
Sex Differences in Cardiovascular Diseases: A Matter of Estrogens, Ceramides, and Sphingosine 1-Phosphate

 , , , and
, , , and 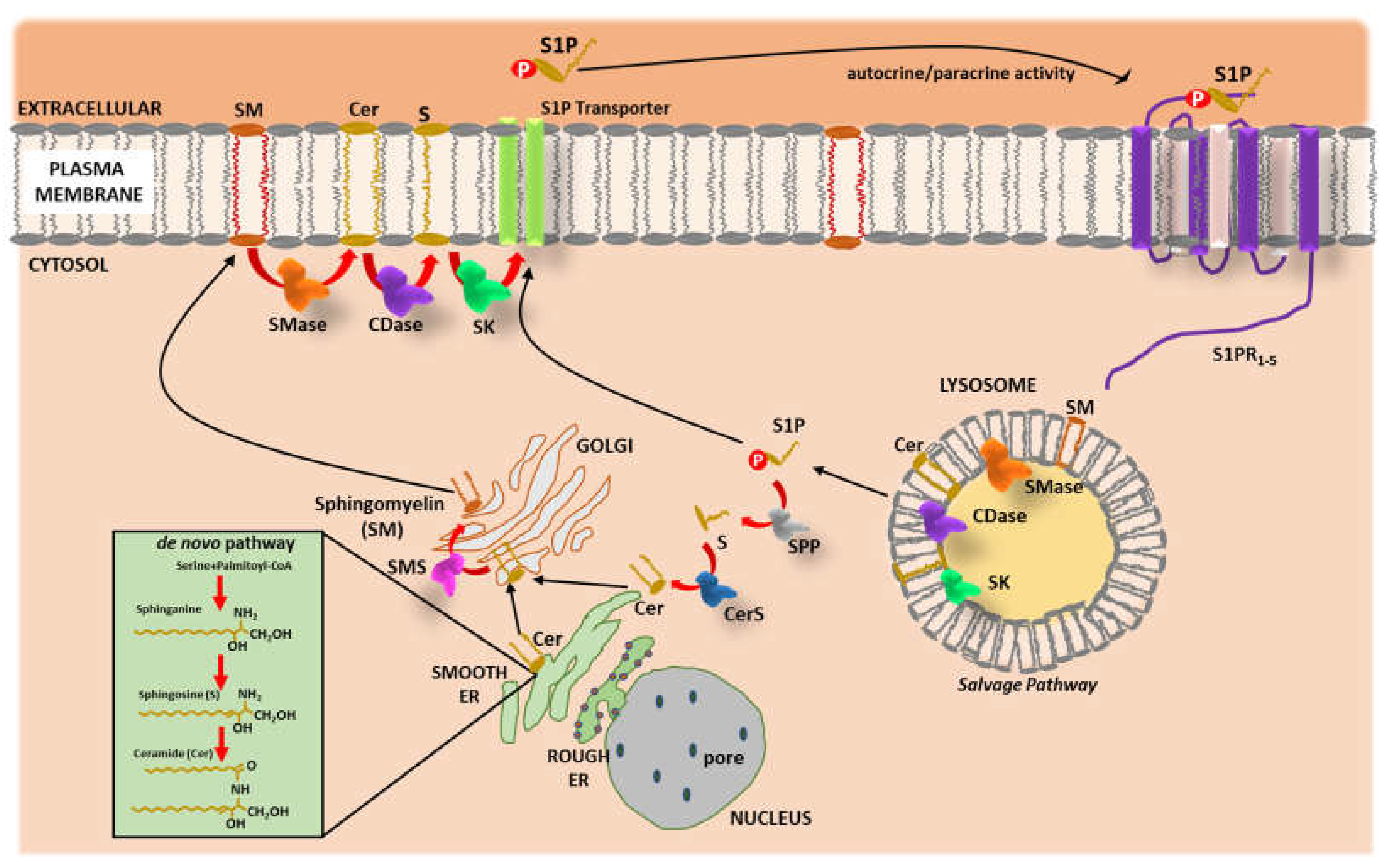{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sphingolipids Metabolism
3. Ceramide and Sphingosine 1-Phosphate in Cardiovascular Physiology and Disease
3.1. Cer
3.2. S1P
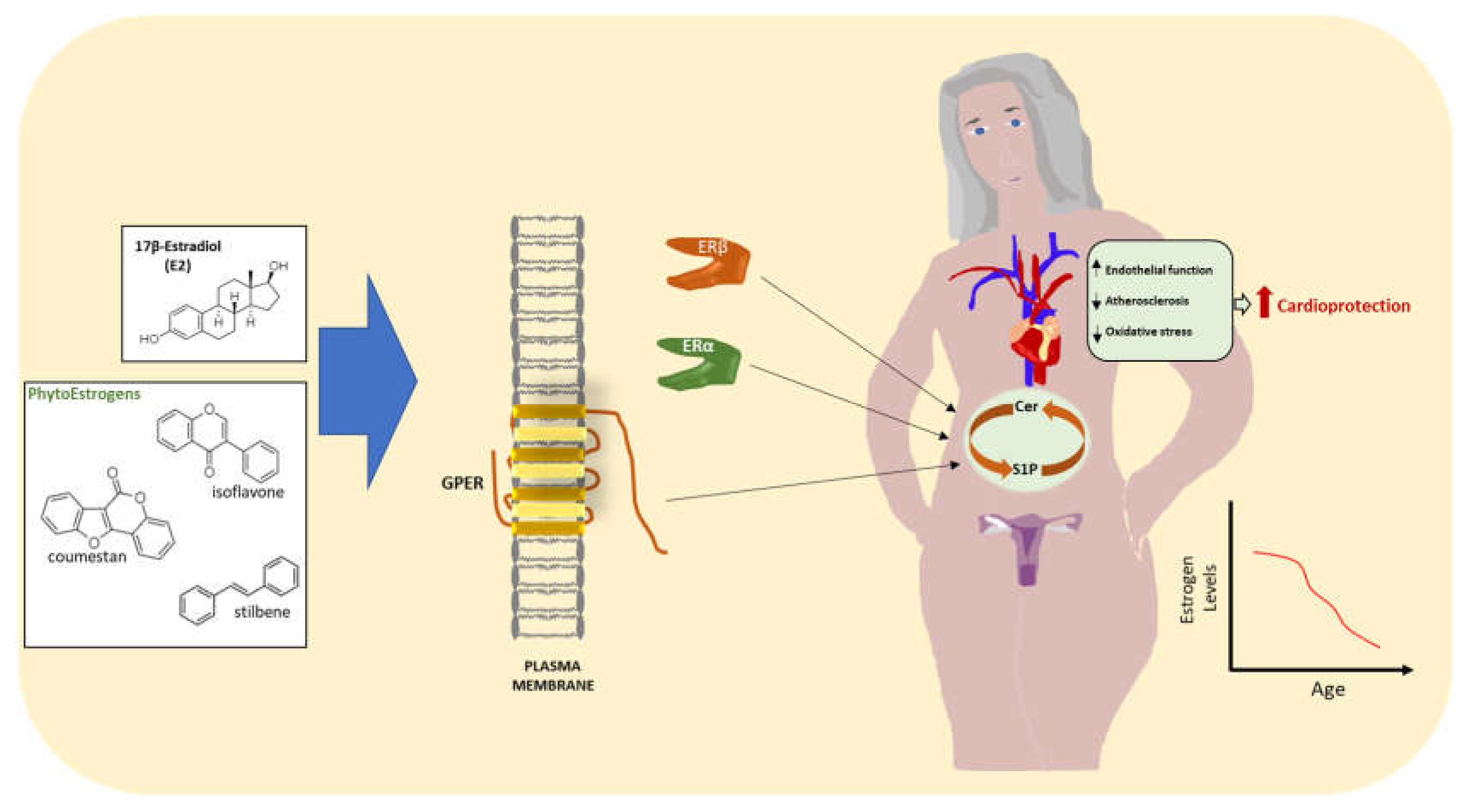
4. Estrogens and CVD
5. Estrogens and Sphingolipids
6. Conclusions and Perspectives
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lucà, F.; Abrignani, M.G.; Parrini, I.; Di Fusco, S.A.; Giubilato, S.; Rao, C.M.; Piccioni, L.; Cipolletta, L.; Passaretti, B.; Giallauria, F.; et al. Update on Management of Cardiovascular Diseases in Women. J. Clin. Med. 2022, 11, 1176. [Google Scholar] [CrossRef] [PubMed]
- Cesaroni, G.; Mureddu, G.F.; Agabiti, N.; Mayer, F.; Stafoggia, M.; Forastiere, F.; Latini, R.; Masson, S.; Davoli, M.; Boccanelli, A.; et al. Sex differences in factors associated with heart failure and diastolic left ventricular dysfunction: A cross-sectional population-based study. BMC Public Health 2021, 21, 415. [Google Scholar] [CrossRef] [PubMed]
- Pullen, A.B.; Kain, V.; Serhan, C.N.; Halade, G.V. Molecular and Cellular Differences in Cardiac Repair of Male and Female Mice. J. Am. Heart Assoc. 2020, 9, e015672. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y.; Mouton, A.J.; Ero, O.K.; Ma, Y.; Padmanabhan Iyer, R.; Flynn, E.R.; Espinoza, I.; Musani, S.K.; Vasan, R.S.; Hall, M.E.; et al. LXR/RXR signaling and neutrophil phenotype following myocardial infarction classify sex differences in remodeling. Basic Res. Cardiol. 2018, 113, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Chen, X.; McClusky, R.; Ruiz-Sundstrom, M.; Itoh, Y.; Umar, S.; Arnold, A.P.; Eghbali, M. The number of X chromosomes influences protection from cardiac ischaemia/reperfusion injury in mice: One X is better than two. Cardiovasc. Res. 2014, 102, 375–384. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Williams, R.; Healy, C.L.; Wright, C.D.; Wu, S.C.; O’Connell, T.D. An association between gene expression and better survival in female mice following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 49, 801–811. [Google Scholar] [CrossRef] [Green Version]
- Ueda, K.; Fukuma, N.; Adachi, Y.; Numata, G.; Tokiwa, H.; Toyoda, M.; Otani, A.; Hashimoto, M.; Liu, P.Y.; Takimoto, E. Sex Differences and Regulatory Actions of Estrogen in Cardiovascular System. Front. Physiol. 2021, 12, 738218. [Google Scholar] [CrossRef]
- Crescioli, C. The Role of Estrogens and Vitamin D in Cardiomyocyte Protection: A Female Perspective. Biomolecules 2021, 11, 1815. [Google Scholar] [CrossRef]
- Matarrese, P.; Maccari, S.; Vona, R.; Gambardella, L.; Stati, T.; Marano, G. Role of β-Adrenergic Receptors and Estrogen in Cardiac Repair after Myocardial Infarction: An Overview. Int. J. Mol. Sci. 2021, 22, 8957. [Google Scholar] [CrossRef]
- Xu, S.; Xie, F.; Tian, L.; Fallah, S.; Babaei, F.; Manno, S.H.C.; Manno, F.A.M.; Zhu, L.; Wong, K.F.; Liang, Y.; et al. Estrogen accelerates heart regeneration by promoting the inflammatory response in zebrafish. J. Endocrinol. 2020, 245, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Sabbatini, A.R.; Kararigas, G. Menopause-Related Estrogen Decrease and the Pathogenesis of HFpEF: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2020, 75, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Shahbazian, M.; Jafarynezhad, F.; Yadeghari, M.; Farhadi, Z.; Samani, S.L.; Esmailidehaj, M.; Safari, F.; Azizian, H. The effects of G protein-coupled receptor 30 (GPR30) on cardiac glucose metabolism in diabetic ovariectomized female rats. J. Basic Clin. Physiol. Pharmacol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.J.W.; Ornatsky, O.; Stewart, D.J.; Picard, P.; Dawood, F.; Wen, W.-H.; Liu, P.P.; Webb, D.J.; Monge, J.C. Effects of estrogen replacement on infarct size, cardiac remodeling, and the endothelin system after myocardial infarction in ovariectomized rats. Circulation 2000, 102, 2983–2989. [Google Scholar] [CrossRef] [Green Version]
- da Silva, J.S.; Montagnoli, T.L.; Rocha, B.S.; Tacco, M.L.C.A.; Marinho, S.C.P.; Zapata-Sudo, G. Estrogen Receptors: Therapeutic Perspectives for the Treatment of Cardiac Dysfunction after Myocardial Infarction. Int. J. Mol. Sci. 2021, 22, 525. [Google Scholar] [CrossRef] [PubMed]
- Grodstein, F.; Manson, J.E.; Colditz, G.A.; Willett, W.C.; Speizer, F.E.; Stampfer, M.J. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann. Intern. Med. 2000, 133, 933–941. [Google Scholar] [CrossRef]
- Gersh, F.L.; O’Keefe, J.H.; Lavie, C.J. Postmenopausal hormone therapy for cardiovascular health: The evolving data. Heart 2021, 107, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Prabakaran, S.; Vitter, S.; Lundberg, G. Cardiovascular Disease in Women Update: Ischemia, Diagnostic Testing, and Menopause Hormone Therapy. Endocr. Pract. 2022, 28, 199–203. [Google Scholar] [CrossRef]
- Pan, M.; Pan, X.; Zhou, J.; Wang, J.; Qi, Q.; Wang, L. Update on hormone therapy for the management of postmenopausal women. Biosci. Trends 2022, 16, 46–57. [Google Scholar] [CrossRef]
- Yang, X.P.; Reckelhoff, J.F. Estrogen, hormonal replacement therapy and cardiovascular disease. Curr. Opin. Nephrol. Hypertens. 2011, 20, 133–138. [Google Scholar] [CrossRef] [Green Version]
- Rozenberg, S.; Vandromme, J.; Antoine, C. Postmenopausal hormone therapy: Risks and benefits. Nat. Rev. Endocrinol. 2013, 9, 216–227. [Google Scholar] [CrossRef]
- Whayne, T.F.; Mukherjee, D. Women, the menopause, hormone replacement therapy and coronary heart disease. Curr. Opin. Cardiol. 2015, 30, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Sukocheva, O.; Wadham, C. Role of sphingolipids in oestrogen signalling in breast cancer cells: An update. J. Endocrinol. 2014, 220, R25–R35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruett, S.T.; Bushnev, A.; Hagedorn, K.; Adiga, M.; Haynes, C.A.; Sullards, M.C.; Liotta, D.C.; Merrill, A.H., Jr. Biodiversity of sphingoid bases (“sphingosines”) and related amino alcohols. J. Lipid Res. 2008, 49, 1621–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Cuvillier, O.; Edsall, L.; Kohama, T.; Menzeleev, R.; Olivera, A.; Thomas, D.; Tu, Z.; Van Brocklyn, J.; Wang, F. Roles of sphingosine-1-phosphate in cell growth, differentiation, and death. Biochem. Biokhimiia 1998, 63, 69–73. [Google Scholar]
