
Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation
by
 , ,
, ,
Jonathan Mandel
1 ,
,
Martina Casari
1,
Maria Stepanyan
1,2,3,4,
Alexey Martyanov
2,4,5 and
Carsten Deppermann
1,* 1
Center for Thrombosis and Hemostasis, University Medical Center of the Johannes Gutenberg-University, 55131 Mainz, Germany
2
Center For Theoretical Problems of Physico-Chemical Pharmacology, 109029 Moscow, Russia
3
Physics Faculty, Lomonosov Moscow State University, 119991 Moscow, Russia
4
Dmitriy Rogachev National Medical Research Center of Pediatric Hematology, Oncology Immunology Ministry of Healthcare of Russian Federation, 117198 Moscow, Russia
5
N.M. Emanuel Institute of Biochemical Physics RAS (IBCP RAS), 119334 Moscow, Russia
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(7), 3868; https://doi.org/10.3390/ijms23073868
Submission received: 2 March 2022
/
Revised: 29 March 2022
/
Accepted: 29 March 2022
/
Published: 31 March 2022
(This article belongs to the Special Issue Molecular Mechanisms of Hemostasis, Thrombosis and Thrombo-Inflammation)
Abstract
:There is accumulating evidence that platelets play roles beyond their traditional functions in thrombosis and hemostasis, e.g., in inflammatory processes, infection and cancer, and that they interact, stimulate and regulate cells of the innate immune system such as neutrophils, monocytes and macrophages. In this review, we will focus on platelet activation in hemostatic and inflammatory processes, as well as platelet interactions with neutrophils and monocytes/macrophages. We take a closer look at the contributions of major platelet receptors GPIb, αIIbβ3, TLT-1, CLEC-2 and Toll-like receptors (TLRs) as well as secretions from platelet granules on platelet–neutrophil aggregate and neutrophil extracellular trap (NET) formation in atherosclerosis, transfusion-related acute lung injury (TRALI) and COVID-19. Further, we will address platelet–monocyte and macrophage interactions during cancer metastasis, infection, sepsis and platelet clearance.
Keywords:
platelets; hemostasis; thrombosis; neutrophils; monocytes; macrophages; inflammation; NETs; COVID-19; atherosclerosis; cancer1. Platelets
Platelets are key drivers of hemostasis and thrombosis. Dysfunction in platelet adhesion, production and clearance lead to cardiovascular diseases (CVD). According to the World Health Organization (WHO), CVDs are the leading cause of death worldwide, with an estimated 17.9 million deaths in 2021 [1]. After vessel injury, platelets adhere to the endothelial matrix and become activated, which leads to thrombus formation and restoration of vascular integrity. In recent years, there has been accumulating evidence that platelets play roles beyond thrombosis and hemostasis, e.g., in inflammatory processes, infection and cancer [2]. In this review, we will focus on the role of platelet activation in hemostatic and inflammatory processes, as well as their interactions with innate immune cells (neutrophils and macrophages) in disease.
1.1. Platelet Adhesion
After being released from megakaryocytes in the bone marrow, platelets circulate for 7–10 days in the vasculature before they are cleared, mainly by macrophages in the liver [3]. Their adhesion receptors and biomechanical properties make platelets efficient guardians of hemostasis.
Platelets possess three different types of storage granules (α-granules, δ-granules and lysosomes), which contain receptors and soluble proteins as well as bioactive molecules, which play important roles not only in hemostasis, but also during inflammation and immunity [4]. Hydrodynamic shear forces generated by the blood flow cause erythrocytes to flow in the center of the vessel and push platelets to the vessel wall. After vascular injury, interactions between adhesion receptors on platelets and extracellular matrix (ECM) proteins exposed by the vascular injury decrease the velocity of platelets and allow thrombus formation.
Under low shear conditions present in the venous circulation, integrin α2β1, α5β1 and α6β1 mediate platelet binding to the ECM proteins collagen, fibronectin and laminin. At higher shear rates present in arteries and arterioles, efficient platelet adhesion depends on the von Willebrand factor (vWF) [5]. The vWF is a large glycoprotein produced by megakaryocytes and endothelial cells that circulates in the vasculature in a complex with coagulation factor VIII and is stored in the α-granules of platelets or Weibel–Palade bodies of endothelial cells [6,7]. A shear-induced conformational change of vWF leads to exposure of the A1, A2 and A3 domains and initiates binding of collagen to the A3 domain [8]. Subsequent binding of A3 to collagen further triggers the exposure of the A1 domain. Under lower shear rates, adhesion is mediated through integrin αIIbβ3 on the surface of platelets binding to immobilized vWF on collagen via its C1 domain [9,10,11]. Under higher shear rates, the adhesion of platelets is mainly mediated by vWF binding to glycoprotein Ib (GPIb). The A1 domain on vWF binds to a leucine-rich repeat on the N-terminal domain of GPIb. After vWF-GPIb binding, the stalk region of GPIα unfolds, tensile stress is generated and downstream signaling is initiated [11]. Intracellular signaling leads to granule exocytosis and the activation of αIIbβ3-integrin. vWF binding is characterized by a rapid on–off binding to αIIbβ3 and GPIb, allowing transient interactions [12,13,14]. A stable connection between platelets and collagen is achieved by collagen–GPVI binding, leading to full platelet activation. Soluble agonists, for instance, fibrinogen, ADP, thromboxane A2 (TxA2) and thrombin, further support platelet activation and aggregation [9,15].
1.2. GPIb Is a Key Receptor for Platelet Activation and Aggregation
GPIb is exclusively expressed on the surface of platelets and megakaryocytes and is part of the GPIb-IX-V complex, which consists of four type-I transmembrane polypeptides: GPIbα, GPIbβ, GPIX and GPV. GPIb-IX-V transduces signals via interactions with the cytoskeleton and without a catalytic intracellular domain [16]. Using an optical tweezer setup with single-molecule force measurement, Zhang and colleagues discovered a mechanosensitive domain in GPIbα, which might be responsible for shear force sensing [17]. Downstream of GPIb, Src/Syk kinases are activated as well as phospholipase C (PLCγ2). This leads to Ca2+ release, phosphoinositide 3-kinase (PI3K) and protein kinase C (PKC) activation [14,18]. Consequently, the binding of the tyrosine kinase Syk leads to the phosphorylation of the linker for activation of T cells (LAT) and the further recruitment of phosphoinositide 3-kinase (PI3K). Phosphatidylinositol (4,5)-bisphosphate (PIP2) is converted by PI3K into phosphatidylinositol (3,4,5)-triphosphate (PIP3). Subsequently, PLCγ2 cleaves PIP3, which generates inositol-1,4,5-triphosphate (IP3) and diacylglycerol (DAG). DAG initiates Ca2+ release, which, in turn, activates PKC. This ultimately results in granule secretion and the upregulation of activated integrin with increased ligand affinity to further enhance platelet aggregation [18,19].
In 1985, Harmon and Jamieson found that thrombin can bind to platelets via GPIb [20]. A study by Dörmann, Clemetson and Kehrel showed that, together with protease-activated receptor-1 (PAR-1) activation, platelet–platelet interaction and αIIbβ3-fibrinogen binding, GPIb binding to thrombin is an essential part in platelet pro-coagulation activity [21]. An analysis of the protein crystal structure revealed that thrombin shares a binding site with vWF [22,23]. It was hypothesized that thrombin interferes with vWF–GPIb binding, thereby also contributing to an anti-coagulant activity, but studies providing definitive proof are missing [24].
P-selectin is a transmembrane protein stored in α-granules and Weibel–Palade bodies within platelets and endothelial cells, respectively. The translocation of P-selectin to the surface of activated platelets mediates inflammatory processes by interacting with P-selectin glycoprotein ligand-1 (PSGL-1) on leukocytes and endothelial cells, but also on platelets [25]. Interestingly, PSGL-1 and GPIb share structural similarities. Both are sialomucins with a cluster of O-linked carbohydrates and express a cluster of anionic amino acids with sulfated tyrosine [26]. Studies suggest that P-selectin on platelets binds to PSGL-1 on endothelial cells after vessel destruction and promotes adhesion [27].
Filamin A is an actin-binding protein and is essential in platelet adhesion and aggregation. It modulates vWF–GPIb-IX-V interactions by binding to the cytoplasmic tail of GPIbα and supporting the interaction between GPIb-IX-V and actin [28]. Experiments using platelets from mice with a transgenic GPIbα containing a defective filamin A binding site revealed impaired platelet adhesion and loss of membrane integrity under high shear conditions [29].
Interactions between GPIb and coagulation factors such as factor XI, factor XII, factor IX, factor VII, kininogen, protein C and vitamin K have been described. It is assumed that GPIb localizes factor IX to the platelet surface to support thrombin formation [30]. These factors not only support platelet aggregation but are essential in hemostasis [31].
1.3. CLEC-2 in Platelet Activation and Immunity
C-type lectin-like receptor 2 (CLEC-2) is a type-II transmembrane receptor expressed as a dimer on platelets, neutrophils, dendritic cells (DCs) and Kupffer cells with an extracellular lectin-like domain. CLEC-2 contains an immunoreceptor tyrosine-based activation motif (ITAM) with a single YXXL motif (hemITAM) necessary for direct signal transduction through the tyrosine kinase Syk [19,32].
Podoplanin is the main binding partner of CLEC-2, expressed on lymphatic endothelial cells and also on inflammatory macrophages. The snake venom rhodocytin is another ligand for CLEC-2 and is frequently used to study its signaling [33]. CLEC-2–podoplanin binding initiates platelet activation and aggregation similar to GPVI through Src, LAT, PLCγ2 and DAG signaling [34,35]. Experiments with CLEC-2-deficient mice showed defective thrombus formation as well as prolonged tail bleeding time in vivo, indicating that CLEC-2 plays an essential role in hemostasis and thrombosis [36].
CLEC-2 also contributes to thromboinflammation; e.g., podoplanin–CLEC-2 interactions on neutrophils or DCs lead to increased protrusions and motility [34]. CLEC-2 activation leads to DC migration to the lymph node and T cell zone to initiate an immune response [37]. Studies showed that CLEC-2 is also involved in hematogenous tumor metastasis by mediating platelet aggregation at tumor sites. Platelet aggregates protect cancer cells and secrete factors to promote tumor growth and proliferation [38,39].
1.4. TLT1
The TREM-like transcript 1 (TLT-1) is part of the triggering receptor expressed on the myeloid cells (TREM) protein family. It has a single Ig superfamily V-set domain and signals downstream via two immunoreceptor tyrosine-based inhibitory motifs (ITIM). During platelet production, TLT-1 is stored in α-granules. After platelet activation, it is translocated to the plasma membrane [40] and also released in soluble form. Soluble TLT-1 (sTLT-1) supports the aggregation of platelets during vascular injury [41] and is also involved in inflammatory diseases and sepsis where sTLT-1 levels are increased [42].
1.5. Toll-like Receptors (TLRs) and Thromboinflammation
Through their Toll-like receptors (TLRs), platelets can sense pathogens such as bacteria and viruses. For instance, lipopolysaccharides (LPS) or viral infections can trigger a pro-inflammatory platelet response with the release TNF and IL-1 [43,44]. Platelets express all nine TLRs. Platelets TLR2 and TLR4 play a major role by binding gram-positive and gram-negative bacteria, respectively [43]. TLR4 is expressed as a homodimer, whereas TLR2 forms heterodimers with TLR1 or TLR6. To a lower extent, platelets also express endosomal TLRs (TLR3, TLR7 and TLR9), which allow for the recognition of viruses [2,45]. The activation of platelet TLRs leads to aggregation but also a pro-inflammatory response [46]. TLR1/TLR2 stimulation with a synthetic agonist (Pam3CSK4), for instance, triggers platelet aggregation. Moreover, the secretion of pro-inflammatory cytokines and chemokines such as RANTES (regulated upon activation, normal T cell expressed and presumably secreted), PDGF, PF4 and neutrophil extracellular trap (NET) formation was observed [47]. The TLR4 response to LPS is well established and leads to the release of cytokines, IL-1β synthesis, platelet–neutrophil aggregate formation and NET formation [45,48]. TLR4 stimulation also potentiates platelet activation in response to hemostatic agonists such as collagen through Akt and ERK signaling [49]. In conclusion, TLRs on platelets have an impact on platelet aggregation but also contribute to thromboinflammation [45].
1.6. Platelet Granules
Platelets communicate with their environment not only via membrane receptors but also via soluble factors released from granules, which also contain receptors that are recruited to the platelet surface upon stimulation. Human platelets contain around 40–80 α-granules, 4–8 dense granules and a few lysosomes [42]. α-granules contain more than 300 different proteins, whereas dense granules contain bioactive amines and nucleotides, such as ADP and ATP, as well as magnesium and calcium (Table 1). Granule development starts during the maturation of platelets by megakaryocytes within the bone marrow [50,51].
The packaging of α-granules takes place after protein synthesis in megakaryocytes but also mature platelets in the multivesicular bodies that are derived from the trans-Golgi network [50]. In addition, platelets are also able to pick up plasma proteins and pack them into granules by endocytosis [54]. These processes are mediated by adapter proteins (AP1, AP2 and AP3), soluble N-ethylmaleimide-sensitive factor (NSF) attachment protein receptor (SNARE) proteins and regulators, clathrin molecules, biogenesis of lysosome-related organelles complex (BLOC-1 and BLOC-2) as well as vacuolar protein sorting-associated proteins (VPS33B and VPS16B). A summary of major proteins involved in packaging as well as the factors stored in α-granules can be found in Table 1, as discussing all of them would go beyond the scope of this review [42,50,51,55,56,57].
Upon platelet activation and following Src kinase and PKC signaling, dense granules release soluble factors such as calcium, ATP, ADP and 5-hydroxytryptophan (5-HT). These factors directly activate platelets via receptors such as the purinergic receptors P2Yx in an autocrine and paracrine way [2,50,52].
Defects in platelet granules can lead to serious diseases. One example for an α-granule defect is the gray platelet syndrome (GPS). Mutations in NBEAL2 cause GPS, leading to impaired granule packaging in megakaryocytes and the absence of α-granules in platelets. Symptoms range from a reduced platelet count to bruising susceptibility, myelofibrosis and epistaxis [57,58,59]. One example of a defect in dense granules is the Hermansky–Pudlak syndrome. This is caused by the mutation of HPS and CHS proteins, such as the AP-3 and Rab38, and leads to a bleeding disorder, recurrent infections and ceroid disposition [60].
2. Platelets and Neutrophils Interactions in Homeostasis and Pathology
Neutrophils are granulocytic cells that are part of the innate immune system and are the first leukocytes to infiltrate the wound site [61]. Interactions between platelets and neutrophils are crucial for inflammation and immunity [61]. Currently, the platelet–neutrophil crosstalk is well characterized (Figure 1), both in physiologic and pathologic conditions, where the thromboinflammatory response is often described as “a vicious cycle” [62]. Upon vessel wall damage, platelets are activated, adhere, change their shape, secrete their granule contents and start to form aggregates [63]. Activated platelets are characterized by P-selectin exposure, which is critical for both hemostasis and platelet–immune cell interactions [64].
2.1. Platelet Activation in Thrombi and/or Circulation Can Cause Neutrophil Incorporation
Platelet P-selectin is a ligand for non-activated neutrophil PSGL1 and the P-selectin–PSGL1 bond enables neutrophil attraction to sites of thrombus formation under low shear stress [64]. PSGL1 is constantly associated with the ITAM-receptor FcRɣ, which is phosphorylated by SFK upon PSGL1 association with P-selectin [65,66]. This results in Syk tyrosine kinase activation, which phosphorylates ADAP adaptor protein and leads to the initiation of PLCɣ2-mediated IP3 production and calcium signaling [65]. On the other hand, ADAP phosphorylation by Syk also results in PI3K activation, which produces PIP3 [67]. PIP3 and calcium synergistically activate the small GTPase Rap1 by inhibiting Rap1 GAP and activating Rap1 GEF, correspondingly [68]. Rap1 activation is the key step in the “inside-out” activation of neutrophil β2-integrins Mac1 and LFA1 [66]. Mac1 and LFA1 then reinforce neutrophil attachment to platelets upon interaction with JAM3 [69], ICAM-2 [70], GPIbɑ [71] and fibrinogen, bound to platelet ɑIIbβ3 integrin [72]. Recently, a new player in platelet–neutrophil interactions, SLC44A2, was identified. This protein is found on neutrophils and can bind to platelet ɑIIbβ3 integrins under flow [73]. Neutrophil integrin activation, ligation and clustering results in “outside-in” signaling, possibly mediated by Syk and SFK tyrosine kinases, which enhances neutrophil activation [65,66].
The capability of platelets to induce neutrophil activation is not limited to P-selectin–PSGL1 interaction. Platelet ɑ-granules contain a set of chemokines, which are potent stimulators of neutrophil activity. Among the most abundant are CXCL4 (PF4) [74] and CXCL7 (NAP2) [75]. PF4 has been shown to mediate neutrophil Mac1 and LFA1 integrin activation [76,77]. However, no direct evidence for the presence of a PF4 receptor on the neutrophil surface has been presented to date. It has been proposed that chondroitin-sulfate proteoglycan could serve as a PF4 receptor; final proof, however, remains lacking [78]. On the contrary, mechanisms of NAP-2-induced neutrophil activation are well understood. NAP-2 is produced from its precursor β-thromboglobulin upon release by neutrophil cathepsin-G [79]. NAP-2 acts through CXCR1 and CXCR2—GPCR receptors which regulate both neutrophil activation and chemotaxis [80,81]. It is noteworthy that NAP-2 can cause CXCR2 internalization upon activation, which attenuates neutrophil responses [82]. In addition to NAP-2, platelets also contain CXCL1, which similarly activates CXCR1 and CXCR2 [83]. Both CXCR1 and CXCR2 cause the Gβɣ-dependent initiation of PLCβ activation and further calcium signaling [84,85]. Interestingly, chemokines can form heterodimers and, thus, can enhance and modulate the activity of each other [86]. For example, PF4 can form pairs with RANTES (CCL5) [87]. Whether such pairing significantly affects platelet–neutrophil interactions has yet to be fully elucidated. Platelets also secrete HMGB1, which can activate both RAGE and TLR4 receptors on neutrophils and, thus, initiate NET formation [88,89]. Finally, upon platelet δ-granule secretion, ADP and ATP are released, which can activate neutrophil purinergic receptors [90]. Other molecules which can mediate neutrophil activation upon platelet granule secretion include TGFβ, CD40L and IL-1β [91]. However, these substances appear to be more potent activators of monocytes instead of neutrophils and their role in platelet–neutrophil interactions is minor [91].
