
Functional Characterization of the MYO6 Variant p.E60Q in Non-Syndromic Hearing Loss Patients

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Results
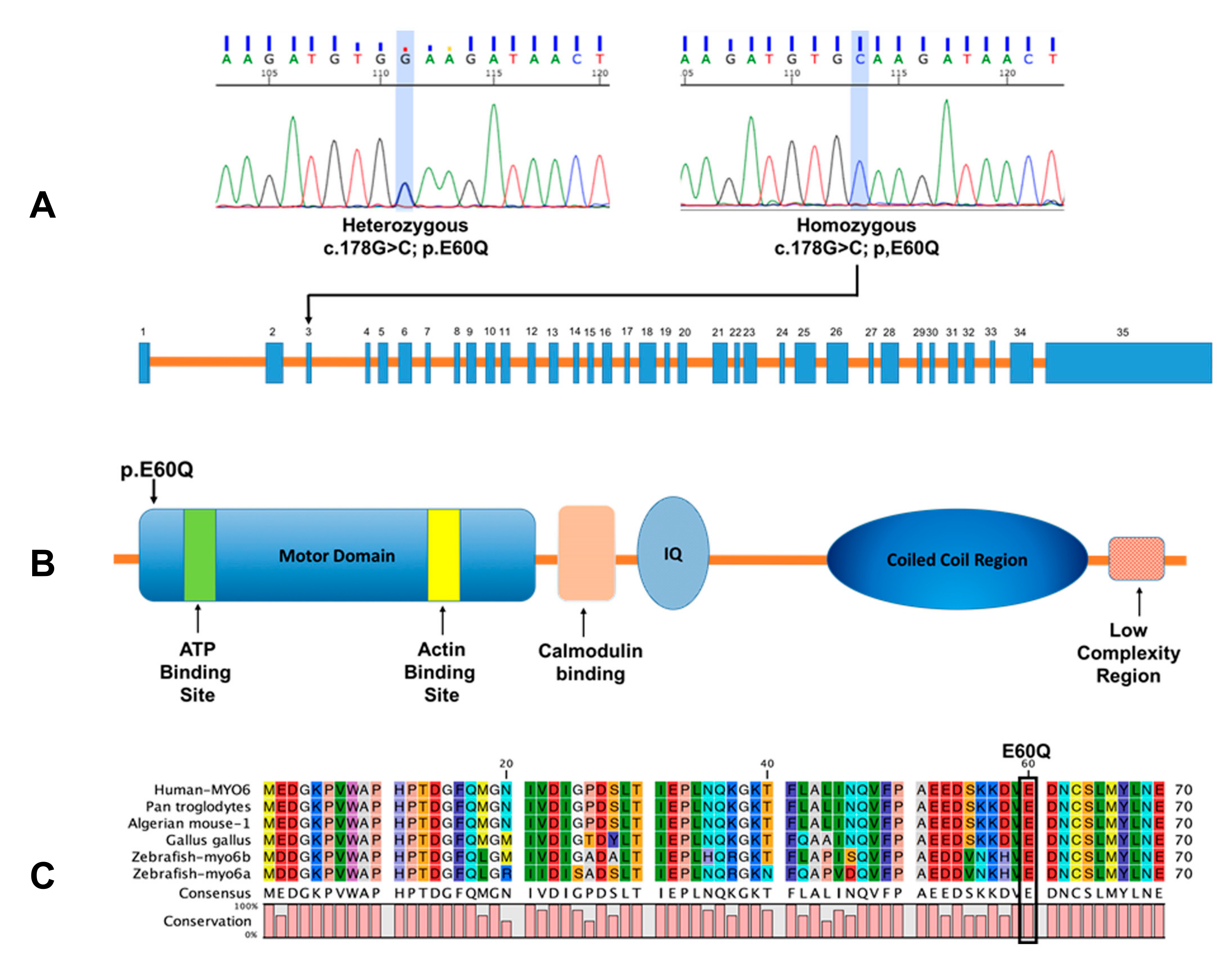
2.1. Prevalence of the Novel MYO6 Variant p.E60Q in Qatari Population
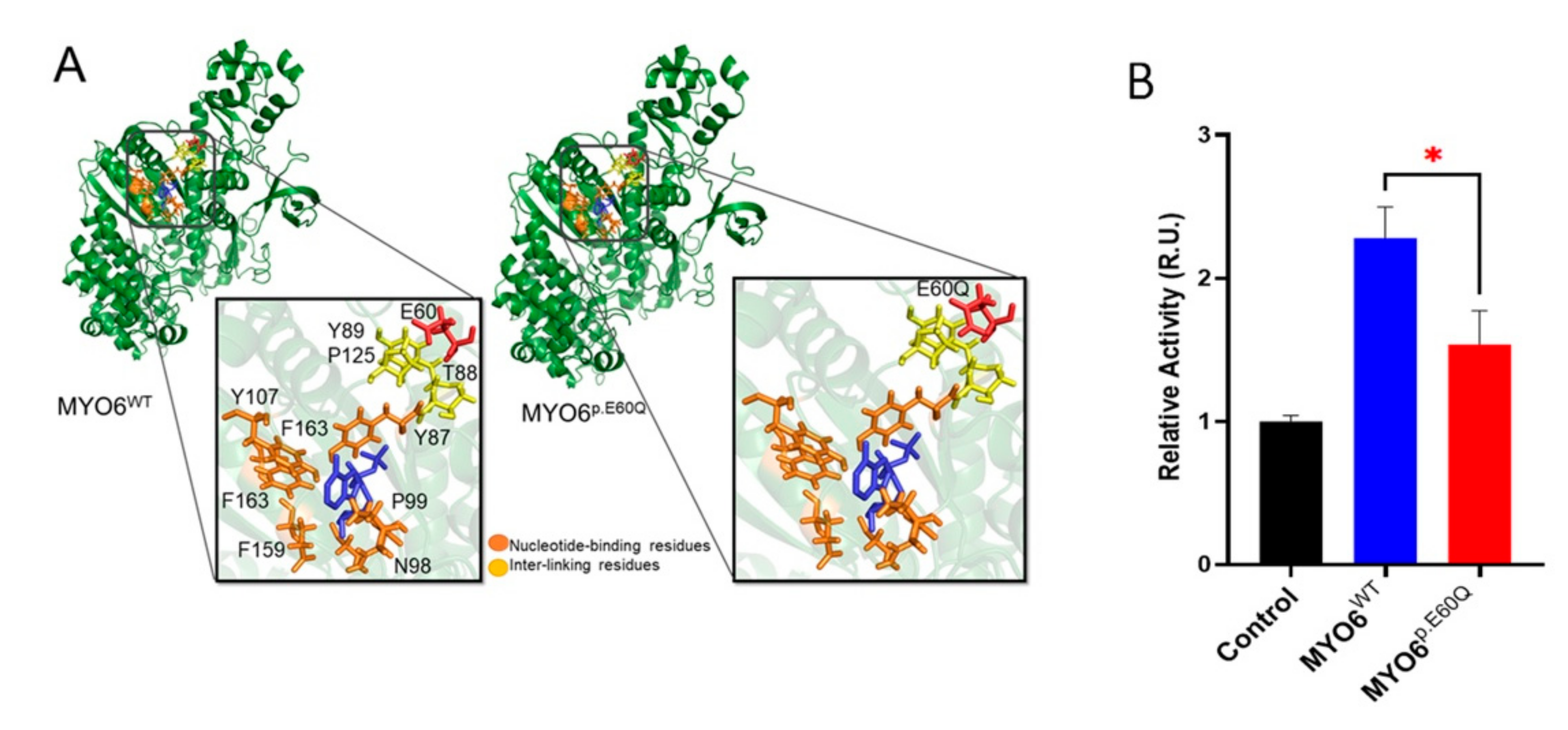