- Zelnik, I.D.; Kim, J.L.; Futerman, A.H. The Complex Tail of Circulating Sphingolipids in Atherosclerosis and Cardiovascular Disease. J. Lipid Atheroscler. 2021, 10, 268–281. [Google Scholar] [CrossRef]
- Jozefczuk, E.; Guzik, T.J.; Siedlinski, M. Significance of sphingosine-1-phosphate in cardiovascular physiology and pathology. Pharmacol. Res. 2020, 156, 104793. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, W.; Li, J.; Sun, Y.; Yang, Q.; Wang, S.; Luo, X.; Wang, W.; Wang, K.; Bai, W.; et al. The imbalance in the aortic ceramide/sphingosine-1-phosphate rheostat in ovariectomized rats and the preventive effect of estrogen. Lipids Health Dis. 2020, 19, 95. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Williams, J.B. The control of the balance between ceramide and sphingo-sine-1-phosphate by sphingosine kinase: Oxidative stress and the seesaw of cell survival and death. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2012, 163, 26–36. [Google Scholar] [CrossRef]
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of Brain, Bailliere, Tindall, and Cox. Glasg. Med. J. 1884, 22, 363–364. [Google Scholar]
- Thudichum, J.L.W. A Treatise on the Chemical Constitution of the Brain; Archon Books: North Haven, CT, USA, 1962. [Google Scholar]
- Carter, H.E.; Glick, F.J.; Norris, W.P.; Phillips, G.E. Biochemistry of the sphingolipids. III. Structure of sphingosine. J. Biol. Chem. 1947, 170, 285–294. [Google Scholar] [CrossRef]
- Sasset, L.; Zhang, Y.; Dunn, T.M.; Di Lorenzo, A. Sphingolipid De Novo Biosynthesis: A Rheostat of Cardiovascular Homeostasis. Trends Endocrinol. Metab. 2016, 27, 807–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, P.J.; Dunn, T.M.; Campopiano, D.J. Sphingolipid biosynthesis in man and microbes. Nat. Prod. Rep. 2018, 35, 921–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanada, K. Discovery of the molecular machinery CERT for endoplasmic reticulum-to-Golgi trafficking of ceramide. Mol. Cell. Biochem. 2006, 286, 23–31. [Google Scholar] [CrossRef]
- Hanada, K. Intracellular trafficking of ceramide by ceramide transfer protein. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 426–437. [Google Scholar] [CrossRef] [Green Version]
- Bikman, B.J.; Summers, S.A. Ceramides as modulators of cellular and whole-body metabolism. J. Clin. Investig. 2011, 121, 4222–4230. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Liccardo, D.; Komici, K.; Corbi, G.; de Lucia, C.; Femminella, G.D.; Elia, A.; Bencivenga, L.; Ferrara, N.; Koch, W.J.; et al. Sphingosine Kinases and Sphingosine 1-Phosphate Receptors: Signaling and Actions in the Cardiovascular System. Front. Pharmacol. 2017, 8, 556. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Maceyka, M.; Spiegel, S. Sphingolipid metabolites in inflammatory disease. Nature 2014, 510, 58–67. [Google Scholar] [CrossRef] [Green Version]
- Merrill, A.H., Jr.; Carman, G.M. Introduction to Thematic Minireview Series: Novel Bioactive Sphingolipids. J. Biol. Chem. 2015, 290, 15362–15364. [Google Scholar] [CrossRef] [Green Version]
- Dunn, T.M.; Tifft, C.J.; Proia, R.L. A perilous path: The inborn errors of sphingolipid metabolism. J. Lipid Res. 2019, 60, 475–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merscher, S.; Fornoni, A. Podocyte pathology and nephropathy—Sphingolipids in glomerular diseases. Front. Endocrinol. 2014, 5, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandhoff, R. Very long chain sphingolipids: Tissue expression, function and synthesis. FEBS Lett. 2010, 584, 1907–1913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettus, B.J.; Chalfant, C.E.; Hannun, Y.A. Ceramide in apoptosis: An overview and current perspectives. Biochim. Biophys. Acta 2002, 1585, 114–125. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Li, N.; Zhou, N.; Wang, D.W. Emerging Roles of Ceramide in Cardiovascular Diseases. Aging Dis. 2022, 13, 232–245. [Google Scholar] [CrossRef]
- Gaggini, M.; Ndreu, R.; Michelucci, E.; Rocchiccioli, S.; Vassalle, C. Ceramides as Mediators of Oxidative Stress and Inflammation in Cardiometabolic Disease. Int. J. Mol. Sci. 2022, 23, 2719. [Google Scholar] [CrossRef]
- Makdessi, S.A.; Sweidan, H.; Schmid, E.; Weimar, U.; Gulbins, E.; Lang, F. Quantitative Determination of Ceramide Molecular Species in Dendritic Cells. Cell Physiol. Biochem. 2016, 39, 1608–1617. [Google Scholar] [CrossRef]
- McGurk, K.A.; Keavney, B.D.; Nicolaou, A. Circulating ceramides as biomarkers of cardiovascular disease: Evidence from phenotypic and genomic studies. Atherosclerosis 2021, 327, 18–30. [Google Scholar] [CrossRef]
- Kurz, J.; Parnham, M.J.; Geisslinger, G.; Schiffmann, S. Ceramides as Novel Disease Biomarkers. Trends Mol. Med. 2019, 25, 20–32. [Google Scholar] [CrossRef]
- Van Echten-Deckert, G. Sphingolipid extraction and analysis by thin-layer chromatography. Methods Enzymol. 2000, 312, 64–79. [Google Scholar]
- Turpin, S.M.; Nicholls, H.T.; Willmes, D.M.; Mourier, A.; Brodesser, S.; Wunderlich, C.M.; Mauer, J.; Xu, E.; Hammerschmidt, P.; Brönneke, H.S.; et al. Obesity-induced CerS6-dependent C16:0 ceramide production promotes weight gain and glucose intolerance. Cell Metab. 2014, 20, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raichur, S.; Wang, S.T.; Chan, P.W.; Li, Y.; Ching, J.; Chaurasia, B.; Dogra, S.; Öhman, M.K.; Takeda, K.; Sugii, S.; et al. CerS2 haploinsufficiency inhibits beta-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab. 2014, 20, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grösch, S.; Schiffmann, S.; Geisslinger, G. Chain length-specific properties of Cers. Prog. Lipid Res. 2012, 51, 50–62. [Google Scholar] [CrossRef] [PubMed]
