
Cell Senescence and Central Regulators of Immune Response

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
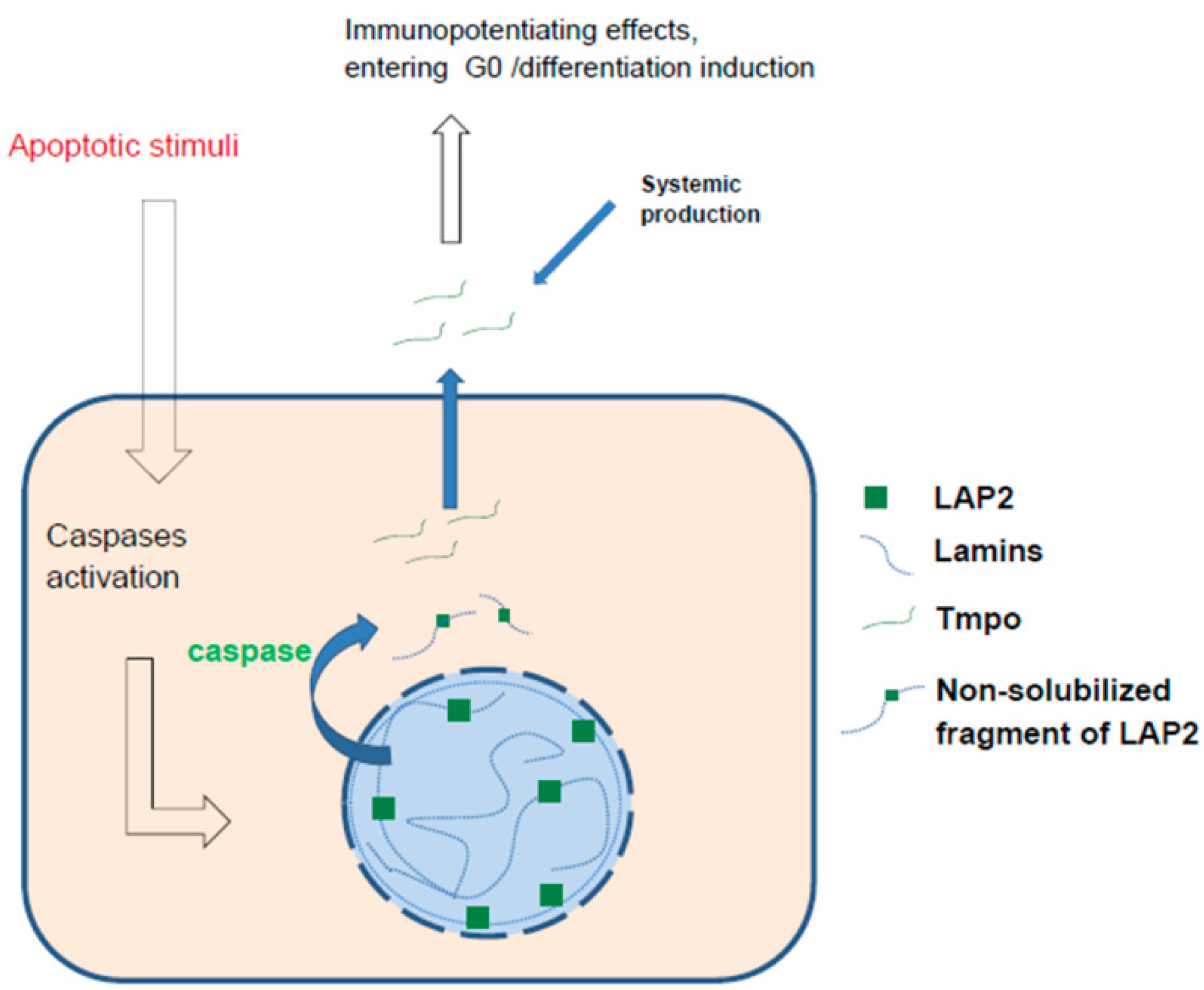{kind=link}
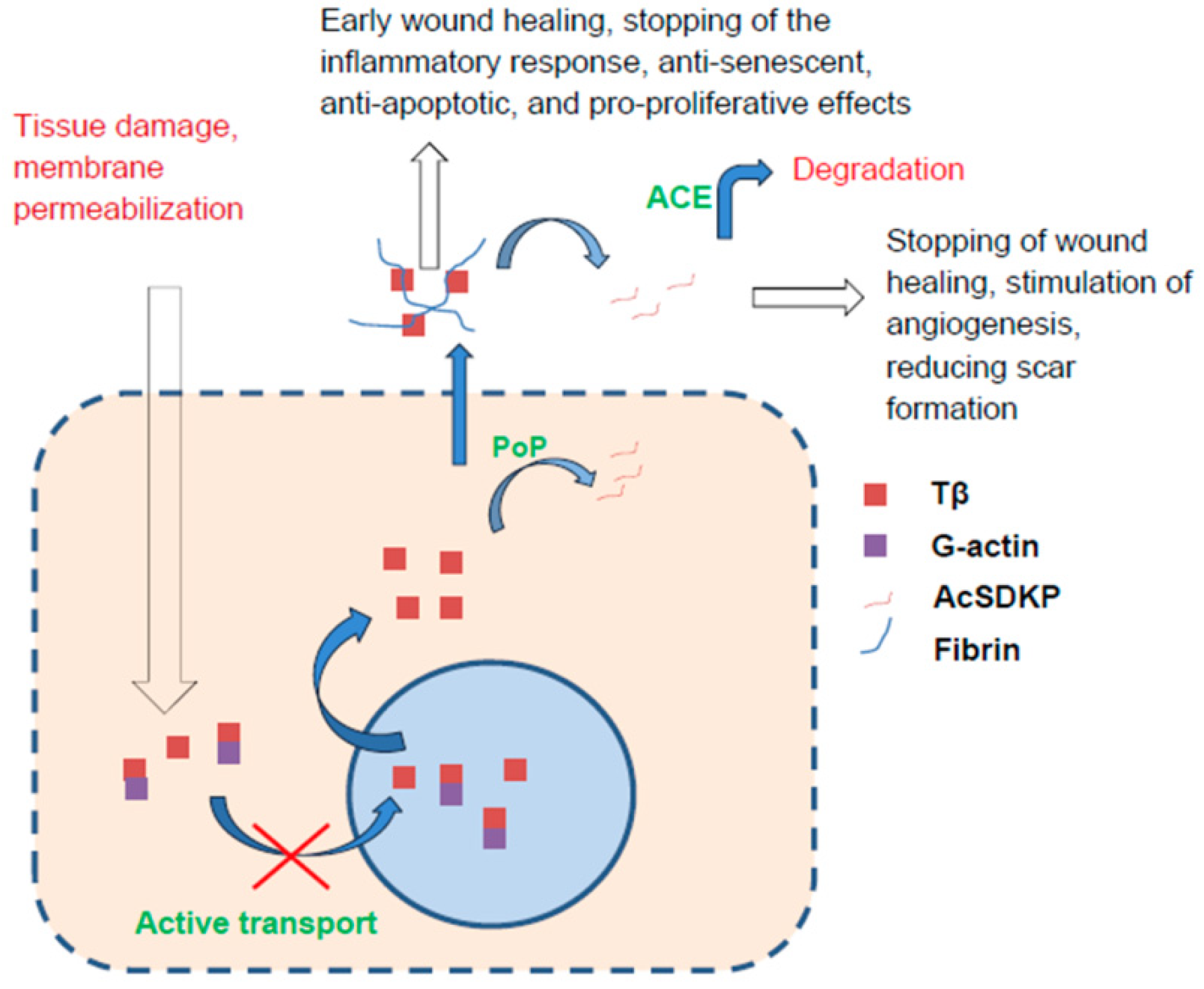{kind=link}
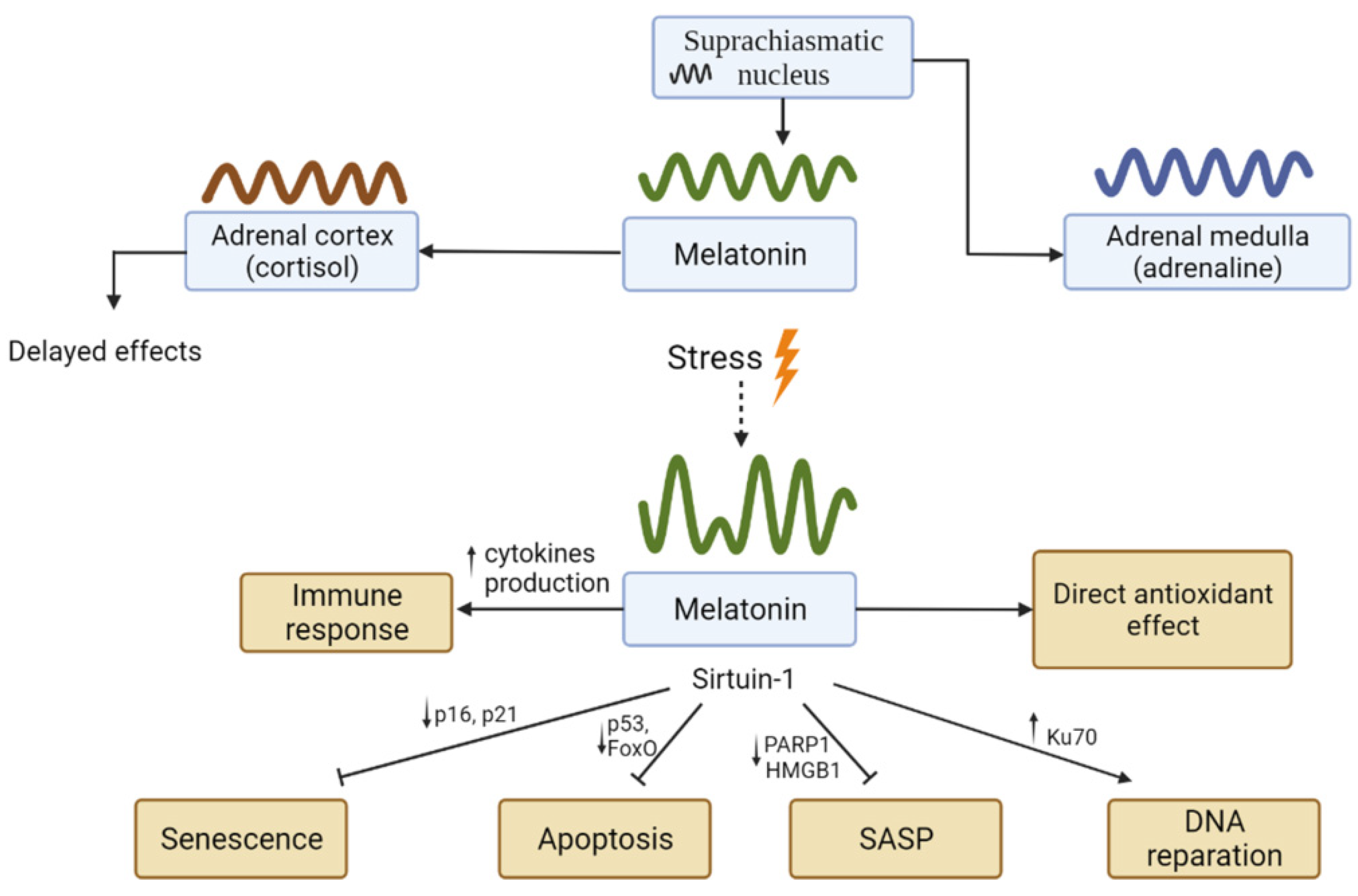{kind=link}
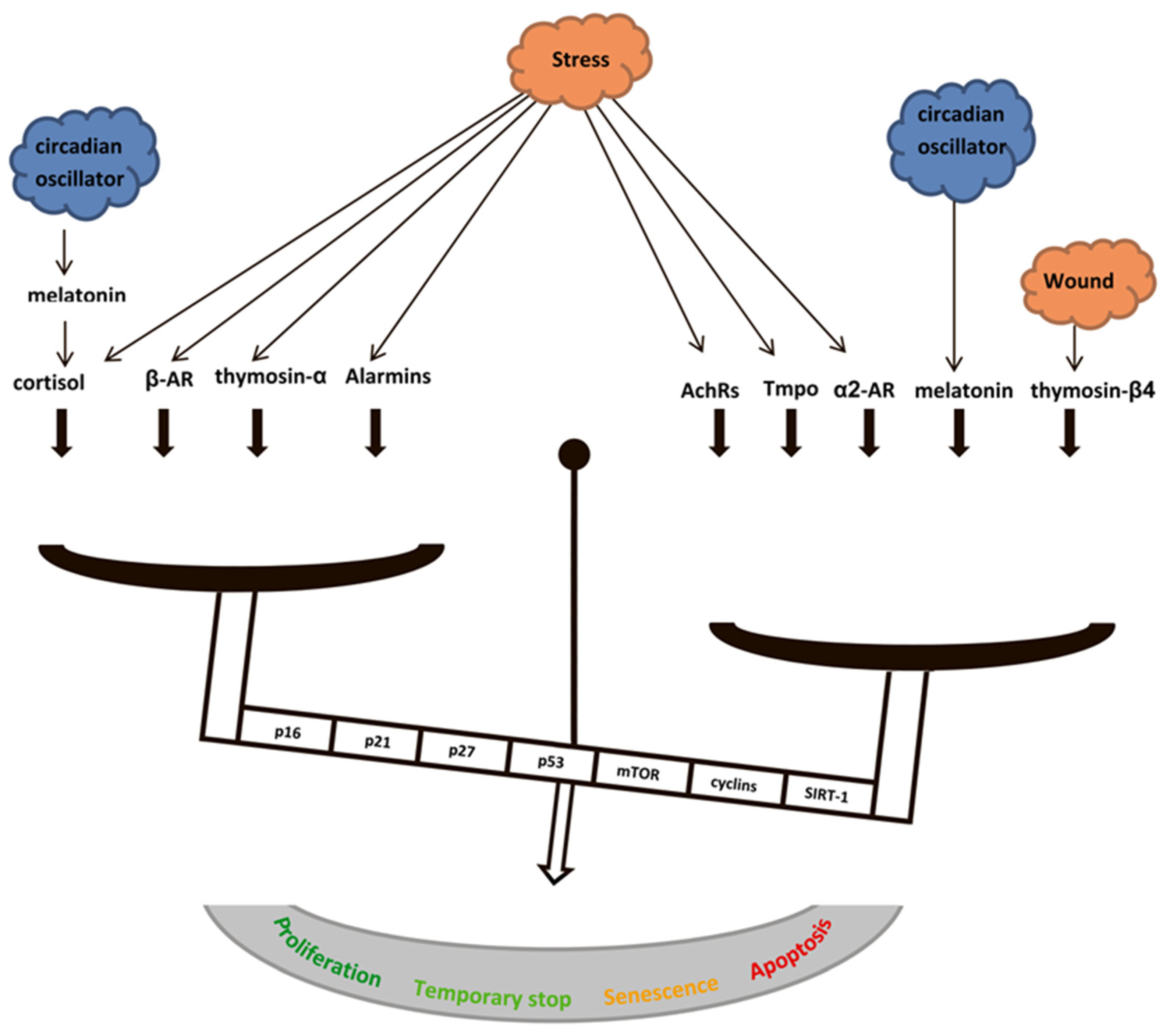{kind=link}
Abstract
:1. Introduction
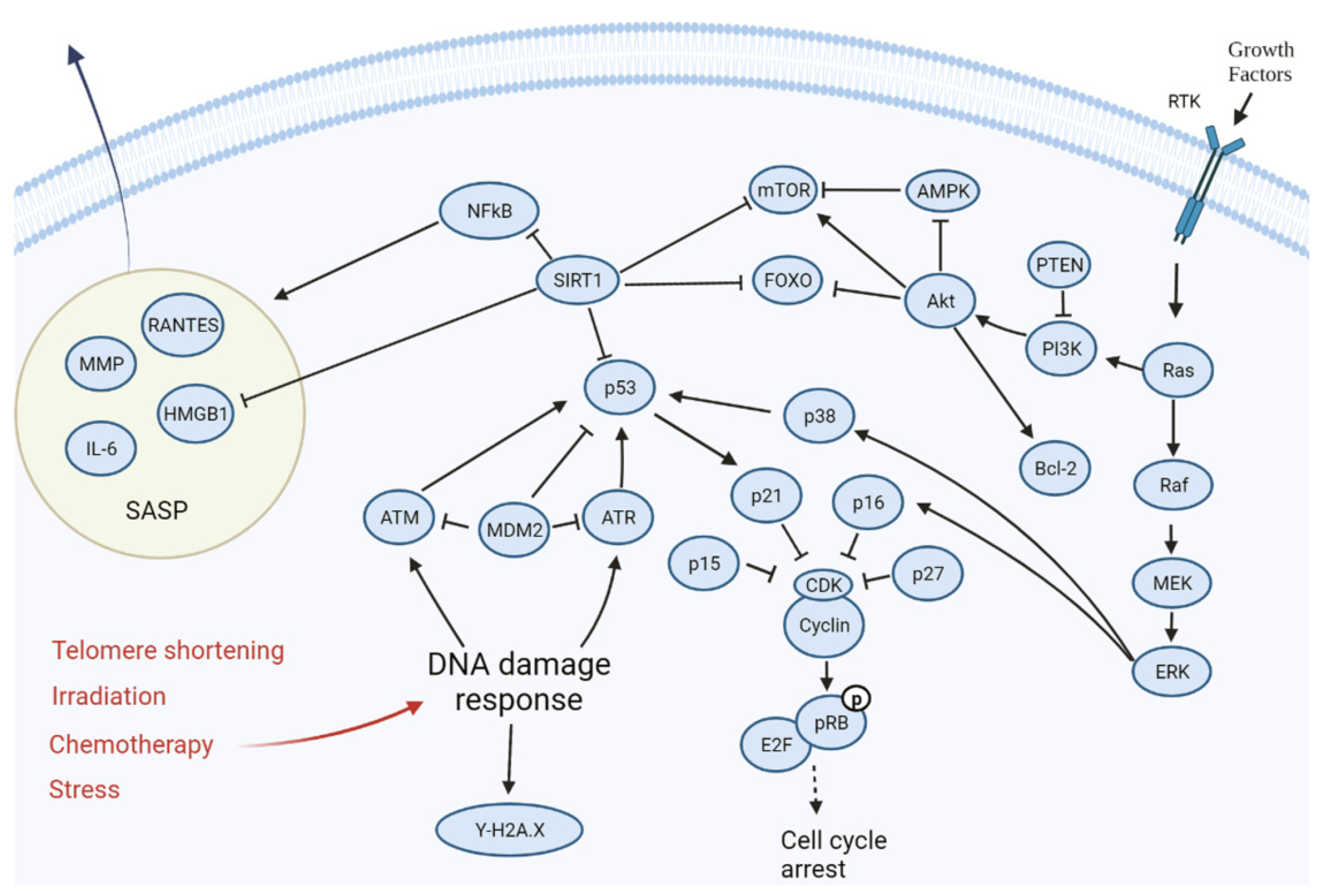
2. Causes of Cell Senescence
3. Cell Cycle Arrest as a Response to Stress Exposure
4. Neuroendocrine Regulators of Immune Response and Cell Senescence
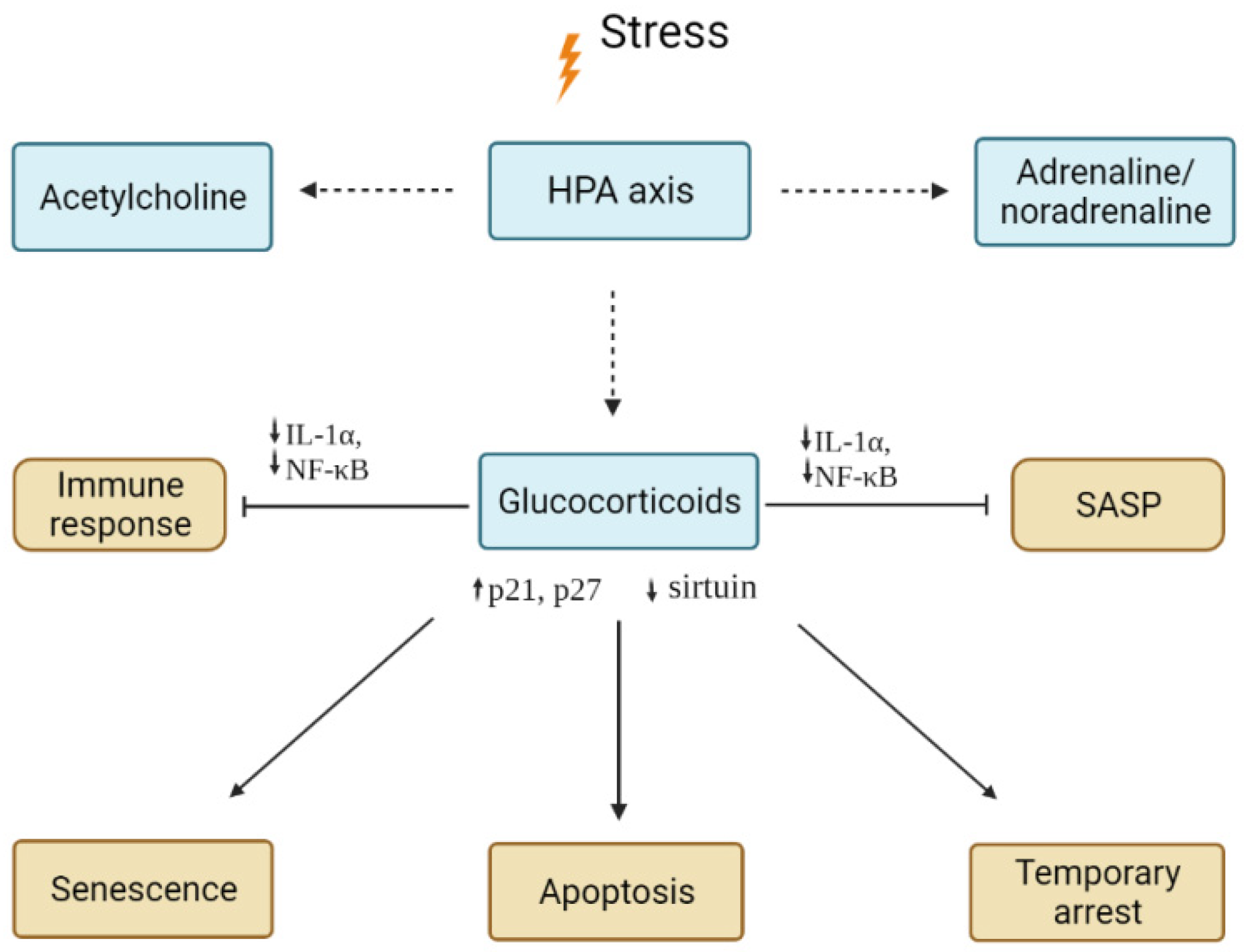
4.1. Glucocorticoids
4.2. Neurotransmitters of the Sympathetic and the Parasympathetic Systems
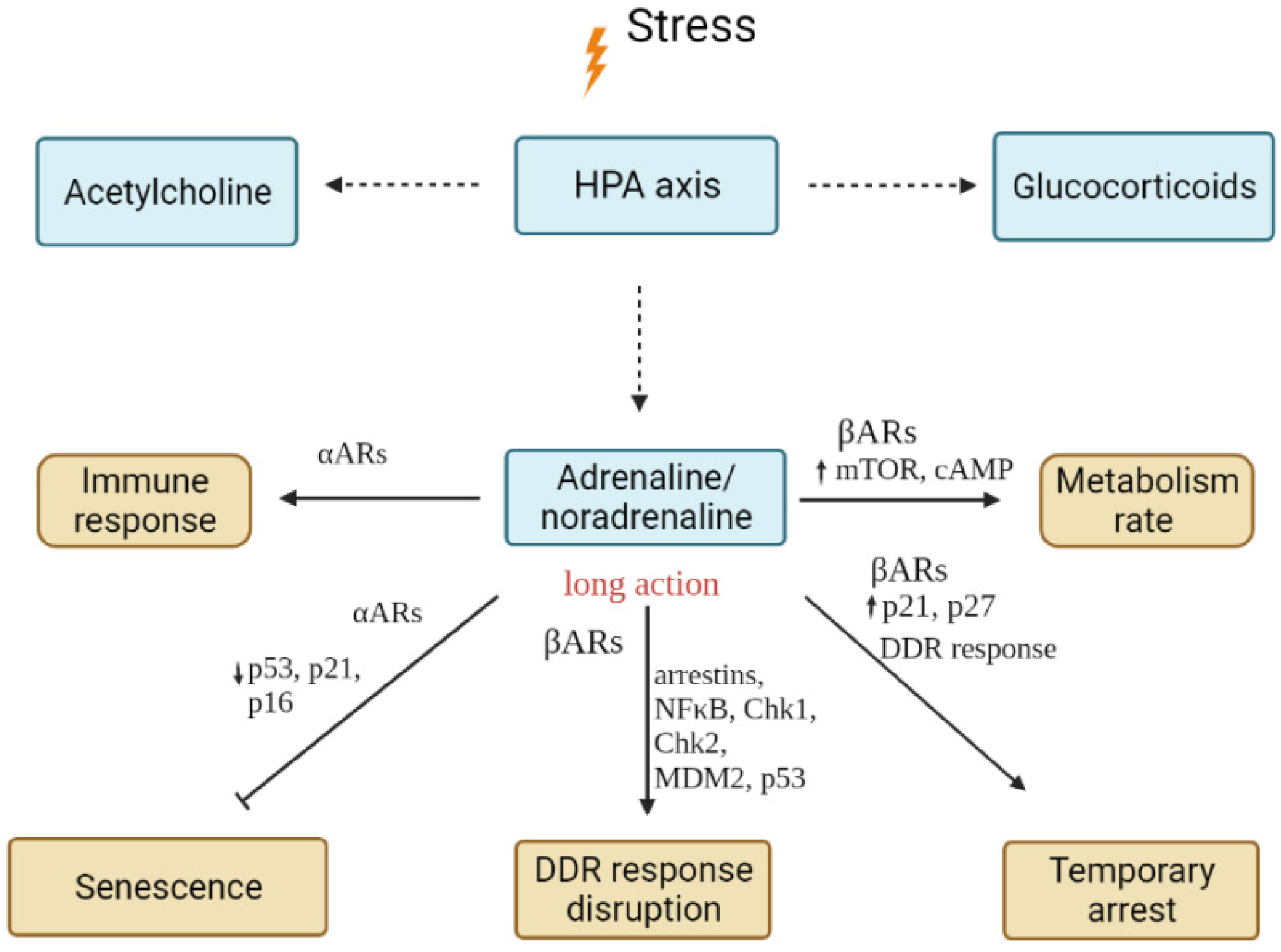
4.2.1. Adrenalin and Noradrenalin
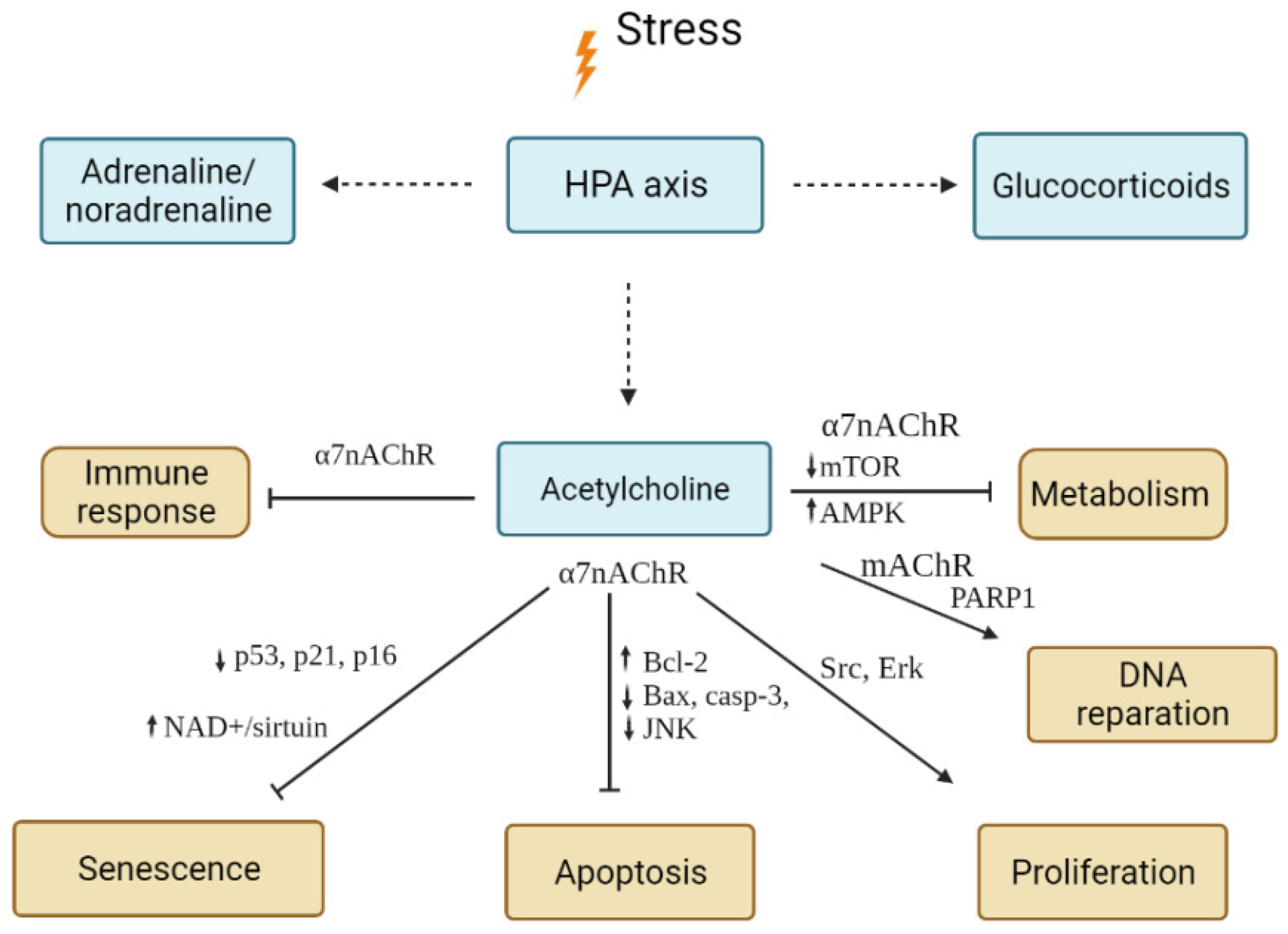
4.2.2. Acetylcholine
4.3. Alarmins
4.4. Thymic Hormones
4.4.1. Thymosin-α
4.4.2. Thymopoietin
4.4.3. Thymosin-β4
4.4.4. Melatonin
5. Conclusions
- The level of systemic stress, because it may induce activation of DDR-response after prolonged adrenalin exposure, or the cell cycle arrest after prolonged glucocorticoid exposure.
- Stage of stress response, because it may lead to oppositely directed changes in senescence-related pathways after adrenaline/acetylcholine/cortisol release during the stress response development, release of ProTα/Tα on the different stages of cellular stresses, or Tβ4/AcSDKP release on different stages of wound healing.
- Energy capacities of the body, because it may shift cell cycle and metabolism regulation, depending of diurnal oscillations of melatonin/cortisol concentrations. In addition, given the differently directed effects on immune and non-immune cells, this system may participate in the redistribution of body resources between an energy-consuming immune response and other functions.
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and Apoptosis: Dueling or Complementary Cell Fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular Senescence and Its Effector Programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rezzani, R.; Franco, C.; Hardeland, R.; Rodella, L.F. Thymus-Pineal Gland Axis: Revisiting Its Role in Human Life and Ageing. Int. J. Mol. Sci. 2020, 21, E8806. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.-P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- di Fagagna, D.F.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Giovanni Nuciforo, P.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Michaloglou, C.; Vredeveld, L.C.W.; Soengas, M.S.; Denoyelle, C.; Kuilman, T.; van der Horst, C.M.A.M.; Majoor, D.M.; Shay, J.W.; Mooi, W.J.; Peeper, D.S. BRAFE600-Associated Senescence-like Cell Cycle Arrest of Human Naevi. Nature 2005, 436, 720–724. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C.; Chen, Z.; Clohessy, J.G.; Carracedo, A.; Trotman, L.C.; Cheng, K.; Varmeh, S.; Kozma, S.C.; Thomas, G.; et al. A Novel Type of Cellular Senescence That Can Be Enhanced in Mouse Models and Human Tumor Xenografts to Suppress Prostate Tumorigenesis. J. Clin. Invest. 2010, 120, 681–693. [Google Scholar] [CrossRef]
- Wang, Z. Regulation of Cell Cycle Progression by Growth Factor-Induced Cell Signaling. Cells 2021, 10, 3327. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the P53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-Y.; Chang, K.-W.; Liu, C.-J.; Tseng, Y.-H.; Lu, H.-H.; Lee, S.-Y.; Lin, S.-C. Ripe Areca Nut Extract Induces G 1 Phase Arrests and Senescence-Associated Phenotypes in Normal Human Oral Keratinocyte. Carcinogenesis 2006, 27, 1273–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, D.; Lebedeva, I.V.; Emdad, L.; Kang, D.; Baldwin, A.S.; Fisher, P.B. Human Polynucleotide Phosphorylase (hPNPaseold-35): A Potential Link between Aging and Inflammation. Cancer Res. 2004, 64, 7473–7478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.W.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-Induced Senescence Relayed by an Interleukin-Dependent Inflammatory Network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Davalos, A.R.; Coppe, J.-P.; Campisi, J.; Desprez, P.-Y. Senescent Cells as a Source of Inflammatory Factors for Tumor Progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Wang, S. Characterization Of IGFBP-3, PAI-1 and SPARC MRNA Expression in Senescent Fibroblasts. Mech. Ageing Dev. 1996, 92, 121–132. [Google Scholar] [CrossRef]
- Grillari, J.; Hohenwarter, O.; Grabherr, R.M.; Katinger, H. Subtractive Hybridization of MRNA from Early Passage and Senescent Endothelial Cells. Exp. Gerontol. 2000, 35, 187–197. [Google Scholar] [CrossRef]
- López-Bermejo, A.; Buckway, C.K.; Devi, G.R.; Hwa, V.; Plymate, S.R.; Oh, Y.; Rosenfeld, R.G. Characterization of Insulin-Like Growth Factor-Binding Protein-Related Proteins (IGFBP-RPs) 1, 2, and 3 in Human Prostate Epithelial Cells: Potential Roles for IGFBP-RP1 and 2 in Senescence of the Prostatic Epithelium. Endocrinology 2000, 141, 4072–4080. [Google Scholar] [CrossRef]
- Kim, K.-H.; Park, G.-T.; Lim, Y.-B.; Rue, S.-W.; Jung, J.-C.; Sonn, J.-K.; Bae, Y.-S.; Park, J.-W.; Lee, Y.-S. Expression of Connective Tissue Growth Factor, a Biomarker in Senescence of Human Diploid Fibroblasts, Is up-Regulated by a Transforming Growth Factor-β-Mediated Signaling Pathway. Biochem. Biophys. Res. Commun. 2004, 318, 819–825. [Google Scholar] [CrossRef]
- Liu, D.; Hornsby, P.J. Senescent Human Fibroblasts Increase the Early Growth of Xenograft Tumors via Matrix Metalloproteinase Secretion. Cancer Res. 2007, 67, 3117–3126. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, S.; Coppe, J.-P.; Krtolica, A.; Campisi, J. Stromal-Epithelial Interactions in Aging and Cancer: Senescent Fibroblasts Alter Epithelial Cell Differentiation. J. Cell Sci. 2005, 118, 485–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, M.D.; Pereira-Smith, O.M.; Smith, J.R. Replicative Senescence of Human Skin Fibroblasts Correlates with a Loss of Regulation and Overexpression of Collagenase Activity. Exp. Cell Res. 1989, 184, 138–147. [Google Scholar] [CrossRef]
- Millis, A.J.T.; Hoyle, M.; McCue, H.M.; Martini, H. Differential Expression of Metalloproteinase and Tissue Inhibitor of Metalloproteinase Genes in Aged Human Fibroblasts. Exp. Cell Res. 1992, 201, 373–379. [Google Scholar] [CrossRef]
- Zeng, G.; Millis, A.J.T. Differential Regulation of Collagenase and Stromelysin MRNA in Late Passage Cultures of Human Fibroblasts. Exp. Cell Res. 1996, 222, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Carmeliet, P. UPAR: A Versatile Signalling Orchestrator. Nat. Rev. Mol. Cell Biol. 2002, 3, 932–943. [Google Scholar] [CrossRef] [PubMed]
- Sato, I.; Morita, I.; Kaji, K.; Ikeda, M.; Nagao, M.; Murota, S. Reduction of Nitric Oxide Producing Activity Associated with in Vitro Aging in Cultured Human Umbilical Vein Endothelial Cell. Biochem. Biophys. Res. Commun. 1993, 195, 1070–1076. [Google Scholar] [CrossRef]
- Lee, A.C.; Fenster, B.E.; Ito, H.; Takeda, K.; Bae, N.S.; Hirai, T.; Yu, Z.-X.; Ferrans, V.J.; Howard, B.H.; Finkel, T. Ras Proteins Induce Senescence by Altering the Intracellular Levels of Reactive Oxygen Species. J. Biol. Chem. 1999, 274, 7936–7940. [Google Scholar] [CrossRef] [Green Version]