2.2. MYO6 Variant Is Predicted to Reduce ATPase Activity by In Silico Protein Modeling
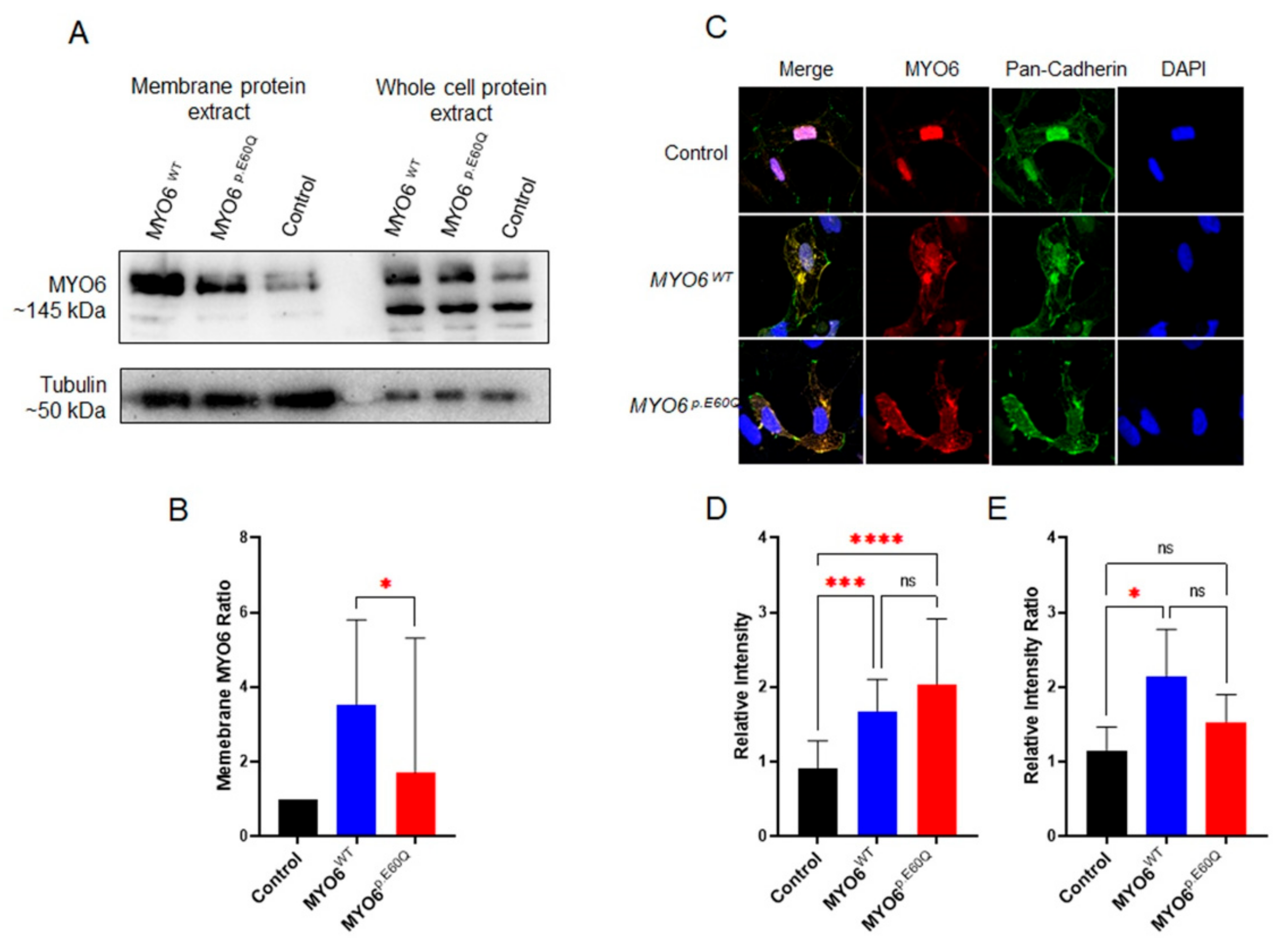
2.3. MYO6p.E60Q Variant Alters Cellular Protein Trafficking
2.4. Functional Validation of MYO6 Variant in the Zebrafish Model
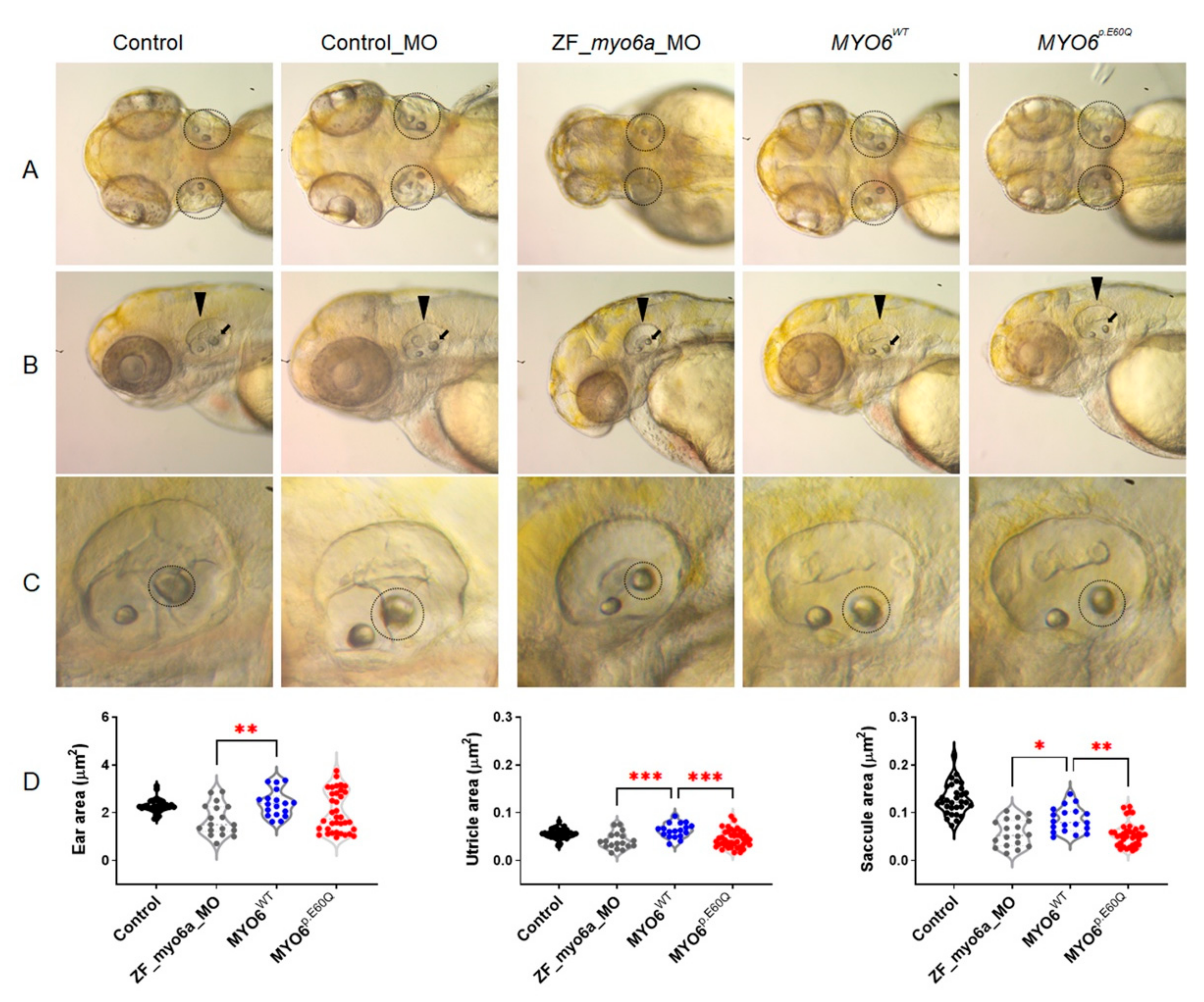
