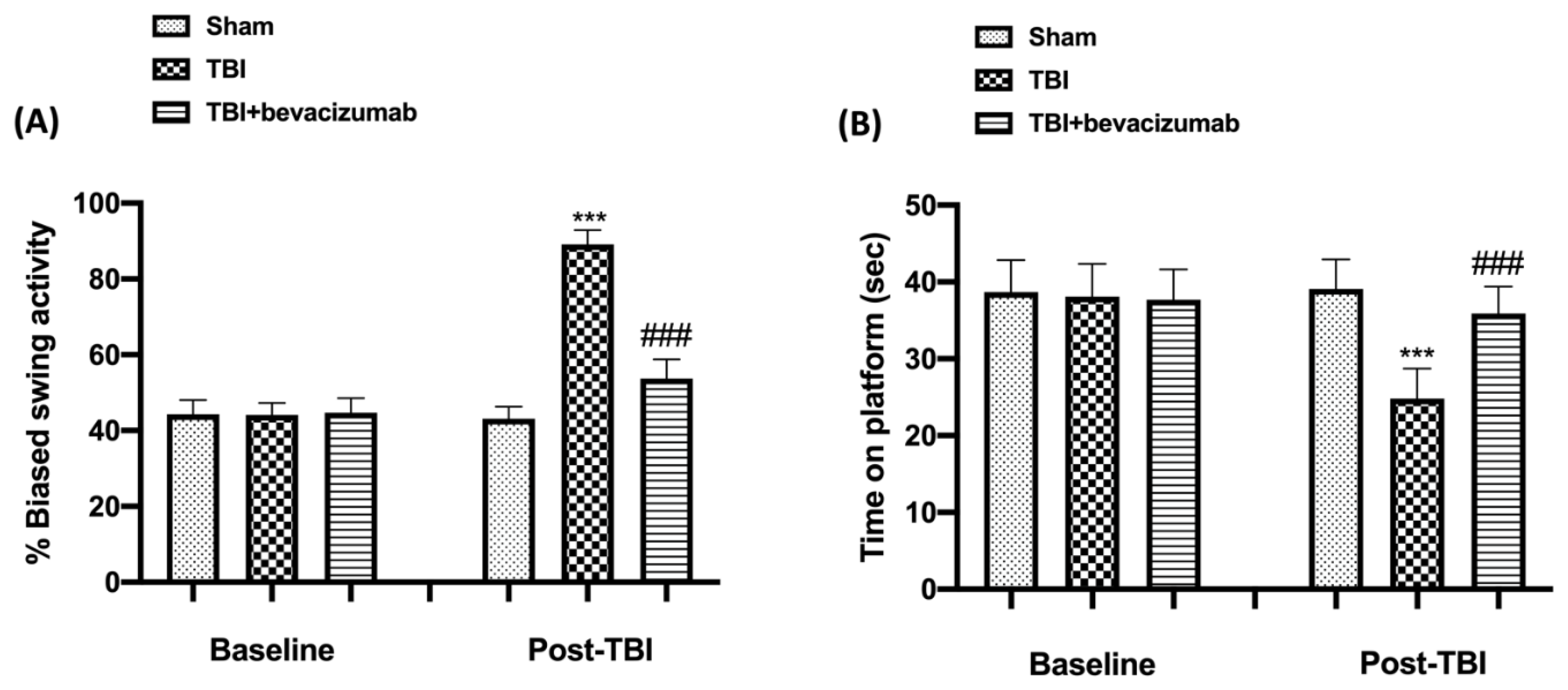{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Bevacizumab on Behavioral Function after TBI
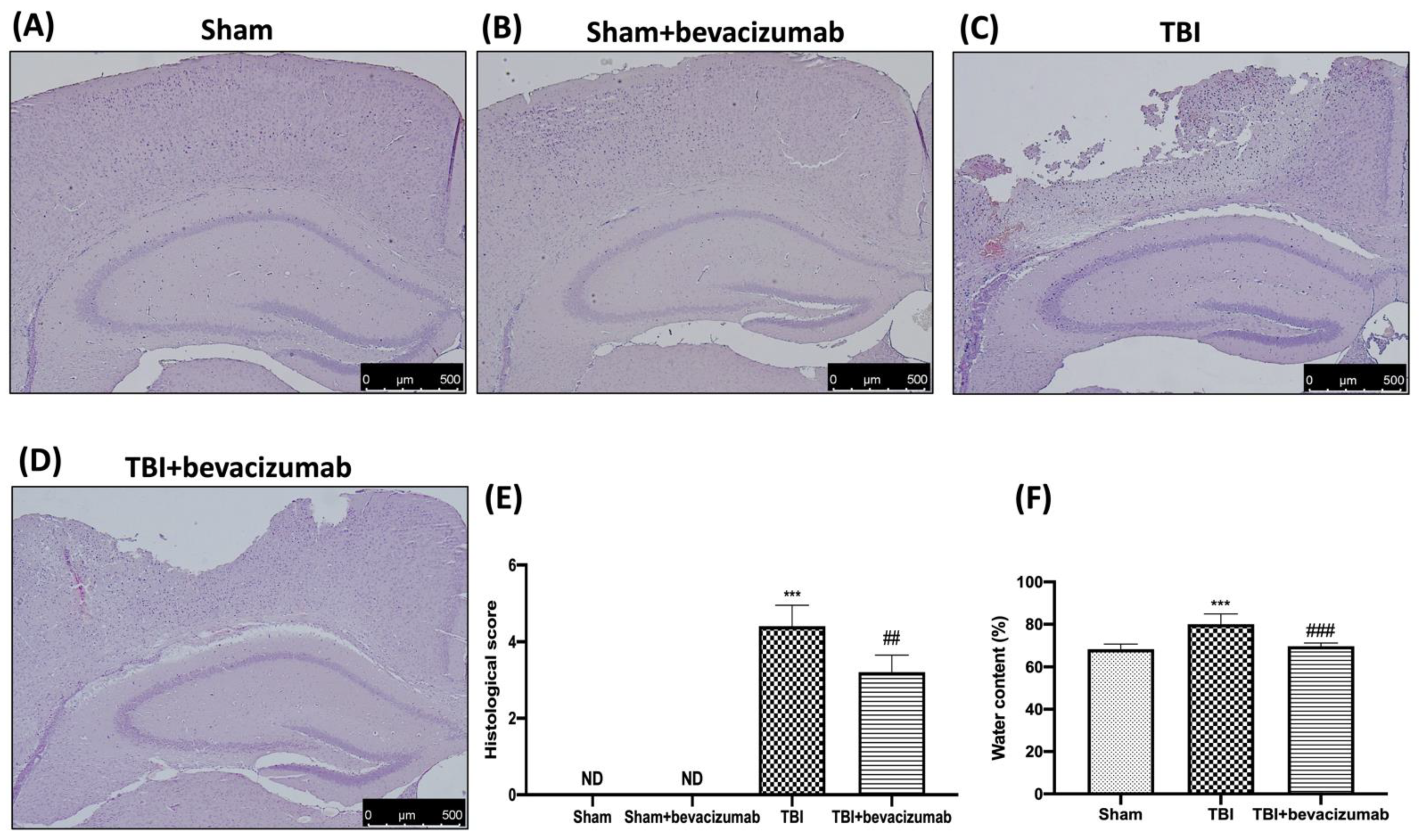
2.2. Bevacizumab Reduces Edema and the Severity of Brain Trauma
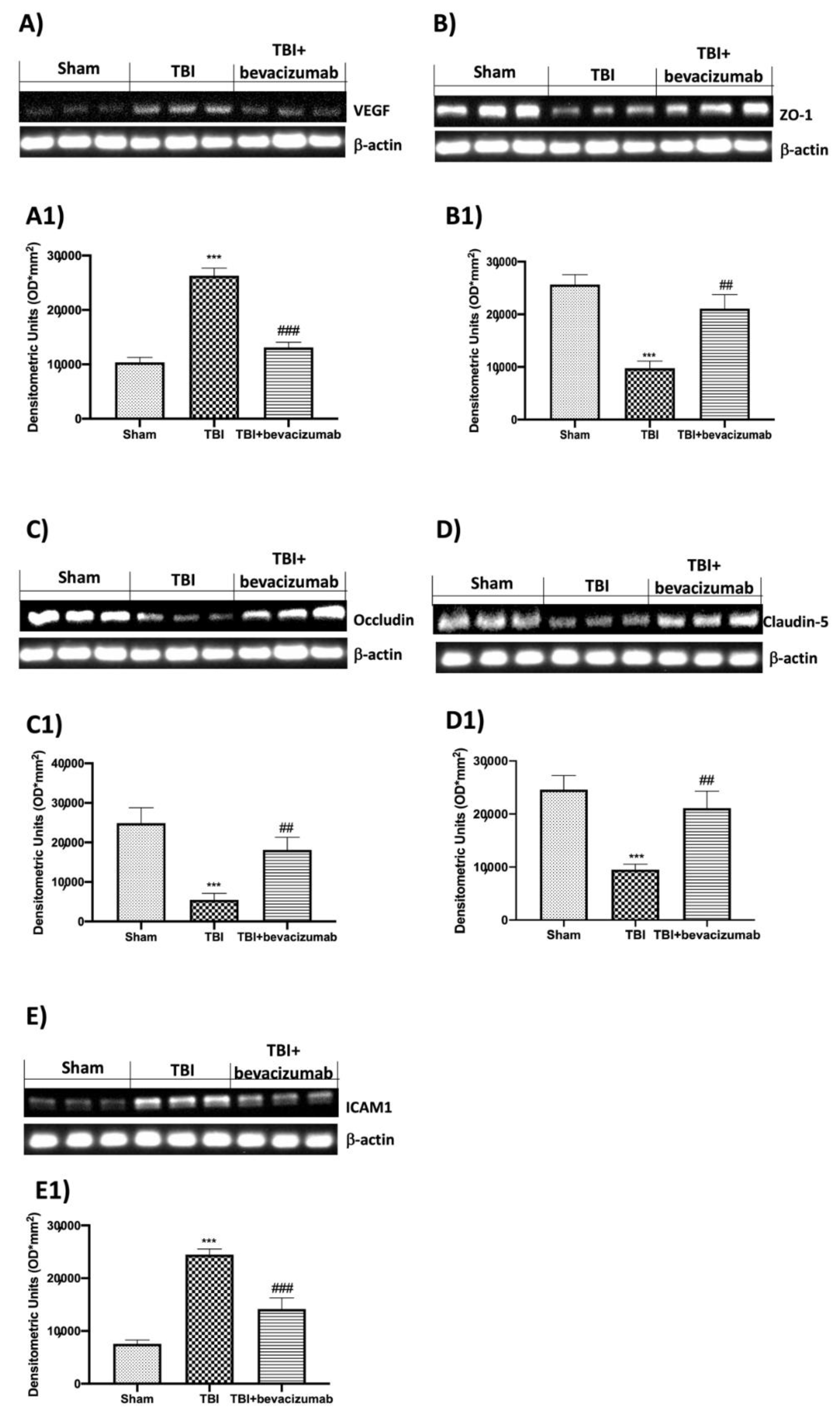
2.3. Effect of Bevacizumab on VEGF and BBB Integrity
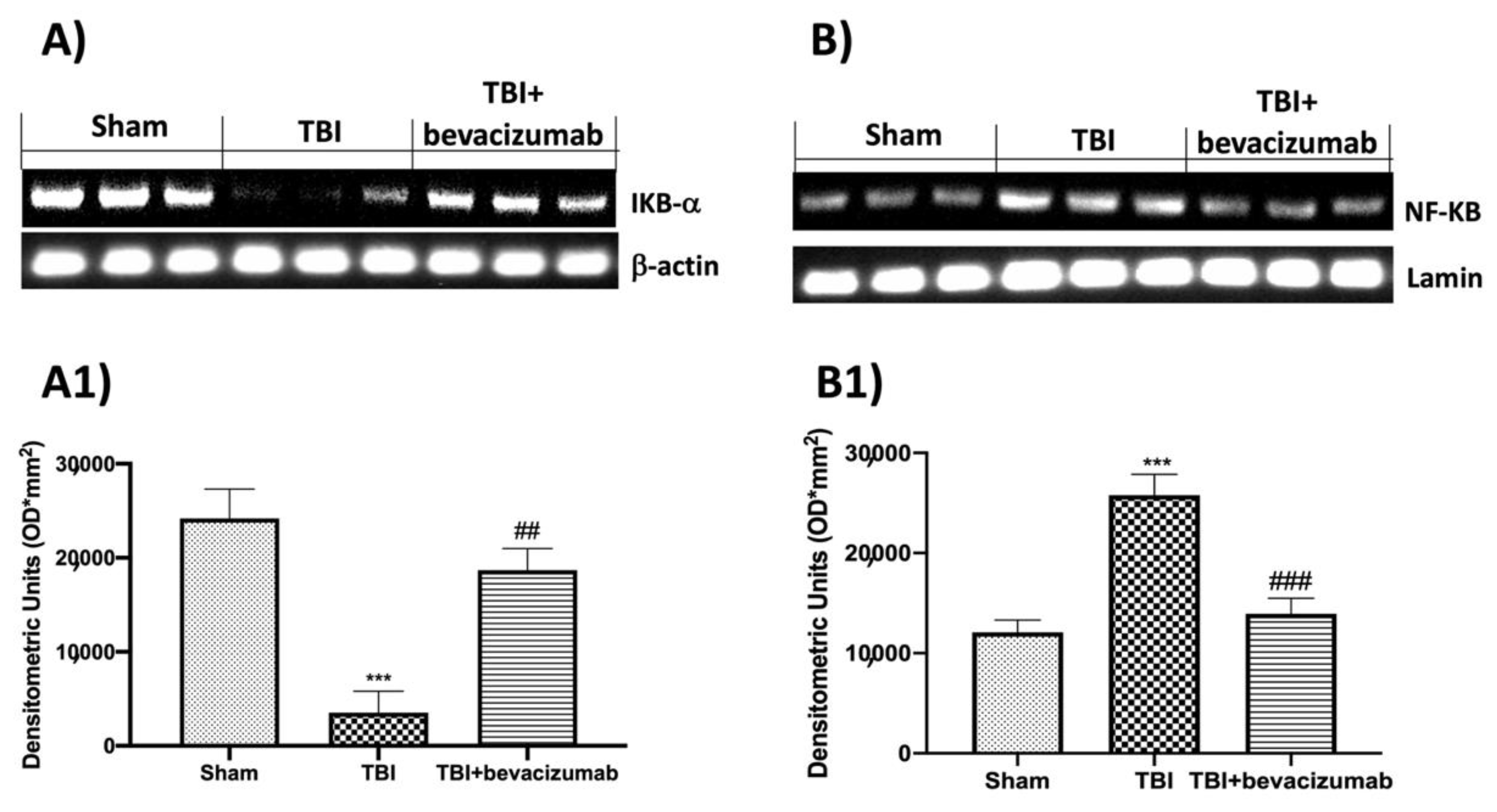
2.4. Bevacizumab Modulates NF-kB Pathway after TBI
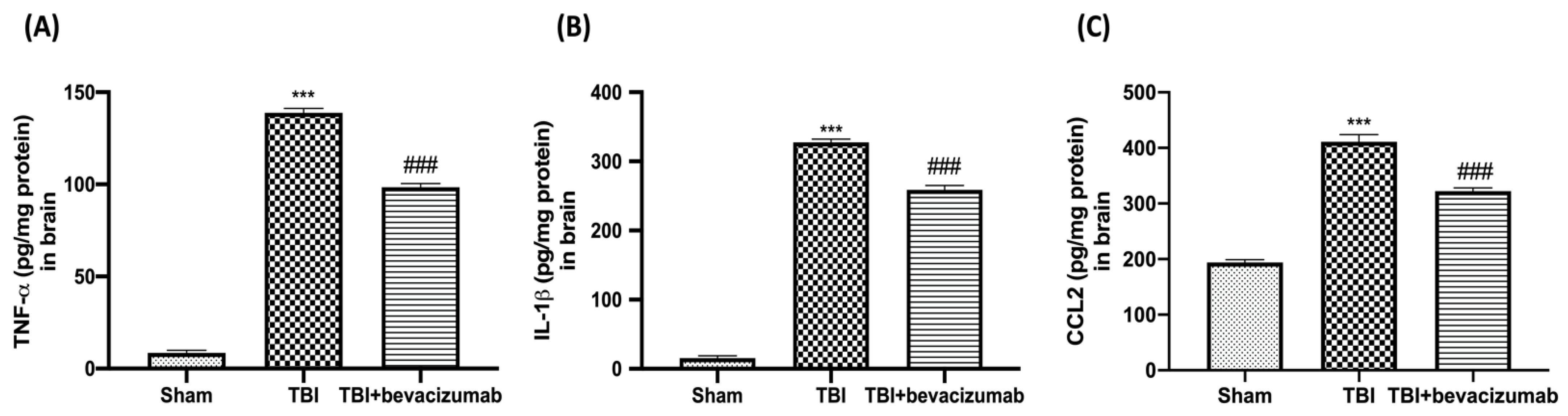
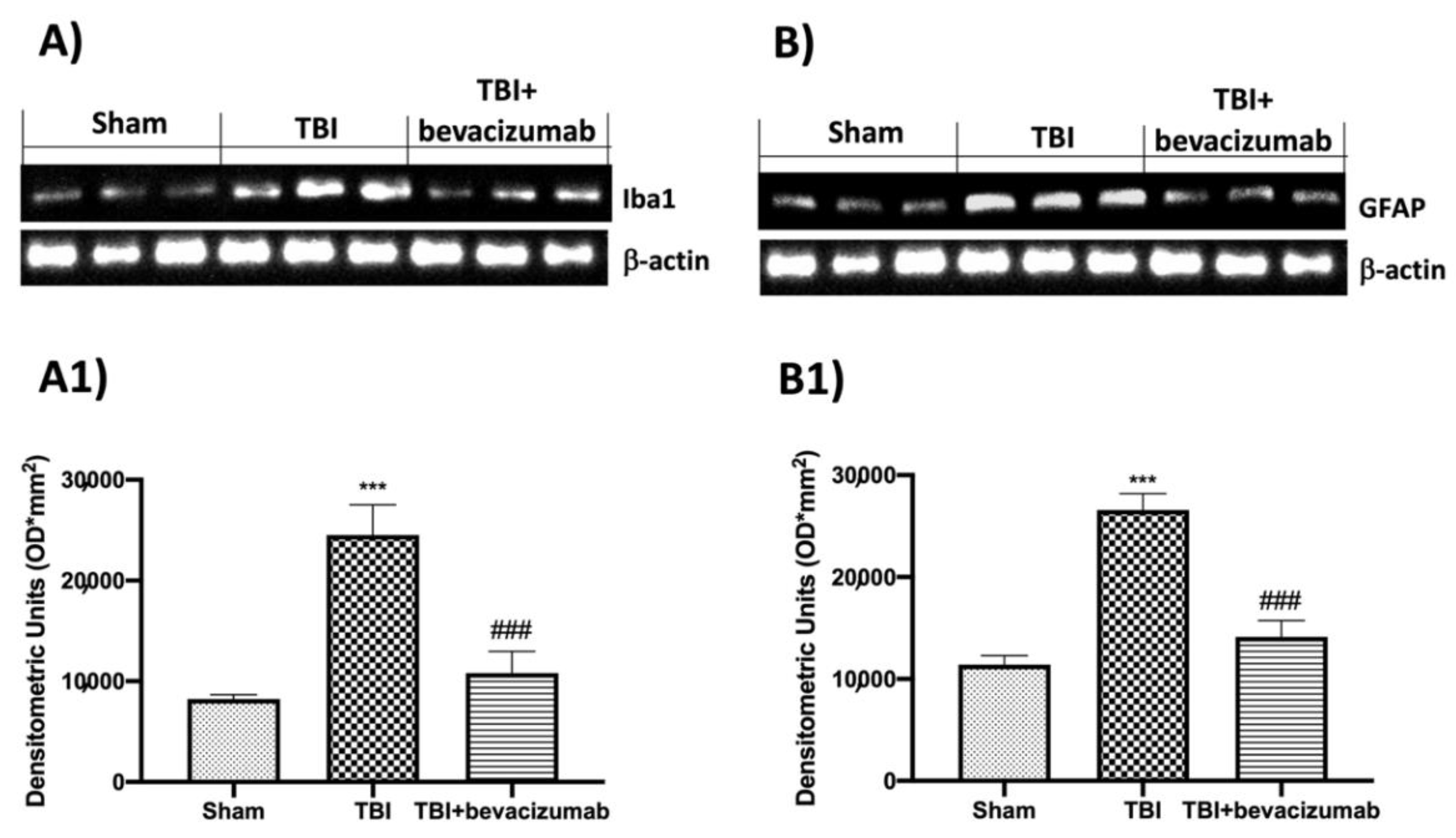
2.5. Bevacizumab Reduced Inflammatory Response and Astrocytes and Microglia Activation
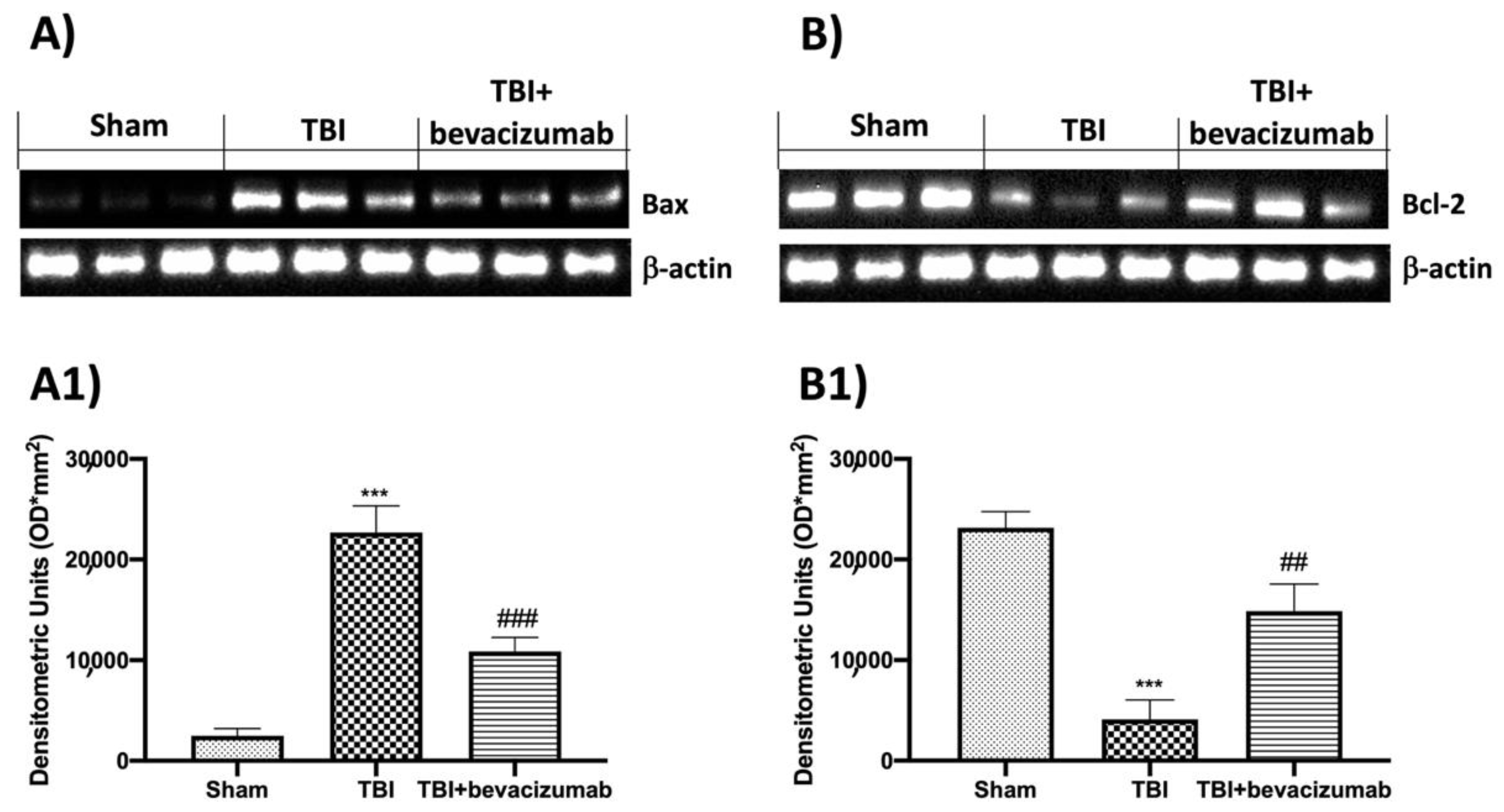
2.6. Effects of Bevacizumab Treatment on Apoptosis Pathway after TBI
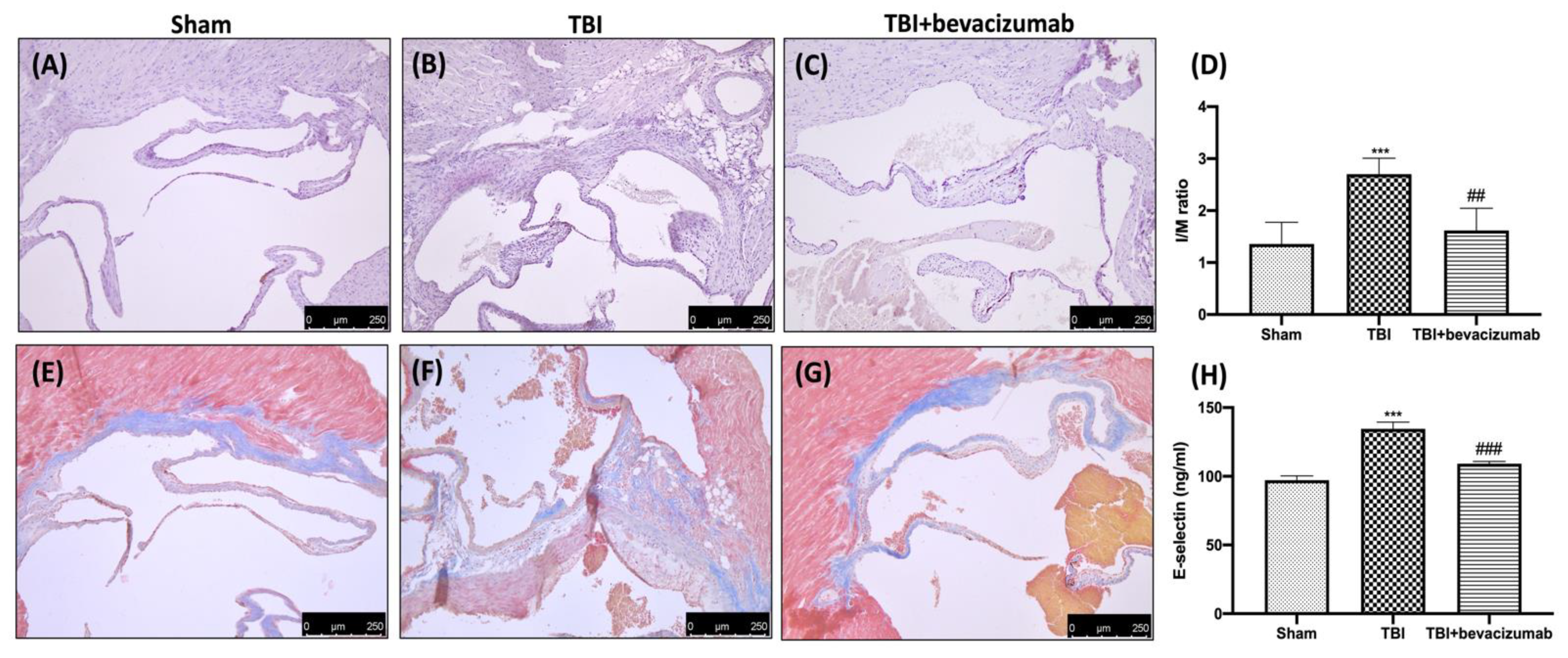
2.7. Effects of Bevacizumab on Atherosclerosis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Controlled Cortical Impact (CCI) Experimental Traumatic Brain Injury TBI
4.3. Experimental Groups
4.4. Behavioral Testing
4.4.1. Rotarod Test (RT)
4.4.2. Elevated Body Swing Test (EBST)
4.5. Measurement of Edema (Brain Water Content)
4.6. Histological Analysis
4.7. Western Blot Analysis
4.8. Cytokine and Soluble E-Selectin Measurements
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dewan, M.C.; Rattani, A.; Gupta, S.; Baticulon, R.E.; Hung, Y.C.; Punchak, M.; Agrawal, A.; Adeleye, A.O.; Shrime, M.G.; Rubiano, A.M.; et al. Estimating the global incidence of traumatic brain injury. J. Neurosurg. 2018, 130, 1080–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kureshi, N.; Erdogan, M.; Thibault-Halman, G.; Fenerty, L.; Green, R.S.; Clarke, D.B. Long-Term Trends in the Epidemiology of Major Traumatic Brain Injury. J. Community Health 2021, 46, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Trovato Salinaro, A.; Fusco, R.; Cordaro, M.; Impellizzeri, D.; Scuto, M.; Ontario, M.L.; Lo Dico, G.; Cuzzocrea, S.; Di Paola, R.; et al. Hericium erinaceus and Coriolus versicolor Modulate Molecular and Biochemical Changes after Traumatic Brain Injury. Antioxidants 2021, 10, 898. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Wu, J.; Stoica, B.A.; Loane, D.J. Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br. J. Pharmacol. 2016, 173, 681–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unterberg, A.W.; Stover, J.; Kress, B.; Kiening, K.L. Edema and brain trauma. Neuroscience 2004, 129, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Alluri, H.; Wiggins-Dohlvik, K.; Davis, M.L.; Huang, J.H.; Tharakan, B. Blood-brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis. 2015, 30, 1093–1104. [Google Scholar] [CrossRef]
- Thal, S.C.; Neuhaus, W. The blood-brain barrier as a target in traumatic brain injury treatment. Arch. Med. Res. 2014, 45, 698–710. [Google Scholar] [CrossRef]
- Jha, R.M.; Kochanek, P.M.; Simard, J.M. Pathophysiology and treatment of cerebral edema in traumatic brain injury. Neuropharmacology 2019, 145, 230–246. [Google Scholar] [CrossRef]
- Michinaga, S.; Kimura, A.; Hatanaka, S.; Minami, S.; Asano, A.; Ikushima, Y.; Matsui, S.; Toriyama, Y.; Fujii, M.; Koyama, Y. Delayed Administration of BQ788, an ETB Antagonist, after Experimental Traumatic Brain Injury Promotes Recovery of Blood-Brain Barrier Function and a Reduction of Cerebral Edema in Mice. J. Neurotrauma 2018, 35, 1481–1494. [Google Scholar] [CrossRef]
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef]
- Muradashvili, N.; Benton, R.L.; Saatman, K.E.; Tyagi, S.C.; Lominadze, D. Ablation of matrix metalloproteinase-9 gene decreases cerebrovascular permeability and fibrinogen deposition post traumatic brain injury in mice. Metab. Brain Dis. 2015, 30, 411–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, W.; Zhao, Z.; Yu, G.; Zhou, Z.; Zhou, Y.; Hu, T.; Jiang, R.; Zhang, J. VEGI attenuates the inflammatory injury and disruption of blood-brain barrier partly by suppressing the TLR4/NF-kappaB signaling pathway in experimental traumatic brain injury. Brain Res. 2015, 1622, 230–239. [Google Scholar] [CrossRef] [PubMed]