2.2. Immunothrombosis Pathways
Not only platelets can be the initiators of platelet–neutrophil association. Inflammation can cause endothelial dysfunction due to high levels of pro-inflammatory cytokines (IL-1, IL-6 and TNF) as well as ferritin [92]. This results in the activation of endothelial cells and expression of P-selectin [92,93]. Neutrophils interact with endothelial P-selectin and become activated in the same manner as upon interaction with platelet P-selectin [65,66]. On the other hand, bacterial infection can activate neutrophils via PAMP/DAMP receptors TLR2, TLR4, Siglec-14, RAGE and CR3 [94]. Neutrophils can also trap viruses into their endosomes. This leads to the decapsulation of the virus and neutrophil TLR7 and TLR8 receptor activation [94]. Activated neutrophils secrete contents of their azurophilic granules, including cathepsin G [95] and NE [96]. Cathepsin G can activate platelet PAR1 receptor in a thrombin-like manner [97]. The same has been assumed for NE; however, no direct experimental evidence is available [91].
Upon hetero-aggregate formation, platelets and neutrophils can activate each other reciprocally. One of the most elegant pathways of such mutual influence is the transcellular synthesis of eicosanoids. Arachidonic acid (AA) is a component of membrane phospholipids, which is released by cytosolic phospholipase 2 (PLA2) upon cell activation [98]. In platelets, AA metabolism by COX1 results in the formation of thromboxane A2 (TxA2)—an essential secondary mediator of platelet activation [63]. However, upon AA release from platelet phospholipids, it can be transferred to neutrophils [91]. Neutrophils are abundant in 5-lipoxygenase (5LO), which is an integral nuclear membrane protein. 5LO activity results in the transition of AA into leukotriene A4 (LTA4), which is further metabolized into leukotriene B4 (LTB4) [91,99]. Alternatively, it can be metabolized into leukotriene C4 (LTC4). Both LTB4 and LTA4 are important pro-inflammatory agents, capable of both activating neutrophils and causing smooth muscle cell contraction and edema [91]. In both activated neutrophils and platelets, reactive oxygen species (ROS) are formed, which enhance the overall activation of both cell types [100]. Additionally, ROS induce NF𝜿B transcription factor activation in neutrophils [101]. NF𝜿B activation can also be triggered by prolonged interaction with platelets via PSGL1 and β2 integrins [102]. This leads to the acquisition of a pro-inflammatory phenotype by neutrophils, characterized by pro-inflammatory cytokine production and, in some cases, tissue factor (TF) surface expression [102,103]. Neutrophils can also activate platelets by releasing antimicrobial cathelicidins via degranulation or as part of NETs. For example, cathelicidin LL-37 and its mouse homologue CRAMP can bind GPVI on the platelet surface, further stimulating platelet and neutrophil activation [104].
In addition to direct platelet activation, activated neutrophils can trigger the activation of the plasma coagulation cascade via TF expression on their surface as well as the secretion of TF-positive microvesicles [92]. This results in the production of thrombin and fibrin generation, which significantly enhances platelet–neutrophil bond formation and mutual activation. It has been suggested that TF-expressing neutrophils and monocytes are key drivers of immunothrombosis [92,103] underlying microvascular thromboinflammation in severe conditions such as ARDS [92,105]. Another distinct feature of platelet–neutrophil interactions in immunothrombotic conditions is the increased potency of neutrophils to form NETs, thus facilitating coagulation and forming a positive feedback loop.
2.3. Platelet–Neutrophil Interactions Result in NETosis
Among the most fascinating features of neutrophils is their capability to release neutrophil extracellular traps (NETs)—cobweb-like chromatin structures, whose prime function is to entangle pathogens (specifically gram-negative bacteria) and to remove them from the circulation [94]. NETosis can be initiated upon neutrophil interaction with fungi, bacteria, immune complexes or activated platelets [94]. The role of platelets in NETosis as well as the overall role of platelets in immunity has emerged only in recent years. Platelet P-selectin interaction with neutrophil PSGL-1 not only results in neutrophil integrin activation by PKC-dependent signaling in neutrophils [66,94]; activated platelets also release HMGB1—which activates the neutrophil RAGE receptors [88]. In addition to HMGB1, activated platelets also secrete the C3a component of the complement system, which, in turn, activates neutrophil C3a receptor [106]. Combined, these stimuli significantly enhance PSGL-1-induced neutrophil activation and can result in the generation of ROS due to NADP oxidase activity in neutrophils [89,94]. ROS stimulate the myeloperoxidase (MPO)-mediated transition of NE from azurophilic granules to the nucleus [94]. NE synergizes with MPO to initiate chromatin decondensation. The next step in NET formation is chromatin histone citrullination through PAD4 [107]. PAD4 activation depends on ROS [108] as well as the cytosolic calcium concentration [109]. PAD4 is also activated downstream of PKC [110,111,112]. The type of NETosis inducer defines the histone citrullination pattern [110,112]. Thus, it can be speculated that different isoforms of PKC are activated downstream of different activators and differentially regulate PAD4 activation. As a consequence, citrullinated neutrophil chromatin, especially H3-histones, are secreted from neutrophils in complex with MPO and NE, and a NET is formed [94]. Citrullinated H3 histones, MPO and NE triple staining have been accepted as the most reliable markers of ongoing NETosis [113].
NETs are potent inducers of thrombus formation, serving as a scaffold for platelet and coagulation factor binding and activation [114]. It has been shown that platelets directly interact with NETs via several ways. Platelets can be activated by H4 histones both in vitro and in vivo [115,116]. Furthermore, citrullinated H3 histones, one of the key markers of ongoing NETosis, have also been shown to activate platelets. Moreover, platelets can interact with C3b attached to NETs [117]. In addition to NE and MPO, NETs also contain cathepsin G. On the other hand, because of their negative charge, NETs can also activate FXII and, thus, activate the contact pathway of coagulation [118]. In line with this, FXII on NETs is able to activate kallikrein [119]. These events manifest in thrombin generation and fibrin deposition among NETs, which attract new platelets from circulation. Finally, NE and other NET-associated proteins such as cathepsin G may also contribute to coagulation by degrading TFPI, which is the key negative regulator of blood coagulation [114,120]. In line with this, NET–fibrin structures are significantly more resistant to plasmin-induced fibrinolysis. Therefore, NETs can either initiate or contribute to the ongoing thrombosis as well as to the tissue damage due to NE and MPO activity [114,121]. It is assumed that the capability of NETs to induce thrombosis is an important mechanism required for pathogen containment. However, excessive NETosis is a potent driver of thromboinflammation and subsequent thromboembolic complications [92].
2.4. Endothelium
An analysis of the platelet–neutrophil interactions cannot be complete without mentioning the contribution of endothelial cells. Indeed, blood vessel endothelial cells orchestrate the activation of neutrophils, platelets and the coagulation cascade. Upon activation, endothelial cells secrete the content of their Weibel–Palade bodies including vWF [122]. vWF interacts with platelet GPIbɑ, which also enables the interaction of platelets with neutrophil β2 integrins [66,91], and it has been proposed that vWF potentiates this interaction. vWF can attract neutrophils to endothelium independently of platelets, and this process depends on the activity of ADAMTS-13—a metalloproteinase required for the conversion of ultra-large vWF multimers into functional vWF [123]. Other contributions of endothelial cells to platelet–neutrophil heteroaggregate formation are based on their ability to bind pro-inflammatory chemokines to their surface [124]. It has been demonstrated that CCL5 and CXCL4 become immobilized on the endothelium and thereby attract neutrophils and monocytes downstream of the site of ongoing thromboinflammation [87]. Finally, upon neutrophil extravasation from the blood flow, a complex interplay between endothelial cells and platelets maintains blood vessel wall integrity, and this process is mediated by platelet CLEC-2 and GPVI receptors as well as granule release [125,126].
2.5. Platelet–Neutrophil Interactions in Pathologic States
Platelet–neutrophil interactions are crucial in host defense and hemostasis [127]. The number of NETs or platelet–neutrophil complexes (PNCs) in blood has been used as a marker of disease severity [128,129,130,131]. Increased amounts of platelet–neutrophil complexes have been observed in autoimmune diseases [74,132,133], acute and chronic inflammatory conditions [134,135] and cardiovascular diseases [136,137].
Ongoing systemic inflammation in immunological diseases may result in increased platelet–neutrophil interactions and a “vicious cycle” of thromboinflammatory responses [134]. For example, patients with ulcerative colitis show a significantly higher number of PNCs and percentage of activated platelets [128]. Platelet activation was proposed to be associated with excessive NET formation in antineutrophil cytoplasmic antibody-associated vasculitis via TLR-CXCL4 signaling [74]. In systemic sclerosis, platelets interact with neutrophils in vitro and in a mouse model and promote neutrophil autophagy via the HMGB1 pathway [132]. Systemic lupus erythematosus (SLE), the most common type of lupus, is a systemic autoimmune disease characterized by a wide range of autoantibodies and clinical manifestations [133]. It was shown that neutrophil-to-lymphocyte and platelet-to-lymphocyte ratios are significantly increased in case of SLE nephritis as well as neutrophil and platelet size and number. These indicate neutrophil and platelet involvement in the disease progress as well as platelet pre-activation and/or enhanced production [138,139]. NETs were also shown to mediate pathology in SLE and antiphospholipid syndrome [140].
Thromboinflammation is also observed in acute inflammation. Early acute myocardial infarction (AMI) and unstable angina are associated with intense neutrophil activation, in contrast to systemic inflammatory syndromes (stable angina, giant cell arteritis, acute bone fracture). Furthermore, the highest amounts of degranulated neutrophils (measured by MPO presence in neutrophils) were observed 4 h post AMI [135]. MPO depletion was associated with platelet activation and platelet–neutrophil complex formation, and a similar pattern was observed when activated platelets were injected in mice [135]. In a transfusion-related acute lung injury (TRALI) mouse model, an increase in platelet–neutrophil interactions and NETs formation was shown, with less NETs present upon anti-platelet therapy [141]. Increased amounts of NETs were observed in the vessels (post mortem) and plasma of patients with TRALI and ALI [141]. In cases of severe sepsis, only 2% of patients showed neutrophils with unaltered MPO levels, suggesting widespread neutrophil activation [135]. NETs are formed during sepsis and their levels correlate with the severity of organ damage and mortality [142]. A mouse model of sepsis demonstrated that gram-positive bacteria can induce neutrophil activation, PNC formation and platelet activation and aggregation in the infected host [143].
Cardiovascular diseases go hand-in-hand with platelet–neutrophil interactions. For example, high levels of circulating DNA and chromatin, which are evidence of NETosis, were shown to be associated with severe atherosclerosis [136]. Composite biomarker score, calculated using both platelet activation and NETosis markers, was shown to be a risk predictor of acute myocardial infarction [137]. HMGB1 was proposed to play a major role in NETosis during myocardial infarction [144]. A review of platelet–neutrophil interactions in cardiovascular diseases was recently published, which provides more details of the mechanisms involved in different cardiovascular conditions [145]. The authors highlight the importance of procoagulant platelets in ischemic stroke [146] and discuss P-selectin and CD40 involvement in atherosclerosis [147,148] and platelet–neutrophil interplay in thrombus formation.
A hyperinflammatory response of the body to infection (cytokine storm) and infiltration of the lungs by neutrophils is observed in COVID-19 [105,121]. Increased NETosis in COVID-19 patients was thoroughly analyzed and the relative number of NETs per neutrophil was calculated to exclude the influence of neutropenia [121,149]. Active COVID-19 virions as well as platelet-rich plasma of COVID-19 patients induced NETosis in neutrophils of healthy donors [149,150]. Microthrombosis was observed in the lungs of COVID-19 patients and NETs were often found in blood clots surrounded by platelets [151,152]. However, NETosis markers only marginally correlate with clinical markers of disease progression [153]. Changes in D-dimer, fibrinogen and platelet count in COVID-19 indicate the activation of the coagulation cascade [154]. Platelet–leukocyte complexes were elevated in COVID-19 patients, with even higher numbers observed in ICU patients [155]. Platelets were pre-activated: changes in P-selectin exposure and GPIb shedding and elevated sizes and higher levels of sCD40L and sP-selectin were shown [156,157,158]. A recent review of COVID-19-associated thrombosis, focusing on all potential contributors, including platelets and neutrophils, was published [159]. At present, the exact contribution of neutrophil–platelet interplay to COVID-19 pathogenesis remains to be studied more thoroughly.
3. Monocytes
Monocytes are part of the ‘‘mononuclear phagocyte system’’ (MPS), together with macrophages and conventional dendritic cells (cDCs) [160]. Circulating monocytes constitute 10% of the total leukocyte population in humans and 4% in mice [161]. Monocytes play key roles in tissue homeostasis and immunity, representing a versatile and dynamic cell population, composed of multiple subsets, which differ in phenotype, size, morphology and transcriptional profiles [162]. In humans, three different monocyte populations were first identified by morphology and differential expressions of surface markers CD14 and CD16 [163]. The major monocyte subset, accounting for approximately 90% of the total monocyte population, expresses high levels of CD14 and no CD16 (CD14+CD16−) and are referred to as classical monocytes. The remaining 10% are shared by CD14+CD16+ intermediate and CD14lowCD16+ ‘‘non-classical’’ monocytes [163,164]. Similarly, in mice, two populations of monocytes can be described based on the expression of Ly6C, CCR2 and CX3CR1. In mice, ‘‘classical’’ monocytes are characterized by the surface marker combination Ly6CHiCX3CR1lowCCR2+ (previously termed inflammatory monocytes), while Ly6ClowCX3CR1HiCCR2− monocytes are defined as ‘‘non-classical’’ (also termed patrolling monocytes) [165].
Specific functions have been attributed to the different subsets: non-classical monocytes are thought to “patrol” the vessel walls and to have endothelial cell-supporting functions [166,167], while classical monocytes have the capability to cross the endothelium and enter tissues in response to appropriate signals, such as monocyte chemoattractant protein-1 and 3 (MCP-1/MCP-3), contributing to immunological responses [166,168,169,170]. Historically, monocytes were thought to be mainly precursor cells of macrophages and dendritic cells. However, currently, it is appreciated that in steady-state conditions, most of the tissue-resident macrophages undergo self-renewal [171,172] and, for this reason, the monocyte’s role has been reassessed. It has become clear that blood monocytes can develop in several ways, acquiring a particular antigen-presenting capability or maturing into macrophages [173].
Even though these monocytes show a tissue-resident macrophage signature, it appears that they preserve a degree of their monocyte identity, e.g., Ms4a7 expression [174], and respond differently during inflammation [175]. In addition, Ly6Chi monocytes can migrate into tissues and retain their monocyte-like state without differentiation into macrophages. These extravascular Ly6Chi monocytes constitute a local monocyte pool with minimal transcriptional alterations [176]. Monocyte emigration takes place constitutively in steady-state conditions and is increased during inflammation, when they gain pro-inflammatory properties. Under these circumstances, macrophages that develop from circulating monocytes exhibit first a pro-inflammatory and later an anti-inflammatory phenotype [173].
3.1. Platelet Interactions with Monocytes/Macrophages
As mentioned before, platelets are equipped with receptors that enable them to sense vessel damage, inflammation or infection to become activated. Once activated, platelets can recruit and interact with leukocytes, including monocytes and macrophages, stimulating mutual activation and the release of cytokines (Figure 2). Direct platelet–monocyte interactions that lead to the formation of heterocellular complexes was discovered decades ago [177]. Upon activation, P-selectin (CD62P) is exposed on the platelet surface, where it can bind to its counterreceptor on myeloid cells PSGL1 [178]. P-selectin/PSGL-1 interactions seem to be the first step in platelet–monocyte aggregation. In support of this, a study demonstrated that the conjugation between platelets and monocytes is abolished by blocking P-selectin [179]. This first association increases the expression of membrane-activated complex 1 (Mac-1) (CD11b/CD18 integrin αMβ2) on the monocyte surface [180], further supporting their interactions with platelets, e.g., through the platelet receptor GPIb, thanks to its I domain, which is homologous to the vWF A1 domain [181] and JAM-C [69]. Platelets and monocytes also come in contact through CD40L–CD40 [182] interactions and monocyte-triggering receptors expressed on myeloid cell 1 (TREM-1) to TLT-1 on platelets [183,184]. Upon activation, platelets adhere to monocytes through bridging molecules such as fibrinogen and thrombospondin [185,186]. Fibrinogen, in this context, has been reported to assist with complex formation, linking Mac-1 on the monocyte surface and integrin αIIbβ3 on platelets [185,187]. In addition to direct interactions, platelet-derived chemokines influence monocyte recruitment and endothelial adhesion and behavior. Platelet CCL5 (RANTES) deposition on inflamed endothelium is relevant for monocyte recruitment [188]. It was also shown experimentally that platelets release large amounts of CXCL4 (PF4) [51,189]. This cytokine enhances monocyte phagocytosis and triggers respiratory bursts [190] via the activation of phosphoinositol-3-kinase PI3K, Syk and p38 MAP kinase [191]. Moreover, platelet CXCL4 induces extracellular signal kinase 1 and 2 (ERK1/2) phosphorylation, which mediates monocyte survival and differentiation as well as Janus kinase (JNK) signaling, which leads to the production and release of cytokines and chemokines [191]. It was also observed that PF4, in collaboration with IL-4, provokes a rapid differentiation of monocytes into specialized antigen-presenting cells (APCs) with unique phenotypical and functional characteristics that are different from macrophages or cDCs [192]. These cells preferentially stimulated the proliferation of lymphocytes and lytic natural killer (NK) cell activity, while they induced only moderate cytokine responses. Recently, Mac-1 and proteoglycans were identified to mediate the binding of PF4 to leukocytes [193].
Once deposited, platelet chemokines can form homophilic as well as heterophilic aggregates, resulting in the further stimulation of their biological activity. For example, RANTES can increase PF4 binding to the monocyte surface, while PF4 drastically enhances RANTES-induced monocyte arrest on endothelial cells [87], predominantly mediated by CCR1 [194]. Platelets also associate with neutrophils to promote monocyte recruitment: platelet CCL5 can form heterodimers with neutrophil HNP1 (alpha-defensin), stimulating monocyte adhesion through CCR5 [195]. Alard and colleagues demonstrated that the disruption of HNP1–CCL5 interactions attenuated monocyte and macrophage recruitment in a mouse model of myocardial infarction.
Activated platelets were also observed to stimulate monocyte chemotactic protein-1 (MCP-1) secretion and intercellular adhesion molecule-1 (ICAM-1) surface expression on endothelial cells by activating the NF-κB pathway, thereby indirectly causing monocyte recruitment [196]. Thus, platelet adhesion to the endothelium and chemokines secreted by platelets greatly contributes to subsequent monocyte adhesion to the vascular wall.
So far, we have provided a general overview of the events resulting in platelet monocyte aggregation, showing how this interaction can lead to changes in cell markers and phenotype and induce mutual cell activation and cytokine production. However, what are the roles of these aggregates? Multiple studies have investigated the mechanism and impact of platelet–monocyte aggregates in a variety of experimental and pathological contexts. Elevated numbers of circulating blood monocyte–platelet complexes were proposed to play a role in the pathogenesis of acute coronary syndromes and atherosclerosis [197,198] and were detected in different diseases, including rheumatoid arthritis and systemic lupus erythematosus [199], type 1 diabetes [200] and end-stage renal disease [201] suggesting an important role in the pathogenesis of inflammatory diseases.