- de Mello, V.D.; Lankinen, M.; Schwab, U.; Kolehmainen, M.; Lehto, S.; Seppänen-Laakso, T.; Oresic, M.; Pulkkinen, L.; Uusitupa, M.; Erkkilä, A.T. Link between plasma ceramides, inflammation and insulin resistance: Association with serum IL-6 concentration in patients with coronary heart disease. Diabetologia 2009, 52, 2612–2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spijkers, L.J.; van den Akker, R.F.; Janssen, B.J.; Debets, J.J.; De Mey, J.G.; Stroes, E.S.; van den Born, B.J.; Wijesinghe, D.S.; Chalfant, C.E.; MacAleese, L.; et al. Hypertension is associated with marked alterations in sphingolipid biology: A potential role for ceramide. PLoS ONE 2011, 6, e21817. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Yu, J.; Shi, R.; Yan, L.; Yang, T.; Li, Y.; Zhang, Z.; Yu, G.; Bai, Y.; Schuchman, E.H.; et al. Elevation of ceramide and activation of secretory acid sphingomyelinase in patients with acute coronary syndromes. Coron. Artery Dis. 2014, 25, 230–235. [Google Scholar] [CrossRef]
- Yu, J.; Pan, W.; Shi, R.; Yang, T.; Li, Y.; Yu, G.; Bai, Y.; Schuchman, E.H.; He, X.; Zhang, G. Ceramide is upregulated and associated with mortality in patients with chronic heart failure. Can. J. Cardiol. 2015, 31, 357–363. [Google Scholar] [CrossRef]
- Havulinna, A.S.; Sysi-Aho, M.; Hilvo, M.; Kauhanen, D.; Hurme, R.; Ekroos, K.; Salomaa, V.; Laaksonen, R. Circulating Ceramides Predict Cardiovascular Outcomes in the Population-Based FINRISK 2002 Cohort. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2424–2430. [Google Scholar] [CrossRef] [Green Version]
- Laaksonen, R.; Ekroos, K.; Sysi-Aho, M.; Hilvo, M.; Vihervaara, T.; Kauhanen, D.; Suoniemi, M.; Hurme, R.; März, W.; Scharnagl, H.; et al. Plasma ceramides predict cardiovascular death in patients with stable coronary artery disease and acute coronary syndromes beyond LDL-cholesterol. Eur. Heart J. 2016, 37, 1967–1976. [Google Scholar] [CrossRef]
- Bismuth, J.; Lin, P.; Yao, Q.; Chen, C. Ceramide: A common pathway for atherosclerosis? Atherosclerosis 2008, 196, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Sun, M.; Wu, J.; Dong, H.; Liu, J.; Gao, R.; Fang, S.; Xing, L.; Hu, S.; Yu, B. Relationship between elevated plasma ceramides and plaque rupture in patients with ST-segment elevation myocardial infarction. Atherosclerosis 2020, 302, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Nwabuo, C.C.; Duncan, M.; Xanthakis, V.; Peterson, L.R.; Mitchell, G.F.; McManus, D.; Cheng, S.; Vasan, R.S. Association of Circulating Ceramides With Cardiac Structure and Function in the Community: The Framingham Heart Study. J. Am. Heart Assoc. 2019, 8, e013050. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Lunardi, G.; Mantovani, A.; Meessen, J.; Bonapace, S.; Temporelli, P.L.; Nicolis, E.; Novelli, D.; Conti, A.; Tavazzi, L.; et al. Relation between plasma ceramides and cardiovascular death in chronic heart failure: A subset analysis of the GISSI-HF trial. ESC Heart Fail. 2020, 7, 3288–3297. [Google Scholar] [CrossRef]
- Lemaitre, R.N.; Jensen, P.N.; Hoofnagle, A.; McKnight, B.; Fretts, A.M.; King, I.B.; Siscovick, D.S.; Psaty, B.M.; Heckbert, S.R.; Mozaffarian, D.; et al. Plasma Ceramides and Sphingomyelins in Relation to Heart Failure Risk. Circ. Heart Fail. 2019, 12, e005708. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, X.; Pang, J.; Zhang, Y.; Zhang, H.; Xu, Z.; Chen, Q.; Ling, W. Associations between plasma Cers and mortality in patients with coronary artery disease. Atherosclerosis 2020, 314, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Tani, M.; Sano, T.; Ito, M.; Igarashi, Y. Mechanisms of sphingosine and sphingosine 1-phosphate generation in human platelets. J. Lipid Res. 2005, 46, 2458–2467. [Google Scholar] [CrossRef] [Green Version]
- Kacimi, R.; Vessey, D.A.; Honbo, N.; Karliner, J.S. Adult cardiac fibroblasts null for sphingosine kinase-1 exhibit growth dysregulation and an enhanced proinflammatory response. J. Mol. Cell. Cardiol. 2007, 43, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Pappu, R.; Schwab, S.R.; Cornelissen, I.; Pereira, J.P.; Regard, J.B.; Xu, Y.; Camerer, E.; Zheng, Y.W.; Huang, Y.; Cyster, J.G.; et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science 2007, 316, 295–298. [Google Scholar] [CrossRef]
- Venkataraman, K.; Thangada, S.; Michaud, J.; Oo, M.L.; Ai, Y.; Lee, Y.M.; Wu, M.; Parikh, N.S.; Khan, F.; Proia, P.L.; et al. Extracellular export of sphingosine kinase-1a contributes to the vascular S1P gradient. Biochem. J. 2006, 397, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar] [CrossRef] [Green Version]
- Takabe, K.; Paugh, S.W.; Milstien, S.; Spiegel, S. “Inside-out” signaling of sphingo-sine-1-phosphate: Therapeutic targets. Pharmacol. Rev. 2008, 60, 181–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hla, T.; Brinkmann, V. Sphingosine 1-phosphate (S1P): Physiology and the effects of S1P receptor modulation. Neurology 2011, 76 (Suppl. 3), S3–S8. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, S.; Milstien, S. Sphingosine-1-phosphate: Signaling inside and out. FEBS Lett. 2000, 476, 55–57. [Google Scholar] [CrossRef] [Green Version]
- Usui, S.; Sugimoto, N.; Takuwa, N.; Sakagami, S.; Takata, S.; Kaneko, S.; Takuwa, Y. Blood lipid mediator sphingosine 1-phosphate potently stimulates platelet-derived growth factor-A and -B chain expression through S1P1-Gi-Ras-MAPK-dependent induction of Kruppel-like factor 5. J. Biol. Chem. 2004, 279, 12300–12311. [Google Scholar] [CrossRef] [Green Version]
- Chun, J.; Goetzl, E.J.; Hla, T.; Igarashi, Y.; Lynch, K.R.; Moolenaar, W.; Pyne, S.; Tigyi, G. International Union of Pharmacology. XXXIV. Lysophospholipid receptor nomenclature. Pharmacol. Rev. 2002, 54, 265–269. [Google Scholar] [CrossRef]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pun, A.; Gao, E.; George, A.J.; Gambino, G.; Rapacciuolo, A.; Leosco, D.; Ibanez, B.; et al. β1-Blockade Prevents Post-Ischemic Myocardial Decompensation Via β3AR-Dependent Protective Sphingosine-1 Phosphate Signaling. J. Am. Coll. Cardiol. 2017, 70, 182–192. [Google Scholar] [CrossRef]
- Cannavo, A.; Rengo, G.; Liccardo, D.; Pagano, G.; Zincarelli, C.; De Angelis, M.C.; Puglia, R.; Di Pietro, E.; Rabinowitz, J.E.; Barone, M.V.; et al. β1-adrenergic receptor and sphingosine-1-phosphate receptor 1 (S1PR1) reciprocal downregulation influences cardiac hypertrophic response and progression to heart failure: Protective role of S1PR1 cardiac gene therapy. Circulation 2013, 128, 1612–1622. [Google Scholar] [CrossRef] [Green Version]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Jeffery, D.R.; Markowitz, C.E.; Reder, A.T.; Weinstock-Guttman, B.; Tobias, K. Fingolimod for the treatment of relapsing multiple sclerosis. Expert Rev. Neurother. 2011, 11, 165–183. [Google Scholar] [CrossRef]
