
Crosstalk between p38 MAPK and GR Signaling


Department of Pharmacy, UiT-The Arctic University of Norway, 9037 Tromsø, Norway
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2022, 23(6), 3322; https://doi.org/10.3390/ijms23063322
Submission received: 17 February 2022
/
Revised: 13 March 2022
/
Accepted: 16 March 2022
/
Published: 19 March 2022
(This article belongs to the Special Issue P38 Signaling Pathway 2.0)
{kind=link}
Abstract
:The p38 MAPK is a signaling pathway important for cells to respond to environmental and intracellular stress. Upon activation, the p38 kinase phosphorylates downstream effectors, which control the inflammatory response and coordinate fundamental cellular processes such as proliferation, apoptosis, and differentiation. Dysregulation of this signaling pathway has been linked to inflammatory diseases and cancer. Secretion of glucocorticoids (GCs) is a classical endocrine response to stress. The glucocorticoid receptor (GR) is the primary effector of GCs and plays an important role in the regulation of cell metabolism and immune response by influencing gene expression in response to hormone-dependent activation. Its ligands, the GCs or steroids, in natural or synthetic variation, are used as standard therapy for anti-inflammatory treatment, severe asthma, autoimmune diseases, and several types of cancer. Several years ago, the GR was identified as one of the downstream targets of p38, and, at the same time, it was shown that glucocorticoids could influence p38 signaling. In this review, we discuss the role of the crosstalk between the p38 and GR in the regulation of gene expression in response to steroids and comprehend the importance and potential of this interplay in future clinical applications.
Keywords:
p38; MAPK; signaling; inflammation; cancer; glucocorticoid receptor; crosslink; inhibitor; drug discovery; glucocorticoids1. Introduction
Some of the most distinct molecular features and essential parts of mammalian cells are the mitogen-activated protein kinase (MAPK) signaling pathways structured in a three-tire of kinases—a MAPK, a MAPK kinase (MAP2K), and a MAPK kinase kinase (MAP3K). These pathways allow the cell to react to different internal and external stimuli such as stress factors, hormones, and cytokines to regulate proliferation, differentiation, and apoptosis, among other fundamental processes. Dysfunctions due to mutations within the MAPK pathways have been correlated to several clinical outcomes and health issues like Alzheimer’s disease, Parkinson’s disease, inflammation, and multiple types of cancer [1,2,3]. Thus, a deepened insight into the mechanisms of MAPK signaling is of great interest when it comes to understanding the pathology and to developing new treatments for major human diseases.
The p38 MAPK pathway is one of three major MAPK signaling pathways present in human cells, next to the extracellular signal-regulated kinase (ERK) and c-Jun NH(2)-terminal kinase (JNK) MAPK pathways. There are five different kinases known to activate p38 signaling, the MAP3Ks ASK1/2 (MAP3K5/6), TAK1 (MAP3K7), TAO1 (MAPK16), and TAO2 (MAP3K17). Downstream of these are the two MAP2Ks, MKK3, and MKK6, and their target protein isoforms p38 MAPK α, β, γ, and δ [4,5]. The p38 kinase, and particularly the isoforms α and β, further activate several substrates like Ser/Thr activated protein kinases MK2/3, MNK1/2, and MSK1/2, as well as transcription factors like ATF2, leading to effective, context-dependent functions such as inflammatory response and cell apoptosis, as well as tumor-promoting or -suppressing effects [6,7].
Acknowledging its clinical value, the research on p38 has been intensified over the years and specific inhibitors mainly targeting p38α and -β are generated and characterized thoroughly. Early inhibitors like SB203580 and SB202190 act on the ATP binding site, while newer ones like BIRB 796/Doramapimod interfere indirectly with ATP binding, preventing p38’s catalytic activity. Nowadays, these tools are widely used for in vitro and in vivo studies, as well as being investigated in pre-clinical trials and offering new host-modulating therapy forms for various diseases including rheumatoid arthritis, Crohn’s disease, tuberculosis, and cancer [8,9].