3.2. Platelet–Monocyte/Macrophages Interaction in Atherosclerosis
Atherosclerosis is a chronic inflammatory disease, comprising an intricated vascular injury, which represents the initial pathological event for several cardiovascular diseases [202]. In this inflammatory environment, circulating lipoproteins, mainly low-density lipoproteins (LDL), become oxidized (oxLDL). Circulating monocytes are recruited to the atherosclerotic lesion site through P-selectin and E-selectin, intracellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1). Monocytes migrate into the subendothelial space under the influence of chemoattractant molecules, where they differentiate into macrophages in response to locally produced factors such as monocyte colony-stimulating factor (M-CSF) [203]. This differentiation includes the upregulation of scavenger receptor A (SR-A) and CD36, which can mediate the uptake of oxLDL [204]. Once macrophages start to phagocytose oxLDL and minimally modified LDL (mmLDL), they transform into foam cells. Foam cells support smooth muscle proliferation, angiogenesis and plaque formation [205]. Finally, the rupture or erosion of vulnerable atherosclerotic plaques leads to arterial thromboembolic events, which may lead to skeletal muscle ischemia or fatal outcomes such as myocardial infarction and stroke [63].
Monocytes and platelets are key players in atherosclerosis [206]. Platelets are considered important drivers of pathogenesis and disease progression thanks to their ability to interact with immune and endothelial cells and through the uptake of LDL [207]. At the site of atherosclerotic plaque formation, platelet activation can be triggered by DAMPs such as oxLDL [208] or podoplanin [209] upregulated on inflammatory macrophages, Th17 T cells and fibroblasts [34]. Specifically, oxLDL has been observed to mediate platelet activation via several pathways, comprising scavenger receptors and platelet-activating factor receptor (PAFR) [208,210]. Platelet PAFR activation by oxLDL occurs simultaneously with classical agonists, ADP and thrombin for example, initiating prothrombotic responses [208]. It has been observed that platelet adhesion to the endothelium precedes leukocyte recruitment and that blocking platelet adhesion with anti-GPIb antibodies or through the genetic deletion of P-selectin reduces leukocyte recruitment to atherosclerotic lesions and plaque development [211,212].
CD40L-CD40 mediated platelet–monocyte interactions facilitate monocyte arrest on inflamed endothelium, accelerating atherosclerosis [147,213]. Platelets support the migration and activation of monocytes, dendritic cells and neutrophils through the release of soluble inflammatory mediators such as CCL5 and PF4, contributing to the progression of atherosclerosis [214]. CXCL4 promotes phenotypic changes in macrophages, resulting in a proatherogenic state with increased susceptibility to foam cell formation [215,216]. Secondly, PF4 may directly aggravate the atherogenic actions of hypercholesterolemia by promoting the retention of lipoproteins. Sachais and colleagues have recently shown that PF4 facilitates the retention of LDL on cell surfaces by inhibiting its degradation through the LDL receptor [217]. Moreover, PF4 facilitates the esterification and uptake of oxLDL by macrophages contributing to foam cell development [218].
3.3. Platelet–Monocyte/Macrophage Interaction during Infectious Diseases
Platelets recognize molecular features of microbes, show bacteriocidic activity and contain immunomodulatory mediators essential for alerting and recruiting cells of the immune system [219]. In the following paragraph, platelet–monocyte/macrophage interactions during viral and bacterial infections will be described with a specific focus on platelets and liver macrophages.
Circulating platelet–monocyte and platelet–lymphocyte aggregates have been reported in increased numbers in HIV-infected subjects [220,221]. Liang and colleagues found that platelet aggregates with CD16+ inflammatory monocytes are more frequent in HIV-infected patients and associate with increased levels of sCD163, a marker of monocyte activation [222,223]. Platelet aggregates with monocytes, lymphocytes and neutrophils have also been observed during dengue virus infection [224,225]. In 2014, Hottz and colleagues observed that platelet–monocyte complexes correlate with increased vascular permeability in dengue patients. They detected that dengue-activated platelets were able to reprogram monocyte responses ex vivo, inducing the secretion of IL-1β, IL-10 and IL-8. [225]. They also found that the interaction of monocytes with apoptotic platelets mediates IL-10 secretion through phosphatidylserine recognition in platelet–monocyte aggregates. Together, their results demonstrated that activated platelets aggregate with monocytes during dengue infection and signal specific cytokine responses that may contribute to the pathogenesis of dengue. Related to SARS-CoV-2, a study by Hottz and colleagues, performed in 2020, found increased platelet activation and platelet–monocyte aggregates formation in severe COVID-19 patients, compared to those with mild disease. They observed that, in COVID-19 patients admitted to the intensive care unit, platelet–monocyte interaction was strongly associated with tissue factor (TF) expression by monocytes as well as with markers of coagulation exacerbation, such as fibrinogen and D-dimers, and all together correlated with a worse outcome [226].
An increasing number of studies confirms the role of interactions between platelets and monocytes/macrophages in immune responses to bacterial infections. Scull and colleagues demonstrated that the incubation of human monocyte-derived macrophages (hMDMs) and autologous activated platelets results in platelet uptake through scavenger-receptors [227]. Following the co-incubation of LPS-activated hMDM with activated autologous platelets, they also observed an increased level of proinflammatory TNF-α, IL-6 and IL-23 cytokines, suggesting that the presence of activated platelets at sites of inflammation may intensify pro-inflammatory macrophage activation. Controversially, other in vitro studies showed the inhibition of pro-inflammatory cytokines during the co-culture of platelets with monocytes–macrophages following bacterial stimulation [228,229,230]. In agreement with this, Hasselbalch and Nielsen observed that activated platelets enhance IL-10 and inhibit TNF-α release from monocytes in a CD40L-dependent manner upon the stimulation of peripheral blood mononuclear cells (PBMCs) with tetanus toxoid (TT), human thyroglobulin (TG), Escherichia coli LPS or Porphyromonas gingivalis [228]. In vivo studies also showed inconclusive results: studies reported that the formation of circulating platelet–monocyte aggregates could amplify the inflammatory response and predict mortality in older septic patients [231,232]. However, others have demonstrated that platelets contribute to bacterial clearance and resolution of inflammation by regulating macrophage responses [229,233,234,235]. Xiang and colleagues used a murine model of septic shock and showed that platelets inhibit the release of macrophage-derived pro-inflammatory TNF-α and IL-6 and reduce mortality via the COX-1-PGE2-EP4 pathway [229]. Carestia and colleagues revealed that the co-culture of human platelets with monocytes stimulated with LPS reduced not only pro-inflammatory TNF-α and IL-6, but also anti-inflammatory IL-10 release [230]. This was also associated with platelet sequestration of monocyte-derived cytokines. In addition, they evaluated whether platelets could modulate the polarization of monocyte-derived macrophages (M-DMs) and found that monocytes incubated with platelets were predominantly polarized towards the M1 phenotype in a cell-contact-dependent manner by the GPIb-CD11b axis. Lastly, they showed that platelet transfusion in septic mice models, at early time points after E. coli infection, increased survival, possibly due to an increase in iNOS+ macrophages, which contribute to efficient bacterial clearance. In summary, although some studies provide information on platelet–monocyte aggregation, more investigation is necessary to gain a better understanding of the biological mechanisms, importance and effects of platelet–monocyte aggregates formed during viral or bacterial infection and sepsis.
The liver is the largest organ in the body, able to filter one-third of the body’s total blood volume/minute with important roles in metabolism, detoxification and defense against bloodstream pathogens [236]. Kupffer cells, discovered by Karl Wilhelm von Kupffer in 1876, are liver-resident macrophages, positioned within the lumen of liver sinusoids. Kupffer cells represent the first line of defense in the circulation and constantly survey the blood to phagocytose and remove pathogens [237,238]. Lee and colleagues showed that Kupffer cells can trap intravascular Borrelia burgdorferi [239]. Moreover, blood-borne methicillin-resistant Staphylococcus aureus (MRSA) can be captured and killed by Kupffer cells through an interaction of the complement receptor of the immunoglobulin superfamily (CRIg) with Lipoteichoic acid (LTA), a gram-positive bacteria cell wall polymer [240]. However, a minority of Staphylococci survive and eventually start to proliferate inside the Kupffer cells [241]. Wong and colleagues discovered that cooperation between platelets and liver macrophages is needed to eradicate blood-borne infections with Bacillus cereus and MRSA [242]. Particularly, they observed through intravital spinning-disk confocal microscopy that platelets regularly perform “touch-and-go” interactions under basal conditions. This binding was shown to be mediated by platelet-adhesion receptor GPIb and vWF constitutively expressed on Kupffer cells. Moreover, throughout the infection, bacteria were caught by Kupffer cells and platelets promptly formed aggregates around bacteria on Kupffer cells’ surfaces to contain them in an integrin αIIbβ3- and complement-dependent manner. Using intravital microscopy of the bloodstream of mice infected with Listeria monocytogenes, another group showed that bacterial clearance is not a uniform process but a “dual-track” mechanism consisting of parallel “fast” and “slow” pathways. They suggested that slow bacterial clearance is controlled through bacterial opsonization, platelet GPIb binding and the capture of bacteria–platelet complexes via CRIg. The fast clearance of free bacteria, instead, requires Kupffer cell scavenger receptors and is independent of complement and platelets [243,244].
3.4. Platelet–Leukocyte Interplay in Cancer Development and Metastasis
Cancer cells can invade nearby tissues and spread throughout the body, resulting in metastasis [245,246]. The tumor microenvironment provides the necessary milieu and blood supply to tumor cells, profoundly influencing the interactions of cancer cells with their surroundings, ultimately determining the fate of the primary tumor: eradication, metastasis or the establishment of dormant micrometastases [247].
Cancer patients frequently present with a hypercoagulable state with an elevated risk for thromboembolic events [248,249]. Tumor cells have been shown to interact with platelets manipulating their physiology, while, on the other side, activated platelets seem to support tumor growth and invasion [250,251]. In particular, tumor cells can activate platelets through the secretion of factors such as ADP [252] or via direct interaction. Platelet ITAM receptors GPVI and CLEC-2, which bind podoplanin expressed at the invasive front of many tumors, participate in cancer pathogenesis [253,254]. Platelet aggregation and secretion of PDGF and TGF-β through CLEC-2 activation [35,255,256] supports tumor cell proliferation and epithelial–mesenchymal transition. High-mobility group box 1 (HMGB1), released by dying tumor cells, on the other hand, interacts with TLR4 on platelets and mediates platelet–tumor cell interaction, promoting metastasis [257]. Through these interactions, platelets become activated and secrete factors that modulate the tumor microenvironment. Vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and transforming growth factor (TGF) are released from α-granules of activated platelets and stimulate tumor angiogenesis [255,258,259].
Pavlović and colleagues analyzed the contribution of platelets to the progression of hepatocellular carcinoma (HCC) [256]. HCC is the second leading cause of cancer-related death worldwide [260,261]. In more than 90% of cases, this cancer follows an underlying chronic inflammation, fibrosis or cirrhosis [261,262]. Upon liver damage, stellate cells can adopt an activated myofibroblast-like phenotype [262]. This results in an increased deposition of ECM proteins and a release of cytokines, growth factors and products of oxidative stress and, later on, cancer progression [263]. Platelets are an important source of PDGF-β and TGF-β, well-described activators of hepatic stellate cells, with key roles in pro-fibrotic signaling [264].The authors showed that activated platelets support HCC progression in vivo by stimulating tumor cell proliferation and modulating the surrounding hepatic microenvironment. In addition, they reported that platelets mediate macrophage accumulation in the liver and modulate the hepatic inflammatory cell population phenotype towards a pro-tumoral state. Furthermore, they show that activated platelets induce pro-fibrotic signaling and hepatic stellate cell activation, which is in line with previous data showing that platelet CXCL4 and serotonin increase in fibrosis progression [265].
Metastases remain the primary cause for cancer-related death. Hematogenous metastasis comprises multiple steps, including tumor cell dissemination through the circulation, arrest in the microvasculature and, ultimately, the colonization of distant organs [245,246]. Platelets can help the process of cancer metastasis through multiple mechanisms: they enhance epithelial to mesenchymal transition; moreover, they promote tumor cell survival in the circulation, extravasation and colonization at the secondary site [266,267]. Platelets contain a huge reservoir of adhesion molecules such as selectins, integrins and immunoglobulin superfamily proteins, which are essential for promoting platelet adhesion. Cancer cells take advantage of these molecules on the platelet surface to form platelet–cancer cell conjugates through carbohydrate–protein recognition by P-selectin, protein bridging (e.g., by fibrinogen) between integrins as well as PECAM-1–integrin binding [268,269,270]. Cancer cells interacting with platelets can escape NK and other immune cells in circulation [269,271]. Furthermore, cancer cell-bound platelets increase cancer cell adhesion on endothelium and facilitate cancer metastasis [266,271,272]. Interactions between platelets and tumor cells are necessary also for the recruitment of granulocytes to the metastatic site. Labelle et al. showed that cancer cell-bound platelets secrete CXCL5 and CXCL7. Released CXCL7 synergized with CXCR2 to recruit neutrophils from the bloodstream. With the additional help of granulocytes, cancer cells can form an early metastatic niche to establish metastatic foci [273]. Platelet depletion completely suppressed neutrophil recruitment to the metastatic site, strongly indicating that platelets play a critical role in attracting neutrophils.
Gil-Bernabé and colleagues observed that platelet–cancer cell conjugates also recruit monocytes into the early metastatic niche [274], through chemokines released from platelets such as RANTES [275]. Following this, the recruited monocytes/macrophages improve the extravasation of cancer cells by producing VEGF that increases vessel permeability [276] and enhances the formation of metastatic foci [275,277].
This evidence shows that platelets contribute to cancer progression, metastasis and angiogenesis in multiple ways. Leukocytes take part in the interaction with cancer cells and can promote metastases formation. Future research is needed to further investigate the mechanisms involved in platelet–cancer cell–leukocyte interactions.
3.5. Platelet Clearance
Hemostatic balance is achieved by the constant clearance of aged, activated or defective platelets in the liver and spleen and the ongoing production of new platelets by megakaryocytes in the bone marrow. How platelet clearance is linked to platelet production is incompletely understood. Desialylation is a marker for aged platelets and the removal of desialylated platelets is an important mechanism of platelet clearance [278,279,280].
The highly glycosylated GPIb-IX-V complex plays an important role in platelet clearance, as the majority of desialylation occurs on GPIbα [281]. The clustering of GPIbα not only mediates platelet aggregation but also clearance. Studies showed that the Fc-independent antibody-mediated activation of GPIbα leads to rapid clearance through hepatic macrophages [282,283,284]. GPIb clustering is one of the main reasons for the rapid clearance of cold-stored platelets after transfusion [285,286], which has a major impact on storage of platelets for transfusion [287].
3.5.1. Platelets Apoptosis
In addition to desialylation, apoptosis is another mechanism involved in the removal of aged platelets and the maintenance of a healthy platelet population. Proteins of the Bcl2-family (BCL-2, BCL-XL and BCL-W) keep platelets viable, whereas pro-death proteins, such as Bax and Bak, are responsible for mitochondrial damage and subsequent apoptosis [288]. Under healthy conditions, pro-survival proteins inhibit Bax/Bak. Cellular stress activates the BH3-only proteins (Bid, Bim, Bad and Bik), which interrupts pro-survival BCL-2-induced Bak/Bax inhibition [289,290]. Activated Bax/Bak generates pores into the mitochondrial membrane, which triggers cytochrome C release, which, in turn, causes the accumulation of Caspase-3 and Caspase-7, leading to DNA damage and the inhibition of transcription [288,291]. How these apoptotic platelets are removed from circulation, however, remains unclear.
A study by Chen and colleagues discovered that the activation of GPIb and subsequent downstream signaling via PI3K initiates Akt-mediated apoptosis. Additionally, opsonization with anti-GPIb antibodies in immune thrombocytopenia (ITP) patients leads to rapid clearance by macrophages through the Fc-receptor [292,293].
3.5.2. Desialylated Platelets Are Rapidly Cleared by Macrophages in the Liver
The glycan composition on the platelet surface plays an important role in platelet clearance. Sialic acid is usually the terminal glycan residue on N- and O-glycans directly connected to β-galactose (β-gal). GPIb-IX-V accounts for ~80% of sialic acid residues on the platelet surface [286]. Platelets with reduced levels of α2,3-linked sialic acids expose β-gal and are cleared through the Ashwell–Morell receptor (AMR), which is expressed on hepatocytes [294]. The AMR exists as a heterooligomeric receptor with two subunits, asialoglycoprotein receptors 1 and 2 (ASGR1/2) [295,296].
AMR-mediated platelet clearance was investigated in AMR knock-out mice that showed reduced platelet clearance as well as platelets with endogenous sialylation defects using St3Gal4−/− mice, which lack this specific sialyltransferase. [294,297]. In response to platelet clearance, hepatocytes stimulate platelet production by the release of thrombopoietin (TPO) [280]. After the binding of desialylated platelets to the AMR, TPO mRNA expression is upregulated via JAK2 phosphorylation and STAT3 [298].
Experiments on GPIbα−/− mice showed a significant but not exclusive contribution of GPIb to platelet clearance through desialylation [299]. Studies with anti-GPIb antibodies showed that downstream signaling of activated GPIb leads to the release of granules and the subsequent surface expression of sialidases such as neuraminidase 1 (Neu1) and 3 (Neu3). These enzymes trigger desialylation of adjacent platelets and ultimately lead to increased platelet clearance [280,283,284]. Bacteria-derived neuraminidase also cause desialylation and thrombocytopenia [300]. Shear-induced activation of GPIb-IX-V triggers desialylation: unfolding of the mechanosensory domain of GPIbα leads to platelet activation and increased expression of Neu1 in vivo [301]. Phagocytic αMβ2 integrin is also partially involved in platelet clearance by recognizing exposed N-Acetlyglucosamin on GPIbα [284,302].
Li and colleagues showed that the recognition of desialylated platelets via the AMR also mediates phagocytosis by Kupffer cells via the C-type lectin receptor CLEC4F in mice [303]. However, CLEC4F is not expressed in humans [304], and it is assumed that other lectins adopt the function of CLEC4F in humans [305]. Indeed, macrophage galactose lectin (MGL), together with AMR, was shown to be involved in Kupffer cell-mediated clearance. Intravital microscopy revealed that desialylated platelets directly bind to Kupffer cells, leading to a rapid clearance. Kupffer cell depletion as well as blocking AMR and MGL resulted in an accumulation of desialylated platelets in circulation, suggesting an essential role of Kupffer cells in platelet clearance [306].
Funding
This study was supported by the German Research Foundation (SFB1292 TP08).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Cardiovascular Diseases. Available online: https://www.who.int/health-topics/cardiovascular-diseases#tab=tab_1 (accessed on 22 November 2021).