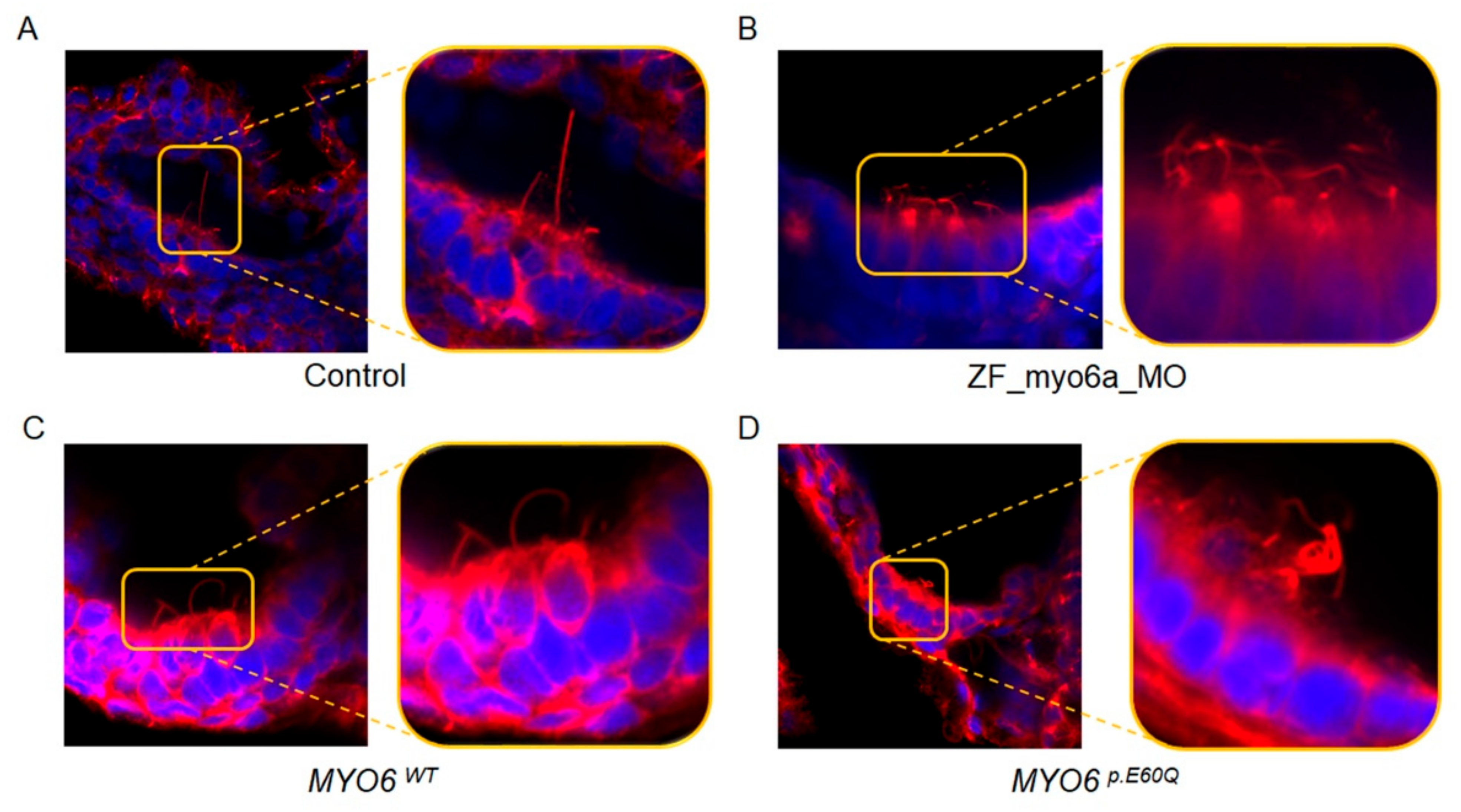
2.4.1. Human MYO6p.E60Q Variant Results in Zebrafish Ear Morphological Defects and Abnormal Inner Ear Hair Cell Development
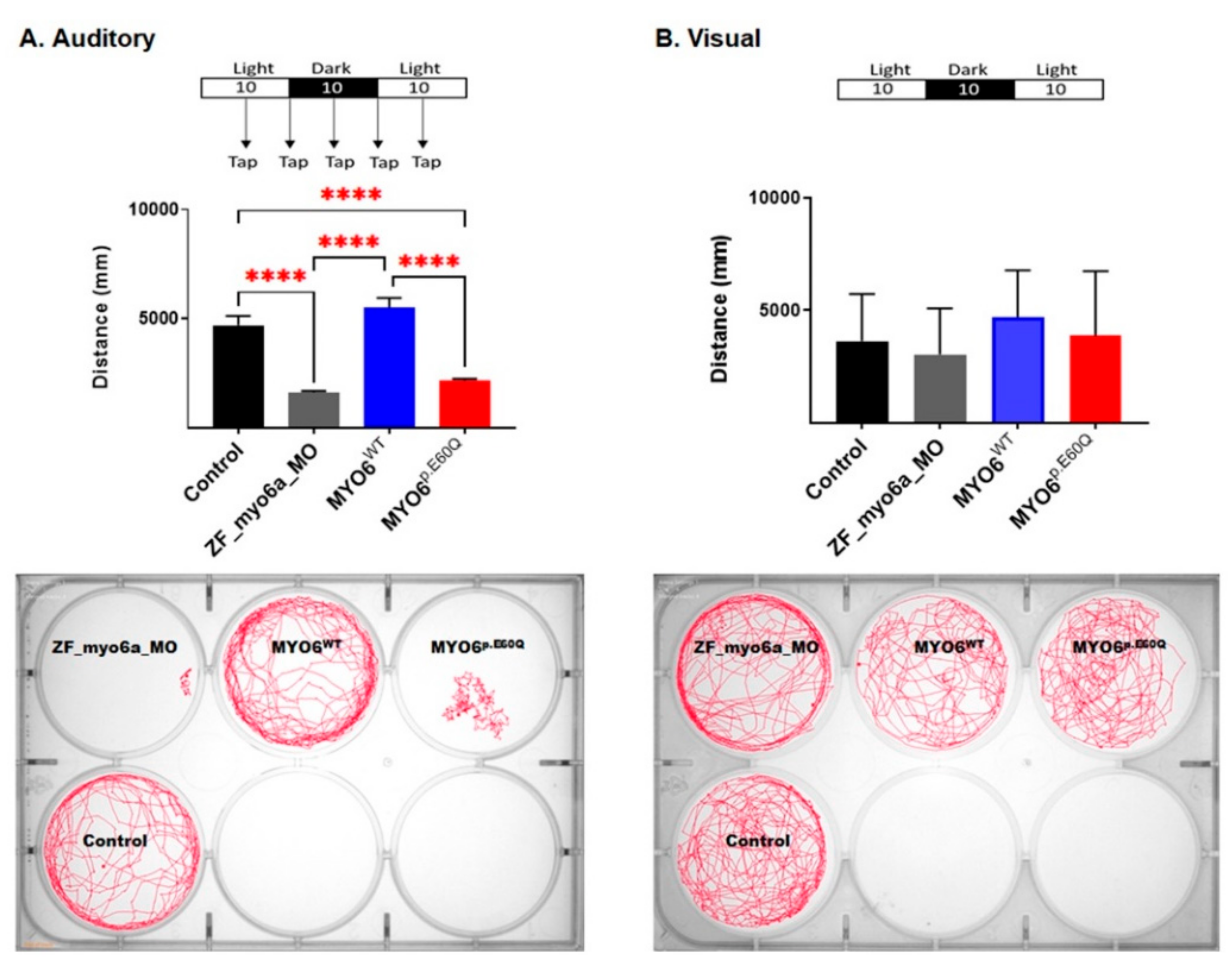
2.4.2. Human MYO6p.E60Q Variant Resulted in Altered Zebrafish Auditory Responses
3. Discussion
4. Materials and Methods
4.1. Sanger Sequencing
4.2. Cell Culture and In Vitro Analysis
4.3. Immunofluorescent Staining, Confocal Microscopy Imaging, and Quantification
4.4. Zebrafish Care and Husbandry
4.5. Morpholino Design and Synthetic mRNA Injection
4.6. Head and Ear Imaging of Zebrafish Larvae
4.7. Zebrafish Staining and Imaging
4.8. Zebrafish Locomotor Behavior Measurements
4.9. Protein Modeling
4.10. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Van Camp, G.S.R. Hereditary Hearing Loss Homepage. 2018. Available online: https://hereditaryhearingloss.org (accessed on 12 October 2021).