The glucocorticoid receptor (GR) is expressed in all cells of the human body and its ligands, the glucocorticoids, display important functions within development, differentiation, and homeostasis. GR affects glucose and fat metabolism, the immune system, the nervous system, and the musculoskeletal system [10]. As a member of the nuclear receptor superfamily, GR influences and controls gene expression via hormone-dependent activation, causing conformational changes and translocation. The receptor cycles between the cytoplasm and nucleus are dependent on the ligand-bound and activation state. The modular receptor protein consists of an N-terminal transactivation domain (AF1), a central DNA-binding domain consisting of two zinc fingers, and a C-terminal ligand-binding and activation domain (AF2). GR can bind to specific sites in the genome either as a dimer or a monomer or interact with other transcription factors (TFs) to bind composite elements. It can also interact indirectly with DNA by binding TFs that are already bound to promoters (tethering) [11]. The DNA sequences responsive to GR are so-called glucocorticoid response elements (GREs) that are distributed widely throughout the genome. Gene regulation by GR is highly context-dependent and subject to the accessibility of chromatin, post-translational modifications, and association with other TFs and co-factors [11,12,13]. Due to alternative splicing and the use of alternative translational start codons, there are several receptor isoforms generated from the gene (NR3C1) encoding GR [14]. GRα and GRβ are the main isoforms, and they differ in their C-termini including the ligand-binding domain. GRα fulfills the classic GR transcriptional function upon activation via corticosteroids and is the isoform referred to when one talks about GR in general. The GRβ isoform lacks ligand binding activity and has been shown to reside in the nucleus where it counteracts the GRα function [15,16], but is also reported to have intrinsic activities regulating gene expression independent of GRα [17]. The way GR regulates transcription is complex, involving the cooperation of numerous transcription factors and co-factors, leading to both activation and repression of genes in a tissue-specific manner. Several post-translational modifications are also known to affect GR function [11].
The GR ligands, corticosteroids, and glucocorticoids (GCs) are steroid hormones released by the adrenal cortex that regulate major biological processes such as development, metabolism, and inflammation. Due to their immunosuppressive effects, synthetic GCs like dexamethasone are widely used in clinics for the treatment of autoimmune and inflammatory diseases [18,19]. They are the first-line treatment for lymphoid cancers causing apoptosis [20], whereas, for patients with solid tumors undergoing chemotherapy or radiation, they are used to treat side effects such as nausea, bone pain, and edema. However, an emerging problem regarding the GC treatment of cancer and other diseases like chronic obstructive pulmonary disease (COPD) and severe asthma, is the development of insensitivity and/or resistance, which prevents effective therapy [21].
2. Crosstalk between p38 and GR
That the MAPK signaling pathways and glucocorticoid signaling could affect each other mutually was already reported more than 20 years ago. The use of dexamethasone showed to cause an inhibition of the MAPK pathway in mast cells [26], while MAPKs were concurrently identified as some of the kinases capable of phosphorylating GR at specific sites in vitro [27]. The first report of GCs inhibiting MAPK activity only involved ERK1/2 [26], however, they were soon reported to inhibit the stress-activated JNK and p38 signaling pathways as well [28,29]. Further on, the specificity of MAPK’s phosphorylation of GR was investigated, and JNK was shown to directly phosphorylate rat GR at Serine (S) 246, corresponding to human GR S226 [30]. Activated ERK2 was further characterized to indirectly affect this phosphorylation site, while p38 had no effect [30]. Since then, it has successfully been established that p38 can regulate phosphorylation of GR at specific sites as well. The activation of the p38 kinase and GR phosphorylation are shown to correlate within several diseases, and this interaction is thought to contribute to the GC resistance observed in the clinic. Below, we attempt to summarize the current research data concerning the crosstalk between the p38 and the GR signaling pathways and provide examples of corresponding biological consequences and the clinical relevance of this crosstalk.