- van der Loo, B.; Labugger, R.; Skepper, J.N.; Bachschmid, M.; Kilo, J.; Powell, J.M.; Palacios-Callender, M.; Erusalimsky, J.D.; Quaschning, T.; Malinski, T.; et al. Enhanced Peroxynitrite Formation Is Associated with Vascular Aging. J. Exp. Med. 2000, 192, 1731–1744. [Google Scholar] [CrossRef] [Green Version]
- Macip, S. Inhibition of P21-Mediated ROS Accumulation Can Rescue P21-Induced Senescence. EMBO J. 2002, 21, 2180–2188. [Google Scholar] [CrossRef]
- Xin, M.-G.; Zhang, J.; Block, E.R.; Patel, J.M. Senescence-Enhanced Oxidative Stress Is Associated with Deficiency of Mitochondrial Cytochrome c Oxidase in Vascular Endothelial Cells. Mech. Ageing Dev. 2003, 124, 911–919. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming in Vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef] [PubMed]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Song, Y.S.; Lee, B.Y.; Hwang, E.S. Dinstinct ROS and Biochemical Profiles in Cells Undergoing DNA Damage-Induced Senescence and Apoptosis. Mech. Ageing Dev. 2005, 126, 580–590. [Google Scholar] [CrossRef]
- Probin, V.; Wang, Y.; Bai, A.; Zhou, D. Busulfan Selectively Induces Cellular Senescence but Not Apoptosis in WI38 Fibroblasts via a P53-Independent but Extracellular Signal-Regulated Kinase-P38 Mitogen-Activated Protein Kinase-Dependent Mechanism. J. Pharm. Exp. 2006, 319, 551–560. [Google Scholar] [CrossRef]
- Debacq-Chainiaux, F.; Borlon, C.; Pascal, T.; Royer, V.; Eliaers, F.; Ninane, N.; Carrard, G.; Friguet, B.; de Longueville, F.; Boffe, S.; et al. Repeated Exposure of Human Skin Fibroblasts to UVB at Subcytotoxic Level Triggers Premature Senescence through the TGF-Β1 Signaling Pathway. J. Cell Sci. 2005, 118, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Ames, B.N. Senescence-like Growth Arrest Induced by Hydrogen Peroxide in Human Diploid Fibroblast F65 Cells. Proc. Natl. Acad. Sci. USA 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or Senescence-like Growth Arrest: Influence of Cell-Cycle Position, P53, P21 and Bax in H2O2 Response of Normal Human Fibroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar] [CrossRef]
- Wang, G.; Cheng, X.; Zhang, J.; Liao, Y.; Jia, Y.; Qing, C. Possibility of Inducing Tumor Cell Senescence during Therapy (Review). Oncol. Lett. 2021, 22, 496. [Google Scholar] [CrossRef] [PubMed]
- Demidenko, Z.N.; Blagosklonny, M.V. Growth Stimulation Leads to Cellular Senescence When the Cell Cycle Is Blocked. Cell Cycle 2008, 7, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Korotchkina, L.G.; Leontieva, O.V.; Bukreeva, E.I.; Demidenko, Z.N.; Gudkov, A.V.; Blagosklonny, M.V. The Choice between P53-Induced Senescence and Quiescence Is Determined in Part by the MTOR Pathway. Aging 2010, 2, 344–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, I.-H.; Bachmann, R.; Shirazi, H.; Chen, J. Regulation of Ribosomal S6 Kinase 2 by Mammalian Target of Rapamycin. J. Biol. Chem. 2002, 277, 31423–31429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubica, N.; Crispino, J.L.; Gallagher, J.W.; Kimball, S.R.; Jefferson, L.S. Activation of the Mammalian Target of Rapamycin Complex 1 Is Both Necessary and Sufficient to Stimulate Eukaryotic Initiation Factor 2Bɛ MRNA Translation and Protein Synthesis. Int. J. Biochem. Cell Biol. 2008, 40, 2522–2533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubica, N.; Bolster, D.R.; Farrell, P.A.; Kimball, S.R.; Jefferson, L.S. Resistance Exercise Increases Muscle Protein Synthesis and Translation of Eukaryotic Initiation Factor 2Bϵ MRNA in a Mammalian Target of Rapamycin-Dependent Manner. J. Biol. Chem. 2005, 280, 7570–7580. [Google Scholar] [CrossRef] [Green Version]
- Blagosklonny, M.V. Cell Senescence: Hypertrophic Arrest beyond the Restriction Point. J. Cell. Physiol. 2006, 209, 592–597. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Cell Cycle Arrest Is Not Senescence. Aging 2011, 3, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.X.; He, S.H.; Liang, X.; Li, W.; Li, T.F.; Li, D.F. Aging, Cell Senescence, the Pathogenesis and Targeted Therapies of Osteoarthritis. Front. Pharm. 2021, 12, 728100. [Google Scholar] [CrossRef]
- Jiang, C.; Sun, Z.-M.; Hu, J.-N.; Jin, Y.; Guo, Q.; Xu, J.-J.; Chen, Z.-X.; Jiang, R.-H.; Wu, Y.-S. Cyanidin Ameliorates the Progression of Osteoarthritis via the Sirt6/NF-ΚB Axis in Vitro and in Vivo. Food Funct. 2019, 10, 5873–5885. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Chrousos, G.P.; Gold, P.W. The Concepts of Stress and Stress System Disorders. Overview of Physical and Behavioral Homeostasis. JAMA 1992, 267, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Boumpas, D.T.; Chrousos, G.P.; Wilder, R.L.; Cupps, T.R.; Balow, J.E. Glucocorticoid Therapy for Immune-Mediated Diseases: Basic and Clinical Correlates. Ann. Intern. Med. 1993, 119, 1198–1208. [Google Scholar] [CrossRef] [Green Version]
- Chrousos, G.P. The Hypothalamic–Pituitary–Adrenal Axis and Immune-Mediated Inflammation. N. Engl. J. Med. 1995, 332, 1351–1363. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.-S.; Kapahi, P.; et al. Glucocorticoids Suppress Selected Components of the Senescence-Associated Secretory Phenotype: Glucocorticoids Suppress the SASP. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, R.C.; Watts, A.C.; Murphy, R.J.; Snelling, S.J.; Carr, A.J.; Hulley, P.A. Glucocorticoids Induce Senescence in Primary Human Tenocytes by Inhibition of Sirtuin 1 and Activation of the P53/P21 Pathway: In Vivo and in Vitro Evidence. Ann. Rheum. Dis. 2014, 73, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Zager, R.A.; Johnson, A.C.M. Acute Kidney Injury Induces Dramatic P21 Upregulation via a Novel, Glucocorticoid-Activated, Pathway. Am. J. Physiol. -Ren. Physiol. 2019, 316, F674–F681. [Google Scholar] [CrossRef]
- Patki, M.; Gadgeel, S.; Huang, Y.; McFall, T.; Shields, A.F.; Matherly, L.H.; Bepler, G.; Ratnam, M. Glucocorticoid Receptor Status Is a Principal Determinant of Variability in the Sensitivity of Non–Small-Cell Lung Cancer Cells to Pemetrexed. J. Thorac. Oncol. 2014, 9, 519–526. [Google Scholar] [CrossRef] [Green Version]
- Patki, M.; McFall, T.; Rosati, R.; Huang, Y.; Malysa, A.; Polin, L.; Fielder, A.; Wilson, M.R.; Lonardo, F.; Back, J.; et al. Chronic P27Kip1 Induction by Dexamethasone Causes Senescence Phenotype and Permanent Cell Cycle Blockade in Lung Adenocarcinoma Cells Over-Expressing Glucocorticoid Receptor. Sci. Rep. 2018, 8, 16006. [Google Scholar] [CrossRef]