- Holinstat, M. Normal platelet function. Cancer Metastasis Rev. 2017, 36, 195–198. [Google Scholar] [CrossRef] [PubMed]
- Quach, M.E.; Chen, W.; Wang, Y.; Deckmyn, H.; Lanza, F.; Nieswandt, B.; Li, R. Differential regulation of the platelet GPIb-IX complex by anti-GPIbβ antibodies. J. Thromb. Haemost. 2021, 19, 2044–2055. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Clancy, L.; Corkrey, H.A.; Freedman, J.E. Circulating Platelets as Mediators of Immunity, Inflammation, and Thrombosis. Circ. Res. 2018, 122, 337–351. [Google Scholar] [CrossRef] [PubMed]
- Weiss, H.J.; Turitto, V.T.; Baumgartner, H.R. Effect of shear rate on platelet interaction with subendothelium in citrated and native blood. I. Shear rate–dependent decrease of adhesion in von Willebrand’s disease and the Bernard-Soulier syndrome. J. Lab. Clin. Med. 1978, 92, 750–764. [Google Scholar]
- Maxwell, M.J.; Westein, E.; Nesbitt, W.S.; Giuliano, S.; Dopheide, S.M.; Jackson, S.P. Identification of a 2-stage platelet aggregation process mediating shear-dependent thrombus formation. Blood 2007, 109, 566–576. [Google Scholar] [CrossRef]
- Sadler, J.E. Biochemistry and genetics of von Willebrand factor. Annu. Rev. Biochem. 1998, 67, 395–424. [Google Scholar] [CrossRef]
- Schneider, S.W.; Nuschele, S.; Wixforth, A.; Gorzelanny, C.; Alexander-Katz, A.; Netz, R.R.; Schneider, M.F. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc. Natl. Acad. Sci. USA 2007, 104, 7899–7903. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.P.; Nesbitt, W.S.; Westein, E. Dynamics of platelet thrombus formation. J. Thromb. Haemost. 2009, 7 (Suppl. 1), 17–20. [Google Scholar] [CrossRef]
- Jackson, S.P. The growing complexity of platelet aggregation. Blood 2007, 109, 5087–5095. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Ciciliano, J.; Myers, D.R.; Tran, R.; Lam, W.A. Platelets and physics: How platelets “feel” and respond to their mechanical microenvironment. Blood Rev. 2015, 29, 377–386. [Google Scholar] [CrossRef]
- Arya, M.; López, J.A.; Romo, G.M.; Cruz, M.A.; Kasirer-Friede, A.; Shattil, S.J.; Anvari, B. Glycoprotein Ib-IX-mediated activation of integrin alpha(IIb)beta(3): Effects of receptor clustering and von Willebrand factor adhesion. J. Thromb. Haemost. 2003, 1, 1150–1157. [Google Scholar] [CrossRef] [PubMed]
- Delaney, M.K.; Liu, J.; Zheng, Y.; Berndt, M.C.; Du, X. The role of Rac1 in glycoprotein Ib-IX-mediated signal transduction and integrin activation. Arter. Thromb. Vasc. Biol. 2012, 32, 2761–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasirer-Friede, A.; Cozzi, M.R.; Mazzucato, M.; De Marco, L.; Ruggeri, Z.M.; Shattil, S.J. Signaling through GP Ib-IX-V activates alpha IIb beta 3 independently of other receptors. Blood 2004, 103, 3403–3411. [Google Scholar] [CrossRef] [PubMed]
- Folie, B.J.; McIntire, L.V. Mathematical analysis of mural thrombogenesis. Concentration profiles of platelet-activating agents and effects of viscous shear flow. Biophys. J. 1989, 56, 1121–1141. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.K.; Fox, J.E. Interaction of purified actin-binding protein with the platelet membrane glycoprotein Ib-IX complex. J. Biol. Chem. 1991, 266, 7144–7147. [Google Scholar] [CrossRef]
- Zhang, W.; Deng, W.; Zhou, L.; Xu, Y.; Yang, W.; Liang, X.; Wang, Y.; Kulman, J.D.; Zhang, X.F.; Li, R. Identification of a juxtamembrane mechanosensitive domain in the platelet mechanosensor glycoprotein Ib-IX complex. Blood 2015, 125, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Gibbins, J.M. Platelet adhesion signalling and the regulation of thrombus formation. J. Cell Sci. 2004, 117, 3415–3425. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Ehrenberg, A.; Toska, L.M.; Metz, L.M.; Klier, M.; Krueger, I.; Reusswig, F.; Elvers, M. Molecular Drivers of Platelet Activation: Unraveling Novel Targets for Anti-Thrombotic and Anti-Thrombo-Inflammatory Therapy. Int. J. Mol. Sci. 2020, 21, 7906. [Google Scholar] [CrossRef]
- Harmon, J.T.; Jamieson, G.A. The glycocalicin portion of platelet glycoprotein Ib expresses both high and moderate affinity receptor sites for thrombin. A soluble radioreceptor assay for the interaction of thrombin with platelets. J. Biol. Chem. 1986, 261, 13224–13229. [Google Scholar] [CrossRef]
- Dörmann, D.; Clemetson, K.J.; Kehrel, B.E. The GPIb thrombin-binding site is essential for thrombin-induced platelet procoagulant activity. Blood 2000, 96, 2469–2478. [Google Scholar] [CrossRef]
- Dumas, J.J.; Kumar, R.; Seehra, J.; Somers, W.S.; Mosyak, L. Crystal structure of the GpIbalpha-thrombin complex essential for platelet aggregation. Science 2003, 301, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Andrews, R.K.; López, J.A.; Berndt, M.C. Molecular mechanisms of platelet adhesion and activation. Int. J. Biochem. Cell Biol. 1997, 29, 91–105. [Google Scholar] [CrossRef]
- Ruggeri, Z.M.; Zarpellon, A.; Roberts, J.R.; Mc Clintock, R.A.; Jing, H.; Mendolicchio, G.L. Unravelling the mechanism and significance of thrombin binding to platelet glycoprotein Ib. Thromb. Haemost. 2010, 104, 894–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frenette, P.S.; Denis, C.V.; Weiss, L.; Jurk, K.; Subbarao, S.; Kehrel, B.; Hartwig, J.H.; Vestweber, D.; Wagner, D.D. P-Selectin glycoprotein ligand 1 (PSGL-1) is expressed on platelets and can mediate platelet-endothelial interactions in vivo. J. Exp. Med. 2000, 191, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Ward, C.M.; Andrews, R.K.; Smith, A.I.; Berndt, M.C. Mocarhagin, a novel cobra venom metalloproteinase, cleaves the platelet von Willebrand factor receptor glycoprotein Ibalpha. Identification of the sulfated tyrosine/anionic sequence Tyr-276-Glu-282 of glycoprotein Ibalpha as a binding site for von Willebrand factor and alpha-thrombin. Biochemistry 1996, 35, 4929–4938. [Google Scholar] [CrossRef]
- Furie, B.; Furie, B.C.; Flaumenhaft, R. A journey with platelet P-selectin: The molecular basis of granule secretion, signalling and cell adhesion. Thromb. Haemost. 2001, 86, 214–221. [Google Scholar] [CrossRef]
- Nakamura, F.; Pudas, R.; Heikkinen, O.; Permi, P.; Kilpeläinen, I.; Munday, A.D.; Hartwig, J.H.; Stossel, T.P.; Ylänne, J. The structure of the GPIb-filamin A complex. Blood 2006, 107, 1925–1932. [Google Scholar] [CrossRef] [Green Version]
- Cranmer, S.L.; Ashworth, K.J.; Yao, Y.; Berndt, M.C.; Ruggeri, Z.M.; Andrews, R.K.; Jackson, S.P. High shear-dependent loss of membrane integrity and defective platelet adhesion following disruption of the GPIbα-filamin interaction. Blood 2011, 117, 2718–2727. [Google Scholar] [CrossRef] [Green Version]
- Lisman, T.; de Groot, P.G. The interaction of recombinant factor VIIa with platelet glycoprotein Ib. Thromb. Res. 2010, 125 (Suppl. 1), S13–S15. [Google Scholar] [CrossRef]
- Bradford, H.N.; Pixley, R.A.; Colman, R.W. Human factor XII binding to the glycoprotein Ib-IX-V complex inhibits thrombin-induced platelet aggregation. J. Biol. Chem. 2000, 275, 22756–22763. [Google Scholar] [CrossRef] [Green Version]
- Meng, D.; Luo, M.; Liu, B. The Role of CLEC-2 and Its Ligands in Thromboinflammation. Front. Immunol. 2021, 12, 688643. [Google Scholar] [CrossRef] [PubMed]
- Kerrigan, A.M.; Dennehy, K.M.; Mourão-Sá, D.; Faro-Trindade, I.; Willment, J.A.; Taylor, P.R.; Eble, J.A.; Reis e Sousa, C.; Brown, G.D. CLEC-2 is a phagocytic activation receptor expressed on murine peripheral blood neutrophils. J. Immunol. 2009, 182, 4150–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astarita, J.L.; Acton, S.E.; Turley, S.J. Podoplanin: Emerging functions in development, the immune system, and cancer. Front. Immunol. 2012, 3, 283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayes, J.; Watson, S.P.; Nieswandt, B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J. Clin. Investig. 2019, 129, 12–23. [Google Scholar] [CrossRef] [Green Version]
- May, F.; Hagedorn, I.; Pleines, I.; Bender, M.; Vögtle, T.; Eble, J.; Elvers, M.; Nieswandt, B. CLEC-2 is an essential platelet-activating receptor in hemostasis and thrombosis. Blood 2009, 114, 3464–3472. [Google Scholar] [CrossRef] [Green Version]
- Acton, S.E.; Astarita, J.L.; Malhotra, D.; Lukacs-Kornek, V.; Franz, B.; Hess, P.R.; Jakus, Z.; Kuligowski, M.; Fletcher, A.L.; Elpek, K.G.; et al. Podoplanin-rich stromal networks induce dendritic cell motility via activation of the C-type lectin receptor CLEC-2. Immunity 2012, 37, 276–289. [Google Scholar] [CrossRef] [Green Version]
- Suzuki-Inoue, K. Platelets and cancer-associated thrombosis: Focusing on the platelet activation receptor CLEC-2 and podoplanin. Blood 2019, 134, 1912–1918. [Google Scholar] [CrossRef]
- Lowe, K.L.; Navarro-Nunez, L.; Watson, S.P. Platelet CLEC-2 and podoplanin in cancer metastasis. Thromb. Res. 2012, 129 (Suppl. 1), S30–S37. [Google Scholar] [CrossRef]
- Washington, A.V.; Schubert, R.L.; Quigley, L.; Disipio, T.; Feltz, R.; Cho, E.H.; McVicar, D.W. A TREM family member, TLT-1, is found exclusively in the alpha-granules of megakaryocytes and platelets. Blood 2004, 104, 1042–1047. [Google Scholar] [CrossRef] [Green Version]
- Morales, J.; Villa, K.; Gattis, J.; Castro, W.; Colon, K.; Lubkowski, J.; Sanabria, P.; Hunter, R.; Washington, A.V. Soluble TLT-1 modulates platelet-endothelial cell interactions and actin polymerization. Blood Coagul. Fibrinolysis 2010, 21, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Kerris, E.W.J.; Hoptay, C.; Calderon, T.; Freishtat, R.J. Platelets and platelet extracellular vesicles in hemostasis and sepsis. J. Investig. Med. 2020, 68, 813–820. [Google Scholar] [CrossRef] [PubMed]
- DAtri, L.P.; Schattner, M. Platelet toll-like receptors in thromboinflammation. Front. Biosci. 2017, 22, 1867–1883. [Google Scholar] [CrossRef] [Green Version]
- Aslam, R.; Speck, E.R.; Kim, M.; Crow, A.R.; Bang, K.W.A.; Nestel, F.P.; Ni, H.; Lazarus, A.H.; Freedman, J.; Semple, J.W. Platelet Toll-like receptor expression modulates lipopolysaccharide-induced thrombocytopenia and tumor necrosis factor-α production In Vivo. Blood 2006, 107, 637–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marín Oyarzún, C.P.; Glembotsky, A.C.; Goette, N.P.; Lev, P.R.; De Luca, G.; Baroni Pietto, M.C.; Moiraghi, B.; Castro Ríos, M.A.; Vicente, A.; Marta, R.F.; et al. Platelet Toll-Like Receptors Mediate Thromboinflammatory Responses in Patients With Essential Thrombocythemia. Front. Immunol. 2020, 11, 705. [Google Scholar] [CrossRef] [PubMed]
- Cognasse, F.; Nguyen, K.A.; Damien, P.; McNicol, A.; Pozzetto, B.; Hamzeh-Cognasse, H.; Garraud, O. The Inflammatory Role of Platelets via Their TLRs and Siglec Receptors. Front. Immunol. 2015, 6, 83. [Google Scholar] [CrossRef] [Green Version]
- Niklaus, M.; Klingler, P.; Weber, K.; Koessler, A.; Boeck, M.; Kobsar, A.; Koessler, J. The involvement of toll-like receptors 2 and 4 in human platelet signalling pathways. Cell Signal. 2020, 76, 109817. [Google Scholar] [CrossRef]
- Carestia, A.; Kaufman, T.; Rivadeneyra, L.; Landoni, V.I.; Pozner, R.G.; Negrotto, S.; D’Atri, L.P.; Gómez, R.M.; Schattner, M. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J. Leukoc. Biol. 2016, 99, 153–162. [Google Scholar] [CrossRef]
- Lopes Pires, M.E.; Clarke, S.R.; Marcondes, S.; Gibbins, J.M. Lipopolysaccharide potentiates platelet responses via toll-like receptor 4-stimulated Akt-Erk-PLA2 signalling. PLoS ONE 2017, 12, e0186981. [Google Scholar] [CrossRef]
- Sharda, A.; Flaumenhaft, R. The life cycle of platelet granules. F1000Research 2018, 7, 236. [Google Scholar] [CrossRef]
- Blair, P.; Flaumenhaft, R. Platelet α-granules: Basic biology and clinical correlates. Blood Rev. 2009, 23, 177–189. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Yuan, Y.; Li, W. Sorting machineries: How platelet-dense granules differ from α-granules. Biosci. Rep. 2018, 38, BSR20180458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pokrovskaya, I.D.; Yadav, S.; Rao, A.; McBride, E.; Kamykowski, J.A.; Zhang, G.; Aronova, M.A.; Leapman, R.D.; Storrie, B. 3D ultrastructural analysis of α-granule, dense granule, mitochondria, and canalicular system arrangement in resting human platelets. Res. Pract. Thromb. Haemost. 2020, 4, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battinelli, E.M.; Thon, J.N.; Okazaki, R.; Peters, C.G.; Vijey, P.; Wilkie, A.R.; Noetzli, L.J.; Flaumenhaft, R.; Italiano, J.E. Megakaryocytes package contents into separate α-granules that are differentially distributed in platelets. Blood Adv. 2019, 3, 3092–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moebius, J.; Zahedi, R.P.; Lewandrowski, U.; Berger, C.; Walter, U.; Sickmann, A. The human platelet membrane proteome reveals several new potential membrane proteins. Mol. Cell. Proteom. 2005, 4, 1754–1761. [Google Scholar] [CrossRef] [Green Version]
- Park, S.Y.; Guo, X. Adaptor protein complexes and intracellular transport. Biosci. Rep. 2014, 34, e00123. [Google Scholar] [CrossRef] [PubMed]
- Sims, M.C.; Mayer, L.; Collins, J.H.; Bariana, T.K.; Megy, K.; Lavenu-Bombled, C.; Seyres, D.; Kollipara, L.; Burden, F.S.; Greene, D.; et al. Novel manifestations of immune dysregulation and granule defects in gray platelet syndrome. Blood 2020, 136, 1956–1967. [Google Scholar] [CrossRef] [PubMed]
- Gunay-Aygun, M.; Falik-Zaccai, T.C.; Vilboux, T.; Zivony-Elboum, Y.; Gumruk, F.; Cetin, M.; Khayat, M.; Boerkoel, C.F.; Kfir, N.; Huang, Y.; et al. NBEAL2 is mutated in gray platelet syndrome and is required for biogenesis of platelet α-granules. Nat. Genet. 2011, 43, 732–734. [Google Scholar] [CrossRef] [Green Version]
- Deppermann, C.; Cherpokova, D.; Nurden, P.; Schulz, J.N.; Thielmann, I.; Kraft, P.; Vogtle, T.; Kleinschnitz, C.; Dutting, S.; Krohne, G.; et al. Gray platelet syndrome and defective thrombo-inflammation in Nbeal2-deficient mice. J. Clin. Investig. 2013, 123, 3331–3342. [Google Scholar] [CrossRef] [Green Version]
- Ambrosio, A.L.; Boyle, J.A.; Di Pietro, S.M. Mechanism of platelet dense granule biogenesis: Study of cargo transport and function of Rab32 and Rab38 in a model system. Blood 2012, 120, 4072–4081. [Google Scholar] [CrossRef] [Green Version]
- Silvestre-Roig, C.; Braster, Q.; Ortega-Gomez, A.; Soehnlein, O. Neutrophils as regulators of cardiovascular inflammation. Nat. Rev. Cardiol. 2020, 17, 327–340. [Google Scholar] [CrossRef]
- Caillon, A.; Trimaille, A.; Favre, J.; Jesel, L.; Morel, O.; Kauffenstein, G. Role of neutrophils, platelets, and extracellular vesicles and their interactions in COVID-19-associated thrombopathy. J. Thromb. Haemost. 2022, 20, 17–31. [Google Scholar] [CrossRef]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; Reis, E.D.; Zhang, W.; Fallon, J.T.; Gordon, R.E.; Coller, B.S. Beta(3)-integrin-deficient mice but not P-selectin-deficient mice develop intimal hyperplasia after vascular injury: Correlation with leukocyte recruitment to adherent platelets 1 hour after injury. Circulation 2001, 103, 2501–2507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futosi, K.; Fodor, S.; Mócsai, A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int. Immunopharmacol. 2013, 17, 638–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefort, C.T.; Ley, K. Neutrophil arrest by LFA-1 activation. Front. Immunol. 2012, 3, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Li, C.; Xu, T.; Liu, W.; Ba, X.; Wang, X.; Zeng, X. PI3K is involved in β1 integrin clustering by PSGL-1 and promotes β1 integrin-mediated Jurkat cell adhesion to fibronectin. Mol. Cell. Biochem. 2014, 385, 287–295. [Google Scholar] [CrossRef]
- M’Rabet, L.; Coffer, P.; Zwartkruis, F.; Franke, B.; Segal, A.W.; Koenderman, L.; Bos, J.L. Activation of the small GTPase rap1 in human neutrophils. Blood 1998, 92, 2133–2140. [Google Scholar] [CrossRef] [Green Version]
- Santoso, S.; Sachs, U.J.; Kroll, H.; Linder, M.; Ruf, A.; Preissner, K.T.; Chavakis, T. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J. Exp. Med. 2002, 196, 679–691. [Google Scholar] [CrossRef]
- Kuijper, P.H.; Gallardo Tores, H.I.; Lammers, J.W.; Sixma, J.J.; Koenderman, L.; Zwaginga, J.J. Platelet associated fibrinogen and ICAM-2 induce firm adhesion of neutrophils under flow conditions. Thromb. Haemost. 1998, 80, 443–448. [Google Scholar] [CrossRef]
- Li, J.; Kim, K.; Jeong, S.Y.; Chiu, J.; Xiong, B.; Petukhov, P.A.; Dai, X.; Li, X.; Andrews, R.K.; Du, X.; et al. Platelet Protein Disulfide Isomerase Promotes Glycoprotein Ibα-Mediated Platelet-Neutrophil Interactions Under Thromboinflammatory Conditions. Circulation 2019, 139, 1300–1319. [Google Scholar] [CrossRef]
- Weber, C.; Springer, T.A. Neutrophil accumulation on activated, surface-adherent platelets in flow is mediated by interaction of Mac-1 with fibrinogen bound to alphaIIbbeta3 and stimulated by platelet-activating factor. J. Clin. Investig. 1997, 100, 2085–2093. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu-Bercu, A.; Grassi, L.; Frontini, M.; Salles, C., II.; Woollard, K.; Crawley, J.T. Activated α(IIb)β(3) on platelets mediates flow-dependent NETosis via SLC44A2. Elife 2020, 9, e53353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumoto, K.; Yasuoka, H.; Yoshimoto, K.; Suzuki, K.; Takeuchi, T. Platelet CXCL4 mediates neutrophil extracellular traps formation in ANCA-associated vasculitis. Sci. Rep. 2021, 11, 222. [Google Scholar] [CrossRef] [PubMed]