2.1. p38-Directed GR Phosphorylation and Regulation
One of the first hints towards a clinically relevant crosstalk between p38 and GR signaling was the study by Irusen et al. [31], which demonstrated that the p38 inhibitor SB203580 potentially reverses the GC insensitivity in patients with severe asthma. The authors isolated peripheral blood mononuclear cells from asthma patients and could show that stimulation with a combination of IL2 and IL4 activated p38, which then resulted in increased GR phosphorylation. This phosphorylation and an altered ligand binding affinity could be inhibited when applying the p38 inhibitor SB203580, but not by an ERK1/2 pathway inhibitor [31].
In the following years of research, the development of phospho-specific antibodies made it easier to study individual GR phosphorylation sites, and the S211 phosphorylation (hGR) was suggested as a biomarker for GR activation in response to hormone treatment [32]. Additionally, S211 was identified as a direct substrate of p38 in lymphoid cells [33]. The application of p38 inhibitors hereby caused a decrease in dexamethasone-induced apoptosis, demonstrating a correlation between p38 activation and glucocorticoid-driven apoptosis [33]. The phosphorylation of S211 was also shown to play a part in GR’s regulation of carbohydrate metabolism. Here, the AMP-activated protein kinase (AMPK), induced by AICAR, was shown to activate p38, which then mediated the phosphorylation of GR at S211 [34]. This event caused tissue- and gene-specific alterations of GR-induced target gene expression by changing the attraction of transcriptional coregulators to DNA-bound GR [34]. The p38-mediated GR phosphorylation seems to vary greatly dependent on cell type, stimulus, and function. In airway smooth muscle (ASM) cells, inhibition of p38 activity decreased basal and fluticasone propionate-induced GR S203 phosphorylation, while increasing basal and fluticasone propionate-induced S211 phosphorylation [35]. Notably, no effect was observed on S226 phosphorylation. The p38-induced S203 phosphorylation had an inhibitory effect on GR translocation to the nucleus, GRE-reporter gene expression, and mRNA expression of the Glucocorticoid-induced leucine zipper (GILZ) gene. This was verified by the use of siRNA against p38, whereas co-transfection with a vector expressing a constitutive active MKK3 mutant activating p38 had the opposite effect. The usage of an S203A mutant hereby confirmed that the phosphorylation of this residue was inhibitory for GR function. The authors concluded that basal p38 MAPK activity in ASM cells was important to keep unliganded GR in an inactive state [35].
The residue S226 of GR was shown to be phosphorylated in vitro by both JNK and ERK2, but not by the p38 MAP kinase [30]. In these studies, the activation of either ERK1/2 or JNK resulted in the repression of GC-mediated transcription. However, a functional S226 was only required for JNK’s repression of GC-induced transcription. This phosphorylation seems to play an important part in the nuclear export of GR [36], and a few studies have also reported that p38 is responsible for the GR S226 phosphorylation in peripheral blood mononuclear cells derived from asthmatic patients. However, these observations are solely based on the quantitation of phospho-S226 western blots, where a minute reduction of S226 phosphorylation is observed in activated cells treated with p38 inhibitors [37,38,39]. Despite the convincing results regarding a connection between p38 activation and a general phosphorylation of GR in the cells and cell lines used, a direct connection between p38 and the GR S226 phosphorylation is not definitively proven. Additional experiments would be required before the S226 of GR can be confirmed as a bona fide p38 phosphorylation site.
The phosphorylation of S134 in GR was shown to be dependent on p38 activity and independent of GC ligand binding [40]. It is shown to be induced by stressors such as H2O2, UV light, and hypoxia, and abolished by the use of a p38 inhibitor in various mammalian cells [40,41]. Furthermore, TGFβ1 can induce phosphorylation of S134 via activation of p38 in triple-negative breast cancer (TNBC) cells, and a functional S134 phospho-site was shown to be required for migration, growth in soft agar, and formation of tumor spheres [42]. Notably, the TGFβ1, as well as the stress-induced phosphorylation of S134, were independent of hormone stimulation, and the TGFβ1-stimulated S134 phosphorylation resulted in a positive feedback loop stimulating the p38 pathway [42]. Phosphorylation of S134 is further shown to enhance GR’s interaction with 14-3-3ζ and PELP1 and induce altered gene expression [40,41,42]. Interestingly, phosphorylation of S134 by AKT1 is reported to be inhibitory by preventing the nuclear translocation of GR [43], and the mechanism here is reported to be interaction with another member of the 14-3-3 family in the cytoplasm [44,45]. The application of an S134A mutant cell line additionally identified a specific TGFβ1-induced pS134 gene signature, indicating that this phosphorylation is necessary for the specific transcription of genes critical for MAPK signaling (e.g., MAP2K5) and cell migration [42].
