
Understanding the Role of LFA-1 in Leukocyte Adhesion Deficiency Type I (LAD I): Moving towards Inflammation?


Abstract
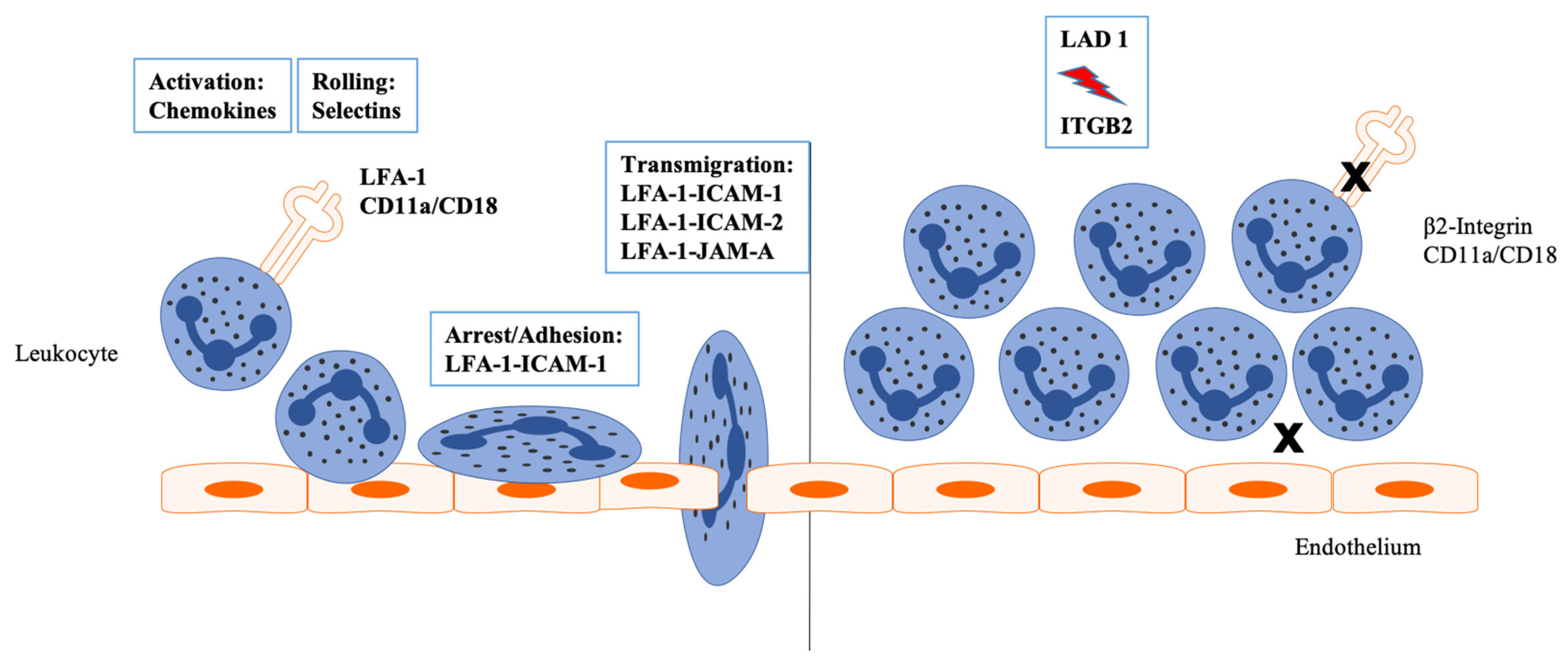
:1. LFA-1 and Primary Immunodeficiency Syndromes
2. Structure of LFA-1 and Important Mutations in LAD I
3. LFA-1 and Its Interacting Partners
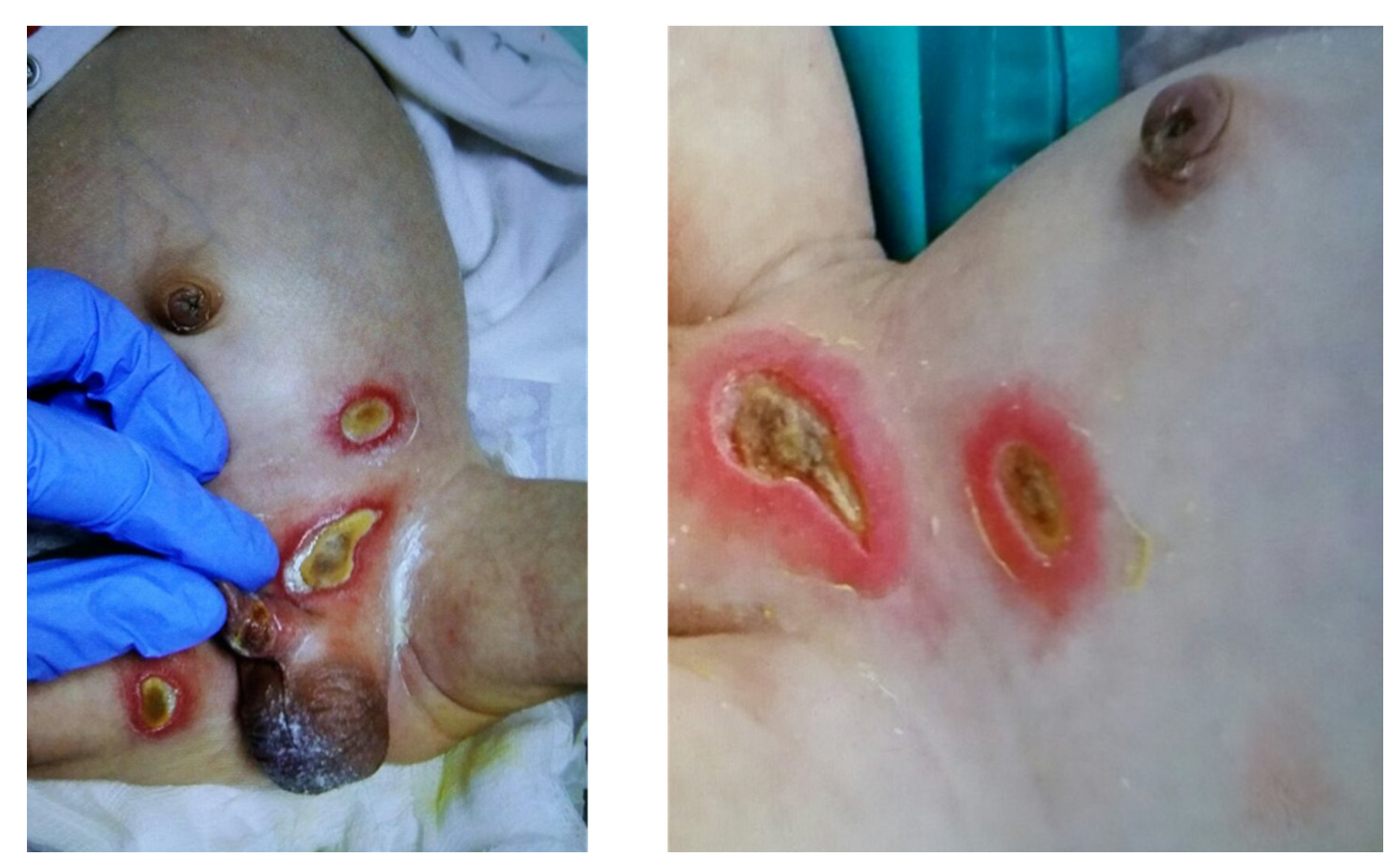
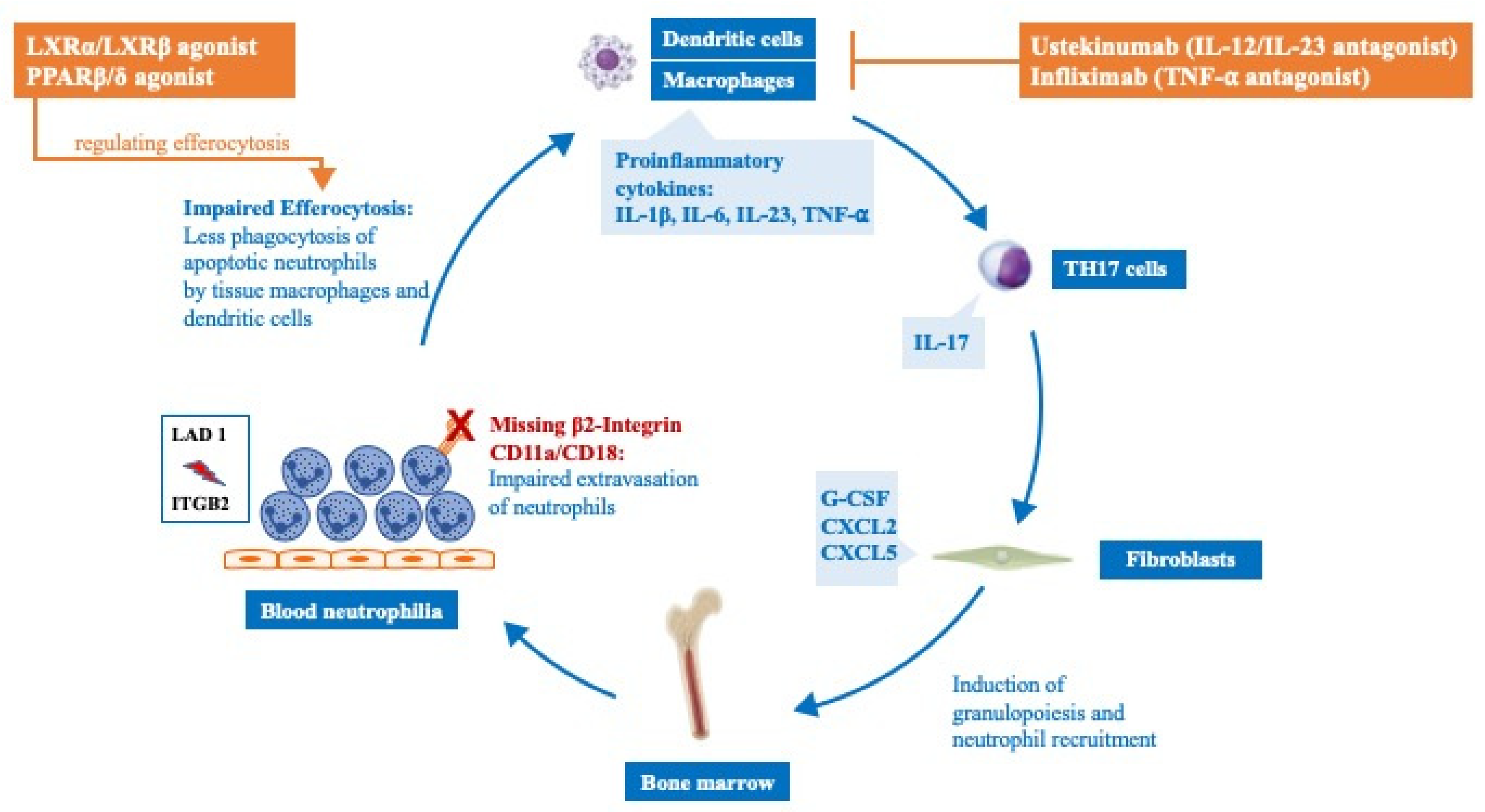
4. LFA-I and Neutrophil Function beyond the Antimicrobial Defense
5. LFA-I and the IL-12/IL-23 Pathway
6. Druggable Targets in LAD I
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Etzioni, A. Genetic etiologies of leukocyte adhesion defects. Curr. Opin. Immunol. 2009, 21, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.C.; Schmalsteig, F.C.; Finegold, M.J.; Hughes, B.J.; Rothlein, R.; Miller, L.J.; Kohl, S.; Tosi, M.F.; Jacobs, R.L.; Waldrop, T.C. The severe and moderate phenotypes of heritable Mac-1, LFA-1 deficiency: Their quantitative definition and relation to leukocyte dysfunction and clinical features. J. Infect. Dis. 1985, 152, 668–689. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-M. The leucocyte β2 (CD18) integrins: The structure, functional regulation and signalling properties. Biosci. Rep. 2012, 32, 241–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishimoto, T.K.; Hollander, N.; Roberts, T.M.; Anderson, D.C.; Springer, T.A. Heterogeneous mutations in the beta subunit common to the LFA-1, Mac-1, and p150,95 glycoproteins cause leukocyte adhesion deficiency. Cell 1987, 50, 193–202. [Google Scholar] [CrossRef]
- Bailly, P.; Tontti, E.; Hermand, P.; Cartron, J.P.; Gahmberg, C.G. The red cell LW blood group protein is an intercellular adhesion molecule which binds to CD11/CD18 leukocyte integrins. Eur. J. Immunol. 1995, 25, 3316–3320. [Google Scholar] [CrossRef]
- Mizuno, T.; Yoshihara, Y.; Inazawa, J.; Kagamiyama, H.; Mori, K. cDNA cloning and chromosomal localization of the human telencephalin and its distinctive interaction with lymphocyte function-associated antigen-1. J. Biol. Chem. 1997, 272, 1156–1163. [Google Scholar] [CrossRef] [Green Version]
- Wojcikiewicz, E.P.; Koenen, R.R.; Fraemohs, L.; Minkiewicz, J.; Azad, H.; Weber, C.; Moy, V.T. LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Biophys. J. 2009, 96, 285–293. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Yang, H.; Wang, M.; Lü, S.; Zhang, Y.; Long, M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. MBoC 2018, 29, 408–418. [Google Scholar] [CrossRef]