- Lu, Q.; Li, J.; Zhang, M. Cargo recognition and cargo-mediated regulation of unconventional myosins. Acc. Chem. Res. 2014, 47, 3061–3070. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.A.; Spudich, J.A. The myosin superfamily at a glance. J. Cell Sci. 2012, 125 Pt 7, 1627–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellers, J.R. Myosins: A diverse superfamily. Biochim. Biophys. Acta 2000, 1496, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Pylypenko, O.; Song, L.; Shima, A.; Yang, Z.; Houdusse, A.M.; Sweeney, H.L. Myosin VI deafness mutation prevents the initiation of processive runs on actin. Proc. Natl. Acad. Sci. USA 2015, 112, E1201–E1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, Z.M.; Morell, R.J.; Riazuddin, S.; Gropman, A.; Shaukat, S.; Ahmad, M.M.; Mohiddin, S.A.; Fananapazir, L.; Caruso, R.C.; Husnain, T.; et al. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 2003, 72, 1315–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchionda, S.; Ahituv, N.; Bisceglia, L.; Sobe, T.; Glaser, F.; Rabionet, R.; Arbones, M.L.; Notarangelo, A.; Di Iorio, E.; Carella, M.; et al. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 2001, 69, 635–640. [Google Scholar] [CrossRef]
- Hertzano, R.; Shalit, E.; Rzadzinska, A.K.; Dror, A.A.; Song, L.; Ron, U.; Tan, J.T.; Shitrit, A.S.; Fuchs, H.; Hasson, T.; et al. A MYO6 mutation destroys coordination between the myosin heads, revealing new functions of myosin VI in the stereocilia of mammalian inner ear hair cells. PLoS Genet. 2008, 4, e1000207. [Google Scholar] [CrossRef] [Green Version]
- Self, T.; Sobe, T.; Copeland, N.G.; Jenkins, N.A.; Avraham, K.B.; Steel, K.P. Role of myosin VI in the differentiation of cochlear hair cells. Dev. Biol. 1999, 214, 331–341. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.Z.; Walsh, J.; Mburu, P.; Kendrick-Jones, J.; Cope, M.J.; Steel, K.P.; Brown, S.D. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 1997, 16, 188–190. [Google Scholar] [CrossRef]
- Liu, X.Z.; Walsh, J.; Tamagawa, Y.; Kitamura, K.; Nishizawa, M.; Steel, K.P.; Brown, S.D. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 1997, 17, 268–269. [Google Scholar] [CrossRef]
- Wang, A.; Liang, Y.; Fridell, R.A.; Probst, F.J.; Wilcox, E.R.; Touchman, J.W.; Morton, C.C.; Morell, R.J.; Noben-Trauth, K.; Camper, S.A.; et al. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 1998, 280, 1447–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kellerman, K.A.; Miller, K.G. An unconventional myosin heavy chain gene from Drosophila melanogaster. J. Cell Biol. 1992, 119, 823–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avraham, K.B.; Hasson, T.; Steel, K.P.; Kingsley, D.M.; Russell, L.B.; Mooseker, M.S.; Copeland, N.G.; Jenkins, N.A. The mouse Snell’s waltzer deafness gene encodes an unconventional myosin required for structural integrity of inner ear hair cells. Nat. Genet. 1995, 11, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Seiler, C.; Ben-David, O.; Sidi, S.; Hendrich, O.; Rusch, A.; Burnside, B.; Avraham, K.B.; Nicolson, T. Myosin VI is required for structural integrity of the apical surface of sensory hair cells in zebrafish. Dev. Biol. 2004, 272, 328–338. [Google Scholar] [CrossRef] [Green Version]
- Avraham, K.B.; Hasson, T.; Sobe, T.; Balsara, B.; Testa, J.R.; Skvorak, A.B.; Morton, C.C.; Copeland, N.G.; Jenkins, N.A. Characterization of unconventional MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice. Hum. Mol. Genet. 1997, 6, 1225–1231. [Google Scholar] [CrossRef] [Green Version]
- Hegan, P.S.; Lanahan, A.A.; Simons, M.; Mooseker, M.S. Myosin VI and cardiomyopathy: Left ventricular hypertrophy, fibrosis, and both cardiac and pulmonary vascular endothelial cell defects in the Snell’s waltzer mouse. Cytoskeleton 2015, 72, 373–387. [Google Scholar] [CrossRef] [Green Version]