The p38-dependent phosphorylation of GR seems to be induced by different stressors like nutrient deprivation, H2O2, hypoxia, and ROS [34,40,41,42], as well as signals associated with inflammation such as LPS, IL2/IL4, and Ilα [31,46,47]. The outcome of this phosphorylation is cell- and tissue-specific and dependent on the phospho-site(s) affected. A summary of the effects of the individual p38-mediated GR phosphorylation events is given in Figure 1. The phosphorylated S211, in general, seems to be associated with the activation of GR, supported by the fact that this site is regulated in a ligand-dependent manner [32,33]. However, it is also shown that the proportion of phosphorylated S211 compared to both pS203 [32,35] and pS226 [48,49] influences GR activity, and that differential phosphorylation influences the structure and activity of the AF1 domain [50] and the interaction with the coactivator GRIP-1 [51]. S203 phosphorylation may prevent nuclear translocation and is associated with decreased GR activity [35], whereas S211 phosphorylation increases nuclear translocation and interaction with transcriptional cofactors [34,49], thus promoting GR-mediated transcription. The phosphorylation status of GR influences which promoters it is recruited to [48], and the p38-dependent phosphorylation of GR is also reported to affect ligand binding affinity [31] and binding to scaffolding proteins such as the 14-3-3 family [40,42].
Finally, it should be mentioned that the p38 MAP kinase may also affect GR activity by indirect mechanisms. GAL4DBD fusion experiments performed in HeLa cells showed that the negative effect that activated p38 had on GR activity depended on the LBD/AF2 region of GR, where there are no phosphorylation sites for p38 [52].
2.2. GR Regulated p38 MAPK Phosphorylation and Dephosphorylation
It is well known that GCs can influence the MAPK signaling pathways, both by genomic and non-genomic means, and that this contributes to their anti-inflammatory attributes [53,54]. Studies show that ERK1/2, JNK, and p38 MAPK phosphorylation and activation can be affected after stimulation with glucocorticoids. Although the mechanism of this regulation is not fully elucidated, the GC-mediated increase in transcription of the MAPK phosphatase 1/Dual-specificity phosphatase 1 (MKP-1/DUSP-1) seems to play a major part [55]. DUSP1 belongs to a family of phosphatases dedicated to an inhibitory regulation of MAP kinase activity by dephosphorylation of the threonine and tyrosine residues in their activation loop [56]. Glucocorticoids can induce the abundance of DUSP1 through induction of mRNA expression, as well as prevention of proteasomal protein degradation [55]. The MAP kinases JNK and p38 are major substrates of DUSP1, and GCs may negatively regulate the inflammatory effect of both the p38 and the JNK signaling pathway in macrophages via induction of DUSP1 expression [57,58].
However, there are also reports indicating that GCs not only repress MAPK signaling, but that they are able to stimulate p38 activity as well. One of these mechanisms involves a GR-dependent increase in expression of the upstream p38 activators MKK3 [33] and MAP3K5 [42]. Dexamethasone stimulation of several lymphoid cell lines hereby caused a modest increase in MKK3 mRNA, and the positive effect on p38 activation was detected after 8 h or more. Notably, neither MKK6 nor MKK4 mRNA was affected [33]. Interestingly, in TNBC cell lines, upon stimulation with TGFβ1, unliganded GR was activated by a p38-mediated phosphorylation of S134 [42]. This activation caused an increase in both MAP3K5 mRNA and protein levels, and it was shown that the utilization of an S134A mutant cell line causes a decrease in MAP3K5 protein expression. The observations of these studies pointed towards the existence of a positive feed-forward loop between the p38 and the GR signaling pathways in TNBC cells [42].