- Jia, F.; Yin, Y.H.; Gao, G.Y.; Wang, Y.; Cen, L.; Jiang, J.Y. MMP-9 inhibitor SB-3CT attenuates behavioral impairments and hippocampal loss after traumatic brain injury in rat. J. Neurotrauma 2014, 31, 1225–1234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Investig. 2003, 111, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Peak, S.J.; Levin, V.A. Role of bevacizumab therapy in the management of glioblastoma. Cancer Manag. Res. 2010, 2, 97–104. [Google Scholar] [PubMed]
- Strickler, J.H.; Hurwitz, H.I. Bevacizumab-based therapies in the first-line treatment of metastatic colorectal cancer. Oncologist 2012, 17, 513–524. [Google Scholar] [CrossRef] [Green Version]
- Diabetic Retinopathy Clinical Research, N.; Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Aiello, L.P.; Antoszyk, A.N.; Arnold-Bush, B.; Baker, C.W.; Bressler, N.M.; et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N Engl. J. Med. 2015, 372, 1193–1203. [Google Scholar] [CrossRef] [Green Version]
- Bourdages, M.; Bigras, J.L.; Farrell, C.A.; Hutchison, J.S.; Lacroix, J.; Canadian Critical Care Trials, G. Cardiac arrhythmias associated with severe traumatic brain injury and hypothermia therapy. Pediatr Crit. Care Med. 2010, 11, 408–414. [Google Scholar] [CrossRef]
- Riera, M.; Llompart-Pou, J.A.; Carrillo, A.; Blanco, C. Head injury and inverted Takotsubo cardiomyopathy. J. Trauma 2010, 68, E13–E15. [Google Scholar] [CrossRef]
- Krishnamoorthy, V.; Prathep, S.; Sharma, D.; Gibbons, E.; Vavilala, M.S. Association between electrocardiographic findings and cardiac dysfunction in adult isolated traumatic brain injury. Indian J. Crit. Care Med. 2014, 18, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Ahmadi, N.; Hajsadeghi, F.; Yehuda, R.; Anderson, N.; Garfield, D.; Ludmer, C.; Vaidya, N. Traumatic brain injury, coronary atherosclerosis and cardiovascular mortality. Brain Inj. 2015, 29, 1635–1641. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Young, P.R.; Barone, F.C.; Feuerstein, G.Z.; Smith, D.H.; McIntosh, T.K. Experimental brain injury induces differential expression of tumor necrosis factor-alpha mRNA in the CNS. Brain Res. Mol. Brain Res. 1996, 36, 287–291. [Google Scholar] [CrossRef]