- Bdeir, K.; Gollomp, K.; Stasiak, M.; Mei, J.; Papiewska-Pajak, I.; Zhao, G.; Worthen, G.S.; Cines, D.B.; Poncz, M.; Kowalska, M.A. Platelet-Specific Chemokines Contribute to the Pathogenesis of Acute Lung Injury. Am. J. Respir. Cell Mol. Biol. 2017, 56, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Kasper, B.; Brandt, E.; Bulfone-Paus, S.; Petersen, F. Platelet factor 4 (PF-4)-induced neutrophil adhesion is controlled by src-kinases, whereas PF-4-mediated exocytosis requires the additional activation of p38 MAP kinase and phosphatidylinositol 3-kinase. Blood 2004, 103, 1602–1610. [Google Scholar] [CrossRef]
- Kasper, B.; Brandt, E.; Ernst, M.; Petersen, F. Neutrophil adhesion to endothelial cells induced by platelet factor 4 requires sequential activation of Ras, Syk, and JNK MAP kinases. Blood 2006, 107, 1768–1775. [Google Scholar] [CrossRef]
- Petersen, F.; Bock, L.; Flad, H.D.; Brandt, E. A chondroitin sulfate proteoglycan on human neutrophils specifically binds platelet factor 4 and is involved in cell activation. J. Immunol. 1998, 161, 4347–4355. [Google Scholar]
- Cohen, A.B.; Stevens, M.D.; Miller, E.J.; Atkinson, M.A.; Mullenbach, G. Generation of the neutrophil-activating peptide-2 by cathepsin G and cathepsin G-treated human platelets. Am. J. Physiol. 1992, 263, L249–L256. [Google Scholar] [CrossRef]
- Brandt, E.; Petersen, F.; Ludwig, A.; Ehlert, J.E.; Bock, L.; Flad, H.D. The beta-thromboglobulins and platelet factor 4: Blood platelet-derived CXC chemokines with divergent roles in early neutrophil regulation. J. Leukoc. Biol. 2000, 67, 471–478. [Google Scholar] [CrossRef]
- Gleissner, C.A.; von Hundelshausen, P.; Ley, K. Platelet chemokines in vascular disease. Arter. Thromb. Vasc. Biol. 2008, 28, 1920–1927. [Google Scholar] [CrossRef] [Green Version]
- Mishra, H.K.; Long, C.; Bahaie, N.S.; Walcheck, B. Regulation of CXCR2 expression and function by a disintegrin and metalloprotease-17 (ADAM17). J. Leukoc. Biol. 2015, 97, 447–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flad, H.D.; Brandt, E. Platelet-derived chemokines: Pathophysiology and therapeutic aspects. Cell. Mol. Life Sci. 2010, 67, 2363–2386. [Google Scholar] [CrossRef] [PubMed]
- Nasser, M.W.; Raghuwanshi, S.K.; Grant, D.J.; Jala, V.R.; Rajarathnam, K.; Richardson, R.M. Differential activation and regulation of CXCR1 and CXCR2 by CXCL8 monomer and dimer. J. Immunol. 2009, 183, 3425–3432. [Google Scholar] [CrossRef] [Green Version]
- Raghuwanshi, S.K.; Su, Y.; Singh, V.; Haynes, K.; Richmond, A.; Richardson, R.M. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J. Immunol. 2012, 189, 2824–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proudfoot, A.E.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.; Kosco-Vilbois, M.H. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schäffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.J.; Jenne, C.N. Role of platelets in neutrophil extracellular trap (NET) production and tissue injury. Semin. Immunol. 2016, 28, 546–554. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Borea, P.A.; Illes, P. P2 receptors meet the immune system. Trends Pharmacol. Sci. 2001, 22, 5–7. [Google Scholar] [CrossRef]
- Li, Z.; Smyth, S.S. 16—Interactions Between Platelets, Leukocytes, and the Endothelium. In Platelets, 4th ed.; Alan, D.M., Ed.; Academic Press: Cambridge, MA, USA, 2019; pp. 295–310. [Google Scholar]
- Bonaventura, A.; Vecchie, A.; Dagna, L.; Martinod, K.; Dixon, D.L.; Van Tassell, B.W.; Dentali, F.; Montecucco, F.; Massberg, S.; Levi, M.; et al. Endothelial dysfunction and immunothrombosis as key pathogenic mechanisms in COVID-19. Nat. Rev. Immunol. 2021, 21, 319–329. [Google Scholar] [CrossRef]
- Gotsch, U.; Jäger, U.; Dominis, M.; Vestweber, D. Expression of P-selectin on endothelial cells is upregulated by LPS and TNF-alpha In Vivo. Cell Adhes. Commun. 1994, 2, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Zamolodchikova, T.S.; Tolpygo, S.M.; Svirshchevskaya, E.V. Cathepsin G-Not Only Inflammation: The Immune Protease Can Regulate Normal Physiological Processes. Front. Immunol. 2020, 11, 411. [Google Scholar] [CrossRef] [Green Version]
- Taylor, S.; Dirir, O.; Zamanian, R.T.; Rabinovitch, M.; Thompson, A.A.R. The Role of Neutrophils and Neutrophil Elastase in Pulmonary Arterial Hypertension. Front. Med. 2018, 5, 217. [Google Scholar] [CrossRef] [PubMed]
- Molino, M.; Blanchard, N.; Belmonte, E.; Tarver, A.P.; Abrams, C.; Hoxie, J.A.; Cerletti, C.; Brass, L.F. Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J. Biol. Chem. 1995, 270, 11168–11175. [Google Scholar] [CrossRef] [Green Version]
- Tithof, P.K.; Schiamberg, E.; Peters-Golden, M.; Ganey, P.E. Phospholipase A2 is involved in the mechanism of activation of neutrophils by polychlorinated biphenyls. Environ. Health Perspect. 1996, 104, 52–58. [Google Scholar] [CrossRef]
- McDonald, P.P.; McColl, S.R.; Naccache, P.H.; Borgeat, P. Activation of the human neutrophil 5-lipoxygenase by leukotriene B4. Br. J. Pharmacol. 1992, 107, 226–232. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Li, J.; Tseng, A.; Andrews, R.K.; Cho, J. NOX2 is critical for heterotypic neutrophil-platelet interactions during vascular inflammation. Blood 2015, 126, 1952–1964. [Google Scholar] [CrossRef] [Green Version]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-κB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef] [Green Version]
- Darbousset, R.; Thomas, G.M.; Mezouar, S.; Frère, C.; Bonier, R.; Mackman, N.; Renné, T.; Dignat-George, F.; Dubois, C.; Panicot-Dubois, L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood 2012, 120, 2133–2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pircher, J.; Czermak, T.; Ehrlich, A.; Eberle, C.; Gaitzsch, E.; Margraf, A.; Grommes, J.; Saha, P.; Titova, A.; Ishikawa-Ankerhold, H.; et al. Cathelicidins prime platelets to mediate arterial thrombosis and tissue inflammation. Nat. Commun. 2018, 9, 1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alon, R.; Sportiello, M.; Kozlovski, S.; Kumar, A.; Reilly, E.C.; Zarbock, A.; Garbi, N.; Topham, D.J. Leukocyte trafficking to the lungs and beyond: Lessons from influenza for COVID-19. Nat. Rev. Immunol. 2021, 21, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Koupenova, M.; Corkrey, H.A.; Vitseva, O.; Manni, G.; Pang, C.J.; Clancy, L.; Yao, C.; Rade, J.; Levy, D.; Wang, J.P.; et al. The role of platelets in mediating a response to human influenza infection. Nat. Commun. 2019, 10, 1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Wysocka, J.; Sayegh, J.; Lee, Y.H.; Perlin, J.R.; Leonelli, L.; Sonbuchner, L.S.; McDonald, C.H.; Cook, R.G.; Dou, Y.; et al. Human PAD4 regulates histone arginine methylation levels via demethylimination. Science 2004, 306, 279–283. [Google Scholar] [CrossRef]
- Li, P.; Li, M.; Lindberg, M.R.; Kennett, M.J.; Xiong, N.; Wang, Y. PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 2010, 207, 1853–1862. [Google Scholar] [CrossRef]
- Vossenaar, E.R.; Radstake, T.R.; van der Heijden, A.; van Mansum, M.A.; Dieteren, C.; de Rooij, D.J.; Barrera, P.; Zendman, A.J.; van Venrooij, W.J. Expression and activity of citrullinating peptidylarginine deiminase enzymes in monocytes and macrophages. Ann. Rheum. Dis. 2004, 63, 373–381. [Google Scholar] [CrossRef] [Green Version]
- Romero, V.; Fert-Bober, J.; Nigrovic, P.A.; Darrah, E.; Haque, U.J.; Lee, D.M.; van Eyk, J.; Rosen, A.; Andrade, F. Immune-mediated pore-forming pathways induce cellular hypercitrullination and generate citrullinated autoantigens in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 209ra150. [Google Scholar] [CrossRef] [Green Version]
- Bawadekar, M.; Shim, D.; Johnson, C.J.; Warner, T.F.; Rebernick, R.; Damgaard, D.; Nielsen, C.H.; Pruijn, G.J.M.; Nett, J.E.; Shelef, M.A. Peptidylarginine deiminase 2 is required for tumor necrosis factor alpha-induced citrullination and arthritis, but not neutrophil extracellular trap formation. J. Autoimmun. 2017, 80, 39–47. [Google Scholar] [CrossRef]
- Neeli, I.; Radic, M. Opposition between PKC isoforms regulates histone deimination and neutrophil extracellular chromatin release. Front. Immunol. 2013, 4, 38. [Google Scholar] [CrossRef] [Green Version]
- Tsourouktsoglou, T.D.; Warnatsch, A.; Ioannou, M.; Hoving, D.; Wang, Q.; Papayannopoulos, V. Histones, DNA, and Citrullination Promote Neutrophil Extracellular Trap Inflammation by Regulating the Localization and Activation of TLR4. Cell Rep. 2020, 31, 107602. [Google Scholar] [CrossRef] [PubMed]
- de Bont, C.M.; Boelens, W.C.; Pruijn, G.J.M. NETosis, complement, and coagulation: A triangular relationship. Cell. Mol. Immunol. 2019, 16, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Maten, E.; de Bont, C.M.; de Groot, R.; de Jonge, M.I.; Langereis, J.D.; van der Flier, M. Alternative pathway regulation by factor H modulates Streptococcus pneumoniae induced proinflammatory cytokine responses by decreasing C5a receptor crosstalk. Cytokine 2016, 88, 281–286. [Google Scholar] [CrossRef]
- Berends, E.T.; Kuipers, A.; Ravesloot, M.M.; Urbanus, R.T.; Rooijakkers, S.H. Bacteria under stress by complement and coagulation. FEMS Microbiol. Rev. 2014, 38, 1146–1171. [Google Scholar] [CrossRef] [Green Version]
- Oehmcke, S.; Mörgelin, M.; Herwald, H. Activation of the human contact system on neutrophil extracellular traps. J. Innate Immun. 2009, 1, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896. [Google Scholar] [CrossRef]
- Veras, F.P.; Pontelli, M.C.; Silva, C.M.; Toller-Kawahisa, J.E.; de Lima, M.; Nascimento, D.C.; Schneider, A.H.; Caetité, D.; Tavares, L.A.; Paiva, I.M.; et al. SARS-CoV-2-triggered neutrophil extracellular traps mediate COVID-19 pathology. J. Exp. Med. 2020, 217, e20201129. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Blood Vessels and Endothelial Cells. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2003; Volume 91, p. 401. [Google Scholar]
- Chen, J.; Chung, D.W. Inflammation, von Willebrand factor, and ADAMTS13. Blood 2018, 132, 141–147. [Google Scholar] [CrossRef] [Green Version]
- Weber, C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Ho-Tin-Noé, B.; Boulaftali, Y.; Camerer, E. Platelets and vascular integrity: How platelets prevent bleeding in inflammation. Blood 2018, 131, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C. Platelets and vascular integrity. Platelets 2018, 29, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Franks, Z.G.; Campbell, R.A.; Weyrich, A.S.; Rondina, M.T. Platelet-leukocyte interactions link inflammatory and thromboembolic events in ischemic stroke. Ann. N. Y. Acad. Sci. 2010, 1207, 11–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pamuk, G.E.; Vural, O.; Turgut, B.; Demir, M.; Umit, H.; Tezel, A. Increased circulating platelet-neutrophil, platelet-monocyte complexes, and platelet activation in patients with ulcerative colitis: A comparative study. Am. J. Hematol. 2006, 81, 753–759. [Google Scholar] [CrossRef]
- Colón, D.F.; Wanderley, C.W.; Franchin, M.; Silva, C.M.; Hiroki, C.H.; Castanheira, F.V.S.; Donate, P.B.; Lopes, A.H.; Volpon, L.C.; Kavaguti, S.K.; et al. Neutrophil extracellular traps (NETs) exacerbate severity of infant sepsis. Crit. Care 2019, 23, 113. [Google Scholar] [CrossRef] [Green Version]
- Abrams, S.T.; Morton, B.; Alhamdi, Y.; Alsabani, M.; Cheng, Z.; Lane, S.; Welters, I.; Wang, G.; Toh, C.-H. A Novel Assay of Neutrophil Extracellular Traps (NETs) Formation Independently Predicts DIC and Identifies the Rationale for Anti-IL-8 Therapies. Blood 2019, 134, 440. [Google Scholar] [CrossRef]
- Schechter, M.C.; Buac, K.; Adekambi, T.; Cagle, S.; Celli, J.; Ray, S.M.; Mehta, C.C.; Rada, B.; Rengarajan, J. Neutrophil extracellular trap (NET) levels in human plasma are associated with active TB. PLoS ONE 2017, 12, e0182587. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Capobianco, A.; Rovere-Querini, P.; Ramirez, G.A.; Tombetti, E.; Valle, P.D.; Monno, A.; D’Alberti, V.; Gasparri, A.M.; Franchini, S.; et al. Platelet microparticles sustain autophagy-associated activation of neutrophils in systemic sclerosis. Sci. Transl. Med. 2018, 10, eaao3089. [Google Scholar] [CrossRef] [Green Version]
- Ceccarelli, F.; Perricone, C.; Massaro, L.; Cipriano, E.; Alessandri, C.; Spinelli, F.R.; Valesini, G.; Conti, F. Assessment of disease activity in Systemic Lupus Erythematosus: Lights and shadows. Autoimmun. Rev. 2015, 14, 601–608. [Google Scholar] [CrossRef]
- Ramirez, G.A.; Manfredi, A.A.; Maugeri, N. Misunderstandings Between Platelets and Neutrophils Build in Chronic Inflammation. Front. Immunol. 2019, 10, 2491. [Google Scholar] [CrossRef]
- Maugeri, N.; Rovere-Querini, P.; Evangelista, V.; Godino, C.; Demetrio, M.; Baldini, M.; Figini, F.; Coppi, G.; Slavich, M.; Camera, M.; et al. An Intense and Short-Lasting Burst of Neutrophil Activation Differentiates Early Acute Myocardial Infarction from Systemic Inflammatory Syndromes. PLoS ONE 2012, 7, e39484. [Google Scholar] [CrossRef]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Cate, H.t.; Hofstra, L.; et al. Elevated Levels of Circulating DNA and Chromatin Are Independently Associated With Severe Coronary Atherosclerosis and a Prothrombotic State. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2032–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hally, K.E.; Parker, O.M.; Brunton-O’Sullivan, M.M.; Harding, S.A.; Larsen, P.D. Linking Neutrophil Extracellular Traps and Platelet Activation: A Composite Biomarker Score for Predicting Outcomes after Acute Myocardial Infarction. Thromb. Haemost. 2021, 121, 1637–1649. [Google Scholar] [CrossRef] [PubMed]
- Ayna, A.B.; Ermurat, S.; Coşkun, B.N.; Harman, H.; Pehlivan, Y. Neutrophil to Lymphocyte Ratio and Mean Platelet Volume as Inflammatory Indicators in Systemic Lupus Erythematosus Nephritis. Arch. Rheumatol. 2016, 32, 21–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, M.M.; Makarem, Y.S.; El Wafaa, A.A.; Khairallah, M.K.A. Platelet-to-lymphocyte and neutrophil-to-lymphocyte ratios as noninvasive predictors for renal involvement in systemic lupus erythematosus in health clinics. Egypt. J. Intern. Med. 2019, 31, 927–933. [Google Scholar] [CrossRef]
- Wirestam, L.; Arve, S.; Linge, P.; Bengtsson, A.A. Neutrophils—Important Communicators in Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 2734. [Google Scholar] [CrossRef] [Green Version]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef] [PubMed]
- Czaikoski, P.G.; Mota, J.M.S.C.; Nascimento, D.C.; Sônego, F.; Castanheira, F.V.e.S.; Melo, P.H.; Scortegagna, G.T.; Silva, R.L.; Barroso-Sousa, R.; Souto, F.O.; et al. Neutrophil Extracellular Traps Induce Organ Damage during Experimental and Clinical Sepsis. PLoS ONE 2016, 11, e0148142. [Google Scholar] [CrossRef] [Green Version]
- Johansson, D.; Shannon, O.; Rasmussen, M. Platelet and Neutrophil Responses to Gram Positive Pathogens in Patients with Bacteremic Infection. PLoS ONE 2011, 6, e26928. [Google Scholar] [CrossRef] [Green Version]
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088. [Google Scholar] [CrossRef]
- Kaiser, R.; Escaig, R.; Erber, J.; Nicolai, L. Neutrophil-Platelet Interactions as Novel Treatment Targets in Cardiovascular Disease. Front. Cardiovasc. Med. 2022, 8, 824112. [Google Scholar] [CrossRef] [PubMed]
- Denorme, F.; Manne, B.K.; Portier, I.; Eustes, A.S.; Kosaka, Y.; Kile, B.T.; Rondina, M.T.; Campbell, R.A. Platelet necrosis mediates ischemic stroke outcome in mice. Blood 2020, 135, 429–440. [Google Scholar] [CrossRef]
- Gerdes, N.; Seijkens, T.; Lievens, D.; Kuijpers, M.J.E.; Winkels, H.; Projahn, D.; Hartwig, H.; Beckers, L.; Megens, R.T.A.; Boon, L.; et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 482–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeiger, F.; Stephan, S.; Hoheisel, G.; Pfeiffer, D.; Ruehlmann, C.; Koksch, M. P-Selectin expression, platelet aggregates, and platelet-derived microparticle formation are increased in peripheral arterial disease. Blood Coagul. Fibrinolysis 2000, 11, 723–728. [Google Scholar] [CrossRef]
- Arcanjo, A.; Logullo, J.; Cristie, C.; Menezes, B.; Souza, T.; Giangiarulo, C.; Reis, M.; Menezes, G.; Castro, M.; Da, Y.; et al. The emerging role of neutrophil extracellular traps in severe acute respiratory syndrome coronavirus 2 (COVID-19). Sci. Rep. 2020, 10, 19630. [Google Scholar] [CrossRef] [PubMed]
- Skendros, P.; Mitsios, A.; Chrysanthopoulou, A.; Mastellos, D.C.; Metallidis, S.; Rafailidis, P.; Ntinopoulou, M.; Sertaridou, E.; Tsironidou, V.; Tsigalou, C.; et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J. Clin. Investig. 2020, 130, 6151–6157. [Google Scholar] [CrossRef]
- Barnes, B.J.; Adrover, J.M.; Baxter-Stoltzfus, A.; Borczuk, A.; Cools-Lartigue, J.; Crawford, J.M.; Daßler-Plenker, J.; Guerci, P.; Huynh, C.; Knight, J.S.; et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. J. Exp. Med. 2020, 217, e20200652. [Google Scholar] [CrossRef]