- Martin, L.F.; Richardson, L.S.; da Silva, M.G.; Sheller-Miller, S.; Menon, R. Dexamethasone Induces Primary Amnion Epithelial Cell Senescence through Telomere-P21 Associated Pathway. Biol. Reprod. 2019, 100, 1605–1616. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.; L’Hôte, V.; Courbeyrette, R.; Kratassiouk, G.; Pinna, G.; Cintrat, J.-C.; Denby-Wilkes, C.; Derbois, C.; Olaso, R.; Deleuze, J.-F.; et al. Glucocorticoids Delay RAF-Induced Senescence Promoted by EGR1. J. Cell Sci. 2019, 132, jcs.230748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiuchi, Z.; Nishibori, Y.; Kutsuna, S.; Kotani, M.; Hada, I.; Kimura, T.; Fukutomi, T.; Fukuhara, D.; Ito-Nitta, N.; Kudo, A.; et al. GLCCI1 Is a Novel Protector against Glucocorticoid-induced Apoptosis in T Cells. FASEB J. 2019, 33, 7387–7402. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune Regulation by Glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Gruver-Yates, A.; Cidlowski, J. Tissue-Specific Actions of Glucocorticoids on Apoptosis: A Double-Edged Sword. Cells 2013, 2, 202–223. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Lee, K.-Y.; Jeong, D.-C.; Kim, H.-K. Effect of P16 on Glucocorticoid Response in a B-Cell Lymphoblast Cell Line. Korean J. Pediatr. 2010, 53, 753. [Google Scholar] [CrossRef] [Green Version]
- Estrela, J.M.; Salvador, R.; Marchio, P.; Valles, S.L.; López-Blanch, R.; Rivera, P.; Benlloch, M.; Alcácer, J.; Pérez, C.L.; Pellicer, J.A.; et al. Glucocorticoid Receptor Antagonism Overcomes Resistance to BRAF Inhibition in BRAFV600E-Mutated Metastatic Melanoma. Am. J. Cancer Res. 2019, 9, 2580–2598. [Google Scholar]
- Block, T.S.; Murphy, T.I.; Munster, P.N.; Nguyen, D.P.; Lynch, F.J. Glucocorticoid Receptor Expression in 20 Solid Tumor Types Using Immunohistochemistry Assay. Cancer Manag. Res. 2017, 9, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Prekovic, S.; Schuurman, K.; Mayayo-Peralta, I.; Manjón, A.G.; Buijs, M.; Yavuz, S.; Wellenstein, M.D.; Barrera, A.; Monkhorst, K.; Huber, A.; et al. Glucocorticoid Receptor Triggers a Reversible Drug-Tolerant Dormancy State with Acquired Therapeutic Vulnerabilities in Lung Cancer. Nat. Commun. 2021, 12, 4360. [Google Scholar] [CrossRef]
- Han, D.; Gao, J.; Gu, X.; Hengstler, J.G.; Zhang, L.; Shahid, M.; Ali, T.; Han, B. P21Waf1/Cip1 Depletion Promotes Dexamethasone-Induced Apoptosis in Osteoblastic MC3T3-E1 Cells by Inhibiting the Nrf2/HO-1 Pathway. Arch. Toxicol. 2018, 92, 679–692. [Google Scholar] [CrossRef]
- Avitsur, R.; Padgett, D.A.; Sheridan, J.F. Social Interactions, Stress, and Immunity. Neurol. Clin. 2006, 24, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Christian, L.M.; Graham, J.E.; Padgett, D.A.; Glaser, R.; Kiecolt-Glaser, J.K. Stress and Wound Healing. Neuroimmunomodulation 2006, 13, 337–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altemus, M.; Rao, B.; Dhabhar, F.S.; Ding, W.; Granstein, R.D. Stress-Induced Changes in Skin Barrier Function in Healthy Women. J. Investig. Dermatol. 2001, 117, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The Origins of Age-Related Proinflammatory State. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef] [Green Version]
- Forsey, R.J.; Thompson, J.M.; Ernerudh, J.; Hurst, T.L.; Strindhall, J.; Johansson, B.; Nilsson, B.-O.; Wikby, A. Plasma Cytokine Profiles in Elderly Humans. Mech. Ageing Dev. 2003, 124, 487–493. [Google Scholar] [CrossRef]
- Brunner, E.J.; Hemingway, H.; Walker, B.R.; Page, M.; Clarke, P.; Juneja, M.; Shipley, M.J.; Kumari, M.; Andrew, R.; Seckl, J.R.; et al. Adrenocortical, Autonomic, and Inflammatory Causes of the Metabolic Syndrome: Nested Case-Control Study. Circulation 2002, 106, 2659–2665. [Google Scholar] [CrossRef]
- Kulstad, J.J.; McMillan, P.J.; Leverenz, J.B.; Cook, D.G.; Green, P.S.; Peskind, E.R.; Wilkinson, C.W.; Farris, W.; Mehta, P.D.; Craft, S. Effects of Chronic Glucocorticoid Administration on Insulin-Degrading Enzyme and Amyloid-Beta Peptide in the Aged Macaque. J. Neuropathol. Exp. Neurol. 2005, 64, 139–146. [Google Scholar] [CrossRef] [Green Version]
- Cavigelli, S.A.; Ragan, C.M.; Michael, K.C.; Kovacsics, C.E.; Bruscke, A.P. Stable Behavioral Inhibition and Glucocorticoid Production as Predictors of Longevity. Physiol. Behav. 2009, 98, 205–214. [Google Scholar] [CrossRef] [Green Version]
- Madden, K.S.; Sanders, V.M.; Felten, D.L. Catecholamine Influences and Sympathetic Neural Modulation of Immune Responsiveness. Annu. Rev. Pharmacol. Toxicol. 1995, 35, 417–448. [Google Scholar] [CrossRef]
- Vizi, E.S. Receptor-Mediated Local Fine-Tuning by Noradrenergic Innervation of Neuroendocrine and Immune Systemsa. Ann. N. Y. Acad. Sci. 1998, 851, 388–396. [Google Scholar] [CrossRef]
- Tracey, K.J. The Inflammatory Reflex. Nature 2002, 420, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Physiology and Immunology of the Cholinergic Antiinflammatory Pathway. J. Clin. Invest. 2007, 117, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic Acetylcholine Receptor A7 Subunit Is an Essential Regulator of Inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Huston, J.M.; Ochani, M.; Rosas-Ballina, M.; Liao, H.; Ochani, K.; Pavlov, V.A.; Gallowitsch-Puerta, M.; Ashok, M.; Czura, C.J.; Foxwell, B.; et al. Splenectomy Inactivates the Cholinergic Antiinflammatory Pathway during Lethal Endotoxemia and Polymicrobial Sepsis. J. Exp. Med. 2006, 203, 1623–1628. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Liao, H.; Ochani, M.; Justiniani, M.; Lin, X.; Yang, L.; Al-Abed, Y.; Wang, H.; Metz, C.; Miller, E.J.; et al. Cholinergic Agonists Inhibit HMGB1 Release and Improve Survival in Experimental Sepsis. Nat. Med. 2004, 10, 1216–1221. [Google Scholar] [CrossRef]
- Giebelen, I.A.J.; van Westerloo, D.J.; LaRosa, G.J.; de Vos, A.F.; van der Poll, T. Stimulation of A7 Cholinergic Receptors Inhibits Lipopolysaccharide-Induced Neutrophil Recruitment by A Tumor Necrosis Factor A-Independent Mechanism. Shock 2007, 27, 443–447. [Google Scholar] [CrossRef]
- Su, X.; Matthay, M.A.; Malik, A.B. Requisite Role of the Cholinergic A7 Nicotinic Acetylcholine Receptor Pathway in Suppressing Gram-Negative Sepsis-Induced Acute Lung Inflammatory Injury. J. Immunol. 2010, 184, 401–410. [Google Scholar] [CrossRef]
- Richter, J.D.; Sonenberg, N. Regulation of Cap-Dependent Translation by EIF4E Inhibitory Proteins. Nature 2005, 433, 477–480. [Google Scholar] [CrossRef]
- Mayer, C.; Zhao, J.; Yuan, X.; Grummt, I. MTOR-Dependent Activation of the Transcription Factor TIF-IA Links RRNA Synthesis to Nutrient Availability. Genes Dev. 2004, 18, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by MTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metab. 2008, 8, 224–236. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Chen, J. Regulation of Peroxisome Proliferator–Activated Receptor-γ Activity by Mammalian Target of Rapamycin and Amino Acids in Adipogenesis. Diabetes 2004, 53, 2748–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schieke, S.M.; Phillips, D.; McCoy, J.P.; Aponte, A.M.; Shen, R.-F.; Balaban, R.S.; Finkel, T. The Mammalian Target of Rapamycin (MTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. J. Biol. Chem. 2006, 281, 27643–27652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Codogno, P.; Meijer, A.J. Autophagy and Signaling: Their Role in Cell Survival and Cell Death. Cell Death Differ. 2005, 12 (Suppl. S2), 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and Regulation of Akt/PKB by the Rictor-MTOR Complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.-H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a Novel Binding Partner of MTOR, Defines a Rapamycin-Insensitive and Raptor-Independent Pathway That Regulates the Cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, G.; Matthes, H.W.D.; Hachet-Haas, M.; El Baghdadi, K.; de Mey, J.; Pepperkok, R.; Simpson, J.C.; Galzi, J.-L.; Lecat, S. Plasma Membrane Translocation of REDD1 Governed by GPCRs Contributes to MTORC1 Activation. J. Cell Sci. 2014, 127, 773–787. [Google Scholar] [CrossRef] [Green Version]