- Huo, Y.; Hafezi-Moghadam, A.; Ley, K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ. Res. 2000, 87, 153–159. [Google Scholar] [CrossRef] [Green Version]
- Singbartl, K.; Thatte, J.; Smith, M.L.; Wethmar, K.; Day, K.; Ley, K. A CD2-green fluorescence protein-transgenic mouse reveals very late antigen-4-dependent CD8+ lymphocyte rolling in inflamed venules. J. Immunol. 2001, 166, 7520–7526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, A.R.; Harvey, B.A.; Leonard, J.; Greenwood, M.C.; Wood, C.B.; Soothill, J.F. Delayed separation of the umbilical cord, widespread infections, and defective neutrophil mobility. Lancet 1979, 1, 1099–1101. [Google Scholar] [CrossRef]
- Moutsopoulos, N.M.; Konkel, J.; Sarmadi, M.; Eskan, M.A.; Wild, T.; Dutzan, N.; Abusleme, L.; Zenobia, C.; Hosur, K.B.; Abe, T.; et al. Defective Neutrophil Recruitment in Leukocyte Adhesion Deficiency Type I Disease Causes Local IL-17-Driven Inflammatory Bone Loss. Sci. Transl. Med. 2014, 6, ra22940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moutsopoulos, N.M.; Zerbe, C.S.; Wild, T.; Dutzan, N.; Brenchley, L.; DiPasquale, G.; Uzel, G.; Axelrod, K.C.; Lisco, A.; Notarangelo, L.D.; et al. Interleukin-12 and Interleukin-23 Blockade in Leukocyte Adhesion Deficiency Type 1. N. Engl. J. Med. 2017, 376, 1141–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novoa, E.A.; Kasbekar, S.; Thrasher, A.J.; Kohn, D.B.; Sevilla, J.; Nguyen, T.; Schwartz, J.D.; Bueren, J.A. Leukocyte adhesion deficiency-I: A comprehensive review of all published cases. J. Allergy Clin. Immunol. Pract. 2018, 6, 1418–1420.e10. [Google Scholar] [CrossRef] [PubMed]
- Kohn, D.B.; Rao, G.R.; Almarza, E.; Terrazas, D.; Nicoletti, E.; Fernandes, A.; Kuo, C. A Phase 1/2 Study of Lentiviral-Mediated Ex-Vivo Gene Therapy for Pediatric Patients with Severe Leukocyte Adhesion Deficiency-I (LAD-I): Results from Phase 1. Blood 2020, 136, 2932. [Google Scholar] [CrossRef]
- Malý, P.; Thall, A.; Petryniak, B.; Rogers, C.E.; Smith, P.L.; Marks, R.M.; Kelly, R.J.; Gersten, K.M.; Cheng, G.; Saunders, T.L.; et al. The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell 1996, 86, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, A.; Ma, S.; Peired, A.J.; Weiss, L.A.; Cunningham-Rundles, C.; Frenette, P.S. Insights into leukocyte adhesion deficiency type 2 from a novel mutation in the GDP-fucose transporter gene. Blood 2003, 101, 1705–1712. [Google Scholar] [CrossRef] [Green Version]