- Landeen, L.K.; Dederko, D.A.; Kondo, C.S.; Hu, B.S.; Aroonsakool, N.; Haga, J.H.; Giles, W.R. Mechanisms of the negative inotropic effects of sphingosine-1-phosphate on adult mouse ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H736–H749. [Google Scholar] [CrossRef]
- Cannavo, A.; Koch, W.J. Targeting β3-Adrenergic Receptors in the Heart: Selective Agonism and β-Blockade. J. Cardiovasc. Pharmacol. 2017, 69, 71–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.; Ancellin, N.; Fernandez, S.M.; Hla, T.; Sha’afi, R.I. Protein kinase C-alpha and sphingosine 1-phosphate-dependent signaling in endothelial cell. Prostaglandins Other Lipid Mediat. 2006, 80, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Marino, A.; Sakamoto, T.; Robador, P.A.; Tomita, K.; Levi, R. S1P1-mediated anti-RAS cardioprotection: Pivotal role of mast cell ALDH2. J. Pharmacol. Exp. Ther. 2017, 362, 230–242. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of Rac and cell motility by the G12/13-Rho and Gi pathways integrated downstream of a single G protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell. Biol. 2003, 23, 1534–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuchardt, M.; Tölle, M.; Prüfer, J.; van der Giet, M. Pharmacological relevance and potential of sphingosine 1-phosphate in the vascular system. Br. J. Pharmacol. 2011, 163, 1140–1162. [Google Scholar] [CrossRef]
- Kono, M.; Mi, Y.; Liu, Y.; Sasaki, T.; Allende, M.L.; Wu, Y.P.; Yamashita, T.; Proia, R.L. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J. Biol. Chem. 2004, 279, 29367–29373. [Google Scholar] [CrossRef] [Green Version]
- Waeber, C.; Blondeau, N.; Salomone, S. Vascular sphingosine-1-phosphate S1P1 and S1P3 receptors. Drug News Perspect. 2004, 17, 365–382. [Google Scholar] [CrossRef]
- Kerage, D.; Brindley, D.N.; Hemmings, D.G. Review: Novel insights into the regulation of vascular tone by sphingosine 1-phosphate. Placenta 2014, 35, S86–S92. [Google Scholar] [CrossRef]
- Szczepaniak, W.S.; Pitt, B.R.; McVerry, B.J. S1P2 receptor-dependent Rho-kinase activation mediates vasoconstriction in the murine pulmonary circulation induced by sphingosine 1-phosphate. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L137–L145. [Google Scholar] [CrossRef] [Green Version]
- Karliner, J.S. Sphingosine kinase and sphingosine 1-phosphate in cardioprotection. J. Cardiovasc. Pharmacol. 2009, 53, 189–197. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.Q.; Goetzl, E.J.; Karliner, J.S. Sphingosine kinase activation mediates ischemic preconditioning in murine heart. Circulation 2004, 110, 1980–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandhuvula, P.; Honbo, N.; Wang, G.Y.; Jin, Z.Q.; Fyrst, H.; Zhang, M.; Borowsky, A.D.; Dillard, L.; Karliner, J.S.; Saba, J.D. S1P lyase: A novel therapeutic target for ischemia-reperfusion injury of the heart. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H1753–H1761. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Nobes, C.D.; Hall, A.; Spiegel, S. Sphingosine 1-phosphate stimulates Rho-mediated tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 fibroblasts. Biochem. J. 1997, 324, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Olivera, A.; Kohama, T.; Edsall, L.; Nava, V.; Cuvillier, O.; Poulton, S.; Spiegel, S. Sphingosine kinase expression increases intracellular sphingosine-1-phosphate and promotes cell growth and survival. J. Cell Biol. 1999, 147, 545–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, K.; Lehmann, I.; Gräler, M.; Bröcker-Preuss, M.; Erbel, R.; Heusch, G.; Levkau, B. HDL-bound sphingosine 1-phosphate (S1P) predicts the severity of coronary artery atherosclerosis. Cell. Physiol. Biochem. 2014, 34, 172–184. [Google Scholar] [CrossRef]
- Sattler, K.J.; Elbasan, S.; Keul, P.; Elter-Schulz, M.; Bode, C.; Gräler, M.H. Sphingosine 1-phosphate levels in plasma and HDL are altered in coronary artery disease. Basic Res. Cardiol. 2010, 105, 821–832. [Google Scholar] [CrossRef]
- Deutschman, D.H.; Carstens, J.S.; Klepper, R.L.; Smith, W.S.; Page, M.T.; Young, T.R.; Gleason, L.A.; Nakajima, N.; Sabbadini, R.A. Predicting obstructive coronary artery disease with serum sphingosine-1-phosphate. Am. Heart J. 2003, 146, 62–68. [Google Scholar] [CrossRef]
- Argraves, K.M.; Sethi, A.A.; Gazzolo, P.J.; Wilkerson, B.A.; Remaley, A.T.; Tybjaerg-Hansen, A.; Nordestgaard, B.G.; Yeatts, S.D.; Nicholas, K.S.; Barth, J.L.; et al. S1P, dihydro-S1P and C24:1-ceramide levels in the HDL-containing fraction of serum inversely correlate with occurrence of ischemic heart disease. Lipids Health Dis. 2011, 10, 70. [Google Scholar] [CrossRef] [Green Version]
- Egom, E.E.; Mamas, M.A.; Chacko, S.; Stringer, S.E.; Charlton-Menys, V.; El-Omar, M.; Chirico, D.; Clarke, B.; Neyses, L.; Cruickshank, J.K.; et al. Serum sphingolipids level as a novel potential marker for early detection of human myocardial ischaemic injury. Front. Physiol. 2013, 4, 130. [Google Scholar] [CrossRef] [Green Version]
- Egom, E.E.; Mohamed, T.M.; Mamas, M.A.; Shi, Y.; Liu, W.; Chirico, D.; Stringer, S.E.; Ke, Y.; Shaheen, M.; Wang, T.; et al. Activation of Pak1/Akt/eNOS signaling following sphingosine-1-phosphate release as part of a mechanism protecting cardiomyocytes against ischemic cell injury. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1487–H1495. [Google Scholar] [CrossRef]
- Klyachkin, Y.M.; Nagareddy, P.R.; Ye, S.; Wysoczynski, M.; Asfour, A.; Gao, E.; Sunkara, M.; Brandon, J.A.; Annabathula, R.; Ponnapureddy, R.; et al. Pharmacological Elevation of Circulating Bioactive Phosphosphingolipids Enhances Myocardial Recovery After Acute Infarction. Stem Cells Transl. Med. 2015, 4, 1333–1343. [Google Scholar] [CrossRef] [PubMed]