- Streisinger, G.; Walker, C.; Dower, N.; Knauber, D.; Singer, F. Production of clones of homozygous diploid zebra fish (Brachydanio rerio). Nature 1981, 291, 293–296. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Lanahan, A.A.; Hermans, K.; Claes, F.; Kerley-Hamilton, J.S.; Zhuang, Z.W.; Giordano, F.J.; Carmeliet, P.; Simons, M. VEGF receptor 2 endocytic trafficking regulates arterial morphogenesis. Dev. Cell. 2010, 18, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Kappler, J.A.; Starr, C.J.; Chan, D.K.; Kollmar, R.; Hudspeth, A.J. A nonsense mutation in the gene encoding a zebrafish myosin VI isoform causes defects in hair-cell mechanotransduction. Proc. Natl. Acad. Sci. USA 2004, 101, 13056–13061. [Google Scholar] [CrossRef] [Green Version]
- Granato, M.; van Eeden, F.J.; Schach, U.; Trowe, T.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996, 123, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Whitfield, T.T.; Granato, M.; van Eeden, F.J.; Schach, U.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Mutations affecting development of the zebrafish inner ear and lateral line. Development 1996, 123, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Ghysen, A.; Dambly-Chaudiere, C. Development of the zebrafish lateral line. Curr. Opin. Neurobiol. 2004, 14, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Alkowari, M.K.; Vozzi, D.; Bhagat, S.; Krishnamoorthy, N.; Morgan, A.; Hayder, Y.; Logendra, B.; Najjar, N.; Gandin, I.; Gasparini, P.; et al. Targeted sequencing identifies novel variants involved in autosomal recessive hereditary hearing loss in Qatari families. Mutat. Res. 2017, 800–802, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Kwon, T.J.; Oh, S.K.; Park, H.J.; Sato, O.; Venselaar, H.; Choi, S.Y.; Kim, S.; Lee, K.Y.; Bok, J.; Lee, S.H.; et al. The effect of novel mutations on the structure and enzymatic activity of unconventional myosins associated with autosomal dominant non-syndromic hearing loss. Open Biol. 2014, 4, 140107. [Google Scholar] [CrossRef] [Green Version]
- Wong, E.Y.; Xu, C.Y.; Brahmachary, M.; Xu, P.X. A Novel ENU-Induced Mutation in Myo6 Causes Vestibular Dysfunction and Deafness. PLoS ONE 2016, 11, e0154984. [Google Scholar] [CrossRef] [Green Version]
- Mbarek, H.; Gandhi, G.D.; Selvaraj, S.; Al-Muftah, W.; Badji, R.; Al-Sarraj, Y.; Saad, C.; Darwish, D.; Alvi, M.; Fadl, T.; et al. Qatar Genome: Insights on Genomics from the Middle East. Hum. Mutat. 2021. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Scott, E.M.; Halees, A.; Itan, Y.; Spencer, E.G.; He, Y.; Azab, M.A.; Gabriel, S.B.; Belkadi, A.; Boisson, B.; Abel, L.; et al. Characterization of Greater Middle Eastern genetic variation for enhanced disease gene discovery. Nat. Genet. 2016, 48, 1071–1076. [Google Scholar] [CrossRef]
- Jung, E.J.; Liu, G.; Zhou, W.; Chen, X. Myosin VI is a mediator of the p53-dependent cell survival pathway. Mol. Cell. Biol. 2006, 26, 2175–2186. [Google Scholar] [CrossRef] [Green Version]
- Vona, B.; Doll, J.; Hofrichter, M.A.H.; Haaf, T.; Varshney, G.K. Small fish, big prospects: Using zebrafish to unravel the mechanisms of hereditary hearing loss. Hear. Res. 2020, 397, 107906. [Google Scholar] [CrossRef] [PubMed]
- Ernest, S.; Rauch, G.J.; Haffter, P.; Geisler, R.; Petit, C.; Nicolson, T. Mariner is defective in myosin VIIA: A zebrafish model for human hereditary deafness. Hum. Mol. Genet. 2000, 9, 2189–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schibler, A.; Malicki, J. A screen for genetic defects of the zebrafish ear. Mech. Dev. 2007, 124, 592–604. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, J.; Hashimoto, K.; Xiao, R.; Vandenberghe, L.H.; Liberman, M.C. Cochlear gene therapy with ancestral AAV in adult mice: Complete transduction of inner hair cells without cochlear dysfunction. Sci. Rep. 2017, 7, 45524. [Google Scholar] [CrossRef] [Green Version]