- Etzioni, A.; Frydman, M.; Pollack, S.; Avidor, I.; Phillips, M.L.; Paulson, J.C.; Gershoni-Baruch, R. Recurrent Severe Infections Caused by a Novel Leukocyte Adhesion Deficiency. N. Engl. J. Med. 1992, 327, 1789–1792. [Google Scholar] [CrossRef]
- Moser, M.; Nieswandt, B.; Ussar, S.; Pozgajova, M.; Fässler, R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat. Med. 2008, 14, 325–330. [Google Scholar] [CrossRef]
- Manevich-Mendelson, E.; Feigelson, S.W.; Pasvolsky, R.; Aker, M.; Grabovsky, V.; Shulman, Z.; Kilic, S.S.; Rosenthal-Allieri, M.A.; Ben-Dor, S.; Mory, A.; et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood 2009, 114, 2344–2353. [Google Scholar] [CrossRef] [PubMed]
- Robert, P.; Canault, M.; Farnarier, C.; Nurden, A.; Grosdidier, C.; Barlogis, V.; Bongrand, P.; Pierres, A.; Chambost, H.; Alessi, M.-C. A Novel Leukocyte Adhesion Deficiency III Variant: Kindlin-3 Deficiency Results in Integrin- and Nonintegrin-Related Defects in Different Steps of Leukocyte Adhesion. J. Immunol. 2011, 186, 5273–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essa, M.F.; Elbashir, E.; Alroqi, F.; Mohammed, R.; Alsultan, A. Successful hematopoietic stem cell transplant in leukocyte adhesion deficiency type III presenting primarily as malignant infantile osteopetrosis. Clin. Immunol. 2020, 213, 108365. [Google Scholar] [CrossRef] [PubMed]
- Sorio, C.; Montresor, A.; Bolomini-Vittori, M.; Caldrer, S.; Rossi, B.; Dusi, S.; Angiari, S.; Johansson, J.E.; Vezzalini, M.; Leal, T.; et al. Mutations of Cystic Fibrosis Transmembrane Conductance Regulator Gene Cause a Monocyte-Selective Adhesion Deficiency. Am. J. Respir. Crit. Care Med. 2016, 193, 1123–1133. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Ley, K. Leukocyte Adhesion Deficiency IV. Monocyte Integrin Activation Deficiency in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 1075–1077. [Google Scholar] [CrossRef] [Green Version]
- Bakhtiar, S.; Salzmann-Manrique, E.; Blok, H.-J.; Eikema, D.-J.; Hazelaar, S.; Ayas, M.; Toren, A.; Goldstein, G.; Moshous, D.; Locatelli, F.; et al. Allogeneic hematopoietic stem cell transplantation in leukocyte adhesion deficiency type I and III. Blood Adv. 2021, 5, 262–273. [Google Scholar]
- Hynes, R.O. Integrins: Bidirectional, allosteric signaling machines. Cell 2002, 110, 673–687. [Google Scholar] [CrossRef] [Green Version]
- Lefort, C.T.; Ley, K. Neutrophil arrest by LFA-1 activation. Front Immunol. 2012, 3, 157. [Google Scholar] [CrossRef] [Green Version]