- Knapp, M.; Lisowska, A.; Zabielski, P.; Musiał, W.; Baranowski, M. Sustained decrease in plasma sphingosine-1-phosphate concentration and its accumulation in blood cells in acute myocardial infarction. Prostaglandins Other Lipid Mediat. 2013, 106, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Polzin, A.; Piayda, K.; Keul, P.; Dannenberg, L.; Mohring, A.; Gräler, M.; Zeus, T.; Kelm, M.; Levkau, B. Plasma sphingosine-1-phosphate concentrations are associated with systolic heart failure in patients with ischemic heart disease. J. Mol. Cell. Cardiol. 2017, 110, 35–37. [Google Scholar] [CrossRef] [PubMed]
- Soltau, I.; Mudersbach, E.; Geissen, M.; Schwedhelm, E.; Winkler, M.S.; Geffken, M.; Peine, S.; Schoen, G.; Debus, E.S.; Larena-Avellaneda, A.; et al. Serum-Sphingosine-1-Phosphate Concentrations Are Inversely Associated with Atherosclerotic Diseases in Humans. PLoS ONE 2016, 11, e0168302. [Google Scholar] [CrossRef] [PubMed]
- Christoffersen, C.; Obinata, H.; Kumaraswamy, S.B.; Galvani, S.; Ahnström, J.; Sevvana, M.; Egerer-Sieber, C.; Muller, Y.A.; Hla, T.; Nielsen, L.B.; et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc. Natl. Acad. Sci. USA 2011, 108, 9613–9618. [Google Scholar] [CrossRef] [Green Version]
- Levkau, B. HDL-S1P: Cardiovascular functions, disease-associated alterations, and therapeutic applications. Front. Pharmacol. 2015, 6, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torretta, E.; Barbacini, P.; Al-Daghri, N.M.; Gelfi, C. Sphingolipids in Obesity and Correlated Co-Morbidities: The Contribution of Gender, Age and Environment. Int. J. Mol. Sci. 2019, 20, 5901. [Google Scholar] [CrossRef] [Green Version]
- Kowalski, G.M.; Carey, A.L.; Selathurai, A.; Kingwell, B.A.; Bruce, C.R. Plasma sphingosine-1-phosphate is elevated in obesity. PLoS ONE 2013, 8, e72449. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; Iwaki, S.; Koike, K.; Yuda, Y.; Nagasaki, A.; Ohkawa, R.; Yatomi, Y.; Furumoto, T.; Tsutsui, H.; Sobel, B.E.; et al. Increased plasma sphingosine-1-phosphate in obese individuals and its capacity to increase the expression of plasminogen activator inhibitor-1 in adipocytes. Coron. Artery Dis. 2013, 24, 642–650. [Google Scholar] [CrossRef]
- Salerni, S.; Di Francescomarino, S.; Cadeddu, C.; Acquistapace, F.; Maffei, S.; Gallina, S. The different role of sex hormones on female cardiovascular physiology and function: Not only oestrogens. Eur. J. Clin. Investig. 2015, 45, 634–645. [Google Scholar] [CrossRef]
- Levin, E.R. Plasma membrane estrogen receptors. Trends Endocrinol. Metabol. 2009, 20, 477–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niță, A.R.; Knock, G.A.; Heads, R.J. Signalling mechanisms in the cardiovascular protective effects of estrogen: With a focus on rapid/membrane signalling. Curr. Res. Physiol. 2021, 4, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, A.M.; Murphy, E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1806–H1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsey, S.H.; Carver, K.A.; Prossnitz, E.R.; Chappell, M.C. Vasodilation in response to the GPR30 agonist G-1 is not different from estradiol in the mRen2.Lewis female rat. J. Cardiovasc. Pharmacol. 2011, 57, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Koch, W.J. GRK2 as negative modulator of NO bioavailability: Implications for cardiovascular disease. Cell. Signal. 2018, 41, 33–40. [Google Scholar] [CrossRef]
- Haynes, M.P.; Russell, K.S.; Bender, J.R. Molecular mechanisms of estrogen actions on the vasculature. J. Nucl. Cardiol. 2000, 7, 500–508. [Google Scholar] [CrossRef]
- Hisamoto, K.; Ohmichi, M.; Kurachi, H.; Hayakawa, J.; Kanda, Y.; Nishio, Y.; Adachi, K.; Tasaka, K.; Miyoshi, E.; Fujiwara, N.; et al. Estrogen induces the Akt-dependent activation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 2001, 276, 3459–3467. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Hisamoto, K.; Kim, K.H.; Haynes, M.P.; Bauer, P.M.; Sanjay, A.; Collinge, M.; Baron, R.; Sessa, W.C.; Bender, J.R. Variant estrogen receptor-c-Src molecular interdependence and c-Src structural requirements for endothelial NO synthase activation. Proc. Natl. Acad. Sci. USA 2007, 104, 16468–16473. [Google Scholar] [CrossRef] [Green Version]
- Stirone, C.; Boroujerdi, A.; Duckles, S.P.; Krause, D.N. Estrogen receptor activation of phosphoinositide-3 kinase, akt, and nitric oxide signaling in cerebral blood vessels: Rapid and long-term effects. Mol. Pharmacol. 2005, 67, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.K.; Adams, M.R.; Herrington, D.M.; Clarkson, T.B. Short-term administration of estrogen and vascular responses of atherosclerotic coronary arteries. J. Am. Coll. Cardiol. 1992, 20, 452–457. [Google Scholar] [CrossRef] [Green Version]
- Wingrove, C.S.; Garr, E.; Pickar, J.H.; Dey, M.; Stevenson, J.C. Effects of equine oestrogens on markers of vasoactive function in human coronary artery endothelial cells. Mol. Cell. Endocrinol. 1999, 150, 33–37. [Google Scholar] [CrossRef]
- Nakamura, Y.; Suzuki, T.; Miki, Y.; Tazawa, C.; Senzaki, K.; Moriya, T.; Saito, H.; Ishibashi, T.; Takahashi, S.; Yamada, S.; et al. Estrogen receptors in atherosclerotic human aorta: Inhibition of human vascular smooth muscle cell proliferation by estrogens. Mol. Cell. Endocrinol. 2004, 219, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Urabe, M.; Tsuchiya, H.; Iwasa, K.; Yokota, K.; Kikuchi, N.; Yamamoto, T.; Honjo, H. Effect of estrogen and progesterone on gene expression of growth regulatory molecules and proto-oncogene in vascular smooth muscle cells. Endocr. J. 2000, 47, 205–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seeger, H.; Wallwiener, D.; Mueck, A.O. Effect of medroxyprogesterone acetate and norethisterone on serum-stimulated and estradiol-inhibited proliferation of human coronary artery smooth muscle cells. Menopause 2001, 8, 5–9. [Google Scholar] [CrossRef]
- Ortmann, J.; Veit, M.; Zingg, S.; Di Santo, S.; Traupe, T.; Yang, Z.; Völzmann, J.; Dubey, R.K.; Christen, S.; Baumgartner, I. Estrogen receptor-α but not -β or GPER inhibits high glucose-induced human VSMC proliferation: Potential role of ROS and ERK. J. Clin. Endocrinol. Metab. 2011, 96, 220–228. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Tsukahara, F.; Yoshioka, T.; Irie, K.; Ohta, H. Suppression by 17beta-estradiol of monocyte adhesion to vascular endothelial cells is mediated by estrogen receptors. Life Sci. 2004, 75, 599–609. [Google Scholar] [CrossRef]