- Middleton, E.A.; He, X.-Y.; Denorme, F.; Campbell, R.A.; Ng, D.; Salvatore, S.P.; Mostyka, M.; Baxter-Stoltzfus, A.; Borczuk, A.C.; Loda, M.; et al. Neutrophil extracellular traps contribute to immunothrombosis in COVID-19 acute respiratory distress syndrome. Blood 2020, 136, 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, Y.; Tang, N.; Liu, H.; Cao, W. Coagulation Dysfunction. Arch. Pathol. Lab. Med. 2020, 144, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Martyanov, A.A.; Boldova, A.E.; Stepanyan, M.G.; An, O.I.; Gur’ev, A.S.; Kassina, D.V.; Volkov, A.Y.; Balatskiy, A.V.; Butylin, A.A.; Karamzin, S.S.; et al. Longitudinal multiparametric characterization of platelet dysfunction in COVID-19: Effects of disease severity, anticoagulation therapy and inflammatory status. Thromb. Res. 2022, 211, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Goshua, G.; Pine, A.B.; Meizlish, M.L.; Chang, C.H.; Zhang, H.; Bahel, P.; Baluha, A.; Bar, N.; Bona, R.D.; Burns, A.J.; et al. Endotheliopathy in COVID-19-associated coagulopathy: Evidence from a single-centre, cross-sectional study. Lancet Haematol. 2020, 7, e575–e582. [Google Scholar] [CrossRef]
- Campo, G.; Contoli, M.; Fogagnolo, A.; Vieceli Dalla Sega, F.; Zucchetti, O.; Ronzoni, L.; Verri, M.; Fortini, F.; Pavasini, R.; Morandi, L.; et al. Over time relationship between platelet reactivity, myocardial injury and mortality in patients with SARS-CoV-2-associated respiratory failure. Platelets 2021, 32, 560–567. [Google Scholar] [CrossRef]
- Mizurini, D.M.; Hottz, E.D.; Bozza, P.T.; Monteiro, R.Q. Fundamentals in COVID-19-Associated Thrombosis: Molecular and Cellular Aspects. Front. Cardiovasc. Med. 2021, 8, 785738. [Google Scholar] [CrossRef] [PubMed]
- van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. The mononuclear phagocyte system: A new classification of macrophages, monocytes, and their precursor cells. Bull. World Health Organ. 1972, 46, 845–852. [Google Scholar]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [CrossRef] [Green Version]
- Wong, K.L.; Tai, J.J.-Y.; Wong, W.-C.; Han, H.; Sem, X.; Yeap, W.-H.; Kourilsky, P.; Wong, S.-C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissmann, F.; Jung, S.; Littman, D.R. Blood Monocytes Consist of Two Principal Subsets with Distinct Migratory Properties. Immunity 2003, 19, 71–82. [Google Scholar] [CrossRef] [Green Version]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, G.; Tacke, R.; Hedrick, C.C.; Hanna, R.N. Nonclassical Patrolling Monocyte Function in the Vasculature. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1306–1316. [Google Scholar] [CrossRef] [Green Version]
- Jakubzick, C.; Gautier, E.L.; Gibbings, S.L.; Sojka, D.K.; Schlitzer, A.; Johnson, T.E.; Ivanov, S.; Duan, Q.; Bala, S.; Condon, T.; et al. Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity 2013, 39, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Tsou, C.-L.; Peters, W.; Si, Y.; Slaymaker, S.; Aslanian, A.M.; Weisberg, S.P.; Mack, M.; Charo, I.F. Critical roles for CCR2 and MCP-3 in monocyte mobilization from bone marrow and recruitment to inflammatory sites. J. Clin. Investig. 2007, 117, 902–909. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Mildner, A.; Jung, S. Development and Function of Dendritic Cell Subsets. Immunity 2014, 40, 642–656. [Google Scholar] [CrossRef] [Green Version]
- Jakubzick, C.V.; Randolph, G.J.; Henson, P.M. Monocyte differentiation and antigen-presenting functions. Nat. Rev. Immunol. 2017, 17, 349–362. [Google Scholar] [CrossRef]
- Bennett, F.C.; Bennett, M.L.; Yaqoob, F.; Mulinyawe, S.B.; Grant, G.A.; Hayden Gephart, M.; Plowey, E.D.; Barres, B.A. A Combination of Ontogeny and CNS Environment Establishes Microglial Identity. Neuron 2018, 98, 1170–1183.e1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, J.C.; Filiano, A.J.; Louveau, A.; Marin, I.; Marsh, R.; Ji, E.; Goldman, D.H.; Smirnov, I.; Geraci, N.; Acton, S.; et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J. Exp. Med. 2018, 215, 1627–1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.-L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of Splenic Reservoir Monocytes and Their Deployment to Inflammatory Sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [Green Version]
- Rinder, H.; Bonan, J.; Rinder, C.; Ault, K.; Smith, B. Dynamics of leukocyte-platelet adhesion in whole blood. Blood 1991, 78, 1730–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, K.L.; Stults, N.L.; Diaz, S.; Smith, D.F.; Cummings, R.D.; Varki, A.; Mcever, R.P. Identification of a specific glycoprotein ligand for P-selectin (CD62) on myeloid cells. J. Cell Biol. 1992, 118, 445–456. [Google Scholar] [CrossRef]
- Fernandes, L.S.; Conde, I.D.; Wayne Smith, C.; Kansas, G.S.; Snapp, K.R.; Bennet, N.; Ballantyne, C.; McIntire, L.V.; O’Brian Smith, E.; Klem, J.A.; et al. Platelet-monocyte complex formation: Effect of blocking PSGL-1 alone, and in combination with alphaIIbbeta3 and alphaMbeta2, in coronary stenting. Thromb. Res. 2003, 111, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.J.; Zohlnhofer, D.; Fakhoury, L.; Ott, I.; Gawaz, M.; Schomig, A. Effect of glycoprotein IIb/IIIa receptor blockade on platelet-leukocyte interaction and surface expression of the leukocyte integrin Mac-1 in acute myocardial infarction. J. Am. Coll. Cardiol. 1999, 34, 1420–1426. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.I.; Chen, Z.; Xu, H.; Li, C.Q.; Dong, J.-F.; Mcintire, L.V.; Ballantyne, C.M.; Zhang, L.; Furman, M.I.; Berndt, M.C.; et al. Platelet Glycoprotein Ibα Is a Counterreceptor for the Leukocyte Integrin Mac-1 (Cd11b/Cd18). J. Exp. Med. 2000, 192, 193–204. [Google Scholar] [CrossRef] [Green Version]
- Lindmark, E.; Tenno, T.; Siegbahn, A. Role of Platelet P-Selectin and CD40 Ligand in the Induction of Monocytic Tissue Factor Expression. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 2322–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouchon, A.; Dietrich, J.; Colonna, M. Cutting Edge: Inflammatory Responses Can Be Triggered by TREM-1, a Novel Receptor Expressed on Neutrophils and Monocytes. J. Immunol. 2000, 164, 4991–4995. [Google Scholar] [CrossRef]
- Haselmayer, P.; Grosse-Hovest, L.; Von Landenberg, P.; Schild, H.; Radsak, M.P. TREM-1 ligand expression on platelets enhances neutrophil activation. Blood 2007, 110, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Gawaz, M.P.; Loftus, J.C.; Bajt, M.L.; Frojmovic, M.M.; Plow, E.F.; Ginsberg, M.H. Ligand bridging mediates integrin alpha IIb beta 3 (platelet GPIIB-IIIA) dependent homotypic and heterotypic cell-cell interactions. J. Clin. Investig. 1991, 88, 1128–1134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverstein, R.L.; Asch, A.S.; Nachman, R.L. Glycoprotein IV mediates thrombospondin-dependent platelet-monocyte and platelet-U937 cell adhesion. J. Clin. Investig. 1989, 84, 546–552. [Google Scholar] [CrossRef]
- Bennett, J.S. Structure and function of the platelet integrin IIb 3. J. Clin. Investig. 2005, 115, 3363–3369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Hundelshausen, P.; Weber, K.S.C.; Huo, Y.; Proudfoot, A.E.I.; Nelson, P.J.; Ley, K.; Weber, C. RANTES Deposition by Platelets Triggers Monocyte Arrest on Inflamed and Atherosclerotic Endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef] [PubMed]
- Slungaard, A. Platelet factor 4: A chemokine enigma. Int J. Biochem. Cell Biol. 2005, 37, 1162–1167. [Google Scholar] [CrossRef]
- Pervushina, O.; Scheuerer, B.; Reiling, N.; Behnke, L.; Schröder, J.-M.; Kasper, B.; Brandt, E.; Bulfone-Paus, S.; Petersen, F. Platelet Factor 4/CXCL4 Induces Phagocytosis and the Generation of Reactive Oxygen Metabolites in Mononuclear Phagocytes Independently of Gi Protein Activation or Intracellular Calcium Transients. J. Immunol. 2004, 173, 2060–2067. [Google Scholar] [CrossRef]
- Kasper, B.; Brandt, E.; Brandau, S.; Petersen, F. Platelet Factor 4 (CXC Chemokine Ligand 4) Differentially Regulates Respiratory Burst, Survival, and Cytokine Expression of Human Monocytes by Using Distinct Signaling Pathways. J. Immunol. 2007, 179, 2584–2591. [Google Scholar] [CrossRef]
- Fricke, I.; Mitchell, D.; Petersen, F.; Böhle, A.; Bulfone-Paus, S.; Brandau, S. Platelet factor 4 in conjunction with IL-4 directs differentiation of human monocytes into specialized antigen- presenting cells. FASEB J. 2004, 18, 1588–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lishko, V.K.; Yakubenko, V.P.; Ugarova, T.P.; Podolnikova, N.P. Leukocyte integrin Mac-1 (CD11b/CD18, αMβ2, CR3) acts as a functional receptor for platelet factor 4. J. Biol. Chem. 2018, 293, 6869–6882. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.; Weber, K.S.C.; Klier, C.; Gu, S.; Wank, R.; Horuk, R.; Nelson, P.J. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and TH1-like/CD45RO+T cells. Blood 2001, 97, 1144–1146. [Google Scholar] [CrossRef] [PubMed]
- Alard, J.E.; Ortega-Gomez, A.; Wichapong, K.; Bongiovanni, D.; Horckmans, M.; Megens, R.T.; Leoni, G.; Ferraro, B.; Rossaint, J.; Paulin, N.; et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015, 7, 317ra196. [Google Scholar] [CrossRef]
- Gawaz, M.; Neumann, F.-J.; Dickfeld, T.; Koch, W.; Laugwitz, K.-L.; Adelsberger, H.; Langenbrink, K.; Page, S.; Neumeier, D.; SchöMig, A.; et al. Activated Platelets Induce Monocyte Chemotactic Protein-1 Secretion and Surface Expression of Intercellular Adhesion Molecule-1 on Endothelial Cells. Circulation 1998, 98, 1164–1171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarma, J.; Laan, C.A.; Alam, S.; Jha, A.; Fox, K.A.A.; Dransfield, I. Increased Platelet Binding to Circulating Monocytes in Acute Coronary Syndromes. Circulation 2002, 105, 2166–2171. [Google Scholar] [CrossRef] [Green Version]
- Furman, M.I.; Benoit, S.E.; Barnard, M.R.; Valeri, C.R.; Borbone, M.L.; Becker, R.C.; Hechtman, H.B.; Michelson, A.D. Increased platelet reactivity and circulating monocyte-platelet aggregates in patients with stable coronary artery disease. J. Am. Coll. Cardiol. 1998, 31, 352–358. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.E.; Harrison, P.; Mackie, I.J.; Isenberg, D.A.; Machin, S.J. Increased circulating platelet-leucocyte complexes and platelet activation in patients with antiphospholipid syndrome, systemic lupus erythematosus and rheumatoid arthritis. Br. J. Haematol. 2001, 115, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.A.; Sommerfield, A.J.; Sarma, J.; Twomey, P.J.; Newby, D.E.; Frier, B.M.; Fox, K.A. Increased CD40 ligand and platelet-monocyte aggregates in patients with type 1 diabetes mellitus. Atherosclerosis 2004, 176, 321–325. [Google Scholar] [CrossRef]
- Ashman, N. Increased platelet-monocyte aggregates and cardiovascular disease in end-stage renal failure patients. Nephrol. Dial. Transplant. 2003, 18, 2088–2096. [Google Scholar] [CrossRef] [Green Version]
- Poston, R.N. Atherosclerosis: Integration of its pathogenesis as a self-perpetuating propagating inflammation: A review. Cardiovasc. Endocrinol. Metab. 2019, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V. Macrophages and Their Role in Atherosclerosis: Pathophysiology and Transcriptome Analysis. BioMed. Res. Int. 2016, 2016, 9582430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.C.; Glass, C.K. The macrophage foam cell as a target for therapeutic intervention. Nat. Med. 2002, 8, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol. 2006, 6, 508–519. [Google Scholar] [CrossRef]
- Ribeiro, L.S.; Migliari Branco, L.; Franklin, B.S. Regulation of Innate Immune Responses by Platelets. Front. Immunol. 2019, 10, 1320. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chen, X.; Salomon, R.G.; Mcintyre, T.M. Platelet Activation by Low Concentrations of Intact Oxidized LDL Particles Involves the PAF Receptor. Arter. Thromb. Vasc. Biol. 2009, 29, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Hatakeyama, K.; Kaneko, M.K.; Kato, Y.; Ishikawa, T.; Nishihira, K.; Tsujimoto, Y.; Shibata, Y.; Ozaki, Y.; Asada, Y. Podoplanin expression in advanced atherosclerotic lesions of human aortas. Thromb. Res. 2012, 129, e70–e76. [Google Scholar] [CrossRef]
- Chan, H.-C.; Ke, L.-Y.; Chu, C.-S.; Lee, A.-S.; Shen, M.-Y.; Cruz, M.A.; Hsu, J.-F.; Cheng, K.-H.; Chan, H.-C.B.; Lu, J.; et al. Highly electronegative LDL from patients with ST-elevation myocardial infarction triggers platelet activation and aggregation. Blood 2013, 122, 3632–3641. [Google Scholar] [CrossRef] [Green Version]
- Massberg, S.; Brand, K.; GrüNer, S.; Page, S.; MüLler, E.; MüLler, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A Critical Role of Platelet Adhesion in the Initiation of Atherosclerotic Lesion Formation. J. Exp. Med. 2002, 196, 887–896. [Google Scholar] [CrossRef]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- Lievens, D.; Zernecke, A.; Seijkens, T.; Soehnlein, O.; Beckers, L.; Munnix, I.C.A.; Wijnands, E.; Goossens, P.; Van Kruchten, R.; Thevissen, L.; et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010, 116, 4317–4327. [Google Scholar] [CrossRef] [Green Version]
- von Hundelshausen, P.; Schmitt, M.M.N. Platelets and their chemokines in atherosclerosis—Clinical applications. Front. Physiol. 2014, 5, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheuerer, B.; Ernst, M.; Durrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A. Macrophage Phenotype Modulation by CXCL4 in Atherosclerosis. Front. Physiol. 2012, 3, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachais, B.S.; Turrentine, T.; Dawicki McKenna, J.M.; Rux, A.H.; Rader, D.; Kowalska, M.A. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb. Haemost. 2007, 98, 1108–1113. [Google Scholar] [PubMed]
- Nassar, T.; Sachais, B.S.; Akkawi, S.E.; Kowalska, M.A.; Bdeir, K.; Leitersdorf, E.; Hiss, E.; Ziporen, L.; Aviram, M.; Cines, D.; et al. Platelet Factor 4 Enhances the Binding of Oxidized Low-density Lipoprotein to Vascular Wall Cells. J. Biol. Chem. 2003, 278, 6187–6193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef] [PubMed]
- Tunjungputri, R.N.; Van Der Ven, A.J.; Schonsberg, A.; Mathan, T.S.; Koopmans, P.; Roest, M.; Fijnheer, R.; Groot, P.G.; de Mast, Q. Reduced platelet hyperreactivity and platelet-monocyte aggregation in HIV-infected individuals receiving a raltegravir-based regimen. Aids 2014, 28, 2091–2096. [Google Scholar] [CrossRef]
- Green, S.A.; Smith, M.; Hasley, R.B.; Stephany, D.; Harned, A.; Nagashima, K.; Abdullah, S.; Pittaluga, S.; Imamichi, T.; Qin, J.; et al. Activated platelet–T-cell conjugates in peripheral blood of patients with HIV infection. Aids 2015, 29, 1297–1308. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.V.; Davidson, D.C.; Jackson, J.W.; Singh, V.B.; Silva, J.; Ramirez, S.H.; Maggirwar, S.B. Characterization of Platelet–Monocyte Complexes in HIV-1–Infected Individuals: Possible Role in HIV-Associated Neuroinflammation. J. Immunol. 2014, 192, 4674–4684. [Google Scholar] [CrossRef] [Green Version]
- Liang, H.; Duan, Z.; Li, D.; Li, D.; Wang, Z.; Ren, L.; Shen, T.; Shao, Y. Higher levels of circulating monocyte–platelet aggregates are correlated with viremia and increased sCD163 levels in HIV-1 infection. Cell. Mol. Immunol. 2015, 12, 435–443. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.-J.; Jen, Y.-H.; Chang, J.-S.; Hsiao, H.-M.; Noisakran, S.; Perng, G.C. Frequency Alterations in Key Innate Immune Cell Components in the Peripheral Blood of Dengue Patients Detected by FACS Analysis. J. Innate Immun. 2011, 3, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Hottz, E.D.; Medeiros-De-Moraes, I.M.; Vieira-De-Abreu, A.; De Assis, E.F.; Vals-De-Souza, R.; Castro-Faria-Neto, H.C.; Weyrich, A.S.; Zimmerman, G.A.; Bozza, F.A.; Bozza, P.T. Platelet Activation and Apoptosis Modulate Monocyte Inflammatory Responses in Dengue. J. Immunol. 2014, 193, 1864–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pão, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341. [Google Scholar] [CrossRef] [PubMed]
- Scull, C.M.; Hays, W.D.; Fischer, T.H. Macrophage pro-inflammatory cytokine secretion is enhanced following interaction with autologous platelets. J. Inflamm. 2010, 7, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gudbrandsdottir, S.; Hasselbalch, H.C.; Nielsen, C.H. Activated Platelets Enhance IL-10 Secretion and Reduce TNF-α Secretion by Monocytes. J. Immunol. 2013, 191, 4059–4067. [Google Scholar] [CrossRef] [Green Version]
- Xiang, B.; Zhang, G.; Guo, L.; Li, X.-A.; Morris, A.J.; Daugherty, A.; Whiteheart, S.W.; Smyth, S.S.; Li, Z. Platelets protect from septic shock by inhibiting macrophage-dependent inflammation via the cyclooxygenase 1 signalling pathway. Nat. Commun. 2013, 4, 2567. [Google Scholar] [CrossRef] [Green Version]
- Carestia, A.; Mena, H.A.; Olexen, C.M.; Ortiz Wilczyñski, J.M.; Negrotto, S.; Errasti, A.E.; Gómez, R.M.; Jenne, C.N.; Carrera Silva, E.A.; Schattner, M. Platelets Promote Macrophage Polarization toward Pro-inflammatory Phenotype and Increase Survival of Septic Mice. Cell Rep. 2019, 28, 896–908.e895. [Google Scholar] [CrossRef] [Green Version]
- Rondina, M.T.; Carlisle, M.; Fraughton, T.; Brown, S.M.; Miller, R.R.; Harris, E.S.; Weyrich, A.S.; Zimmerman, G.A.; Supiano, M.A.; Grissom, C.K. Platelet-Monocyte Aggregate Formation and Mortality Risk in Older Patients With Severe Sepsis and Septic Shock. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2015, 70, 225–231. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Ren, J.; Hu, D.; Wu, X.; Li, G.; Wang, G.; Gu, G.; Chen, J.; Li, R.; Li, Y.; et al. Monocyte subsets and monocyte-platelet aggregates: Implications in predicting septic mortality among surgical critical illness patients. Biomarkers 2016, 21, 509–516. [Google Scholar] [CrossRef]
- De Stoppelaar, S.F.; Van ’t Veer, C.; Claushuis, T.A.M.; Albersen, B.J.A.; Roelofs, J.J.T.H.; Van Der Poll, T. Thrombocytopenia impairs host defense in gram-negative pneumonia–derived sepsis in mice. Blood 2014, 124, 3781–3790. [Google Scholar] [CrossRef] [Green Version]