- Shear, M.; Insel, P.A.; Melmon, K.L.; Coffino, P. Agonist-Specific Refractoriness Induced by Isoproterenol. Studies with Mutant Cells. J. Biol. Chem. 1976, 251, 7572–7576. [Google Scholar] [CrossRef]
- Benovic, J.L.; Bouvier, M.; Caron, M.G.; Lefkowitz, R.J. Regulation of Adenylyl Cyclase-Coupled Beta-Adrenergic Receptors. Annu. Rev. Cell Biol. 1988, 4, 405–428. [Google Scholar] [CrossRef]
- Leysen, H.; van Gastel, J.; Hendrickx, J.; Santos-Otte, P.; Martin, B.; Maudsley, S. G Protein-Coupled Receptor Systems as Crucial Regulators of DNA Damage Response Processes. IJMS 2018, 19, 2919. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.R.; Kovacs, J.J.; Whalen, E.J.; Rajagopal, S.; Strachan, R.T.; Grant, W.; Towers, A.J.; Williams, B.; Lam, C.M.; Xiao, K.; et al. A Stress Response Pathway Regulates DNA Damage through Β2-Adrenoreceptors and β-Arrestin-1. Nature 2011, 477, 349–353. [Google Scholar] [CrossRef] [Green Version]
- Hara, M.R.; Sachs, B.D.; Caron, M.G.; Lefkowitz, R.J. Pharmacological Blockade of a β2 AR-β-Arrestin-1 Signaling Cascade Prevents the Accumulation of DNA Damage in a Behavioral Stress Model. Cell Cycle 2013, 12, 219–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flint, M.S.; Baum, A.; Chambers, W.H.; Jenkins, F.J. Induction of DNA Damage, Alteration of DNA Repair and Transcriptional Activation by Stress Hormones. Psychoneuroendocrinology 2007, 32, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Flint, M.S.; Baum, A.; Episcopo, B.; Knickelbein, K.Z.; Liegey Dougall, A.J.; Chambers, W.H.; Jenkins, F.J. Chronic Exposure to Stress Hormones Promotes Transformation and Tumorigenicity of 3T3 Mouse Fibroblasts. Stress 2013, 16, 114–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reeder, A.; Attar, M.; Nazario, L.; Bathula, C.; Zhang, A.; Hochbaum, D.; Roy, E.; Cooper, K.L.; Oesterreich, S.; Davidson, N.E.; et al. Stress Hormones Reduce the Efficacy of Paclitaxel in Triple Negative Breast Cancer through Induction of DNA Damage. Br. J. Cancer 2015, 112, 1461–1470. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Fang, L.; Stolz, D.; Tamm, M.; Roth, M. Long Acting Β2-Agonist’s Activation of Cyclic AMP Cannot Halt Ongoing Mitogenic Stimulation in Airway Smooth Muscle Cells. Pulm. Pharmacol. Ther. 2019, 56, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-X.; Kimura, S.; Murao, K.; Yu, X.; Obata, K.; Matsuyoshi, H.; Takaki, M. Effects of Angiotensin Type I Receptor Blockade on the Cardiac Raf/MEK/ERK Cascade Activated via Adrenergic Receptors. J. Pharm. Sci. 2010, 113, 224–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liou, S.-F.; Lin, H.-H.; Liang, J.-C.; Chen, I.-J.; Yeh, J.-L. Inhibition of Human Prostate Cancer Cells Proliferation by a Selective Alpha1-Adrenoceptor Antagonist Labedipinedilol-A Involves Cell Cycle Arrest and Apoptosis. Toxicology 2009, 256, 13–24. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Chang, F.; Lehmann, B.; Terrian, D.M.; Milella, M.; Tafuri, A.; et al. Roles of the Raf/MEK/ERK Pathway in Cell Growth, Malignant Transformation and Drug Resistance. Biochim. Biophys. Acta 2007, 1773, 1263–1284. [Google Scholar] [CrossRef] [Green Version]
- Shenoy, S.K.; Drake, M.T.; Nelson, C.D.; Houtz, D.A.; Xiao, K.; Madabushi, S.; Reiter, E.; Premont, R.T.; Lichtarge, O.; Lefkowitz, R.J. β-Arrestin-Dependent, G Protein-Independent ERK1/2 Activation by the Β2 Adrenergic Receptor. J. Biol. Chem. 2006, 281, 1261–1273. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Chen, C.; Chen, X.; Han, M.; Li, J. Dexmedetomidine Attenuates Renal Fibrosis via A2-Adrenergic Receptor-Dependent Inhibition of Cellular Senescence after Renal Ischemia/Reperfusion. Life Sci. 2018, 207, 1–8. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, S.; Yu, X.; Zhou, S.; Ge, M.; Chi, X.; Cai, J. Dexmedetomidine Protects Rat Liver against Ischemia-Reperfusion Injury Partly by the A2A-Adrenoceptor Subtype and the Mechanism Is Associated with the TLR4/NF-ΚB Pathway. IJMS 2016, 17, 995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Z.-Q.; Zhang, J.-J.; Kong, N.; Zhang, G.-Y.; Ke, P.; Han, T.; Su, D.-F.; Liu, C. Autophagy Is Involved in Neuroprotective Effect of Alpha7 Nicotinic Acetylcholine Receptor on Ischemic Stroke. Front. Pharmacol. 2021, 12, 676589. [Google Scholar] [CrossRef] [PubMed]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK Phosphorylation of Raptor Mediates a Metabolic Checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hua, Y.; Yang, B.; Chen, Q.; Zhang, J.; Hu, J.; Fan, Y. Activation of A7 Nicotinic Acetylcholine Receptor Protects Against 1-Methyl-4-Phenylpyridinium-Induced Astroglial Apoptosis. Front. Cell. Neurosci. 2019, 13, 507. [Google Scholar] [CrossRef]
- Li, D.-J.; Huang, F.; Ni, M.; Fu, H.; Zhang, L.-S.; Shen, F.-M. A7 Nicotinic Acetylcholine Receptor Relieves Angiotensin II–Induced Senescence in Vascular Smooth Muscle Cells by Raising Nicotinamide Adenine Dinucleotide–Dependent SIRT1 Activity. ATVB 2016, 36, 1566–1576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egleton, R.D.; Brown, K.C.; Dasgupta, P. Nicotinic Acetylcholine Receptors in Cancer: Multiple Roles in Proliferation and Inhibition of Apoptosis. Trends Pharm. Sci. 2008, 29, 151–158. [Google Scholar] [CrossRef]
- Schuller, H.M. Neurotransmitter Receptor-Mediated Signaling Pathways as Modulators of Carcinogenesis. In Progress in Experimental Tumor Research; Zänker, K.S., Entschladen, F., Eds.; KARGER: Basel, Switzerland, 2007; Volume 39, pp. 45–63. ISBN 978-3-318-01441-9. [Google Scholar]
- Adamczyk, A.; Jeśko, H.; Strosznajder, R.P. Alzheimer’s Disease Related Peptides Affected Cholinergic Receptor Mediated Poly(ADP-Ribose) Polymerase Activity in the Hippocampus. Folia Neuropathol. 2005, 43, 139–142. [Google Scholar]
- Ahel, D.; Horejsí, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(ADP-Ribose)-Dependent Regulation of DNA Repair by the Chromatin Remodeling Enzyme ALC1. Science 2009, 325, 1240–1243. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jiang, Y.; Chen, J.; Dai, C.; Liu, D.; Pan, W.; Wang, L.; Fasae, M.B.; Sun, L.; Wang, L.; et al. Activation of M3 Muscarinic Acetylcholine Receptors Delayed Cardiac Aging by Inhibiting the Caspase-1/IL-1β Signaling Pathway. Cell Physiol. Biochem. 2018, 49, 1249–1257. [Google Scholar] [CrossRef]
- Arredondo, J.; Hall, L.L.; Ndoye, A.; Chernyavsky, A.A.I.; Jolkovsky, D.L.; Grando, S.A. Muscarinic Acetylcholine Receptors Regulating Cell Cycle Progression Are Expressed in Human Gingival Keratinocytes. J. Periodontal. Res. 2003, 38, 79–89. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and Alarmins: All We Need to Know about Danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Andersson, U.; Yang, H.; Harris, H. High-Mobility Group Box 1 Protein (HMGB1) Operates as an Alarmin Outside as Well as inside Cells. Semin. Immunol. 2018, 38, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High Mobility Group 1 Protein (HMG-1) Stimulates Proinflammatory Cytokine Synthesis in Human Monocytes. J. Exp. Med. 2000, 192, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T. Monocytic Cells Hyperacetylate Chromatin Protein HMGB1 to Redirect It towards Secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef] [Green Version]