- Mikkola, T.S.; St Clair, R.W. Estradiol reduces basal and cytokine induced monocyte adhesion to endothelial cells. Maturitas 2002, 41, 313–319. [Google Scholar] [CrossRef]
- Friedrich, E.B.; Clever, Y.P.; Wassmann, S.; Hess, C.; Nickenig, G. 17Beta-estradiol inhibits monocyte adhesion via down-regulation of Rac1 GTPase. J. Mol. Cell. Cardiol. 2006, 40, 87–95. [Google Scholar] [CrossRef]
- Kurokawa, A.; Azuma, K.; Mita, T.; Toyofuku, Y.; Fujitani, Y.; Hirose, T.; Iwabuchi, K.; Ogawa, H.; Takeda, S.; Kawamori, R.; et al. 2-Methoxyestradiol reduces monocyte adhesion to aortic endothelial cells in ovariectomized rats. Endocr. J. 2007, 54, 1027–1031. [Google Scholar] [CrossRef] [Green Version]
- Simoncini, T.; De Caterina, R.; Genazzani, A.R. Selective estrogen receptor modulators: Different actions on vascular cell adhesion molecule-1 (VCAM-1) expression in human endothelial cells. J. Clin. Endocrinol. Metab. 1999, 84, 815–818. [Google Scholar] [CrossRef]
- Yethon, J.A.; Whitfield, C. Lipopolysaccharide as a target for the development of novel therapeutics in gram-negative bacteria. Curr. Drug Targets Infect. Disord. 2001, 1, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Gorabi, A.M.; Kiaie, N.; Khosrojerdi, A.; Jamialahmadi, T.; Al-Rasadi, K.; Johnston, T.P.; Sahebkar, A. Implications for the role of lipopolysaccharide in the development of atherosclerosis. Trends Cardiovasc. Med. 2021, in press. [Google Scholar] [CrossRef] [PubMed]
- Kiechl, S.; Lorenz, E.; Reindl, M.; Wiedermann, C.J.; Oberhollenzer, F.; Bonora, E.; Willeit, J.; Schwartz, D.A. Toll-like receptor 4 polymorphisms and atherogenesis. N. Engl. J. Med. 2002, 347, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Liccardo, D.; Cannavo, A.; Spagnuolo, G.; Ferrara, N.; Cittadini, A.; Rengo, C.; Rengo, G. Periodontal Disease: A Risk Factor for Diabetes and Cardiovascular Disease. Int. J. Mol. Sci. 2019, 20, 1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Xu, J.; Zheng, S.; Huang, J.; Xiang, Q.; Fu, X.; Wang, T. 17beta-estradiol down-regulates lipopolysaccharide-induced MCP-1 production and cell migration in vascular smooth muscle cells. J. Mol. Endocrinol. 2010, 45, 87–97. [Google Scholar] [CrossRef] [Green Version]
- Spinetti, G.; Wang, M.; Monticone, R.; Zhang, J.; Zhao, D.; Lakatta, E.G. Rat aortic MCP-1 and its receptor CCR2 increase with age and alter vascular smooth muscle cell function. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Wang, Q.; Fei, T.; Han, J.D.; Chen, Y.G. MCP-1 mediates TGF-beta-induced angiogenesis by stimulating vascular smooth muscle cell migration. Blood 2007, 109, 987–994. [Google Scholar] [CrossRef] [Green Version]
- Couse, J.F.; Lindzey, J.; Grandien, K.; Gustafsson, J.A.; Korach, K.S. Tissue distribution and quantitative analysis of estrogen receptor-alpha (ERalpha) and estrogen receptor-beta (ERbeta) messenger ribonucleic acid in the wild-type and ERalpha-knockout mouse. Endocrinology 1997, 138, 4613–4621. [Google Scholar] [CrossRef]
- Grohé, C.; Kahlert, S.; Löbbert, K.; Stimpel, M.; Karas, R.H.; Vetter, H.; Neyses, L. Cardiac myocytes and fibroblasts contain functional estrogen receptors. FEBS Lett. 1997, 416, 107–112. [Google Scholar] [CrossRef]
- Grohé, C.; Kahlert, S.; Löbbert, K.; Vetter, H. Expression of oestrogen receptor alpha and beta in rat heart: Role of local oestrogen synthesis. J. Endocrinol. 1998, 156, R1–R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Zhang, Z.; Hu, F.; Yang, W.; Yuan, J.; Cui, J.; Hao, S.; Hu, J.; Zhou, Y.; Qiao, S. 17β-estradiol prevents cardiac diastolic dysfunction by stimulating mitochondrial function: A preclinical study in a mouse model of a human hypertrophic cardiomyopathy mutation. J. Steroid Biochem. Mol. Biol. 2015, 147, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Devanathan, S.; Whitehead, T.; Schweitzer, G.G.; Fettig, N.; Kovacs, A.; Korach, K.S.; Finck, B.N.; Shoghi, K.I. An animal model with a cardiomyocyte-specific deletion of estrogen receptor alpha: Functional, metabolic, and differential network analysis. PLoS ONE 2014, 9, e101900. [Google Scholar] [CrossRef] [PubMed]
- Fliegner, D.; Schubert, C.; Penkalla, A.; Witt, H.; Kararigas, G.; Dworatzek, E.; Staub, E.; Martus, P.; Ruiz Noppinger, P.; Kintscher, U.; et al. Female sex and estrogen receptor-beta attenuate cardiac remodeling and apoptosis in pressure overload. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1597–R1606. [Google Scholar] [CrossRef]
- Cao, J.; Zhu, T.; Lu, L.; Geng, L.; Wang, L.; Zhang, Q.; Yang, K.; Wang, H.; Shen, W. Estrogen induces cardioprotection in male C57BL/6J mice after acute myocardial infarction via decreased activity of matrix metalloproteinase-9 and increased Akt-Bcl-2 anti-apoptotic signaling. Int. J. Mol. Med. 2011, 28, 231–237. [Google Scholar] [CrossRef]
- Hsieh, D.J.; Kuo, W.W.; Lai, Y.P.; Shibu, M.A.; Shen, C.Y.; Pai, P.; Yeh, Y.L.; Lin, J.Y.; Viswanadha, V.P.; Huang, C.Y. 17β-Estradiol and/or Estrogen Receptor β Attenuate the Autophagic and Apoptotic Effects Induced by Prolonged Hypoxia Through HIF-1α-Mediated BNIP3 and IGFBP-3 Signaling Blockage. Cell. Physiol. Biochem. 2015, 36, 274–284. [Google Scholar] [CrossRef]
- Lin, K.H.; Kuo, W.W.; Shibu, M.A.; Day, C.H.; Hsieh, Y.L.; Chung, L.C.; Chen, R.J.; Wen, S.Y.; Viswanadha, V.P.; Huang, C.Y. E2/ER β Enhances Calcineurin Protein Degradation and PI3K/Akt/MDM2 Signal Transduction to Inhibit ISO-Induced Myocardial Cell Apoptosis. Int. J. Mol. Sci. 2017, 18, 892. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Sun, X.; Chou, J.; Lin, M.; Ferrario, C.M.; Zapata-Sudo, G.; Groban, L. Cardiomyocyte-specific deletion of the G protein-coupled estrogen receptor (GPER) leads to left ventricular dysfunction and adverse remodeling: A sex-specific gene profiling analysis. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1870–1882. [Google Scholar] [CrossRef]
- Li, W.L.; Xiang, W.; Ping, Y. Activation of novel estrogen receptor GPER results in inhibition of cardiocyte apoptosis and cardioprotection. Mol. Med. Rep. 2015, 12, 2425–2430. [Google Scholar] [CrossRef] [Green Version]