- Wuescher, L.M.; Takashima, A.; Worth, R.G. A novel conditional platelet depletion mouse model reveals the importance of platelets in protection against Staphylococcus aureus bacteremia. J. Thromb. Haemost. 2015, 13, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, R.A.; Wuescher, L.M.; Dona, K.R.; Worth, R.G. Platelets Mediate Host Defense againstStaphylococcus aureusthrough Direct Bactericidal Activity and by Enhancing Macrophage Activities. J. Immunol. 2017, 198, 344–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Katz, S.; Jimenez, M.A.; Lehmkuhler, W.E.; Grosfeld, J.L. Liver bacterial clearance following hepatic artery ligation and portacaval shunt. J. Surg. Res. 1991, 51, 267–270. [Google Scholar] [CrossRef]
- Rabinovitch, M. Professional and non-professional phagocytes: An introduction. Trends Cell Biol. 1995, 5, 85–87. [Google Scholar] [CrossRef]
- Lee, W.-Y.; Moriarty, T.J.; Wong, C.H.Y.; Zhou, H.; Strieter, R.M.; Van Rooijen, N.; Chaconas, G.; Kubes, P. An intravascular immune response to Borrelia burgdorferi involves Kupffer cells and iNKT cells. Nat. Immunol. 2010, 11, 295–302. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Surewaard, B.G.; Wong, C.H.; Geoghegan, J.A.; Jenne, C.N.; PKubes, P. CRIg Functions as a Macrophage Pattern Recognition Receptor to Directly Bind and Capture Blood-Borne Gram-Positive Bacteria. Cell Host Microbe 2016, 20, 99–106. [Google Scholar] [CrossRef] [Green Version]
- Surewaard, B.G.J.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 2016, 213, 1141–1151. [Google Scholar] [CrossRef] [Green Version]
- Wong, C.H.Y.; Jenne, C.N.; Petri, B.; Chrobok, N.L.; Kubes, P. Nucleation of platelets with blood-borne pathogens on Kupffer cells precedes other innate immunity and contributes to bacterial clearance. Nat. Immunol. 2013, 14, 785–792. [Google Scholar] [CrossRef] [Green Version]
- Verschoor, A.; Neuenhahn, M.; Navarini, A.A.; Graef, P.; Plaumann, A.; Seidlmeier, A.; Nieswandt, B.; Massberg, S.; Zinkernagel, R.M.; Hengartner, H.; et al. A platelet-mediated system for shuttling blood-borne bacteria to CD8α+ dendritic cells depends on glycoprotein GPIb and complement C3. Nat. Immunol. 2011, 12, 1194–1201. [Google Scholar] [CrossRef]
- Broadley, S.P.; Plaumann, A.; Coletti, R.; Lehmann, C.; Wanisch, A.; Seidlmeier, A.; Esser, K.; Luo, S.; Rämer, P.C.; Massberg, S.; et al. Dual-Track Clearance of Circulating Bacteria Balances Rapid Restoration of Blood Sterility with Induction of Adaptive Immunity. Cell Host Microbe 2016, 20, 36–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaffer, C.L.; Weinberg, R.A. A perspective on cancer cell metastasis. Science 2011, 331, 1559–1564. [Google Scholar] [CrossRef] [PubMed]
- Seyfried, T.N.; Huysentruyt, L.C. On the Origin of Cancer Metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Khorana, A.A.; Connolly, G.C. Assessing Risk of Venous Thromboembolism in the Patient With Cancer. J. Clin. Oncol. 2009, 27, 4839–4847. [Google Scholar] [CrossRef]
- Lyman, G.H.; Khorana, A.A. Cancer, Clots and Consensus: New Understanding of an Old Problem. J. Clin. Oncol. 2009, 27, 4821–4826. [Google Scholar] [CrossRef] [Green Version]
- Heinmöller, E.; Weinel, R.J.; Heidtmann, H.H.; Salge, U.; Seitz, R.; Schmitz, I.; Müller, K.M.; Zirngibl, H. Studies on tumor-cell-induced platelet aggregation in human lung cancer cell lines. J. Cancer Res. Clin. Oncol. 1996, 122, 735–744. [Google Scholar] [CrossRef]
- Han, X.; Guo, B.; Li, Y.; Zhu, B. Tissue factor in tumor microenvironment: A systematic review. J. Hematol. Oncol. 2014, 7, 54. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, M.; Bottsford-Miller, J.; Pradeep, S.; Taylor, M.L.; Choi, H.-J.; Hansen, J.M.; Dalton, H.J.; Stone, R.L.; Cho, M.S.; Nick, A.M.; et al. FAK regulates platelet extravasation and tumor growth after antiangiogenic therapy withdrawal. J. Clin. Investig. 2016, 126, 1885–1896. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Sato, S.; Oh-Hara, T.; Takami, M.; Koike, S.; Mishima, Y.; Hatake, K.; Fujita, N. Platelets Promote Tumor Growth and Metastasis via Direct Interaction between Aggrus/Podoplanin and CLEC-2. PLoS ONE 2013, 8, e73609. [Google Scholar] [CrossRef]
- Volz, J.; Mammadova-Bach, E.; Gil-Pulido, J.; Nandigama, R.; Remer, K.; Sorokin, L.; Zernecke, A.; Abrams, S.I.; Ergün, S.; Henke, E.; et al. Inhibition of platelet GPVI induces intratumor hemorrhage and increases efficacy of chemotherapy in mice. Blood 2019, 133, 2696–2706. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Huang, Z.; Zhang, W.; Jiang, L.; Hultenby, K.; Zhu, L.; Hu, H.; Nilsson, G.P.; Li, N. Distinct platelet packaging, release, and surface expression of proangiogenic and antiangiogenic factors on different platelet stimuli. Blood 2011, 117, 3907–3911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavlović, N.; Kopsida, M.; Gerwins, P.; Heindryckx, F. Activated platelets contribute to the progression of hepatocellular carcinoma by altering the tumor environment. Life Sci. 2021, 277, 119612. [Google Scholar] [CrossRef]
- Yu, L.-X.; Yan, L.; Yang, W.; Wu, F.-Q.; Ling, Y.; Chen, S.-Z.; Tang, L.; Tan, Y.-X.; Cao, D.; Wu, M.-C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef] [PubMed]
- Wojtukiewicz, M.Z.; Sierko, E.; Hempel, D.; Tucker, S.C.; Honn, K.V. Platelets and cancer angiogenesis nexus. Cancer Metastasis Rev. 2017, 36, 249–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, S.; Takemoto, A.; Takami, M.; Oh-Hara, T.; Fujita, N. Platelets promote osteosarcoma cell growth through activation of the platelet-derived growth factor receptor-Akt signaling axis. Cancer Sci. 2014, 105, 983–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maini, M.K.; Schurich, A. Platelets harness the immune response to drive liver cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 12840–12841. [Google Scholar] [CrossRef] [Green Version]
- Ringelhan, M.; Pfister, D.; O’Connor, T.; Pikarsky, E.; Heikenwalder, M. The immunology of hepatocellular carcinoma. Nat. Immunol. 2018, 19, 222–232. [Google Scholar] [CrossRef]
- Heindryckx, F. Targeting the tumor stroma in hepatocellular carcinoma. World J. Hepatol. 2014, 7, 165. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Rudolph, K.L. Hepatocellular carcinoma: Epidemiology and molecular carcinogenesis. Gastroenterology 2007, 132, 2557–2576. [Google Scholar] [CrossRef]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, N.; Rani, B.; Gerwins, P.; Heindryckx, F. Platelets as Key Factors in Hepatocellular Carcinoma. Cancers 2019, 11, 1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gay, L.J.; Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 2011, 11, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Labelle, M.; Begum, S.; Hynes, R.O. Direct Signaling between Platelets and Cancer Cells Induces an Epithelial-Mesenchymal-Like Transition and Promotes Metastasis. Cancer Cell 2011, 20, 576–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsig, L. The role of platelet activation in tumor metastasis. Expert Rev. Anticancer. Ther. 2008, 8, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar]
- Timar, J.; Chopra, H.; Rong, X.; Hatfield, J.S.; Fligiel, S.E.G.; Onoda, J.M.; Taylor, J.D.; Honn, K.V. Calcium channel blocker treatment of tumor cells induces alterations in the cytoskeleton, mobility of the integrin αIIbβ3 and tumor-cell-induced platelet aggregation. J. Cancer Res. Clin. Oncol. 1992, 118, 425–434. [Google Scholar] [CrossRef]
- Janowska-Wieczorek, A.; Wysoczynski, M.; Kijowski, J.; Marquez-Curtis, L.; Machalinski, B.; Ratajczak, J.; Ratajczak, M.Z. Microvesicles derived from activated platelets induce metastasis and angiogenesis in lung cancer. Int. J. Cancer 2005, 113, 752–760. [Google Scholar] [CrossRef]
- Honn, K.V.; Tang, D.G.; Crissman, J.D. Platelets and cancer metastasis: A causal relationship? Cancer Metastasis Rev. 1992, 11, 325–351. [Google Scholar] [CrossRef]
- Labelle, M.; Begum, S.; Hynes, R.O. Platelets guide the formation of early metastatic niches. Proc. Natl. Acad. Sci. USA 2014, 111, E3053–E3061. [Google Scholar] [CrossRef] [Green Version]
- Gil-Bernabé, A.M.; Ferjančič, Š.; Tlalka, M.; Zhao, L.; Allen, P.D.; Im, J.H.; Watson, K.; Hill, S.A.; Amirkhosravi, A.; Francis, J.L.; et al. Recruitment of monocytes/macrophages by tissue factor-mediated coagulation is essential for metastatic cell survival and premetastatic niche establishment in mice. Blood 2012, 119, 3164–3175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Läubli, H.; Spanaus, K.-S.; Borsig, L. Selectin-mediated activation of endothelial cells induces expression of CCL5 and promotes metastasis through recruitment of monocytes. Blood 2009, 114, 4583–4591. [Google Scholar] [CrossRef] [Green Version]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popivanova, B.K.; Kostadinova, F.I.; Furuichi, K.; Shamekh, M.M.; Kondo, T.; Wada, T.; Egashira, K.; Mukaida, N. Blockade of a Chemokine, CCL2, Reduces Chronic Colitis-Associated Carcinogenesis in Mice. Cancer Res. 2009, 69, 7884–7892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Chen, W.; Zhang, W.; Lee-Sundlov, M.M.; Casari, C.; Berndt, M.C.; Lanza, F.; Bergmeier, W.; Hoffmeister, K.M.; Zhang, X.F.; et al. Desialylation of O-glycans on glycoprotein Ibα drives receptor signaling and platelet clearance. Haematologica 2021, 106, 220–229. [Google Scholar] [CrossRef] [Green Version]
- Rivadeneyra, L.; Falet, H.; Hoffmeister, K.M. Circulating platelet count and glycans. Curr. Opin. Hematol. 2021, 28, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Hoffmeister, K.M.; Falet, H. Platelet clearance by the hepatic Ashwell-Morrell receptor: Mechanisms and biological significance. Thromb. Res. 2016, 141 (Suppl. 2), S68–S72. [Google Scholar] [CrossRef] [Green Version]
- Okumura, I.; Lombart, C.; Jamieson, G.A. Platelet glycocalicin. II. Purification and characterization. J. Biol. Chem. 1976, 251, 5950–5955. [Google Scholar] [CrossRef]
- Marini, I.; Zlamal, J.; Faul, C.; Holzer, U.; Hammer, S.; Pelzl, L.; Bethge, W.; Althaus, K.; Bakchoul, T. Autoantibody-mediated desialylation impairs human thrombopoiesis and platelet lifespan. Haematologica 2021, 106, 196–207. [Google Scholar] [CrossRef]
- Li, J.; van der Wal, D.E.; Zhu, G.; Xu, M.; Yougbare, I.; Ma, L.; Vadasz, B.; Carrim, N.; Grozovsky, R.; Ruan, M.; et al. Desialylation is a mechanism of Fc-independent platelet clearance and a therapeutic target in immune thrombocytopenia. Nat. Commun. 2015, 6, 7737. [Google Scholar] [CrossRef] [Green Version]
- Yan, R.; Chen, M.; Ma, N.; Zhao, L.; Cao, L.; Zhang, Y.; Zhang, J.; Yu, Z.; Wang, Z.; Xia, L.; et al. Glycoprotein Ibα clustering induces macrophage-mediated platelet clearance in the liver. Thromb. Haemost. 2015, 113, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liang, X.; Syed, A.K.; Jessup, P.; Church, W.R.; Ware, J.; Josephson, C.D.; Li, R. Inhibiting GPIbα Shedding Preserves Post-Transfusion Recovery and Hemostatic Function of Platelets After Prolonged Storage. Arter. Thromb. Vasc. Biol. 2016, 36, 1821–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solum, N.O.; Hagen, I.; Filion-Myklebust, C.; Stabaek, T. Platelet glycocalicin. Its membrane association and solubilization in aqueous media. Biochim. Biophys. Acta 1980, 597, 235–246. [Google Scholar] [CrossRef]
- Hoffmeister, K.M.; Felbinger, T.W.; Falet, H.; Denis, C.V.; Bergmeier, W.; Mayadas, T.N.; von Andrian, U.H.; Wagner, D.D.; Stossel, T.P.; Hartwig, J.H. The clearance mechanism of chilled blood platelets. Cell 2003, 112, 87–97. [Google Scholar] [CrossRef] [Green Version]
- McArthur, K.; Chappaz, S.; Kile, B.T. Apoptosis in megakaryocytes and platelets: The life and death of a lineage. Blood 2018, 131, 605–610. [Google Scholar] [CrossRef]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Quach, M.E.; Chen, W.; Li, R. Mechanisms of platelet clearance and translation to improve platelet storage. Blood 2018, 131, 1512–1521. [Google Scholar] [CrossRef]
- Julien, O.; Wells, J.A. Caspases and their substrates. Cell Death Differ. 2017, 24, 1380–1389. [Google Scholar] [CrossRef]
- Chen, M.; Yan, R.; Zhou, K.; Li, X.; Zhang, Y.; Liu, C.; Jiang, M.; Ye, H.; Meng, X.; Pang, N.; et al. Akt-mediated platelet apoptosis and its therapeutic implications in immune thrombocytopenia. Proc. Natl. Acad. Sci. USA 2018, 115, E10682–E10691. [Google Scholar] [CrossRef] [Green Version]
- Harrington, W.J.; Minnich, V.; Hollingsworth, J.W.; Moore, C.V. Demonstration of a thrombocytopenic factor in the blood of patients with thrombocytopenic purpura. J. Lab. Clin. Med. 1951, 38, 1–10. [Google Scholar]
- Sørensen, A.L.; Rumjantseva, V.; Nayeb-Hashemi, S.; Clausen, H.; Hartwig, J.H.; Wandall, H.H.; Hoffmeister, K.M. Role of sialic acid for platelet life span: Exposure of beta-galactose results in the rapid clearance of platelets from the circulation by asialoglycoprotein receptor-expressing liver macrophages and hepatocytes. Blood 2009, 114, 1645–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashwell, G.; Morell, A.G. The role of surface carbohydrates in the hepatic recognition and transport of circulating glycoproteins. Adv. Enzymol. Relat. Areas Mol. Biol. 1974, 41, 99–128. [Google Scholar] [CrossRef] [PubMed]
- Grewal, P.K. The Ashwell-Morell receptor. Methods Enzymol. 2010, 479, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Ellies, L.G.; Ditto, D.; Levy, G.G.; Wahrenbrock, M.; Ginsburg, D.; Varki, A.; Le, D.T.; Marth, J.D. Sialyltransferase ST3Gal-IV operates as a dominant modifier of hemostasis by concealing asialoglycoprotein receptor ligands. Proc. Natl. Acad. Sci. USA 2002, 99, 10042–10047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grozovsky, R.; Begonja, A.J.; Liu, K.; Visner, G.; Hartwig, J.H.; Falet, H.; Hoffmeister, K.M. The Ashwell-Morell receptor regulates hepatic thrombopoietin production via JAK2-STAT3 signaling. Nat. Med. 2015, 21, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Grewal, P.K.; Aziz, P.V.; Uchiyama, S.; Rubio, G.R.; Lardone, R.D.; Le, D.; Varki, N.M.; Nizet, V.; Marth, J.D. Inducing host protection in pneumococcal sepsis by preactivation of the Ashwell-Morell receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 20218–20223. [Google Scholar] [CrossRef] [Green Version]
- Grewal, P.K.; Uchiyama, S.; Ditto, D.; Varki, N.; Le, D.T.; Nizet, V.; Marth, J.D. The Ashwell receptor mitigates the lethal coagulopathy of sepsis. Nat. Med. 2008, 14, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Xu, Y.; Chen, W.; Paul, D.S.; Syed, A.K.; Dragovich, M.A.; Liang, X.; Zakas, P.; Berndt, M.C.; Di Paola, J.; et al. Platelet clearance via shear-induced unfolding of a membrane mechanoreceptor. Nat. Commun. 2016, 7, 12863. [Google Scholar] [CrossRef]
- Josefsson, E.C.; Gebhard, H.H.; Stossel, T.P.; Hartwig, J.H.; Hoffmeister, K.M. The macrophage alphaMbeta2 integrin alphaM lectin domain mediates the phagocytosis of chilled platelets. J. Biol. Chem. 2005, 280, 18025–18032. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Fu, J.; Ling, Y.; Yago, T.; McDaniel, J.M.; Song, J.; Bai, X.; Kondo, Y.; Qin, Y.; Hoover, C.; et al. Sialylation on O-glycans protects platelets from clearance by liver Kupffer cells. Proc. Natl. Acad. Sci. USA 2017, 114, 8360–8365. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.E.; Snelling, T.; Smith, D.F.; Drickamer, K. Absence of a human ortholog of rodent Kupffer cell galactose-binding receptor encoded by the CLEC4f gene. Glycobiology 2019, 29, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tang, Y.; Hoover, C.; Kondo, Y.; Huang, D.; Restagno, D.; Shao, B.; Gao, L.; Michael McDaniel, J.; Zhou, M.; et al. Kupffer cell receptor CLEC4F is important for the destruction of desialylated platelets in mice. Cell Death Differ. 2021, 28, 3009–3021. [Google Scholar] [CrossRef] [PubMed]
- Deppermann, C.; Kratofil, R.M.; Peiseler, M.; David, B.A.; Zindel, J.; Castanheira, F.; van der Wal, F.; Carestia, A.; Jenne, C.N.; Marth, J.D.; et al. Macrophage galactose lectin is critical for Kupffer cells to clear aged platelets. J. Exp. Med. 2020, 217, e20190723. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mechanisms of platelet–neutrophil complex formation. Upon endothelial cell disruption, platelets and neutrophils become activated due to exposure to collagen and activated endothelial cells. Platelet activation results in P-selectin exposure and subsequent neutrophil activation via P-selectin–PSGL1 interaction. Neutrophil activation is further enhanced upon platelet granule content release (CXCL4, CXCL7, CXCL1, HMGB1). The synergistic activation of neutrophils by all these agents via Mac-1, PSGL-1, SLC44A2 and CD40 receptors eventually results in platelet–neutrophil complexes and neutrophil DNA-trap secretion—NETosis. NETs contain the proteases cathepsin G, neutrophil elastase and myeloperoxidase, which can both activate platelets and the coagulation cascade. Multiple positive feedback loops are present at all levels of this system and, thus, both platelet and neutrophils become involved in immunothrombosis and subsequent thromboinflammation. Cell activation is shown by yellow lightnings. Dotted lines represent interactions of soluble mediators and receptors. NET—neutrophil extracellular trap, PNC—platelet–neutrophil complex, Plt—platelet, Neu—neutrophil.