- Pan, B.; Askew, C.; Galvin, A.; Heman-Ackah, S.; Asai, Y.; Indzhykulian, A.A.; Jodelka, F.M.; Hastings, M.L.; Lentz, J.J.; Vandenberghe, L.H.; et al. Gene therapy restores auditory and vestibular function in a mouse model of Usher syndrome type 1c. Nat. Biotechnol. 2017, 35, 264–272. [Google Scholar] [CrossRef]
- Da’as, S.I.; Aamer, W.; Hasan, W.; Al-Maraghi, A.; Al-Kurbi, A.; Kilani, H.; AlRayahi, J.; Zamel, K.; Stotland, M.A.; Fakhro, K.A. PGAP3 Associated with Hyperphosphatasia with Mental Retardation Plays a Novel Role in Brain Morphogenesis and Neuronal Wiring at Early Development. Cells 2020, 9, 1782. [Google Scholar] [CrossRef]
- Wang, Y.N.; Hou, Y.Y.; Sun, M.Z.; Zhang, C.Y.; Bai, G.; Zhao, X.; Feng, X.Z. Behavioural screening of zebrafish using neuroactive traditional Chinese medicine prescriptions and biological targets. Sci. Rep. 2014, 4, 5311. [Google Scholar] [CrossRef] [Green Version]
- Menetrey, J.; Bahloul, A.; Wells, A.L.; Yengo, C.M.; Morris, C.A.; Sweeney, H.L.; Houdusse, A. The structure of the myosin VI motor reveals the mechanism of directionality reversal. Nature 2005, 435, 779–785. [Google Scholar] [CrossRef] [Green Version]
- Menetrey, J.; Llinas, P.; Mukherjea, M.; Sweeney, H.L.; Houdusse, A. The structural basis for the large powerstroke of myosin VI. Cell 2007, 131, 300–308. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef]
- Gajendrarao, P.; Krishnamoorthy, N.; Kassem, H.; Moharem-Elgamal, S.; Cecchi, F.; Olivotto, I.; Yacoub, M.H. Molecular modeling of disease causing mutations in domain C1 of cMyBP-C. PLoS ONE 2013, 8, e59206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, A.; Vuckovic, D.; Krishnamoorthy, N.; Rubinato, E.; Ambrosetti, U.; Castorina, P.; Franze, A.; Vozzi, D.; La Bianca, M.; Cappellani, S.; et al. Next-generation sequencing identified SPATC1L as a possible candidate gene for both early-onset and age-related hearing loss. Eur. J. Hum. Genet. 2019, 27, 70–79. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutation Prediction Tools | Allele Frequencies (%) | ||
|---|---|---|---|
| MutationTaster 1 | 1 | Qatar Genome Program | 0.5 |
| SIFT 2 | 0.01 | GME 7 | 0.15 |
| Polyphen-2 3 | 0.994 | genomAD—Exomes | 0.0012 |
| CADD 4 | 24.2 | genomAD—Genomes [29] | 0.0007 |
| PhyloP 5 | 9.444 | ExAC | 0.0008 |
| GERP++ 6 | 5.92 | TopMed | 0.0015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alkowari, M.; Espino-Guarch, M.; Daas, S.; Abdelrahman, D.; Hasan, W.; Krishnamoorthy, N.; Sathappan, A.; Sheehan, P.; Panhuys, N.V.; The Qatar Genome Program Research Consortium; et al. Functional Characterization of the MYO6 Variant p.E60Q in Non-Syndromic Hearing Loss Patients. Int. J. Mol. Sci. 2022, 23, 3369. https://doi.org/10.3390/ijms23063369
Alkowari M, Espino-Guarch M, Daas S, Abdelrahman D, Hasan W, Krishnamoorthy N, Sathappan A, Sheehan P, Panhuys NV, The Qatar Genome Program Research Consortium, et al. Functional Characterization of the MYO6 Variant p.E60Q in Non-Syndromic Hearing Loss Patients. International Journal of Molecular Sciences. 2022; 23(6):3369. https://doi.org/10.3390/ijms23063369
Chicago/Turabian StyleAlkowari, Moza, Meritxell Espino-Guarch, Sahar Daas, Doua Abdelrahman, Waseem Hasan, Navaneethakrishnan Krishnamoorthy, Abbirami Sathappan, Patrick Sheehan, Nicholas Van Panhuys, The Qatar Genome Program Research Consortium, and et al. 2022. "Functional Characterization of the MYO6 Variant p.E60Q in Non-Syndromic Hearing Loss Patients" International Journal of Molecular Sciences 23, no. 6: 3369. https://doi.org/10.3390/ijms23063369