- Rauvala, H.; Rouhiainen, A. RAGE as a Receptor of HMGB1 (Amphoterin): Roles in Health and Disease. Curr. Mol. Med. 2007, 7, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y.; et al. A Critical Cysteine Is Required for HMGB1 Binding to Toll-like Receptor 4 and Activation of Macrophage Cytokine Release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, U.; Ottestad, W.; Tracey, K.J. Extracellular HMGB1: A Therapeutic Target in Severe Pulmonary Inflammation Including COVID-19? Mol. Med. 2020, 26, 42. [Google Scholar] [CrossRef]
- Deng, M.; Tang, Y.; Li, W.; Wang, X.; Zhang, R.; Zhang, X.; Zhao, X.; Liu, J.; Tang, C.; Liu, Z.; et al. The Endotoxin Delivery Protein HMGB1 Mediates Caspase-11-Dependent Lethality in Sepsis. Immunity 2018, 49, 740–753.e7. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Jiang, Y.; Wang, J.; Shi, X.; Liu, Q.; Liu, Z.; Li, Y.; Scott, M.J.; Xiao, G.; Li, S.; et al. Macrophage Endocytosis of High-Mobility Group Box 1 Triggers Pyroptosis. Cell Death Differ. 2014, 21, 1229–1239. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Bhat, O.M.; Lohner, H.; Zhang, Y.; Li, P.-L. Downregulation of Lysosomal Acid Ceramidase Mediates HMGB1-Induced Migration and Proliferation of Mouse Coronary Arterial Myocytes. Front. Cell Dev. Biol. 2020, 8, 111. [Google Scholar] [CrossRef]
- Kang, R.; Chen, R.; Zhang, Q.; Hou, W.; Wu, S.; Cao, L.; Huang, J.; Yu, Y.; Fan, X.-G.; Yan, Z.; et al. HMGB1 in Health and Disease. Mol. Asp. Med. 2014, 40, 1–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, P.; Voronov, E.; Dinarello, C.A.; Apte, R.N.; Cohen, I. Alarmins: Feel the Stress. J. Immunol. 2017, 198, 1395–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moussion, C.; Ortega, N.; Girard, J.-P. The IL-1-Like Cytokine IL-33 Is Constitutively Expressed in the Nucleus of Endothelial Cells and Epithelial Cells In Vivo: A Novel ‘Alarmin’? PLoS ONE 2008, 3, e3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, I.; Rider, P.; Vornov, E.; Tomas, M.; Tudor, C.; Wegner, M.; Brondani, L.; Freudenberg, M.; Mittler, G.; Ferrando-May, E.; et al. Erratum: Corrigendum: IL-1α Is a DNA Damage Sensor Linking Genotoxic Stress Signaling to Sterile Inflammation and Innate Immunity. Sci. Rep. 2016, 6, 19100. [Google Scholar] [CrossRef] [Green Version]
- Jeong, J.; Juhn, K.; Lee, H.; Kim, S.-H.; Min, B.-H.; Lee, K.-M.; Cho, M.-H.; Park, G.-H.; Lee, K.-H. SIRT1 Promotes DNA Repair Activity and Deacetylation of Ku70. Exp. Mol. Med. 2007, 39, 8–13. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.S.; Choi, H.S.; Ham, S.A.; Yoo, T.; Lee, W.J.; Paek, K.S.; Seo, H.G. Deacetylation-Mediated Interaction of SIRT1-HMGB1 Improves Survival in a Mouse Model of Endotoxemia. Sci. Rep. 2015, 5, 15971. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, T.; Maier, B.; Bartke, A.; Scrable, H. Progressive Loss of SIRT1 with Cell Cycle Withdrawal. Aging Cell 2006, 5, 413–422. [Google Scholar] [CrossRef]
- Anwar, T.; Khosla, S.; Ramakrishna, G. Increased Expression of SIRT2 Is a Novel Marker of Cellular Senescence and Is Dependent on Wild Type P53 Status. Cell Cycle 2016, 15, 1883–1897. [Google Scholar] [CrossRef] [Green Version]
- Son, M.J.; Kwon, Y.; Son, T.; Cho, Y.S. Restoration of Mitochondrial NAD+ Levels Delays Stem Cell Senescence and Facilitates Reprogramming of Aged Somatic Cells. Stem Cells 2016, 34, 2840–2851. [Google Scholar] [CrossRef]
- Chen, J.; Xie, J.-J.; Jin, M.-Y.; Gu, Y.-T.; Wu, C.-C.; Guo, W.-J.; Yan, Y.-Z.; Zhang, Z.-J.; Wang, J.-L.; Zhang, X.-L.; et al. Sirt6 Overexpression Suppresses Senescence and Apoptosis of Nucleus Pulposus Cells by Inducing Autophagy in a Model of Intervertebral Disc Degeneration. Cell Death Dis. 2018, 9, 56. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 Is a Central Mediator of Senescent Phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef] [PubMed]
- Lunin, S.M.; Novoselova, E.G. Thymus Hormones as Prospective Anti-Inflammatory Agents. Expert Opin. Targets 2010, 14, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Lunin, S.; Khrenov, M.; Glushkova, O.; Parfenyuk, S.; Novoselova, T.; Novoselova, E. Precursors of Thymic Peptides as Stress Sensors. Expert Opin. Biol. 2020, 20, 1461–1475. [Google Scholar] [CrossRef] [PubMed]
- McClure, J.E.; Lameris, N.; Wara, D.W.; Goldstein, A.L. Immunochemical Studies on Thymosin: Radioimmunoassay of Thymosin Alpha 1. J. Immunol. 1982, 128, 368–375. [Google Scholar]
- Weller, F.E.; Shah, U.; Cummings, G.D.; Chretien, P.B.; Mutchnick, M.G. Serum Levels of Immunoreactive Thymosin Alpha 1 and Thymosin Beta 4 in Large Cohorts of Healthy Adults. Thymus 1992, 19, 45–52. [Google Scholar] [PubMed]
- Gao, D.; Zhang, X.; Zhang, J.; Cao, J.; Wang, F. Expression of Thymosin A1-Thymopentin Fusion Peptide in Pichia Pastoris and Its Characterization. Arch. Pharm. Res. 2008, 31, 1471–1476. [Google Scholar] [CrossRef] [PubMed]
- Milenkovic, L.; McCann, S.M. Effects of Thymosin Alpha-1 on Pituitary Hormone Release. Neuroendocrinology 1992, 55, 14–19. [Google Scholar] [CrossRef]
- Karetsou, Z.; Martic, G.; Tavoulari, S.; Christoforidis, S.; Wilm, M.; Gruss, C.; Papamarcaki, T. Prothymosin α Associates with the Oncoprotein SET and Is Involved in Chromatin Decondensation. FEBS Lett. 2004, 577, 496–500. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, C.; Hasan, S.; Rowe, M.; Hottiger, M.; Orre, R.; Robertson, E.S. Epstein-Barr Virus Nuclear Antigen 3C and Prothymosin Alpha Interact with the P300 Transcriptio.onal Coactivator at the CH1 and CH3/HAT Domains and Cooperate in Regulation of Transcription and Histone Acetylation. J. Virol. 2002, 76, 4699–4708. [Google Scholar] [CrossRef] [Green Version]
- Karetsou, Z.; Kretsovali, A.; Murphy, C.; Tsolas, O.; Papamarcaki, T. Prothymosin Alpha Interacts with the CREB-Binding Protein and Potentiates Transcription. EMBO Rep. 2002, 3, 361–366. [Google Scholar] [CrossRef] [Green Version]
- Orre, R.S.; Cotter, M.A.; Subramanian, C.; Robertson, E.S. Prothymosin Alpha Functions as a Cellular Oncoprotein by Inducing Transformation of Rodent Fibroblasts in Vitro. J. Biol. Chem. 2001, 276, 1794–1799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, T.; Wang, T.; Maezawa, M.; Kobayashi, M.; Ohnishi, S.; Hatanaka, K.; Hige, S.; Shimizu, Y.; Kato, M.; Asaka, M.; et al. Overexpression of the Oncoprotein Prothymosin Alpha Triggers a P53 Response That Involves P53 Acetylation. Cancer Res. 2006, 66, 3137–3144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lal, A.; Kawai, T.; Yang, X.; Mazan-Mamczarz, K.; Gorospe, M. Antiapoptotic Function of RNA-Binding Protein HuR Effected through Prothymosin α. EMBO J. 2005, 24, 1852–1862. [Google Scholar] [CrossRef] [PubMed]
