
CRPC Membrane-Camouflaged, Biomimetic Nanosystem for Overcoming Castration-Resistant Prostate Cancer by Cellular Vehicle-Aided Tumor Targeting

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
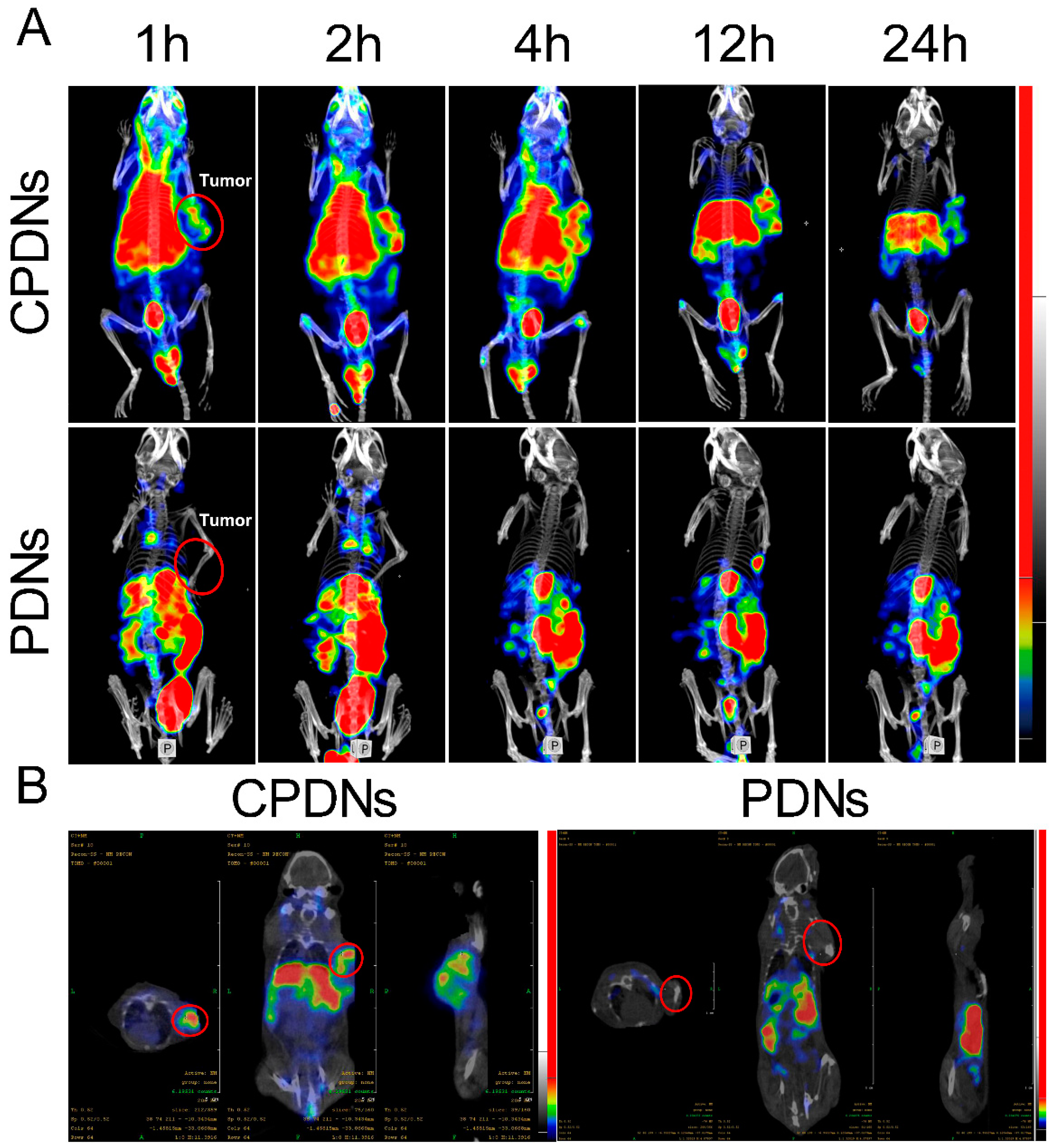
2. Results and Discussion
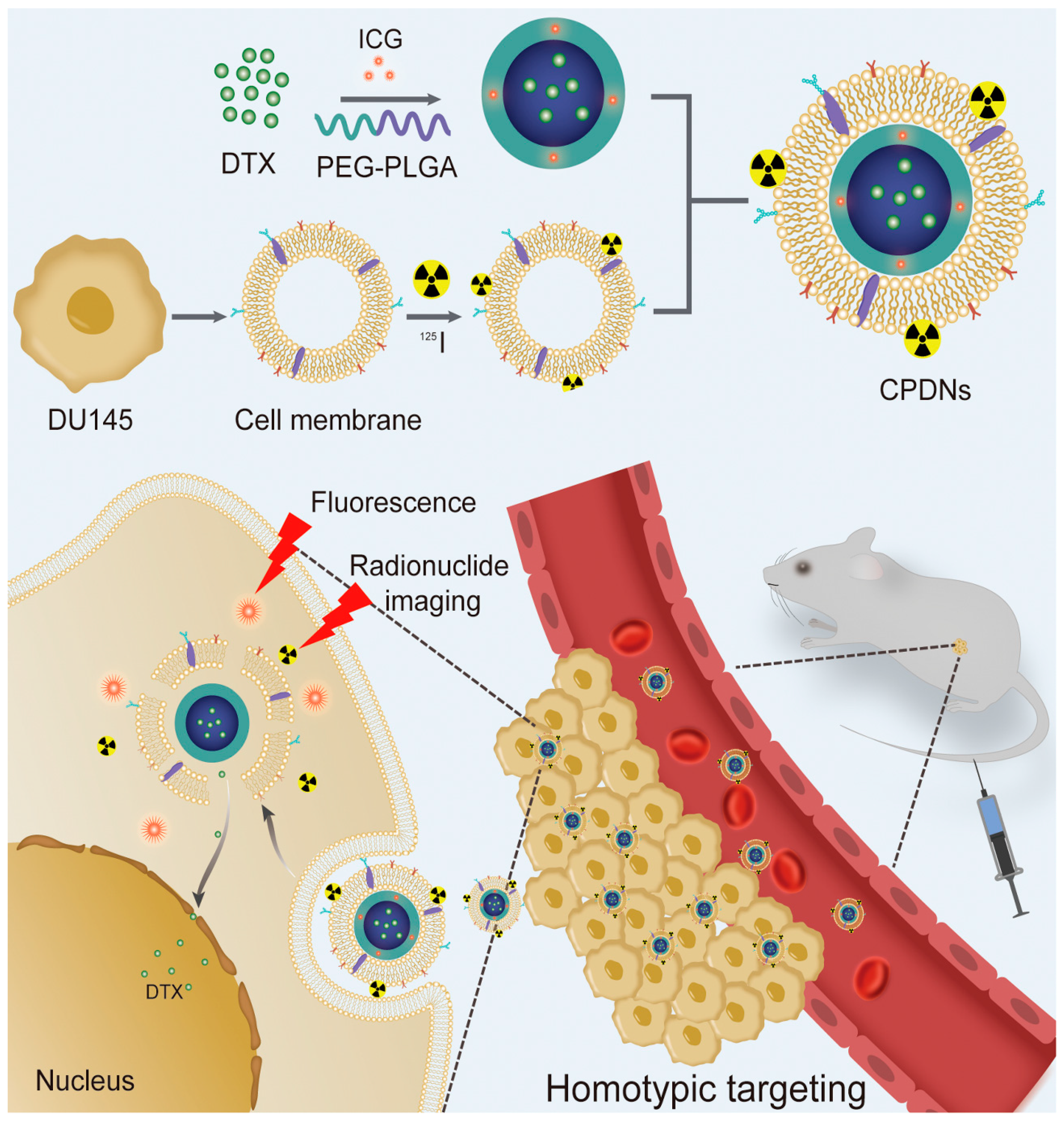
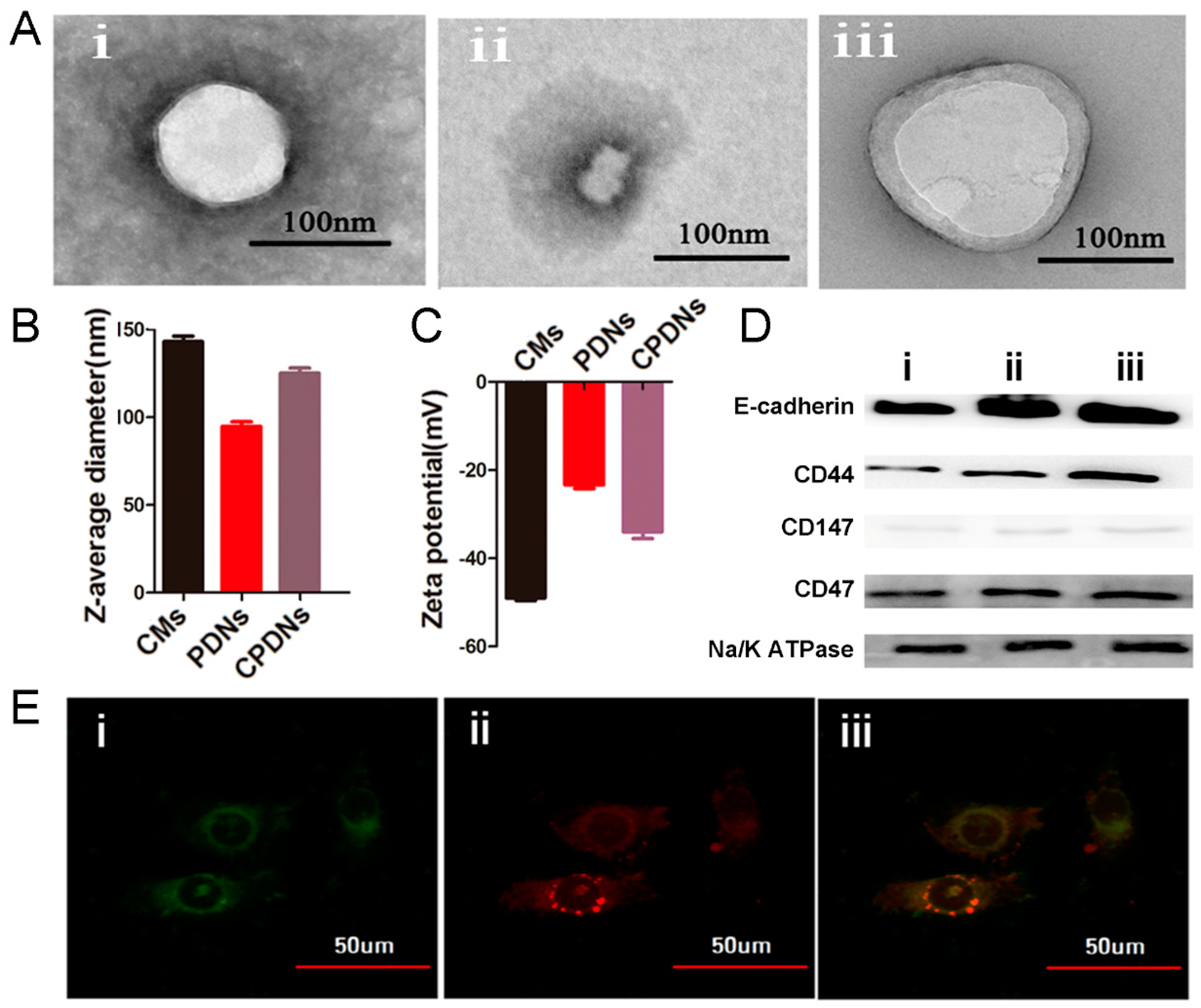
2.1. Preparation and Characterization of DU145 CPDNs
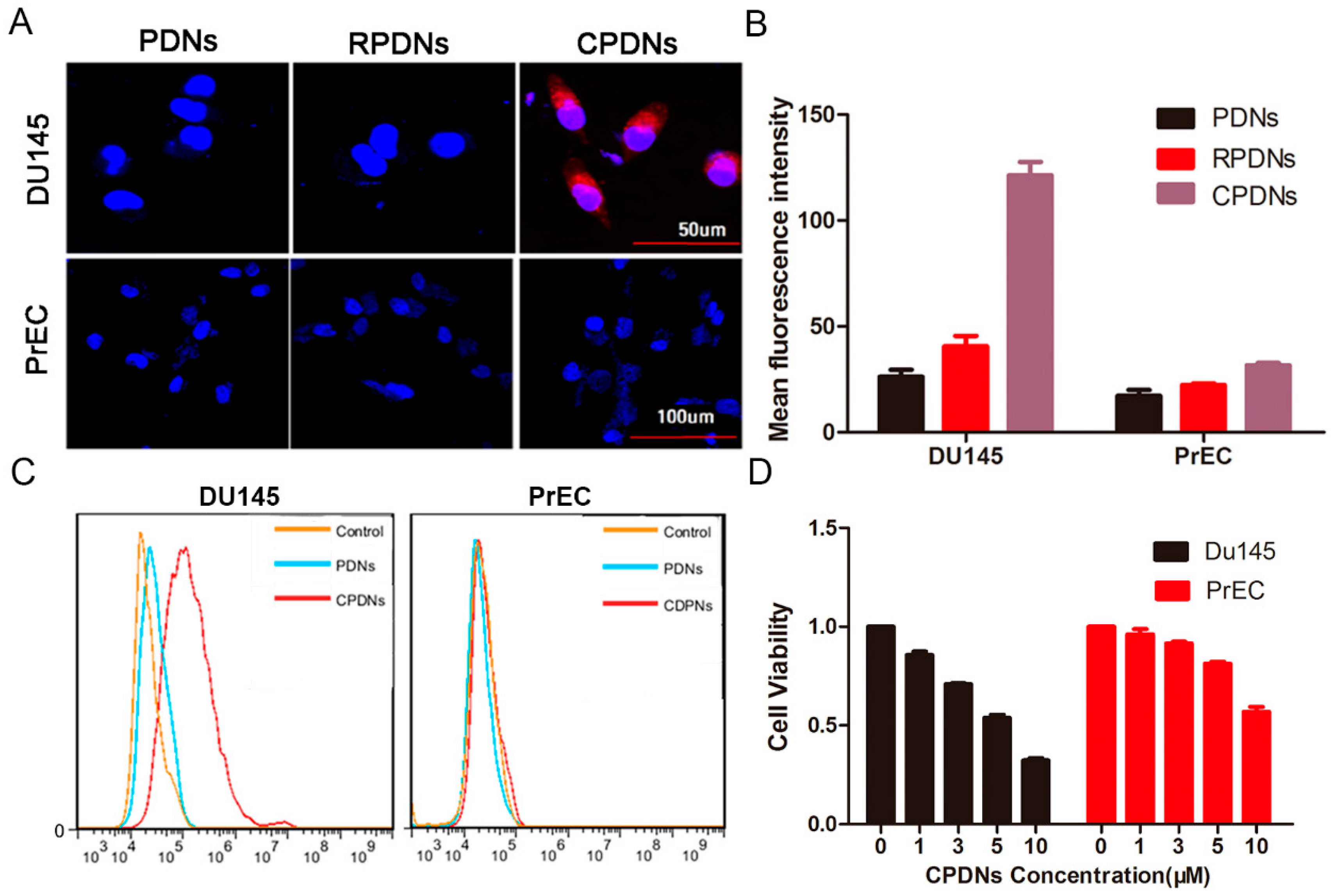
2.2. Cellular Uptake of CPDNs
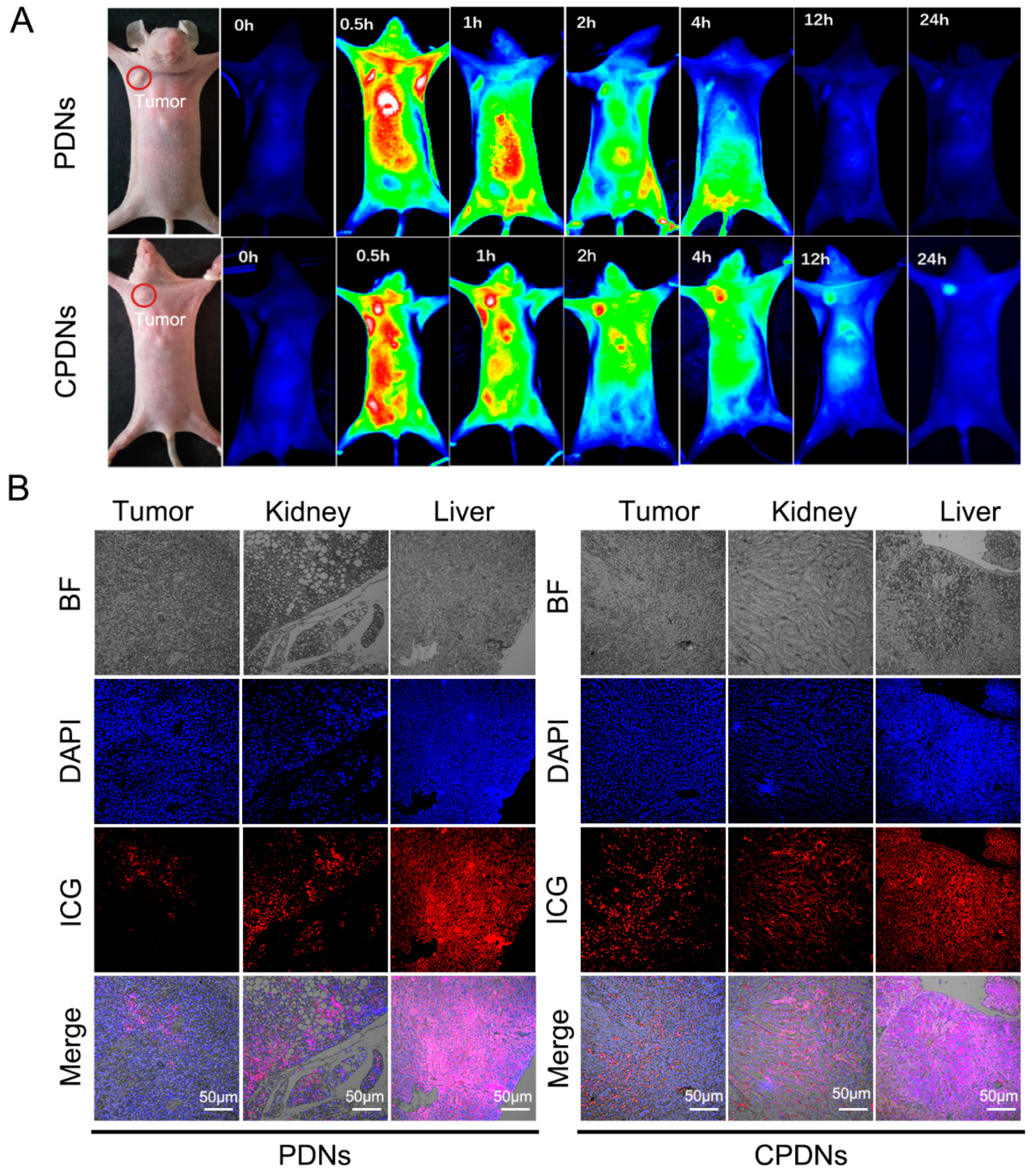
2.3. Biocompatibility Effect In Vivo
Treatment of CRPC
3. Conclusions
4. Experimental Section
4.1. Materials
4.2. Preparation of DU145 Cell Membranes
4.3. Preparation of PDNs
4.4. CPDN Synthesis
4.5. Characterization of CPDNs
4.6. Determination of DTX Encapsulation Rate in CPDNs
4.7. Release Percentage of DTX in CPDNs
4.8. Protein Identification of CPDNs
4.9. Intracellular Localization of PDNs and CPDNs
4.10. Stability Test
4.11. Qualitative Observation of In Vitro Cellular Uptake
4.12. Quantitative Detection of In Vitro Cellular Uptake
4.13. Cytotoxicity Test
4.14. Animal Model and Tumor Formation Assay
4.15. In Vivo Tumor Imaging
4.16. SPECT-CT
4.17. Immunohistochemical Staining (IHC)
4.18. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasool, R.U.; Natesan, R.; Deng, Q.; Aras, S.; Lal, P.; Sander Effron, S.; Mitchell-Velasquez, E.; Posimo, J.M.; Carskadon, S.; Baca, S.C.; et al. CDK7 Inhibition Suppresses Castration-Resistant Prostate Cancer through MED1 Inactivation. Cancer Discov. 