- Mould, A.P.; Humphries, M.J. Regulation of integrin function through conformational complexity: Not simply a knee-jerk reaction? Curr. Opin. Cell Biol. 2004, 16, 544–551. [Google Scholar] [CrossRef]
- Luo, B.-H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Ann. Rev. Immunol. 2007, 25, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Schürpf, T.; Springer, T.A. Regulation of integrin affinity on cell surfaces: Regulation of integrin affinity on cell surfaces. EMBO J. 2011, 30, 4712–4727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimaoka, M.; Xiao, T.; Liu, J.-H.; Yang, Y.; Dong, Y.; Jun, C.-D.; McCormack, A.; Zhang, R.; Joachimiak, A.; Takagi, J.; et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell 2003, 112, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Walling, B.L.; Kim, M. LFA-1 in T Cell Migration and Differentiation. Front Immunol. 2018, 9, 952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van de Vijver, E.; Maddalena, A.; Sanal, Ö.; Holland, S.M.; Uzel, G.; Madkaikar, M.; de Boer, M.; van Leeuwen, K.; Köker, M.Y.; Parvaneh, N.; et al. Hematologically important mutations: Leukocyte adhesion deficiency (first update). Blood Cells Mol. Dis. 2012, 48, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogg, N.; Bates, P.A. Genetic analysis of integrin function in man: LAD-1 and other syndromes. Matrix Biol. 2000, 19, 211–222. [Google Scholar] [CrossRef]
- Kambli, P.M.; Bargir, U.A.; Yadav, R.M.; Gupta, M.R.; Dalvi, A.D.; Hule, G.; Kelkar, M.; Sawant-Desai, S.; Setia, P.; Jodhawat, N.; et al. Clinical and Genetic Spectrum of a Large Cohort of Patients With Leukocyte Adhesion Deficiency Type 1 and 3: A Multicentric Study From India. Front Immunol. 2020, 11, 612703. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Sakurada, S.; Kato, T.; Okamoto, T. Induction of cytokines and ICAM-1 by proinflammatory cytokines in primary rheumatoid synovial fibroblasts and inhibition by N-acetyl-L-cysteine and aspirin. Int. Immunol. 1996, 8, 1483–1493. [Google Scholar] [CrossRef] [Green Version]
- de Fougerolles, A.R.; Stacker, S.A.; Schwarting, R.; Springer, T.A. Characterization of ICAM-2 and evidence for a third counter-receptor for LFA-1. J. Exp. Med. 1991, 174, 253–267. [Google Scholar] [CrossRef] [Green Version]
- Fawcett, J.; Holness, C.L.; Needham, L.A.; Turley, H.; Gatter, K.C.; Mason, D.Y.; Simmons, D.L. Molecular cloning of ICAM-3, a third ligand for LFA-1, constitutively expressed on resting leukocytes. Nature 1992, 360, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, O.D.; Devitt, A.; Bell, E.D.; Simmons, D.L.; Gregory, C.D. Macrophage recognition of ICAM-3 on apoptotic leukocytes. J. Immunol. 1999, 162, 6800–6810. [Google Scholar] [PubMed]