- Karapetian, R.N.; Evstafieva, A.G.; Abaeva, I.S.; Chichkova, N.V.; Filonov, G.S.; Rubtsov, Y.P.; Sukhacheva, E.A.; Melnikov, S.V.; Schneider, U.; Wanker, E.E.; et al. Nuclear Oncoprotein Prothymosin α Is a Partner of Keap1: Implications for Expression of Oxidative Stress-Protecting Genes. Mol. Cell Biol. 2005, 25, 1089–1099. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cui, F.; Lu, S.; Lu, H.; Jiang, T.; Chen, J.; Zhang, X.; Jin, Y.; Peng, Z.; Tang, H. Increased Expression of Prothymosin-α, Independently or Combined with TP53, Correlates with Poor Prognosis in Colorectal Cancer. Int. J. Clin. Exp. Pathol. 2014, 7, 4867–4876. [Google Scholar]
- Matsunaga, H.; Ueda, H. Stress-Induced Non-Vesicular Release of Prothymosin-α Initiated by an Interaction with S100A13, and Its Blockade by Caspase-3 Cleavage. Cell Death Differ. 2010, 17, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Samara, P.; Karachaliou, C.-E.; Ioannou, K.; Papaioannou, N.E.; Voutsas, I.F.; Zikos, C.; Pirmettis, I.; Papadopoulos, M.; Kalbacher, H.; Livaniou, E.; et al. Prothymosin Alpha: An Alarmin and More. Curr. Med. Chem. 2017, 24, 1747–1760. [Google Scholar] [CrossRef]
- Roy, R.; Singh, S.M.; Shanker, A.; Sodhi, A. Mechanism of Thymocyte Apoptosis Induced by Serum of Tumor-Bearing Host: The Molecular Events Involved and Their Inhibition by Thymosin Alpha-1. Int. J. Immunopharmacol. 2000, 22, 309–321. [Google Scholar] [CrossRef]
- Paul, S.; Sodhi, A. Modulatory Role of Thymosin-Alpha-1 in Normal Bone-Marrow Haematopoiesis and Its Effect on Myelosuppression in T-Cell Lymphoma Bearing Mice. Immunol. Lett. 2002, 82, 171–182. [Google Scholar] [CrossRef]
- Fan, Y.; Chang, H.; Yu, Y.; Liu, J.; Wang, R. Thymosin Alpha1 Suppresses Proliferation and Induces Apoptosis in Human Leukemia Cell Lines. Peptides 2006, 27, 2165–2173. [Google Scholar] [CrossRef]
- Guo, Y.; Chang, H.; Li, J.; Xu, X.; Shen, L.; Yu, Z.; Liu, W. Thymosin Alpha 1 Suppresses Proliferation and Induces Apoptosis in Breast Cancer Cells through PTEN-Mediated Inhibition of PI3K/Akt/MTOR Signaling Pathway. Apoptosis 2015, 20, 1109–1121. [Google Scholar] [CrossRef] [PubMed]
- Kharazmi-Khorassani, J.; Asoodeh, A. Thymosin Alpha-1; a Natural Peptide Inhibits Cellular Proliferation, Cell Migration, the Level of Reactive Oxygen Species and Promotes the Activity of Antioxidant Enzymes in Human Lung Epithelial Adenocarcinoma Cell Line (A549). Environ. Toxicol. 2019, 34, 941–949. [Google Scholar] [CrossRef] [PubMed]
- Lao, X.; Liu, M.; Chen, J.; Zheng, H. A Tumor-Penetrating Peptide Modification Enhances the Antitumor Activity of Thymosin Alpha 1. PLoS ONE 2013, 8, e72242. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Zhang, Z.; Jiang, X.; Nie, Y.; Wu, F. Stability evaluation of thymopentin in preparation process. Sichuan Da Xue Xue Bao Yi Xue Ban 2003, 34, 292–294. [Google Scholar] [PubMed]
- Twomey, J.J.; Goldstein, G.; Lewis, V.M.; Bealmear, P.M.; Good, R.A. Bioassay Determinations of Thymopoietin and Thymic Hormone Levels in Human Plasma. Proc. Natl. Acad. Sci. USA 1977, 74, 2541–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angioni, S.; Iori, G.; Cellini, M.; Sardelli, S.; Massolo, F.; Petraglia, F.; Genazzani, A.R. Acute Beta-Interferon or Thymopentin Administration Increases Plasma Growth Hormone and Cortisol Levels in Children. Acta Endocrinol. 1992, 127, 237–241. [Google Scholar] [CrossRef]
- Malaise, M.G.; Hazee-Hagelstein, M.T.; Reuter, A.M.; Vrinds-Gevaert, Y.; Goldstein, G.; Franchimont, P. Thymopoietin and Thymopentin Enhance the Levels of ACTH, β-Endorphin and β-Lipotropin from Rat Pituitary Cells in Vitro. Acta Endocrinol. 1987, 115, 455–460. [Google Scholar] [CrossRef]
- Novoselova, E.G.; Lunin, S.M.; Khrenov, M.O.; Novoselova, T.V.; Fesenko, E.E. Involvement of NF-KappaB Transcription Factor in the Antiinflammatory Activity o.of Thymic Peptides. Dokl. Biol. Sci. 2009, 428, 484–486. [Google Scholar] [CrossRef]
- Lunin, S.M.; Novoselova, T.V.; Khrenov, M.O.; Glushkova, O.V.; Parfeniuk, S.B.; Smolikhina, T.I.; Fesenko, E.E.; Novoselova, E.G. Immunomodulatory effects of thymopentin under acute and chronic inflammations in mice. Biofizika 2009, 54, 260–266. [Google Scholar] [CrossRef]
- Foisner, R.; Gerace, L. Integral Membrane Proteins of the Nuclear Envelope Interact with Lamins and Chromosomes, and Binding Is Modulated by Mitotic Phosphorylation. Cell 1993, 73, 1267–1279. [Google Scholar] [CrossRef]
- Furukawa, K.; Panté, N.; Aebi, U.; Gerace, L. Cloning of a CDNA for Lamina-Associated Polypeptide 2 (LAP2) and Identification of Regions That Specify Targeting to the Nuclear Envelope. EMBO J. 1995, 14, 1626–1636. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, K. LAP2 Binding Protein 1 (L2BP1/BAF) Is a Candidate Mediator of LAP2-Chromatin Interaction. J. Cell Sci. 1999, 112 Pt 15, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Guan, T.; Gerace, L. Lamin-Binding Fragment of LAP2 Inhibits Increase in Nuclear Volume during the Cell Cycle and Progression into S Phase. J. Cell Biol. 1997, 139, 1077–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gant, T.M.; Harris, C.A.; Wilson, K.L. Roles of LAP2 Proteins in Nuclear Assembly and DNA Replication: Truncated LAP2β Proteins Alter Lamina Assembly, Envelope Formation, Nuclear Size, and DNA Replication Efficiency in Xenopus Laevis Extracts. J. Cell Biol. 1999, 144, 1083–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dechat, T.; Korbei, B.; Vaughan, O.A.; Vlcek, S.; Hutchison, C.J.; Foisner, R. Lamina-Associated Polypeptide 2alpha Binds Intranuclear A-Type Lamins. J. Cell Sci. 2000, 113 Pt 19, 3473–3484. [Google Scholar] [CrossRef]
- Dechat, T.; Gajewski, A.; Korbei, B.; Gerlich, D.; Daigle, N.; Haraguchi, T.; Furukawa, K.; Ellenberg, J.; Foisner, R. LAP2alpha and BAF Transiently Localize to Telomeres and Specific Regions on Chromatin during Nuclear Assembly. J. Cell Sci. 2004, 117, 6117–6128. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Jia, J.; Zhou, Z.; Chu, Q.; Lian, W.; Chen, Z. The Emerging Role of Thymopoietin-Antisense RNA 1 as Long Noncoding RNA in the Pathogenesis of Human Cancers. DNA Cell Biol. 2021, 40, 848–857. [Google Scholar] [CrossRef]
- Zhou, H.; Zhang, L.; Tu, H. Downregulation of Thymopoietin by MiR-139-5p Suppresses Cell Proliferation and Induces Cell Cycle Arrest/Apoptosis in Pancreatic Ductal Adenocarcinoma. Oncol. Lett. 2019, 18, 3443–3452. [Google Scholar] [CrossRef] [Green Version]
- Chojnowski, A.; Ong, P.F.; Wong, E.S.M.; Lim, J.S.Y.; Mutalif, R.A.; Navasankari, R.; Dutta, B.; Yang, H.; Liow, Y.Y.; Sze, S.K.; et al. Progerin Reduces LAP2α-Telomere Association in Hutchinson-Gilford Progeria. Elife 2015, 4, e07759. [Google Scholar] [CrossRef]
- Gotzmann, J.; Vlcek, S.; Foisner, R. Caspase-Mediated Cleavage of the Chromosome-Binding Domain of Lamina-Associated Polypeptide 2 Alpha. J. Cell Sci. 2000, 113 Pt 21, 3769–3780. [Google Scholar] [CrossRef]
- Buendia, B.; Santa-Maria, A.; Courvalin, J.C. Caspase-Dependent Proteolysis of Integral and Peripheral Proteins of Nuclear Membranes and Nuclear Pore Complex Proteins during Apoptosis. J. Cell Sci. 1999, 112 Pt 11, 1743–1753. [Google Scholar] [CrossRef] [PubMed]