2019, 9, 1538–1555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Sun, L.; Hao, T.; Zhang, B.; Chen, X.; Li, H.; Zhang, Z.; Zhu, S.; Quan, C.; Niu, Y.; et al. Restoration of FKBP51 protein promotes the progression of castration resistant prostate cancer. Ann. Transl. Med. 2019, 7, 729. [Google Scholar] [CrossRef] [PubMed]
- James, A.; Vincent, B.; Sivadas, A.; Pavithran, K. A Study on the Clinical Outcome of Abiraterone Acetate in Castration Resistant Prostate Cancer Patients. Int. J. Hematol. Oncol. Stem Cell Res. 2018, 12, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men with Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef] [PubMed]
- Saad, F.; Aprikian, A.; Finelli, A.; Fleshner, N.E.; Gleave, M.; Kapoor, A.; Niazi, T.; North, S.A.; Pouliot, F.; Rendon, R.A.; et al. 2019 Canadian Urological Association (CUA)-Canadian Uro Oncology Group (CUOG) guideline: Management of castration-resistant prostate cancer (CRPC). Can. Urol. Assoc. J. 2019, 13, 307–314. [Google Scholar] [CrossRef] [Green Version]
- Mohler, J.L.; Antonarakis, E.S.; Armstrong, A.J.; D’Amico, A.V.; Davis, B.J.; Dorff, T.; Eastham, J.A.; Enke, C.A.; Farrington, T.A.; Higano, C.S.; et al. Prostate Cancer, Version 2.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2019, 17, 479–505. [Google Scholar] [CrossRef] [Green Version]
- Nagao, K.; Matsuyama, H. Docetaxel chemotherapy against CRPC. Jpn. J. Clin. Med. 2016, 74, 619–623. [Google Scholar]