- Ihanus, E.; Uotila, L.M.; Toivanen, A.; Varis, M.; Gahmberg, C.G. Red-cell ICAM-4 is a ligand for the monocyte/macrophage integrin CD11c/CD18: Characterization of the binding sites on ICAM-4. Blood 2007, 109, 802–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, L.; Yoshihara, Y.; Mizuno, T.; Mori, K.; Gahmberg, C.G. The neuronal glycoprotein telencephalin is a cellular ligand for the CD11a/CD18 leukocyte integrin. J. Immunol. 1997, 158, 928–936. [Google Scholar]
- Dustin, M.L. Cell adhesion molecules and actin cytoskeleton at immune synapses and kinapses. Curr. Opin. Cell Biol. 2007, 19, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Burbach, B.J.; Medeiros, R.B.; Mueller, K.L.; Shimizu, Y. T-cell receptor signaling to integrins. Immunol. Rev. 2007, 218, 65–81. [Google Scholar] [CrossRef]
- Eidell, K.P.; Lovy, A.; Sylvain, N.R.; Scangarello, F.A.; Muendlein, H.I.; Ophir, M.J.; Nguyen, K.; Seminario, M.-C.; Bunnell, S.C. LFA-1 and kindlin-3 enable the collaborative transport of SLP-76 microclusters by myosin and dynein motors. J. Cell Sci. 2021, 134, jcs258602. [Google Scholar] [CrossRef]
- Tran, D.Q.; Glass, D.D.; Uzel, G.; Darnell, D.A.; Spalding, C.; Holland, S.M.; Shevach, E.M. Analysis of adhesion molecules, target cells, and role of IL-2 in human FOXP3+ regulatory T cell suppressor function. J. Immunol. 2009, 182, 2929–2938. [Google Scholar] [CrossRef] [Green Version]
- Halle, S.; Keyser, K.A.; Stahl, F.R.; Busche, A.; Marquardt, A.; Zheng, X.; Galla, M.; Heissmeyer, V.; Heller, K.; Boelter, J.; et al. In Vivo Killing Capacity of Cytotoxic T Cells Is Limited and Involves Dynamic Interactions and T Cell Cooperativity. Immunity 2016, 44, 233–245. [Google Scholar] [CrossRef] [Green Version]
- Woodland, D.L.; Kohlmeier, J.E. Migration, maintenance and recall of memory T cells in peripheral tissues. Nat. Rev. Immunol. 2009, 9, 153–161. [Google Scholar] [CrossRef]
- Urlaub, D.; Höfer, K.; Müller, M.-L.; Watzl, C. LFA-1 Activation in NK Cells and Their Subsets: Influence of Receptors, Maturation, and Cytokine Stimulation. J. Immunol. 2017, 198, 1944–1951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- YCarrasco, R.; Fleire, S.J.; Cameron, T.; Dustin, M.L.; Batista, F.D. LFA-1/ICAM-1 interaction lowers the threshold of B cell activation by facilitating B cell adhesion and synapse formation. Immunity 2004, 20, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Camponeschi, A.; Gerasimcik, N.; Wang, Y.; Fredriksson, T.; Chen, D.; Farroni, C.; Thorarinsdottir, K.; Ottsjö, L.S.; Aranburu, A.; Cardell, S.; et al. Dissecting Integrin Expression and Function on Memory B Cells in Mice and Humans in Autoimmunity. Front. Immunol. 2019, 10, 534. [Google Scholar] [CrossRef] [PubMed]