- Perez-Polo, J.R.; Rea, H.C.; Johnson, K.M.; Parsley, M.A.; Unabia, G.C.; Xu, G.; Infante, S.K.; Dewitt, D.S.; Hulsebosch, C.E. Inflammatory consequences in a rodent model of mild traumatic brain injury. J. Neurotrauma 2013, 30, 727–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Battista, A.P.; Rizoli, S.B.; Lejnieks, B.; Min, A.; Shiu, M.Y.; Peng, H.T.; Baker, A.J.; Hutchison, M.G.; Churchill, N.; Inaba, K.; et al. Sympathoadrenal Activation is Associated with Acute Traumatic Coagulopathy and Endotheliopathy in Isolated Brain Injury. Shock 2016, 46, 96–103. [Google Scholar] [CrossRef]
- Devoto, C.; Arcurio, L.; Fetta, J.; Ley, M.; Rodney, T.; Kanefsky, R.; Gill, J. Inflammation Relates to Chronic Behavioral and Neurological Symptoms in Military Personnel with Traumatic Brain Injuries. Cell Transplant. 2017, 26, 1169–1177. [Google Scholar] [CrossRef]
- Lin, W.S.; Lin, C.S.; Liou, J.T.; Lin, W.Y.; Lin, C.L.; Cheng, S.M.; Lin, I.C.; Kao, C.H. Risk of Coronary Artery Disease in Patients With Traumatic Intracranial Hemorrhage: A Nationwide, Population-Based Cohort Study. Medicine 2015, 94, e2284. [Google Scholar] [CrossRef]
- Wang, J.; Su, E.; Wang, H.; Guo, C.; Lawrence, D.A.; Eitzman, D.T. Traumatic Brain Injury Leads to Accelerated Atherosclerosis in Apolipoprotein E Deficient Mice. Sci. Rep. 2018, 8, 5639. [Google Scholar] [CrossRef] [Green Version]
- Mellergard, P.; Sjogren, F.; Hillman, J. Release of VEGF and FGF in the extracellular space following severe subarachnoidal haemorrhage or traumatic head injury in humans. Br. J. Neurosurg. 2010, 24, 261–267. [Google Scholar] [CrossRef]
- Slevin, M.; Krupinski, J.; Slowik, A.; Kumar, P.; Szczudlik, A.; Gaffney, J. Serial measurement of vascular endothelial growth factor and transforming growth factor-beta1 in serum of patients with acute ischemic stroke. Stroke 2000, 31, 1863–1870. [Google Scholar] [CrossRef]