- Park, J.C.; Eisenberger, M. Chemotherapy in Prostate Cancer beyond Metastatic CRPC. Oncology 2015, 29, 782–790. [Google Scholar]
- Awasthi, R.; Roseblade, A.; Hansbro, P.M.; Rathbone, M.J.; Dua, K.; Bebawy, M. Nanoparticles in Cancer Treatment: Opportunities and Obstacles. Curr. Drug Targets 2018, 19, 1696–1709. [Google Scholar] [CrossRef]
- Yang, L.; Li, W.; Huang, Y.; Zhou, Y.; Chen, T. Rational Design of Cancer-Targeted Benzoselenadiazole by RGD Peptide Functionalization for Cancer Theranostics. Macromol. Rapid Commun. 2015, 36, 1559–1565. [Google Scholar] [CrossRef] [PubMed]
- Lei, B.; Wang, M.; Jiang, Z.; Qi, W.; Su, R.; He, Z. Constructing Redox-Responsive Metal-Organic Framework Nanocarriers for Anticancer Drug Delivery. ACS Appl. Mater. Interfaces 2018, 10, 16698–16706. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhao, K.; Bu, W.; Ni, D.; Liu, Y.; Feng, J.; Shi, J. Marriage of scintillator and semiconductor for synchronous radiotherapy and deep photodynamic therapy with diminished oxygen dependence. Angew. Chem. Int. Ed. Engl. 2015, 54, 1770–1774. [Google Scholar] [CrossRef] [PubMed]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H. Toward a full understanding of the EPR effect in primary and metastatic tumors as well as issues related to its heterogeneity. Adv. Drug Deliv. Rev. 2015, 91, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Park, K. Questions on the role of the EPR effect in tumor targeting. J. Control. Release 2013, 172, 391. [Google Scholar] [CrossRef]