Non-genomic and rapid effects of GCs on p38 MAPK activation have also been reported in several cell types [59,60,61]. In rat PC12 cells [59] and in rat primary cultured hippocampal cells [60], the GC-dependent activation of p38 was fast (within 5 min) and proved to be dependent on an active protein kinase C (PKC), since the PKC inhibitor Gø6976 would block this effect. Remarkably, the GR antagonist RU38486 (mifepristone) did not have any effect, indicating that GR is not involved in mediating this rapid GC-dependent activation of p38 in these cell types. However, in muscle cells, rapid activation of p38 by GCs has also been reported. In this case, the effect was mediated by GR localized in the membrane and extracellular matrix (ECM) [61] and was dependent on the muscle fiber type and the duration of the treatment. The GR antagonist RU38486 abolished this effect, as did a monoclonal antibody against the receptor, indicating that it was extracellularly located GR that mediated this. Since a Focal Adhesion Kinase inhibitor (FAK inhibitor 14) abolished the phosphorylation of p38, the signaling from GR to p38 seemed to go through FAK [61]. The authors also showed that GR can interact with laminin in the ECM, hypothesizing that GR being activated by GCs causes a structural change in laminin and an activation of FAK by integrins.
Finally, GR is reported to activate p38 indirectly by causing an increase in reactive oxygen species (ROS). In chondrocytes, treatment with dexamethasone caused higher levels of NOX4 expression, which thereby increased ROS and led to activation of p38. This further affected the expression of matrix metalloproteinase-13 (MMP-13) and induced apoptosis [62].
2.3. Clinical Aspects of p38 and GR Crosstalk
After the confirmation of a direct crosstalk between the p38 MAP kinase and the glucocorticoid receptor, several studies have been focusing on the importance and practical usage of this interaction in the clinic. The interaction between p38 signaling and GR activity may be different depending on the diseases and tissues affected. Because GCs have immunosuppressive and anti-inflammatory functions, synthetic glucocorticoids (e.g., dexamethasone and prednisolone) are widely used for the treatment of chronic inflammatory conditions including respiratory, autoimmune, and cutaneous diseases [63]. Inhibition of the pro-inflammatory effects of p38 activity is one of the mechanisms used by GR to achieve this [54]. The p38 activation seems also to be involved in the induction of apoptosis caused by GCs in leukemic cancer cells [33] and in chondrocytes [62]. However, a major problem with the use of GC in the clinic is GC resistance or decreased GC sensitivity, and several examples indicate that p38 activity contributes to this resistance [38,39,64]. For an extensive review on MAPKs (also including ERK and JNK) and their involvement in GC resistance, the reader is referred to [22]. GCs are the preferred therapy for asthma patients, and corticosteroid insensitivity, leading to the development of severe asthma and poor life quality of the patients, is one of the major limiting factors for successful treatment and prognosis [25,31]. Relevant data of patients with severe asthma suggested that p38 activation leads to the phosphorylation and inactivation of GR, followed by subsequent corticosteroid insensitivity. The application of a p38 inhibitor can restore GC sensitivity and has been shown in peripheral blood mononuclear and bronchial epithelial cells derived from patients with severe asthma and chronic obstructive pulmonary disease (COPD) [38,39,64]. The combination of p38 inhibitor and dexamethasone is also shown to have a beneficial effect unrelated to the restoration of GC sensitivity. This was illustrated in a recent study, where an additive anti-inflammatory effect was observed in a TNF-α stimulated human lung fibroblast cell line and in fibroblasts obtained from human lung tissue [65]. For patients with cystic fibrosis (CF), where GCs are used as anti-inflammatory drugs to reduce lung inflammation, occurring insensitivity is also a problem. Here again, treatment with p38 inhibitors can increase the responsiveness to glucocorticoids in CF bronchial epithelial cells [66].