- Duband-Goulet, I.; Courvalin, J.C.; Buendia, B. LBR, a Chromatin and Lamin Binding Protein from the Inner Nuclear Membrane, Is Proteolyzed at Late Stages of Apoptosis. J. Cell Sci. 1998, 111 Pt 10, 1441–1451. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Jing, F.; Huang, P.; Geng, Z.; Xu, J.; Li, J.; Chen, D.; Zhu, Y.; Wang, Z.; Huang, W.; et al. Thymopentin Alleviates Premature Ovarian Failure in Mice by Activating YY2/Lin28A and Inhibiting the Expression of Let-7 Family MicroRNAs. Cell Prolif. 2021, 54, e13089. [Google Scholar] [CrossRef]
- Kasim, V.; Xie, Y.-D.; Wang, H.-M.; Huang, C.; Yan, X.-S.; Nian, W.-Q.; Zheng, X.-D.; Miyagishi, M.; Wu, S.-R. Transcription Factor Yin Yang 2 Is a Novel Regulator of the P53/P21 Axis. Oncotarget 2017, 8, 54694–54707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Chang, H.; Yu, Y.; Liu, J.; Zhao, L.; Yang, D.; Wang, R. Thymopentin (TP5), an Immunomodulatory Peptide, Suppresses Proliferation and Induces Differentiation in HL-60 Cells. Biochim. Biophys. Acta 2006, 1763, 1059–1066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cao, Y.; Meng, Y.; You, Z.; Liu, X.; Liu, Z. The Novel Role of Thymopentin in Induction of Maturation of Bone Marrow Dendritic Cells (BMDCs). Int. Immunopharmacol. 2014, 21, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Gonser, S.; Crompton, N.E.A.; Folkers, G.; Weber, E. Increased Radiation Toxicity by Enhanced Apoptotic Clearance of HL-60 Cells in the Presence of the Pentapeptide Thymopentin, Which Selectively Binds to Apoptotic Cells. Mutat. Res. 2004, 558, 19–26. [Google Scholar] [CrossRef]
- Naylor, P.H.; Friedman-Kien, A.; Hersh, E.; Erdos, M.; Goldstein, A.L. Thymosin A1 and Thymosin Β4 in Serum: Comparison of Normal, Cord, Homosexual and AIDS Serum. Int. J. Immunopharmacol. 1986, 8, 667–676. [Google Scholar] [CrossRef]
- Weller, F.E.; Mutchnick, M.G.; Goldstein, A.L.; Naylor, P.H. Enzyme Immunoassay Measurement of Thymosin Beta 4 in Human Serum. J. Biol. Response Mod. 1988, 7, 91–96. [Google Scholar]
- Junot, C.; Nicolet, L.; Ezan, E.; Gonzales, M.F.; Menard, J.; Azizi, M. Effect of Angiotensin-Converting Enzyme Inhibition on Plasma, Urine, and Tissue Concentrations of Hemoregulatory Peptide Acetyl-Ser-Asp-Lys-Pro in Rats. J. Pharm. Exp. 1999, 291, 982–987. [Google Scholar]
- Gonzalez-Franquesa, A.; Stocks, B.; Borg, M.L.; Kuefner, M.; Dalbram, E.; Nielsen, T.S.; Agrawal, A.; Pankratova, S.; Chibalin, A.V.; Karlsson, H.K.R.; et al. Discovery of Thymosin Β4 as a Human Exerkine and Growth Factor. Am. J. Physiol. Cell Physiol. 2021, 321, C770–C778. [Google Scholar] [CrossRef] [PubMed]
- Carlier, M.-F.; Pantaloni, D. Actin Assembly in Response to Extracellular Signals: Role of Capping Proteins, Thymosin Β4 and Profilin. Semin. Cell Biol. 1994, 5, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Hannappel, E.; van Kampen, M. Determination of Thymosin Beta 4 in Human Blood Cells and Serum. J. Chromatogr. 1987, 397, 279–285. [Google Scholar] [CrossRef]
- Frohm, M.; Gunne, H.; Bergman, A.-C.; Agerberth, B.; Bergman, T.; Boman, A.; Liden, S.; Jornvall, H.; Boman, H.G. Biochemical and Antibacterial Analysis of Human Wound and Blister Fluid. Eur. J. Biochem. 1996, 237, 86–92. [Google Scholar] [CrossRef]
- Malinda, K.M.; Goldstein, A.L.; Kueinman, H.K. Thymosin β4 Stimulates Directional Migration of Human Umbilical Vein Endothelial Cells. FASEB J. 1997, 11, 474–481. [Google Scholar] [CrossRef]
- Sosne, G.; Christopherson, P.L.; Barrett, R.P.; Fridman, R. Thymosin-Β4 Modulates Corneal Matrix Metalloproteinase Levels and Polymorphonuclear Cell Infiltration after Alkali Injury. Invest. Ophthalmol. Vis. Sci. 2005, 46, 2388. [Google Scholar] [CrossRef]
- Sosne, G.; Qiu, P.; Christopherson, P.L.; Wheater, M.K. Thymosin Beta 4 Suppression of Corneal NFkappaB: A Potential Anti-Inflammatory Pathway. Exp. Eye Res. 2007, 84, 663–669. [Google Scholar] [CrossRef] [Green Version]
- Cavasin, M.A.; Rhaleb, N.-E.; Yang, X.-P.; Carretero, O.A. Prolyl Oligopeptidase Is Involved in Release of the Antifibrotic Peptide Ac-SDKP. Hypertension 2004, 43, 1140–1145. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-M.; Lawrence, F.; Kovacevic, M.; Bignon, J.; Papadimitriou, E.; Lallemand, J.-Y.; Katsoris, P.; Potier, P.; Fromes, Y.; Wdzieczak-Bakala, J. The Tetrapeptide AcSDKP, an Inhibitor of Primitive Hematopoietic Cell Proliferation, Induces Angiogenesis in Vitro and in Vivo. Blood 2003, 101, 3014–3020. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.; Rhaleb, N.-E.; Pokharel, S.; Harding, P.; Rasoul, S.; Peng, H.; Carretero, O.A. Novel Anti-Inflammatory Mechanisms of N-Acetyl-Ser-Asp-Lys-Pro in Hypertension-Induced Target Organ Damage. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1226–H1232. [Google Scholar] [CrossRef]
- Rieger, K.J.; Saez-Servent, N.; Papet, M.P.; Wdzieczak-Bakala, J.; Morgat, J.L.; Thierry, J.; Voelter, W.; Lenfant, M. Involvement of Human Plasma Angiotensin I-Converting Enzyme in the Degradation of the Haemoregulatory Peptide N -Acetyl-Seryl-Aspartyl-Lysyl-Proline. Biochem. J. 1993, 296, 373–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, A.; Hannappel, E.; Kleinman, H. Thymosin β: Actin-Sequestering Protein Moonlights to Repair Injured Tissues. Trends Mol. Med. 2005, 11, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yu, L.; Zhao, Y.; Fu, G.; Zhou, B. Thymosin Β4 Reduces Senescence of Endothelial Progenitor Cells via the PI3K/Akt/ENOS Signal Transduction Pathway. Mol. Med. Rep. 2013, 7, 598–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Qiu, F.; Xu, S.; Yu, L.; Fu, G. Thymosin Β4 Activates Integrin-Linked Kinase and Decreases Endothelial Progenitor Cells Apoptosis under Serum Deprivation. J. Cell Physiol. 2011, 226, 2798–2806. [Google Scholar] [CrossRef]
- Naeem, A.; Harish, V.; Coste, S.; Parasido, E.M.; Choudhry, M.U.; Kromer, L.F.; Ihemelandu, C.; Petricoin, E.F.; Pierobon, M.; Noon, M.S.; et al. Regulation of Chemosensitivity in Human Medulloblastoma Cells by P53 and the PI3 Kinase Signaling Pathway. Mol. Cancer Res. 2022, 20, 114–126. [Google Scholar] [CrossRef]
- Han, H.-J.; Kim, S.; Kwon, J. Thymosin Beta 4-Induced Autophagy Increases Cholinergic Signaling in PrP (106-126)-Treated HT22 Cells. Neurotox. Res. 2019, 36, 58–65. [Google Scholar] [CrossRef]
- Renga, G.; Oikonomou, V.; Moretti, S.; Stincardini, C.; Bellet, M.M.; Pariano, M.; Bartoli, A.; Brancorsini, S.; Mosci, P.; Finocchi, A.; et al. Thymosin Β4 Promotes Autophagy and Repair via HIF-1α Stabilization in Chronic Granulomatous Disease. Life Sci. Alliance 2019, 2, e201900432. [Google Scholar] [CrossRef]
- Yoon, H.J.; Oh, Y.L.; Ko, E.-J.; Kang, A.; Eo, W.K.; Kim, K.H.; Lee, J.Y.; Kim, A.; Chun, S.; Kim, H.; et al. Effects of Thymosin Β4-Derived Peptides on Migration and Invasion of Ovarian Cancer Cells. Genes Genom. 2021, 43, 987–993. [Google Scholar] [CrossRef]
- Moon, E.-Y.; Song, J.-H.; Yang, K.-H. Actin-Sequestering Protein, Thymosin-Beta-4 (TB4), Inhibits Caspase-3 Activation in Paclitaxel-Induced Tumor Cell Death. Oncol. Res. 2007, 16, 507–516. [Google Scholar] [CrossRef]