Figure 1.
Mechanisms of platelet–neutrophil complex formation. Upon endothelial cell disruption, platelets and neutrophils become activated due to exposure to collagen and activated endothelial cells. Platelet activation results in P-selectin exposure and subsequent neutrophil activation via P-selectin–PSGL1 interaction. Neutrophil activation is further enhanced upon platelet granule content release (CXCL4, CXCL7, CXCL1, HMGB1). The synergistic activation of neutrophils by all these agents via Mac-1, PSGL-1, SLC44A2 and CD40 receptors eventually results in platelet–neutrophil complexes and neutrophil DNA-trap secretion—NETosis. NETs contain the proteases cathepsin G, neutrophil elastase and myeloperoxidase, which can both activate platelets and the coagulation cascade. Multiple positive feedback loops are present at all levels of this system and, thus, both platelet and neutrophils become involved in immunothrombosis and subsequent thromboinflammation. Cell activation is shown by yellow lightnings. Dotted lines represent interactions of soluble mediators and receptors. NET—neutrophil extracellular trap, PNC—platelet–neutrophil complex, Plt—platelet, Neu—neutrophil.

Figure 2.
Platelet–monocyte interactions and platelet–monocyte complex (PMC) formation. Several factors such as bacterial or viral infection (1) can contribute to the activation of both platelets and monocytes (2). Once activated, platelets and monocytes can interact directly through receptors expressed on their surface, and soluble factors released from platelets can further modulate monocyte activity (3). This interaction can lead monocytes to extravasate and differentiate towards macrophages (4a) or alternatively lead to platelet–monocyte complex formation (4b). Cell activation is shown by yellow lightnings. Plt—platelet, Mon—monocyte, PMC—platelet–monocyte complex.
Figure 2.
Platelet–monocyte interactions and platelet–monocyte complex (PMC) formation. Several factors such as bacterial or viral infection (1) can contribute to the activation of both platelets and monocytes (2). Once activated, platelets and monocytes can interact directly through receptors expressed on their surface, and soluble factors released from platelets can further modulate monocyte activity (3). This interaction can lead monocytes to extravasate and differentiate towards macrophages (4a) or alternatively lead to platelet–monocyte complex formation (4b). Cell activation is shown by yellow lightnings. Plt—platelet, Mon—monocyte, PMC—platelet–monocyte complex.


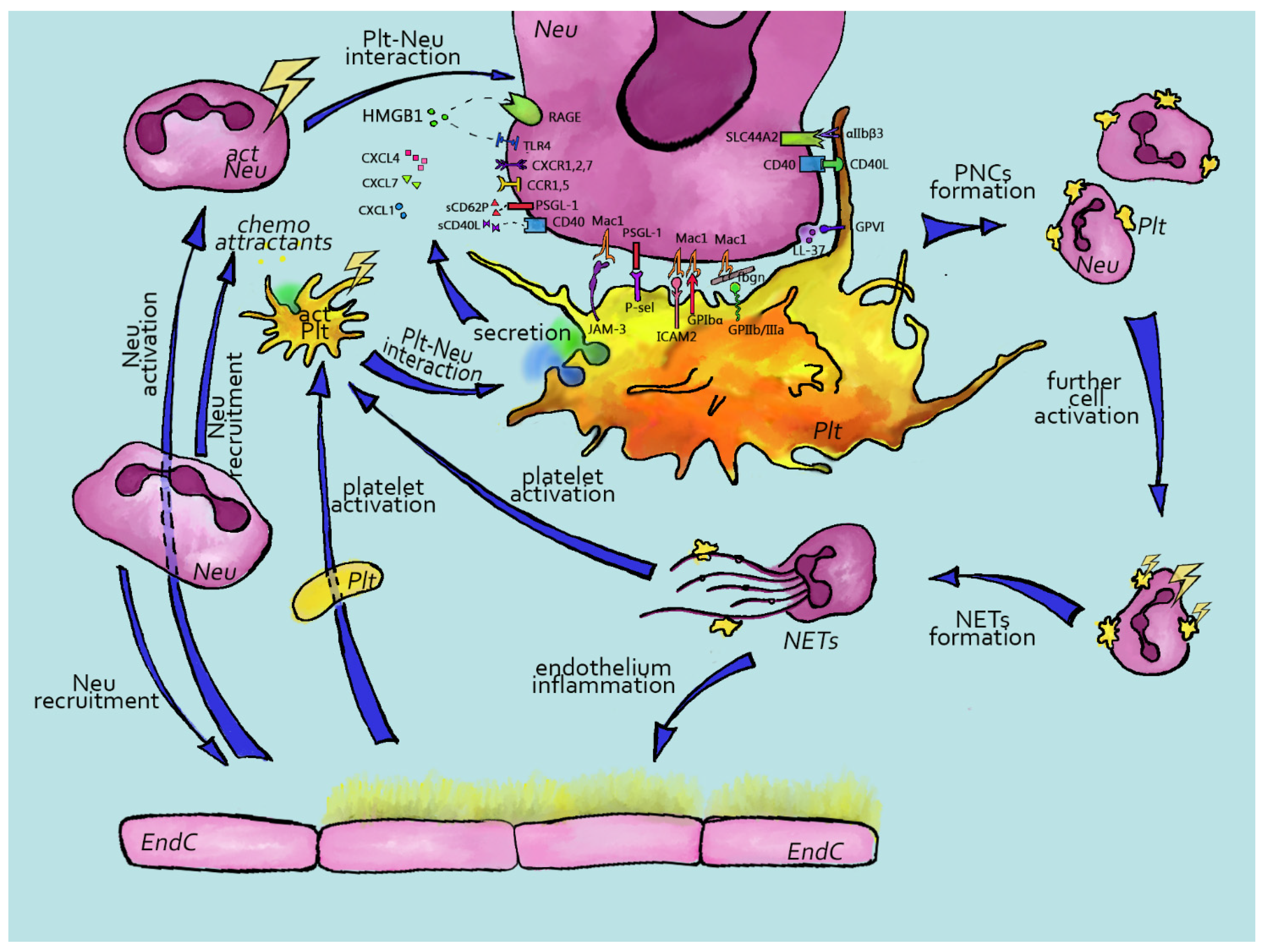{kind=link}
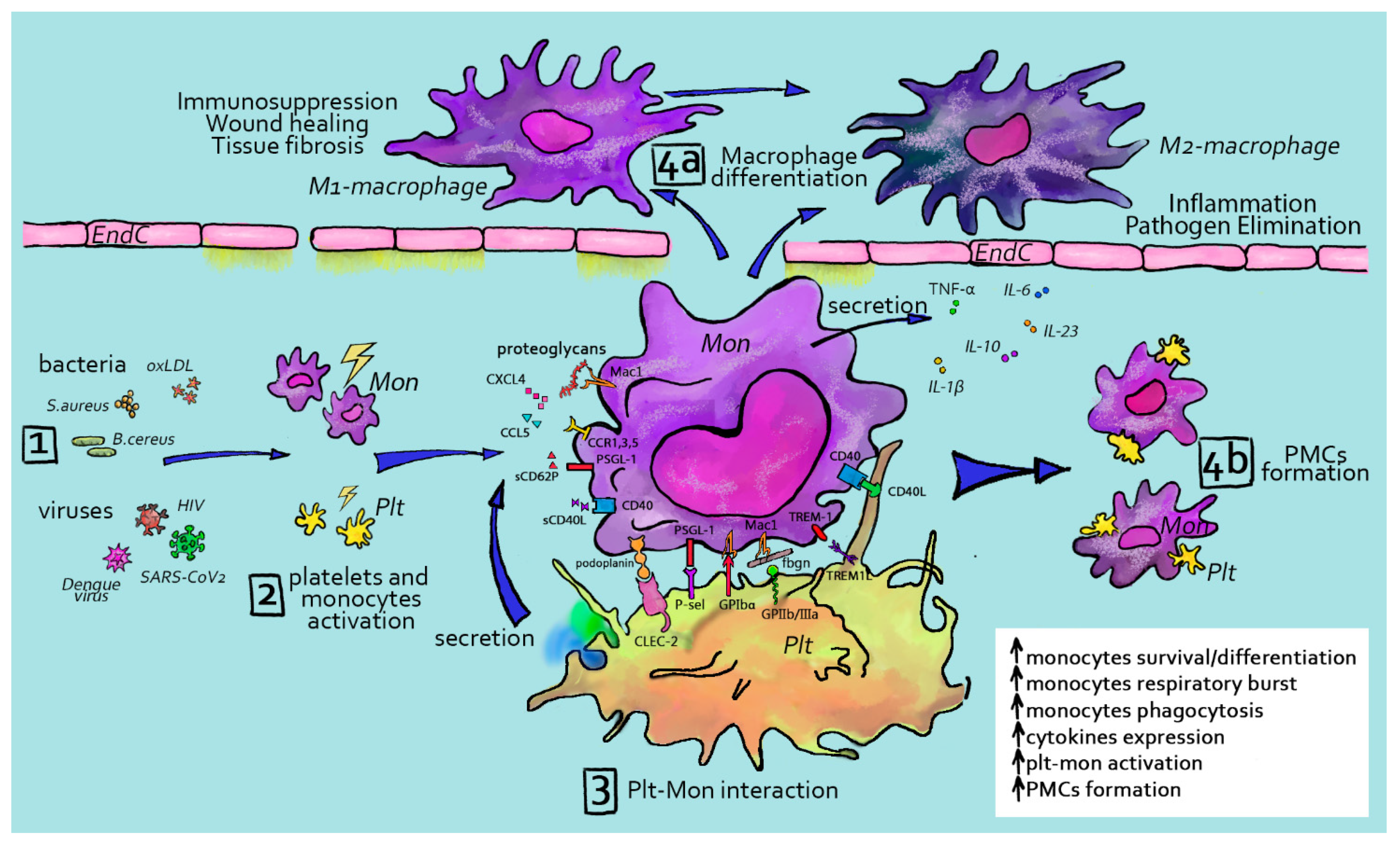{kind=link}
| α-Granules | Dense Granules | |
|---|---|---|
| Structure | Peripheral membrane with an electron-dense nucleoid (chemokines and proteoglycans), an adjacent zone with less electron-dense fibrinogen and a peripheral zone with vWF stored in tubular structures Ovoid 250 nm × 300 nm | Peripherally distributed and electron-dense spherical bodies 150 nm in diameter |
| Content | Membrane proteins: integrins (αIIb, α6, β3), immunoglobulin family receptors (GPVI, Fc receptors, PECAM), leucine-rich family receptors (GPIb-IX-V), tetraspanins, TREM-like transcript-1 (TLT-1), fibrocystin L, CD109, P-selectin and other (CD36, Glut-3) Soluble proteins: platelet factor 4 (PF4), fibrinogen, vWF, factor V, factor XI, factor XIII, plasminogen activator inhibitor-1 (PAI-1), α2-antiplasmin, chemokines, VEGF, endostatin, FGF, EGF, HGF, IGF, TSP-1, PDWHF, antithrombin, tissue factor inhibitor (TFPI), protein S, protease nexin-2 and pro-inflammatory cytokines | Soluble proteins: Bioactive amines (serotonin and histamine), adenine nucleotides (ADP and ATP), Ca2+, Mg2+ and polyphosphates |
| Biogenesis | Multi-vesicular bodies (MVBs) derived from the trans-Golgi network Molecules involved: clathrin, COPII, Adapter proteins (AP-1-3), SNAREs and GTPases | Multi-vesicular bodies (MVBs) derived from the endosomal system Molecules involved: Adapter protein 3 (AP-3) and biogenesis of lysosome-related organelles (BLOC 1-3) |
| Protein sorting | Proteins produced in ER of MKs and sorted via trans-Golgi Sorting varies between molecules Mechanisms involved: Signal sequence (e.g., Chemokines), glycosaminoglycans (e.g., soluble proteins), aggregation of protein monomers (e.g., vWF), sorting receptors, endocytosis (e.g., plasma proteins such as fibrinogen) and pinocytosis (e.g., plasma proteins such as albumin or immunoglobulin) | Transport of molecules via membrane pumps (e.g., vesicular nucleotide transporter (VNUT) and multidrug resistance-associated protein 4 (MRP4)) |
| Transport | Move along microtubules | Near the plasma membrane |
| Stimulation and release | Fusing with the plasma membrane by SNAREs (vesicular SNAREs and target SNAREs) Regulated by Sec1/Munc, CDCrel-1, ATPases, SNAPs and Rab proteins | Fusing with the plasma membrane by SNAREs (vesicular SNAREs and target SNAREs) Regulated by Sec1/Munc, CDCrel-1, ATPases, SNAPs and Rab proteins |
| Function | adhesion and coagulation: fibrinogen, vWF, GPIbα-IX-V, integrin αIIbβ3, GPVI, coagulation factors (V, XI, XIII), precursors of thrombin, prothrombin and kininogen as well as inhibitory proteases (plasminogen activator inhibitor-1 (PAI-1) and α-antiplasmin hemostasis: antithrombin, C1-inhibitor, tissue factor pathway inhibitor (TFPI), protein S, protease nexin-2 and proteinase (plasmin and plasminogen) inflammation: P-selectin, pro-inflammatory factors and chemokines (platelet factor 4 (PF-4), CD40, CD154, CXCL1, CXCL4, CXCL5, CXCL7, CXCL8, CXCL12, CCL2, CCL3 and CCL5) antimicrobial host defense: CXCL4, thymosin-β4, CXCL7, CCL5 and complement factors (C1q, C3 and C4 precursors, C8 and C9) angiogenesis: vascular endothelium growth factor (VEGF), platelet-derived growth factor (PDGF), fibroblast growth factor (FGF), epidermal growth factor (EGF), hepatocyte growth factor (HGF) and insulin-like growth factor (IGF) inhibitors of angiogenesis: thrombospondin-1 (TSP-1) and CXCL-4 wound healing: platelet derived wound healing factor (PDWHF) malignancy: VEGF and adhesive proteins such as P-selectin, vitronectin and fibronectin | Potentiate platelet activation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mandel, J.; Casari, M.; Stepanyan, M.; Martyanov, A.; Deppermann, C. Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation. Int. J. Mol. Sci. 2022, 23, 3868. https://doi.org/10.3390/ijms23073868
AMA Style
Mandel J, Casari M, Stepanyan M, Martyanov A, Deppermann C. Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation. International Journal of Molecular Sciences. 2022; 23(7):3868. https://doi.org/10.3390/ijms23073868
Chicago/Turabian StyleMandel, Jonathan, Martina Casari, Maria Stepanyan, Alexey Martyanov, and Carsten Deppermann. 2022. "Beyond Hemostasis: Platelet Innate Immune Interactions and Thromboinflammation" International Journal of Molecular Sciences 23, no. 7: 3868. https://doi.org/10.3390/ijms23073868
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.