- Pikula, A.; Beiser, A.S.; Chen, T.C.; Preis, S.R.; Vorgias, D.; DeCarli, C.; Au, R.; Kelly-Hayes, M.; Kase, C.S.; Wolf, P.A.; et al. Serum brain-derived neurotrophic factor and vascular endothelial growth factor levels are associated with risk of stroke and vascular brain injury: Framingham Study. Stroke 2013, 44, 2768–2775. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, M.; Crupi, R.; Di Paola, R.; Ontario, M.L.; Bella, R.; Calabrese, E.J.; Crea, R.; Cuzzocrea, S.; Calabrese, V. Inflammasomes, hormesis, and antioxidants in neuroinflammation: Role of NRLP3 in Alzheimer disease. J. Neurosci. Res. 2017, 95, 1360–1372. [Google Scholar] [CrossRef] [PubMed]
- Proescholdt, M.A.; Heiss, J.D.; Walbridge, S.; Muhlhauser, J.; Capogrossi, M.C.; Oldfield, E.H.; Merrill, M.J. Vascular endothelial growth factor (VEGF) modulates vascular permeability and inflammation in rat brain. J. Neuropathol. Exp. Neurol. 1999, 58, 613–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merrill, M.J.; Oldfield, E.H. A reassessment of vascular endothelial growth factor in central nervous system pathology. J. Neurosurg. 2005, 103, 853–868. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.M.; Zhang, C.L.; Chen, A.Q.; Wang, H.L.; Zhou, Y.F.; Li, Y.N.; Hu, B. Immune Cells in the BBB Disruption After Acute Ischemic Stroke: Targets for Immune Therapy? Front. Immunol. 2021, 12, 678744. [Google Scholar] [CrossRef] [PubMed]
- Galea, I. The blood-brain barrier in systemic infection and inflammation. Cell Mol. Immunol. 2021, 18, 2489–2501. [Google Scholar] [CrossRef]
- Hall, E.D.; Bryant, Y.D.; Cho, W.; Sullivan, P.G. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J. Neurotrauma 2008, 25, 235–247. [Google Scholar] [CrossRef]
- Rink, A.; Fung, K.M.; Trojanowski, J.Q.; Lee, V.M.; Neugebauer, E.; McIntosh, T.K. Evidence of apoptotic cell death after experimental traumatic brain injury in the rat. Am. J. Pathol. 1995, 147, 1575–1583. [Google Scholar]
- Deckwerth, T.L.; Elliott, J.L.; Knudson, C.M.; Johnson, E.M., Jr.; Snider, W.D.; Korsmeyer, S.J. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 1996, 17, 401–411. [Google Scholar] [CrossRef] [Green Version]
- Reed, J.C. Bcl-2 family proteins. Oncogene 1998, 17, 3225–3236. [Google Scholar] [CrossRef] [Green Version]