- Du, J.Z.; Du, X.J.; Mao, C.Q.; Wang, J. Tailor-made dual pH-sensitive polymer-doxorubicin nanoparticles for efficient anticancer drug delivery. J. Am. Chem. Soc. 2011, 133, 17560–17563. [Google Scholar] [CrossRef]
- Shen, Z.; Ye, H.; Kroger, M.; Li, Y. Aggregation of polyethylene glycol polymers suppresses receptor-mediated endocytosis of PEGylated liposomes. Nanoscale 2018, 10, 4545–4560. [Google Scholar] [CrossRef] [Green Version]
- Katragadda, U.; Teng, Q.; Rayaprolu, B.M.; Chandran, T.; Tan, C. Multi-drug delivery to tumor cells via micellar nanocarriers. Int. J. Pharm. 2011, 419, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Palivan, C.G.; Goers, R.; Najer, A.; Zhang, X.; Car, A.; Meier, W. Bioinspired polymer vesicles and membranes for biological and medical applications. Chem. Soc. Rev. 2016, 45, 377–411. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Cai, Q.; Tang, Y.; Xu, Y.; Wang, Q.; Li, T.; Xu, H.; Wang, S.; Fan, K.; Liu, Z.; et al. PEGylated Lipid bilayer coated mesoporous silica nanoparticles for co-delivery of paclitaxel and curcumin: Design, characterization and its cytotoxic effect. Int. J. Pharm. 2018, 536, 272–282. [Google Scholar] [CrossRef]
- Prasad, L.K.; O’Mary, H.; Cui, Z. Nanomedicine delivers promising treatments for rheumatoid arthritis. Nanomedicine 2015, 10, 2063–2074. [Google Scholar] [CrossRef] [Green Version]