- Cannavo, A.; Liccardo, D.; Eguchi, A.; Elliott, K.J.; Traynham, C.J.; Ibetti, J.; Eguchi, S.; Leosco, D.; Ferrara, N.; Rengo, G.; et al. Myocardial pathology induced by aldosterone is dependent on non-canonical activities of G protein-coupled receptor kinases. Nat. Commun. 2016, 7, 10877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Sato, P.Y.; Chuprun, J.K.; Peroutka, R.J.; Otis, N.J.; Ibetti, J.; Pan, S.; Sheu, S.S.; Gao, E.; Koch, W.J. Prodeath signaling of G protein-coupled receptor kinase 2 in cardiac myocytes after ischemic stress occurs via extracellular signal-regulated kinase-dependent heat shock protein 90-mediated mitochondrial targeting. Circ. Res. 2013, 112, 1121–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannavo, A.; Komici, K.; Bencivenga, L.; D’amico, M.L.; Gambino, G.; Liccardo, D.; Ferrara, N.; Rengo, G. GRK2 as a therapeutic target for heart failure. Expert Opin. Ther. Targets 2018, 22, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Maning, J.; McCrink, K.A.; Pollard, C.M.; Desimine, V.L.; Ghandour, J.; Perez, A.; Cora, N.; Ferraino, K.E.; Parker, B.M.; Brill, A.R.; et al. Antagonistic Roles of GRK2 and GRK5 in Cardiac Aldosterone Signaling Reveal GRK5-Mediated Cardioprotection via Mineralocorticoid Receptor Inhibition. Int. J. Mol. Sci. 2020, 21, 2868. [Google Scholar] [CrossRef] [PubMed]
- Esposito, G.; Perrino, C.; Cannavo, A.; Schiattarella, G.G.; Borgia, F.; Sannino, A.; Pironti, G.; Gargiulo, G.; Di Serafino, L.; Franzone, A.; et al. EGFR trans-activation by urotensin II receptor is mediated by β-arrestin recruitment and confers cardioprotection in pressure overload-induced cardiac hypertrophy. Basic Res. Cardiol. 2011, 106, 577–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aitken, J.M.; Hart, D.M.; Lindsay, R. Oestrogen replacement therapy for prevention of osteoporosis after oophorectomy. Br. Med. J. 1973, 3, 515–518. [Google Scholar] [CrossRef]
- Nachtigall, L.E.; Nachtigall, R.H.; Nachtigall, R.D.; Beckman, E.M. Estrogen replacement therapy I: A 10-year prospective study in the relationship to osteoporosis. Obstet. Gynecol. 1979, 53, 277–281. [Google Scholar]
- Rossouw, J.E.; Manson, J.E.; Kaunitz, A.M.; Anderson, G.L. Lessons learned from the Women’s Health Initiative trials of menopausal hormone therapy. Obstet. Gynecol. 2013, 121, 172–176. [Google Scholar] [CrossRef]
- Rossouw, J.E.; Anderson, G.L.; Prentice, R.L.; LaCroix, A.Z.; Kooperberg, C.; Stefanick, M.L.; Jackson, R.D.; Beresford, S.A.; Howard, B.V.; Johnson, K.C.; et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA 2002, 288, 321–333. [Google Scholar] [CrossRef] [Green Version]
- Anderson, G.L.; Limacher, M.; Assaf, A.R.; Bassford, T.; Beresford, S.A.; Black, H.; Bonds, D.; Brunner, R.; Brzyski, R.; Caan, B.; et al. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s Health Initiative randomized controlled trial. JAMA 2004, 291, 1701–1712. [Google Scholar] [CrossRef]
- Bhavnani, B.R. Pharmacokinetics and pharmacodynamics of conjugated equine estrogens: Chemistry and metabolism. Proc. Soc. Exp. Biol. Med. 1998, 217, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Smith, N.L.; Blondon, M.; Wiggins, K.L.; Harrington, L.B.; van Hylckama Vlieg, A.; Floyd, J.S.; Hwang, M.; Bis, J.C.; McKnight, B.; Rice, K.M.; et al. Lower risk of cardiovascular events in postmenopausal women taking oral estradiol compared with oral conjugated equine estrogens. JAMA Intern. Med. 2014, 174, 25–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegner, M.S.; Wanger, R.A.; Oertel, S.; Brachtendorf, S.; Hartmann, D.; Schiffmann, S.; Marschalek, R.; Schreiber, Y.; Ferreirós, N.; Geisslinger, G.; et al. Ceramide synthases CerS4 and CerS5 are upregulated by 17β-estradiol and GPER1 via AP-1 in human breast cancer cells. Biochem. Pharmacol. 2014, 92, 577–589. [Google Scholar] [CrossRef] [PubMed]
- Hahnefeld, L.; Gruber, L.; Schömel, N.; Fischer, C.; Mattjus, P.; Gurke, R.; Beretta, M.; Ferreirós, N.; Geisslinger, G.; Wegner, M.-S. Ether lipid and sphingolipid expression patterns are estrogen receptor-dependently altered in breast cancer cells. Int. J. Biochem. Cell Biol. 2020, 127, 105834. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, S.; Sandner, J.; Birod, K.; Wobst, I.; Angioni, C.; Ruckhäberle, E.; Kaufmann, M.; Ackermann, H.; Lötsch, J.; Schmidt, H.; et al. Ceramide synthases and ceramide levels are increased in breast cancer tissue. Carcinogenesis 2009, 30, 745–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelsohn, M.E.; Karas, R.H. The protective effects of estrogen on the cardiovascular system. N. Engl. J. Med. 1999, 340, 1801–1811. [Google Scholar] [CrossRef]
- Guo, S.; Yu, Y.; Zhang, N.; Cui, Y.; Zhai, L.; Li, H.; Zhang, Y.; Li, F.; Kan, Y.; Qin, S. Higher level of plasma bioactive molecule sphingosine 1-phosphate in women is associated with estrogen. Biochim. Biophys. Acta 2014, 1841, 836–846. [Google Scholar] [CrossRef]
- Vozella, V.; Basit, A.; Piras, F.; Realini, N.; Armirotti, A.; Bossù, P.; Assogna, F.; Sensi, S.L.; Spalletta, G.; Piomelli, D. Elevated plasma ceramide levels in post-menopausal women: A cross-sectional study. Aging 2019, 11, 73–88. [Google Scholar] [CrossRef]
- Barbacini, P.; Torretta, E.; Arosio, B.; Ferri, E.; Capitanio, D.; Moriggi, M.; Gelfi, C. Novel Insight into the Serum Sphingolipid Fingerprint Characterizing Longevity. Int. J. Mol. Sci. 2022, 23, 2428. [Google Scholar] [CrossRef]
- D’Angelo, G.; Capasso, S.; Sticco, L.; Russo, D. Glycosphingolipids: Synthesis and functions. FEBS J. 2013, 280, 6338–6353. [Google Scholar] [CrossRef]
- Collaborative Group on Hormonal Factors in Breast Cancer. Menarche, menopause, and breast cancer risk: Individual participant meta-analysis, including 118 964 women with breast cancer from 117 epidemiological studies. Lancet Oncol. 2012, 13, 1141–1151. [Google Scholar] [CrossRef]
- Zhong, G.-C.; Liu, Y.; Chen, N.; Hao, F.B.; Wang, K.; Cheng, J.H.; Gong, J.P.; Ding, X. Reproductive factors, menopausal hormone therapies and primary liver cancer risk: A systematic review and dose-response meta-analysis of observational studies. Hum. Reprod. Update 2016, 23, 126–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanazir, M.; Boricic, I.; Delic, D.; Tepavcevic, D.K.; Knezevic, A.; Jovanovic, T.; Pekmezovic, T. Risk factors for hepatocellular carcinoma: A case-control study in belgrade (serbia). Tumori J. 2010, 96, 911–917. [Google Scholar] [CrossRef]