- Cornelius, C.; Perrotta, R.; Graziano, A.; Calabrese, E.J.; Calabrese, V. Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: Mitochondria as a “chi”. Immun. Ageing 2013, 10, 15. [Google Scholar] [CrossRef] [Green Version]
- Ley, K. Functions of selectins. Results Probl. Cell Differ. 2001, 33, 177–200. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.M.; Chapman, S.M.; Brown, A.A.; Frenette, P.S.; Hynes, R.O.; Wagner, D.D. The combined role of P- and E-selectins in atherosclerosis. J. Clin. Investig. 1998, 102, 145–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Wal, A.C.; Das, P.K.; Tigges, A.J.; Becker, A.E. Adhesion molecules on the endothelium and mononuclear cells in human atherosclerotic lesions. Am. J. Pathol. 1992, 141, 1427–1433. [Google Scholar] [PubMed]
- Bensch, K.W.; Raida, M.; Magert, H.J.; Schulz-Knappe, P.; Forssmann, W.G. hBD-1: A novel beta-defensin from human plasma. FEBS Lett. 1995, 368, 331–335. [Google Scholar] [CrossRef] [Green Version]
- Bodary, P.F.; Homeister, J.W.; Vargas, F.B.; Wickenheiser, K.J.; Cudney, S.S.; Bahrou, K.L.; Ohman, M.; Rabbani, A.B.; Eitzman, D.T. Generation of soluble P- and E-selectins in vivo is dependent on expression of P-selectin glycoprotein ligand-1. J. Thromb. Haemost. 2007, 5, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Novitzky, I.; Marianayagam, N.J.; Weiss, S.; Muhsinoglu, O.; Fridman, M.; Leibovitch, T.A.; Goldenberg-Cohen, N.; Michowiz, S. Comparison of Neuroprotective Effect of Bevacizumab and Sildenafil following Induction of Stroke in a Mouse Model. BioMed. Res. Int. 2016, 2016, 3938523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campolo, M.; Esposito, E.; Ahmad, A.; Di Paola, R.; Paterniti, I.; Cordaro, M.; Bruschetta, G.; Wallace, J.L.; Cuzzocrea, S. Hydrogen sulfide-releasing cyclooxygenase inhibitor ATB-346 enhances motor function and reduces cortical lesion volume following traumatic brain injury in mice. J. Neuroinflam. 2014, 11, 196. [Google Scholar] [CrossRef] [Green Version]