- Raza, F.; Zafar, H.; Zhu, Y.; Ren, Y.; Ullah, A.; Khan, A.U.; He, X.; Han, H.; Aquib, M.; Boakye-Yiadom, K.O.; et al. A Review on Recent Advances in Stabilizing Peptides/Proteins upon Fabrication in Hydrogels from Biodegradable Polymers. Pharmaceutics 2018, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.; Yang, C.; Liu, J.; Wang, W.; Guo, S.; Li, J.; Sun, Y.; Dong, H.; Deng, L.; Zhang, J.; et al. Improving the oral delivery efficiency of anticancer drugs by chitosan coated polycaprolactone-grafted hyaluronic acid nanoparticles. J. Mater. Chem. B 2014, 2, 4021–4033. [Google Scholar] [CrossRef]
- Wei, J.; Sun, J.; Liu, Y. Enhanced targeting of prostate cancer-initiating cells by salinomycin-encapsulated lipid-PLGA nanoparticles linked with CD44 antibodies. Oncol. Lett. 2019, 17, 4024–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, L. Prostate cancer: Radiotherapy induces epigenetic changes. Nat. Rev. Urol. 2016, 13, 241. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Qin, Y.; Hao, F.; Li, Q.; Zhang, W.; Zhao, C.; Chen, S.; Zhao, L.; Wang, L.; Cai, J. CD147 modulates androgen receptor activity through the Akt/Gsk-3beta/beta-catenin/AR pathway in prostate cancer cells. Oncol. Lett. 2016, 12, 1124–1128. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Marin, D.; Jarvis, C.; Nelius, T.; de Riese, W.; Volpert, O.V.; Filleur, S. PEDF increases the tumoricidal activity of macrophages towards prostate cancer cells in vitro. PLoS ONE 2017, 12, e0174968. [Google Scholar] [CrossRef] [Green Version]