The crosstalk between p38 and GR was also shown for conditions like neuropathic pain and pancreatitis. In the spinal cords of rats with spared nerve injury, activation of p38 leads to the downregulation of GR expression, activation of NF-κB signaling and the generation of an inflammatory reaction and neuropathic pain [67]. Furthermore, dexamethasone has been shown to inhibit p38 activity in mild but not in severe acute pancreatitis [68], and hydrocortisone was recently shown to inhibit the observed upregulation and activity of p38 evoked by a co-culture of pancreatic acinar and stellate cells [69]. Achieving deeper insights into the crosstalk between GR and p38 might therefore not only provide means to combat GC resistance but may also provide new therapy options for diseases like neuropathy and acute pancreatitis in the future.
2.4. Cancer-Related Interactions
Glucocorticoids are used as a first-line treatment to induce apoptosis in lymphoid cancers [20] but are also widely applied to treat secondary effects caused by chemotherapy and irradiation in various solid tumors [70,71].
It has long been known that activation of the p38 pathway has been linked to the GC-induced apoptosis in lymphoid cells [33]. In contrast to the treatment of asthma, where p38 activity is inhibited for the restoration of GC function, GC resistance in acute lymphoblastic leukemia (ALL) is relieved by activation of the p38 pathway. Anisomycin, a potent p38/JNK signaling activator, has for instance when used in a low dose in combination with dexamethasone been reported to sensitize the GC-resistant T cell leukemia cell line CEM-C1 [72]. This co-treatment activated GR, JNK, and p38, all of which were inhibited using GR antagonist RU486, suggesting that GR might be upstream of the p38/JNK pathway in this cell line [72].
The effect of GC treatment on solid tumors is disputed. While there are studies showing that utilization of GCs and/or high expression of GR has no effect on tumor development [73] or even a positive effect [74,75], several reports indicate a negative outcome correlated with poor prognosis and increased risk of metastasis in the patients [76,77,78,79]. Several papers by Carol Lange’s research group have shown that p38’s phosphorylation of S134 is more prominent in triple-negative breast cancer (TNBC) than in other breast cancer subtypes and that this phosphorylation mediates the GR/HIF-1 interaction and induced expression of the breast tumor kinase PTK6/Brk gene, which is associated with aggressive breast cancer development [41,80] Further, this group has shown that TGFβ1 is able to activate p38 and phosphorylate S134 and that this phosphorylation is essential for survival, migration, invasion, and stemness properties of the TNBC cancer cells [42]. GCs can interfere with chemotherapeutic treatment [81], and several of those treatments are able to activate the p38 pathway [82,83]. There are indications that the crosstalk between GR and p38 may play a part in chemotherapy resistance [80], and this should be followed up in future investigations.
3. Conclusions
The glucocorticoid receptor and the p38 kinase are both key mediators of mammalian stress responses. GR is directly regulated by the binding of stress hormones, the glucocorticoids, while p38 is regulated as part of a three-tier kinase cascade dependent on input from other stress-sensing receptors and molecules. More and more data indicate that these pathways do not operate independent of each other and that they in fact interact with and influence each other’s actions. GR mediates the regulation of genes that are either positive or negative regulators of the p38 signaling pathway, while p38, on the other hand, can mediate the phosphorylation of GR, influencing its localization and activity. The effects of activation of the p38 signaling pathway on GR-mediated transcription seems to clearly be cell- and tissue-specific. For example, a lack of inhibition of p38 activity by GC in asthmatic cells will result in GC resistance, while a failure of GC to induce activation of p38 will result in treatment resistance in leukemic cells. Thus, in order to treat this GC resistance, inhibition of p38 activity is required for asthmatic cells, whereas in leukemic cells, activation of p38 activity is beneficial.
In retrospect of the herein reviewed data, it can be summarized that the fine-tuned regulation of the stress response through both glucocorticoids and activation of the p38 pathway is important and that dysregulation of this response may be a consequence and cause of a wide range of diseases. Since the p38-GR crosstalk and its molecular effects are cell- and tissue-specific, detailed knowledge of the cell type specific interplay between these two stress-induced signaling pathways will be required for pinpointing the most optimal treatment strategies in the future.