- Zohar, O.; Rubovitch, V.; Milman, A.; Schreiber, S.; Pick, C.G. Behavioral consequences of minimal traumatic brain injury in mice. Acta Neurobiol. Exp. (Wars) 2011, 71, 36–45. [Google Scholar]
- Kawai, T.; Akira, S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007, 13, 460–469. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Neuroprotective Effect of Artesunate in Experimental Model of Traumatic Brain Injury. Front. Neurol. 2018, 9, 590. [Google Scholar] [CrossRef] [Green Version]
- Stubbendorff, M.; Hua, X.; Deuse, T.; Ali, Z.; Reichenspurner, H.; Maegdefessel, L.; Robbins, R.C.; Schrepfer, S. Inducing myointimal hyperplasia versus atherosclerosis in mice: An introduction of two valid models. J. Vis. Exp. 2014, 87, e51459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Amico, R.; Trovato Salinaro, A.; Cordaro, M.; Fusco, R.; Impellizzeri, D.; Interdonato, L.; Scuto, M.; Ontario, M.L.; Crea, R.; Siracusa, R.; et al. Hidrox((R)) and Chronic Cystitis: Biochemical Evaluation of Inflammation, Oxidative Stress, and Pain. Antioxidants 2021, 10, 1046. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Paterniti, I.; Cordaro, M.; Crupi, R.; Bruschetta, G.; Campolo, M.; Cuzzocrea, S.; Esposito, E. Neuroprotective Effects of Temsirolimus in Animal Models of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 2403–2419. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, R.; Fusco, R.; Gugliandolo, E.; Cordaro, M.; Siracusa, R.; Impellizzeri, D.; Peritore, A.F.; Crupi, R.; Cuzzocrea, S.; Di Paola, R. Effects of a new compound containing Palmitoylethanolamide and Baicalein in myocardial ischaemia/reperfusion injury in vivo. Phytomedicine 2019, 54, 27–42. [Google Scholar] [CrossRef]
- Cordaro, M.; Siracusa, R.; Impellizzeri, D.; Ramona, D.A.; Peritore, A.F.; Crupi, R.; Gugliandolo, E.; Fusco, R.; Di Paola, R.; Schievano, C.; et al. Safety and efficacy of a new micronized formulation of the ALIAmide palmitoylglucosamine in preclinical models of inflammation and osteoarthritis pain. Arthritis Res. Ther. 2019, 21, 254. [Google Scholar] [CrossRef] [Green Version]