- Murata, Y.; Saito, Y.; Kotani, T.; Matozaki, T. CD47-signal regulatory protein alpha signaling system and its application to cancer immunotherapy. Cancer Sci. 2018, 109, 2349–2357. [Google Scholar] [CrossRef]
- Elliott, B.; Millena, A.C.; Matyunina, L.; Zhang, M.; Zou, J.; Wang, G.; Zhang, Q.; Bowen, N.; Eaton, V.; Webb, G.; et al. Essential role of JunD in cell proliferation is mediated via MYC signaling in prostate cancer cells. Cancer Lett. 2019, 448, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xie, R.; Yodsanit, N.; Ye, M.; Wang, Y.; Gong, S. Biomimetic fibrin-targeted and H2O2-responsive nanocarriers for thrombus therapy. Nano Today 2020, 35, 100986. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Ran, D.; Lu, L.; Zhan, C.; Ruan, H.; Hu, X.; Xie, C.; Jiang, K.; Li, J.; Zhou, J.; et al. Ligand-Modified Cell Membrane Enables the Targeted Delivery of Drug Nanocrystals to Glioma. Acs Nano 2019, 13, 5591–5601. [Google Scholar] [CrossRef] [PubMed]
- Fang, R.H.; Kroll, A.V.; Gao, W.; Zhang, L. Cell Membrane Coating Nanotechnology. Adv. Mater. 2018, 30, 1706759. [Google Scholar] [CrossRef]
- Li, J.; Zhen, X.; Lyu, Y.; Jiang, Y.; Huang, J.; Pu, K. Cell Membrane Coated Semiconducting Polymer Nanoparticles for Enhanced Multimodal Cancer Phototheranostics. Acs Nano 2018, 12, 8520–8530. [Google Scholar] [CrossRef]
- Hao, Y.; Zhang, B.; Zheng, C.; Ji, R.; Ren, X.; Guo, F.; Sun, S.; Shi, J.; Zhang, H.; Zhang, Z.; et al. The tumor-targeting core-shell structured DTX-loaded PLGA@Au nanoparticles for chemo-photothermal therapy and X-ray imaging. J. Control. Release 2015, 220, 545–555. [Google Scholar] [CrossRef]