- Torres-Farfan, C.; Seron-Ferre, M.; Dinet, V.; Korf, H.-W. Immunocytochemical Demonstration of Day/Night Changes of Clock Gene Protein Levels in the Murine Adrenal Gland: Differences between Melatonin-Proficient (C3H) and Melatonin-Deficient (C57BL) Mice. J. Pineal Res. 2006, 40, 64–70. [Google Scholar] [CrossRef]
- Campino, C.; Valenzuela, F.J.; Torres-Farfan, C.; Reynolds, H.E.; Abarzua-Catalan, L.; Arteaga, E.; Trucco, C.; Guzmán, S.; Valenzuela, G.J.; Seron-Ferre, M. Melatonin Exerts Direct Inhibitory Actions on ACTH Responses in the Human Adrenal Gland. Horm. Metab. Res. 2011, 43, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Farfan, C.; Richter, H.G.; Rojas-García, P.; Vergara, M.; Forcelledo, M.L.; Valladares, L.E.; Torrealba, F.; Valenzuela, G.J.; Serón-Ferré, M. Mt1 Melatonin Receptor in the Primate Adrenal Gland: Inhibition of Adrenocorticotropin-Stimulated Cortisol Production by Melatonin. J. Clin. Endocrinol. Metab. 2003, 88, 450–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranjo, M.C.; Guerrero, J.M.; Rubio, A.; Lardone, P.J.; Carrillo-Vico, A.; Carrascosa-Salmoral, M.P.; Jiménez-Jorge, S.; Arellano, M.V.; Leal-Noval, S.R.; Leal, M.; et al. Melatonin Biosynthesis in the Thymus of Humans and Rats. Cell Mol. Life Sci. 2007, 64, 781–790. [Google Scholar] [CrossRef]
- Hardeland, R. Aging, Melatonin, and the Pro- and Anti-Inflammatory Networks. Int. J. Mol. Sci. 2019, 20, E1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrillo-Vico, A.; Calvo, J.R.; Abreu, P.; Lardone, P.J.; García-Mauriño, S.; Reiter, R.J.; Guerrero, J.M. Evidence of Melatonin Synthesis by Human Lymphocytes and Its Physiological Significance: Possible Role as Intracrine, Autocrine, and/or Paracrine Substance. FASEB J. 2004, 18, 537–539. [Google Scholar] [CrossRef]
- Maestroni, G.J.; Otsa, K.; Cutolo, M. Melatonin Treatment Does Not Improve Rheumatoid Arthritis. Br. J. Clin. Pharm. 2008, 65, 797–798. [Google Scholar] [CrossRef] [Green Version]
- Ghareghani, M.; Dokoohaki, S.; Ghanbari, A.; Farhadi, N.; Zibara, K.; Khodadoust, S.; Parishani, M.; Ghavamizadeh, M.; Sadeghi, H. Melatonin Exacerbates Acute Experimental Autoimmune Encephalomyelitis by Enhancing the Serum Levels of Lactate: A Potential Biomarker of Multiple Sclerosis Progression. Clin. Exp. Pharm. Physiol 2017, 44, 52–61. [Google Scholar] [CrossRef]
- Hardeland, R. Melatonin and Inflammation-Story of a Double-Edged Blade. J. Pineal Res. 2018, 65, e12525. [Google Scholar] [CrossRef] [Green Version]
- Kuklina, E.M.; Glebezdina, N.S.; Nekrasova, I.V. Role of Melatonin in the Regulation of Differentiation of T Cells Producing Interleukin-17 (Th17). Bull. Exp. Biol. Med. 2016, 160, 656–658. [Google Scholar] [CrossRef]
- Molinero, P.; Soutto, M.; Benot, S.; Hmadcha, A.; Guerrero, J.M. Melatonin Is Responsible for the Nocturnal Increase Observed in Serum and Thymus of Thymosin Alpha1 and Thymulin Concentrations: Observations in Rats and Humans. J. Neuroimmunol. 2000, 103, 180–188. [Google Scholar] [CrossRef]
- Reiter, R.J.; Acuña-Castroviejo, D.; Tan, D.X.; Burkhardt, S. Free Radical-Mediated Molecular Damage. Mechanisms for the Protective Actions of Melatonin in the Central Nervous System. Ann. N. Y. Acad. Sci. 2001, 939, 200–215. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wang, X.; Geng, P.; Tang, X.; Xiang, L.; Lu, X.; Li, J.; Ruan, Z.; Chen, J.; Xie, G.; et al. Melatonin Regulates PARP1 to Control the Senescence-Associated Secretory Phenotype (SASP) in Human Fetal Lung Fibroblast Cells. J. Pineal Res. 2017, 63, e12405. [Google Scholar] [CrossRef] [PubMed]
- Bae, W.-J.; Park, J.; Kang, S.-K.; Kwon, I.-K.; Kim, E.-C. Effects of Melatonin and Its Underlying Mechanism on Ethanol-Stimulated Senescence and Osteoclastic Differentiation in Human Periodontal Ligament Cells and Cementoblasts. IJMS 2018, 19, 1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, J.; Yan, Y.; Teng, X.; Wen, X.; Li, N.; Peng, S.; Liu, W.; Donadeu, F.X.; Zhao, S.; Hua, J. Melatonin Prevents Senescence of Canine Adipose-Derived Mesenchymal Stem Cells through Activating NRF2 and Inhibiting ER Stress. Aging 2018, 10, 2954–2972. [Google Scholar] [CrossRef]
- Brooks, C.L.; Gu, W. How Does SIRT1 Affect Metab.bolism, Senescence and Cancer? Nat. Rev. Cancer 2009, 9, 123–128. [Google Scholar] [CrossRef]
- Deng, C.-X. SIRT1, Is It a Tumor Promoter or Tumor Suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Yi, J.; Luo, J. SIRT1 and P53, Effect on Cancer, Senescence and Beyond. Biochim. Biophys. Acta 2010, 1804, 1684–1689. [Google Scholar] [CrossRef] [Green Version]
- Han, N.; Wang, Z.; Li, X. Melatonin Alleviates D-Galactose-Decreased Hyaluronic Acid Production in Synovial Membrane Cells via Sirt1 Signalling. Cell Biochem. Funct. 2021, 39, 488–495. [Google Scholar] [CrossRef]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of FOXOs and P53 by SIRT1 Modulators under Oxidative Stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef]
- Kang, J.-W.; Cho, H.-I.; Lee, S.-M. Melatonin Inhibits MTOR-Dependent Autophagy during Liver Ischemia/Reperfusion. Cell Physiol. Biochem. 2014, 33, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.R.; Yang, J.Q.; Liu, F.; Shen, X.Q.; Zhou, Y.J. Melatonin Attenuates Vascular Calcification by Activating Autophagy via an AMPK/MTOR/ULK1 Signaling Pathway. Exp. Cell Res. 2020, 389, 111883. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.E.; Cole, S.W.; Seeman, T.E.; Breen, E.C.; Witarama, T.; Arevalo, J.M.G.; Ma, J.; Irwin, M.R. Partial Sleep Deprivation Activates the DNA Damage Response (DDR) and the Senescence-Associated Secretory Phenotype (SASP) in Aged Adult Humans. Brain Behav. Immun. 2016, 51, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spinedi, E.; Cardinali, D.P. Neuroendocrine-Metabolic Dysfunction and Sleep Disturbances in Neurodegenerative Disorders: Focus on Alzheimer’s Disease and Melatonin. Neuroendocrinology 2019, 108, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory Action of Glucocorticoids--New Mechanisms for Old Drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, P.A.; Garvin, J.L. Cardiovascular and Renal Control in NOS-Deficient Mouse Models. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 284, R628–R638. [Google Scholar] [CrossRef] [PubMed] [Green Version]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunin, S.M.; Novoselova, E.G.; Glushkova, O.V.; Parfenyuk, S.B.; Novoselova, T.V.; Khrenov, M.O. Cell Senescence and Central Regulators of Immune Response. Int. J. Mol. Sci. 2022, 23, 4109. https://doi.org/10.3390/ijms23084109
Lunin SM, Novoselova EG, Glushkova OV, Parfenyuk SB, Novoselova TV, Khrenov MO. Cell Senescence and Central Regulators of Immune Response. International Journal of Molecular Sciences. 2022; 23(8):4109. https://doi.org/10.3390/ijms23084109
Chicago/Turabian StyleLunin, Sergey M., Elena G. Novoselova, Olga V. Glushkova, Svetlana B. Parfenyuk, Tatyana V. Novoselova, and Maxim O. Khrenov. 2022. "Cell Senescence and Central Regulators of Immune Response" International Journal of Molecular Sciences 23, no. 8: 4109. https://doi.org/10.3390/ijms23084109