- Sumagin, R.; Prizant, H.; Lomakina, E.; Waugh, R.E.; Sarelius, I.H. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J. Immunol. 2010, 185, 7057–7066. [Google Scholar] [CrossRef] [Green Version]
- Furze, R.C.; Rankin, S.M. Neutrophil mobilization and clearance in the bone marrow. Immunology 2008, 125, 281–288. [Google Scholar] [CrossRef]
- Basu, S.; Hodgson, G.; Katz, M.; Dunn, A.R. Evaluation of role of G-CSF in the production, survival, and release of neutrophils from bone marrow into circulation. Blood 2002, 100, 854–861. [Google Scholar] [CrossRef] [Green Version]
- Jakubowski, A.; Gabrilove, J. Granulocyte colony stimulating factor (G-CSF): Biology and clinical status. Cancer Biother. Radiopharm. 1996, 11, 5–20. [Google Scholar] [CrossRef]
- Eskan, M.A.; Jotwani, R.; Abe, T.; Chmelar, J.; Lim, J.-H.; Liang, S.; Ciero, P.A.; Krauss, J.L.; Li, F.; Rauner, M.; et al. The leukocyte integrin antagonist Del-1 inhibits IL-17-mediated inflammatory bone loss. Nat. Immunol. 2012, 13, 465–473. [Google Scholar] [CrossRef] [Green Version]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef] [Green Version]
- Fossiez, F.; Banchereau, J.; Murray, R.; van Kooten, C.; Garrone, P.; Lebecque, S. Interleukin-17. Int. Rev. Immunol. 1998, 16, 541–551. [Google Scholar] [CrossRef]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef] [Green Version]
- Scharffetter-Kochanek, K.; Lu, H.; Norman, K.; van Nood, N.; Munoz, F.; Grabbe, S.; McArthur, M.; Lorenzo, I.; Kaplan, S.; Ley, K.; et al. Spontaneous skin ulceration and defective T cell function in CD18 null mice. J. Exp. Med. 1998, 188, 119–131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forlow, S.B.; Schurr, J.R.; Kolls, J.K.; Bagby, G.J.; Schwarzenberger, P.O.; Ley, K. Increased granulopoiesis through interleukin-17 and granulocyte colony-stimulating factor in leukocyte adhesion molecule-deficient mice. Blood 2001, 98, 3309–3314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvitz, H.R. Genetic control of programmed cell death in the nematode Caenorhabditis elegans. Cancer Res. 1999, 59, 1701s–1706s. [Google Scholar] [PubMed]