- Gao, Y.; Ren, F.; Ding, B.; Sun, N.; Liu, X.; Ding, X.; Gao, S. A thermo-sensitive PLGA-PEG-PLGA hydrogel for sustained release of docetaxel. J. Drug Target. 2011, 19, 516–527. [Google Scholar] [CrossRef]
- Ghinea, N.; Robin, B.; Pichon, C.; Leclere, R.; Nicolas, A.; Chnecker, C.; Cote, J.F.; Guillonneau, B.; Radu, A. Vasa nervorum angiogenesis in prostate cancer with perineural invasion. Prostate 2019, 79, 640–646. [Google Scholar] [CrossRef]
- Xia, Q.; Zhang, Y.; Li, Z.; Hou, X.; Feng, N. Red blood cell membrane-camouflaged nanoparticles: A novel drug delivery system for antitumor application. Acta Pharm. Sin. B 2019, 9, 675–689. [Google Scholar] [CrossRef]
- Feng, Q.; Yang, X.; Hao, Y.; Wang, N.; Feng, X.; Hou, L.; Zhang, Z. Cancer Cell Membrane-Biomimetic Nanoplatform for Enhanced Sonodynamic Therapy on Breast Cancer via Autophagy Regulation Strategy. ACS Appl. Mater. Interfaces 2019, 11, 32729–32738. [Google Scholar] [CrossRef]
- Shao, J.; Pijpers, I.A.B.; Cao, S.; Williams, D.S.; Yan, X.; Li, J.; Abdelmohsen, L.; van Hest, J.C.M. Biomorphic Engineering of Multifunctional Polylactide Stomatocytes toward Therapeutic Nano-Red Blood Cells. Adv. Sci. 2019, 6, 1801678. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.; Deng, G.; Sun, Z.; Peng, X.; Li, J.; Gong, P.; Zhang, P.; Cai, L. Scaffolds biomimicking macrophages for a glioblastoma NIR-Ib imaging guided photothermal therapeutic strategy by crossing Blood-Brain Barrier. Biomaterials 2019, 211, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Rafiei, P.; Haddadi, A. Docetaxel-loaded PLGA and PLGA-PEG nanoparticles for intravenous application: Pharmacokinetics and biodistribution profile. Int. J. Nanomed. 2017, 12, 935–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.M.; Zhang, L.; Aryal, S.; Cheung, C.; Fang, R.H.; Zhang, L. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. USA 2011, 108, 10980–10985. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, K.; Li, Z.; Hu, Q.; Sun, J.; Chen, M. CRPC Membrane-Camouflaged, Biomimetic Nanosystem for Overcoming Castration-Resistant Prostate Cancer by Cellular Vehicle-Aided Tumor Targeting. Int. J. Mol. Sci. 2022, 23, 3623. https://doi.org/10.3390/ijms23073623
Lu K, Li Z, Hu Q, Sun J, Chen M. CRPC Membrane-Camouflaged, Biomimetic Nanosystem for Overcoming Castration-Resistant Prostate Cancer by Cellular Vehicle-Aided Tumor Targeting. International Journal of Molecular Sciences. 2022; 23(7):3623. https://doi.org/10.3390/ijms23073623
Chicago/Turabian StyleLu, Kai, Zheng Li, Qiang Hu, Jianfei Sun, and Ming Chen. 2022. "CRPC Membrane-Camouflaged, Biomimetic Nanosystem for Overcoming Castration-Resistant Prostate Cancer by Cellular Vehicle-Aided Tumor Targeting" International Journal of Molecular Sciences 23, no. 7: 3623. https://doi.org/10.3390/ijms23073623