- Mucci, L.A.; Kuper, H.E.; Tamimi, R.; Lagiou, P.; Spanos, E.; Trichopoulos, D. Age at menarche and age at menopause in relation to hepatocellular carcinoma in women. Br. J. Obstet. Gynaecol. 2001, 108, 291–294. [Google Scholar] [CrossRef]
- Czubowicz, K.; Jęśko, H.; Wencel, P.; Lukiw, W.J.; Strosznajder, R.P. The Role of Ceramide and Sphingosine-1-Phosphate in Alzheimer’s Disease and Other Neurodegenerative Disorders. Mol. Neurobiol. 2019, 56, 5436–5455. [Google Scholar] [CrossRef] [Green Version]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [Green Version]
- Davinelli, S.; Scapagnini, G.; Marzatico, F.; Nobile, V.; Ferrara, N.; Corbi, G. Influence of equol and resveratrol supplementation on health-related quality of life in menopausal women: A randomized, placebo-controlled study. Maturitas 2017, 96, 77–83. [Google Scholar] [CrossRef]
- Zywno, H.; Bzdega, W.; Kolakowski, A.; Kurzyna, P.; Harasim-Symbor, E.; Sztolsztener, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The Influence of Coumestrol on Sphingolipid Signaling Pathway and Insulin Resistance Development in Primary Rat Hepatocytes. Biomolecules 2021, 11, 268. [Google Scholar] [CrossRef]
- Charytoniuk, T.; Iłowska, N.; Berk, K.; Drygalski, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. The effect of enterolactone on sphingolipid pathway and hepatic insulin resistance development in HepG2 cells. Life Sci. 2019, 217, 1–7. [Google Scholar] [CrossRef]
- Charytoniuk, T.; Harasim-Symbor, E.; Polak, A.; Drygalski, K.; Berk, K.; Chabowski, A.; Konstantynowicz-Nowicka, K. Influence of Resveratrol on Sphingolipid Metabolism in Hepatocellular Carcinoma Cells in Lipid Overload State. Anticancer. Agents Med. Chem. 2019, 19, 121–129. [Google Scholar] [CrossRef]
- Moreira, A.C.; Silva, A.M.; Santos, M.S.; Sardão, V.A. Phytoestrogens as alternative hormone replacement therapy in menopause: What is real, what is unknown. J. Steroid Biochem. Mol. Biol. 2014, 143, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Franco, O.H.; Chowdhury, R.; Troup, J.; Voortman, T.; Kunutsor, S.; Kavousi, M.; Oliver-Williams, C.; Muka, T. Use of Plant-Based Therapies and Menopausal Symptoms: A Systematic Review and Meta-analysis. JAMA 2016, 315, 2554–2563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kang, J.Y.; Ko, K.P.; Park, S.K. The Association between Plasma Concentration of Phytoestrogens and Hypertension within the Korean Multicenter Cancer Cohort. Nutrients 2021, 13, 4366. [Google Scholar] [CrossRef] [PubMed]
- Hammad, S.M.; Harden, O.C.; Wilson, D.A.; Twal, W.O.; Nietert, P.J.; Oates, J.C. Plasma Sphingolipid Profile Associated With Subclinical Atherosclerosis and Clinical Disease Markers of Systemic Lupus Erythematosus: Potential Predictive Value. Front. Immunol. 2021, 12, 694318. [Google Scholar] [CrossRef]
- Wolters, M.; Dejanovic, G.M.; Asllanaj, E.; Günther, K.; Pohlabeln, H.; Bramer, W.M.; Ahrens, J.; Nagrani, R.; Pigeot, I.; Franco, O.H.; et al. Effects of phytoestrogen supplementation on intermediate cardiovascular disease risk factors among postmenopausal women: A meta-analysis of randomized controlled trials. Menopause 2020, 27, 1081–1092. [Google Scholar] [CrossRef]
- Bairey Merz, C.N.; Shaw, L.J.; Reis, S.E.; Bittner, V.; Kelsey, S.F.; Olson, M.; Johnson, B.D.; Pepine, C.J.; Mankad, S.; Sharaf, B.L.; et al. Insights from the NHLBI-Sponsored Women’s Ischemia Syndrome Evaluation (WISE) Study: Part II: Gender differences in presentation, diagnosis, and outcome with regard to gender-based pathophysiology of atherosclerosis and macrovascular and microvascular coronary disease. J. Am. Coll. Cardiol. 2006, 47, S21–S29. [Google Scholar] [CrossRef] [Green Version]
- Kunzova, S.; Maugeri, A.; Medina-Inojosa, J.; Lopez-Jimenez, F.; Vinciguerra, M.; Marques-Vidal, P. Determinants of Metabolic Health Across Body Mass Index Categories in Central Europe: A Comparison Between Swiss and Czech Populations. Front. Public Health 2020, 8, 108. [Google Scholar] [CrossRef]
- Medina-Inojosa, J.R.; Vinciguerra, M.; Maugeri, A.; Kunzova, S.; Sochor, O.; Movsisyan, N.; Geda, Y.E.; Stokin, G.B.; Lopez-Jimenez, F. Prevalence of ideal cardiovascular health in a Central European community: Results from the Kardiovize Brno 2030 Project. Eur. J. Prev. Cardiol. 2020, 27, 441–443. [Google Scholar] [CrossRef]
- Kander, M.C.; Cui, Y.; Liu, Z. Gender difference in oxidative stress: A new look at the mechanisms for cardiovascular diseases. J. Cell. Mol. Med 2017, 21, 1024–1032. [Google Scholar] [CrossRef]
- Kim, K.P.; Shin, K.O.; Park, K.; Yun, H.J.; Mann, S.; Lee, Y.M.; Cho, Y.; Vitamin, C. Stimulates Epidermal Ceramide Production by Regulating ItsMetabolic Enzymes. Biomol. Ther. 2015, 23, 525–530. [Google Scholar] [CrossRef] [Green Version]
- Babenko, N.A.; Hassouneh LKh Kharchenko, V.S.; Garkavenko, V.V. Vitamin E prevents the age-dependent and palmitate-induced disturbances of sphingolipid turnover in liver cells. Age 2012, 34, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arain, F.A.; Kuniyoshi, F.H.; Abdalrhim, A.D.; Miller, V.M. Sex/gender medicine. The biological basis for personalized care in cardiovascular medicine. Circ. J. 2009, 73, 1774–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arosio, B.; Corbi, G.; Davinelli, S.; Giordano, V.; Liccardo, D.; Rapacciuolo, A.; Cannavo, A. Sex Differences in Cardiovascular Diseases: A Matter of Estrogens, Ceramides, and Sphingosine 1-Phosphate. Int. J. Mol. Sci. 2022, 23, 4009. https://doi.org/10.3390/ijms23074009
Arosio B, Corbi G, Davinelli S, Giordano V, Liccardo D, Rapacciuolo A, Cannavo A. Sex Differences in Cardiovascular Diseases: A Matter of Estrogens, Ceramides, and Sphingosine 1-Phosphate. International Journal of Molecular Sciences. 2022; 23(7):4009. https://doi.org/10.3390/ijms23074009
Chicago/Turabian StyleArosio, Beatrice, Graziamaria Corbi, Sergio Davinelli, Vienna Giordano, Daniela Liccardo, Antonio Rapacciuolo, and Alessandro Cannavo. 2022. "Sex Differences in Cardiovascular Diseases: A Matter of Estrogens, Ceramides, and Sphingosine 1-Phosphate" International Journal of Molecular Sciences 23, no. 7: 4009. https://doi.org/10.3390/ijms23074009