Author Contributions
Writing—original draft preparation, L.Z., I.M. and O.M.S.; writing—review and editing, L.Z., I.M. and O.M.S.; funding acquisition, O.M.S. and I.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Northern Norway Regional Health Authorities (HelseNord), grant number HNF1547-20, and by UiT—the Arctic University of Norway.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeung, Y.T.; Aziz, F.; Guerrero-Castilla, A.; Argüelles, S. Signaling Pathways in Inflammation and Anti-inflammatory Therapies. Curr. Pharm. Des. 2018, 24, 1449–1484. [Google Scholar] [CrossRef] [PubMed]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Sanz-Ezquerro, J.J. p38γ and p38δ: From Spectators to Key Physiological Players. Trends Biochem. Sci. 2017, 42, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, Y.; Gu, Z.; Li, L.; Liu, Y.; Wang, L.; Su, L. p38 MAPK-MK2 pathway regulates the heat-stress-induced accumulation of reactive oxygen species that mediates apoptotic cell death in glial cells. Oncol. Lett. 2017, 15, 775–782. [Google Scholar] [CrossRef]
- Wang, J.; Wang, G.; Cheng, D.; Huang, S.; Chang, A.; Tan, X.; Wang, Q.; Zhao, S.; Wu, D.; Liu, A.T.; et al. Her2 promotes early dissemination of breast cancer by suppressing the p38-MK2-Hsp27 pathway that is targetable by Wip1 inhibition. Oncogene 2020, 39, 6313–6326. [Google Scholar] [CrossRef]
- Yong, H.-Y.; Koh, M.-S.; Moon, A. The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin. Investig. Drugs 2009, 18, 1893–1905. [Google Scholar] [CrossRef]
- Hölscher, C.; Gräb, J.; Hölscher, A.; Müller, A.L.; Schäfer, S.C.; Rybniker, J. Chemical p38 MAP kinase inhibition constrains tissue inflammation and improves antibiotic activity in Mycobacterium tuberculosis-infected mice. Sci. Rep. 2020, 10, 13629. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Weikum, E.R.; Knuesel, M.T.; Ortlund, E.A.; Yamamoto, K.R. Glucocorticoid receptor control of transcription: Precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 2017, 18, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Vockley, C.M.; D’Ippolito, A.M.; McDowell, I.C.; Majoros, W.H.; Safi, A.; Song, L.; Crawford, G.E.; Reddy, T.E. Direct GR Binding Sites Potentiate Clusters of TF Binding across the Human Genome. Cell 2016, 166, 1269–1281.e19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thormann, V.; Rothkegel, M.C.; Schöpflin, R.; Glaser, L.V.; Djuric, P.; Li, N.; Chung, H.R.; Schwahn, K.; Vingron, M.; Meijsing, S.H. Genomic dissection of enhancers uncovers principles of combinatorial regulation and cell type-specific wiring of enhancer-promoter contacts. Nucleic Acids Res. 2018, 46, 2868–2882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, N.Z.; Cidlowski, J. Glucocorticoid receptor isoforms generate transcription specificity. Trends Cell Biol. 2006, 16, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Oakley, R.H.; Sar, M.; Cidlowski, J.A. The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J. Biol. Chem. 1996, 271, 9550–9559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Ramírez, P.; Tliba, O. Glucocorticoid Receptor β (GRβ): Beyond Its Dominant-Negative Function. Int. J. Mol. Sci. 2021, 22, 3649. [Google Scholar] [CrossRef] [PubMed]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellner, S.; Kocabey, S.; Zhang, T.; Nekolla, K.; Hutten, S.; Krombach, F.; Liedl, T.; Rehberg, M. Dexamethasone-conjugated DNA nanotubes as anti-inflammatory agents in vivo. Biomaterials 2017, 134, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Sionov, R.V.; Spokoini, R.; Kfir-Erenfeld, S.; Cohen, O.; Yefenof, E. Chapter 6 Mechanisms Regulating the Susceptibility of Hematopoietic Malignancies to Glucocorticoid-Induced Apoptosis. Adv. Cancer Res. 2008, 101, 127–248. [Google Scholar] [CrossRef] [PubMed]