- Smith, E.; Zarbock, A.; Stark, M.A.; Burcin, T.L.; Bruce, A.C.; Foley, P.; Ley, K. IL-23 Is Required for Neutrophil Homeostasis in Normal and Neutrophilic Mice. J. Immunol. 2007, 179, 8274–8279. [Google Scholar] [CrossRef] [Green Version]
- Stark, M.A.; Huo, Y.; Burcin, T.L.; Morris, M.A.; Olson, T.S.; Ley, K. Phagocytosis of apoptotic neutrophils regulates granulopoiesis via IL-23 and IL-17. Immunity 2005, 22, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Moutsopoulos, N.M.; Chalmers, N.I.; Barb, J.J.; Abusleme, L.; Greenwell-Wild, T.; Dutzan, N.; Paster, B.J.; Munson, P.J.; Fine, D.H.; Uzel, G.; et al. Subgingival microbial communities in Leukocyte Adhesion Deficiency and their relationship with local immunopathology. PLoS Pathog. 2015, 11, e1004698. [Google Scholar] [CrossRef]
- D’Agata, I.D.; Paradis, K.; Chad, Z.; Bonny, Y.; Seidman, E. Leucocyte adhesion deficiency presenting as a chronic ileocolitis. Gut 1996, 39, 605–608. [Google Scholar] [CrossRef]
- Uzel, G.; Kleiner, D.E.; Kuhns, D.B.; Holland, S.M. Dysfunctional LAD-1 neutrophils and colitis. Gastroenterology 2001, 121, 958–964. [Google Scholar] [CrossRef]
- Dayan, J.R.; Dolinger, M.; Benkov, K.; Dunkin, D.; Jossen, J.; Lai, J.; Phan, B.L.; Pittman, N.; Dubinsky, M.C. Real World Experience With Ustekinumab in Children and Young Adults at a Tertiary Care Pediatric Inflammatory Bowel Disease Center. J. Pediatr. Gastroenterol. Nutr. 2019, 69, 61–67. [Google Scholar] [CrossRef]
- Gonzalez, N.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramirez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 2009, 31, 245–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, Y.-S.; Kim, S.-Y.; Kim, M.-J.; Lim, J.-H.; Cho, M.-S.; Kang, J.L. PPARγ activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal. Immunol. 2015, 8, 1031–1046. [Google Scholar] [CrossRef] [PubMed]
- Kajikawa, T.; Wang, B.; Li, X.; Wang, H.; Chavakis, T.; Moutsopoulos, N.M.; Hajishengallis, G. Frontline Science: Activation of metabolic nuclear receptors restores periodontal tissue homeostasis in mice with leukocyte adhesion deficiency-1. J. Leukoc. Biol. 2020, 108, 1501–1514. [Google Scholar] [CrossRef] [PubMed]
- Marsili, M.; Lougaris, V.; Lucantoni, M.; di Marzio, D.; Baronio, M.; Vitali, M.; Lombardi, G.; Chiarelli, F.; Breda, L. Successful anti-TNF-α treatment in a girl with LAD-1 disease and autoimmune manifestations. J. Clin. Immunol. 2014, 34, 788–791. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Moutsopoulos, N.M.; Hajishengallis, E.; Chavakis, T. Immune and regulatory functions of neutrophils in inflammatory bone loss. Semin. Immunol. 2016, 28, 146–158. [Google Scholar] [CrossRef] [Green Version]