- Peritore, A.F.; D’Amico, R.; Cordaro, M.; Siracusa, R.; Fusco, R.; Gugliandolo, E.; Genovese, T.; Crupi, R.; Di Paola, R.; Cuzzocrea, S.; et al. PEA/Polydatin: Anti-Inflammatory and Antioxidant Approach to Counteract DNBS-Induced Colitis. Antioxidants 2021, 10, 464. [Google Scholar] [CrossRef]
- Peritore, A.F.; Siracusa, R.; Fusco, R.; Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Crupi, R.; Genovese, T.; Impellizzeri, D.; Cuzzocrea, S.; et al. Ultramicronized Palmitoylethanolamide and Paracetamol, a New Association to Relieve Hyperalgesia and Pain in a Sciatic Nerve Injury Model in Rat. Int. J. Mol. Sci. 2020, 21, 3509. [Google Scholar] [CrossRef]
- Fusco, R.; Gugliandolo, E.; Siracusa, R.; Scuto, M.; Cordaro, M.; D’Amico, R.; Evangelista, M.; Peli, A.; Peritore, A.F.; Impellizzeri, D.; et al. Formyl peptide receptor 1 signaling in acute inflammation and neural differentiation induced by traumatic brain injury. Biology 2020, 9, 238. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Genovese, T.; Impellizzeri, D.; D’Amico, R.; Fusco, R.; Peritore, A.F.; Di Paola, D.; Interdonato, L.; Gugliandolo, E.; Crupi, R.; Di Paola, R.; et al. Role of Bevacizumab on Vascular Endothelial Growth Factor in Apolipoprotein E Deficient Mice after Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 4162. https://doi.org/10.3390/ijms23084162
Genovese T, Impellizzeri D, D’Amico R, Fusco R, Peritore AF, Di Paola D, Interdonato L, Gugliandolo E, Crupi R, Di Paola R, et al. Role of Bevacizumab on Vascular Endothelial Growth Factor in Apolipoprotein E Deficient Mice after Traumatic Brain Injury. International Journal of Molecular Sciences. 2022; 23(8):4162. https://doi.org/10.3390/ijms23084162
Chicago/Turabian StyleGenovese, Tiziana, Daniela Impellizzeri, Ramona D’Amico, Roberta Fusco, Alessio Filippo Peritore, Davide Di Paola, Livia Interdonato, Enrico Gugliandolo, Rosalia Crupi, Rosanna Di Paola, and et al. 2022. "Role of Bevacizumab on Vascular Endothelial Growth Factor in Apolipoprotein E Deficient Mice after Traumatic Brain Injury" International Journal of Molecular Sciences 23, no. 8: 4162. https://doi.org/10.3390/ijms23084162