- Papp, K.A.; Caro, I.; Leung, H.M.; Garovoy, M.; Mease, P.J. Efalizumab for the treatment of psoriatic arthritis. J. Cutan. Med. Surg. 2007, 11, 57–66. [Google Scholar] [CrossRef]
- Carson, K.R.; Focosi, D.; Major, E.O.; Petrini, M.; Richey, E.A.; West, D.P.; Bennett, C.L. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: A Review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009, 10, 816–824. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Wajgt, A.; et al. AFFIRM Investigators, A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.; Goldin, E.; Gordon, F.H.; Malchow, H.A.; Rask-Madsen, J.; Rutgeerts, P.; Vyhnálek, P.; Zádorová, Z.; Palmer, T.; Donoghue, S. Natalizumab Pan-European Study Group, Natalizumab for active Crohn’s disease. N. Engl. J. Med. 2003, 348, 24–32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Target | Mechanism | Results |
|---|---|---|---|
| Ustekinumab | IL-23/IL-12 | Binds p40 subunit and blocks IL12/IL23 receptor interaction | Decreased IL-17 levels, resolution of inflammatory lesions in an adult patient |
| LXRα/LXRβ agonist | LXRα/LXRβ | Activation of LXR promotes clearance of apoptotic cells in macrophages | Decreased IL-23 and IL-17, improved bone levels in CD18−/− mice (71) |
| PPARβ/δ agonist | PPARβ/δ | Activation of PPAR leads to regulation of efferocytosis | Decreased IL-23 and IL-17, improved bone levels in CD18−/− mice (72) |
| Infliximab | TNF-α | Binds and neutralizes TNF-α | Improvement of Crohn’s-like colitis and arthritis in LAD 1 patient (74) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fekadu, J.; Modlich, U.; Bader, P.; Bakhtiar, S. Understanding the Role of LFA-1 in Leukocyte Adhesion Deficiency Type I (LAD I): Moving towards Inflammation? Int. J. Mol. Sci. 2022, 23, 3578. https://doi.org/10.3390/ijms23073578
Fekadu J, Modlich U, Bader P, Bakhtiar S. Understanding the Role of LFA-1 in Leukocyte Adhesion Deficiency Type I (LAD I): Moving towards Inflammation? International Journal of Molecular Sciences. 2022; 23(7):3578. https://doi.org/10.3390/ijms23073578
Chicago/Turabian StyleFekadu, Julia, Ute Modlich, Peter Bader, and Shahrzad Bakhtiar. 2022. "Understanding the Role of LFA-1 in Leukocyte Adhesion Deficiency Type I (LAD I): Moving towards Inflammation?" International Journal of Molecular Sciences 23, no. 7: 3578. https://doi.org/10.3390/ijms23073578