- Barnes, P.J. Development of New Drugs for COPD. Curr. Med. Chem. 2013, 20, 1531–1540. [Google Scholar] [CrossRef] [PubMed]
- Sevilla, L.M.; Jiménez-Panizo, A.; Alegre-Martí, A.; Estébanez-Perpiñá, E.; Caelles, C.; Pérez, P. Glucocorticoid Resistance: Interference between the Glucocorticoid Receptor and the MAPK Signalling Pathways. Int. J. Mol. Sci. 2021, 22, 10049. [Google Scholar] [CrossRef] [PubMed]
- Sousa, A.R.; Lane, S.J.; Cidlowski, J.A.; Staynov, D.Z.; Lee, T.H. Glucocorticoid resistance in asthma is associated with elevated in vivo expression of the glucocorticoid receptor β-isoform. J. Allergy Clin. Immunol. 2000, 105, 943–950. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Tao, N.; Zheng, L.; Sun, T. LL-37 restored glucocorticoid sensitivity impaired by virus dsRNA in lung. Int. Immunopharmacol. 2020, 79, 106057. [Google Scholar] [CrossRef] [PubMed]
- Henderson, I.; Caiazzo, E.; McSharry, C.; Guzik, T.J.; Maffia, P. Why do some asthma patients respond poorly to glucocorticoid therapy? Pharmacol. Res. 2020, 160, 105189. [Google Scholar] [CrossRef] [PubMed]
- Rider, L.; Hirasawa, N.; Santini, F.; Beaven, M.A. Activation of the mitogen-activated protein kinase cascade is suppressed by low concentrations of dexamethasone in mast cells. J. Immunol. 1996, 157, 2374–2380. [Google Scholar] [PubMed]
- Krstic, M.D.; Rogatsky, I.; Yamamoto, K.R.; Garabedian, M.J. Mitogen-activated and cyclin-dependent protein kinases selectively and differentially modulate transcriptional enhancement by the glucocorticoid receptor. Mol. Cell. Biol. 1997, 17, 3947–3954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swantek, J.L.; Cobb, M.H.; Geppert, T.D. Jun N-terminal kinase/stress-activated protein kinase (JNK/SAPK) is required for lipopolysaccharide stimulation of tumor necrosis factor alpha (TNF-alpha) translation: Glucocorticoids inhibit TNF-alpha translation by blocking JNK/SAPK. Mol. Cell. Biol. 1997, 17, 6274–6282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasa, M.; Brook, M.; Saklatvala, J.; Clark, A.R. Dexamethasone Destabilizes Cyclooxygenase 2 mRNA by Inhibiting Mitogen-Activated Protein Kinase p38. Mol. Cell. Biol. 2001, 21, 771–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Antagonism of glucocorticoid receptor transcriptional activation by the c-Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 1998, 95, 2050–2055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irusen, E.; Matthews, J.G.; Takahashi, A.; Barnes, P.J.; Chung, K.F.; Adcock, I. p38 Mitogen-activated protein kinase–induced glucocorticoid receptor phosphorylation reduces its activity: Role in steroid-insensitive asthma. J. Allergy Clin. Immunol. 2002, 109, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Frederick, J.; Garabedian, M.J. Deciphering the Phosphorylation “Code” of the Glucocorticoid Receptor In Vivo. J. Biol. Chem. 2002, 277, 26573–26580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.L.; Webb, M.S.; Copik, A.J.; Wang, Y.; Johnson, B.H.; Kumar, R.; Thompson, E.B. p38 Mitogen-Activated Protein Kinase (MAPK) Is a Key Mediator in Glucocorticoid-Induced Apoptosis of Lymphoid Cells: Correlation between p38 MAPK Activation and Site-Specific Phosphorylation of the Human Glucocorticoid Receptor at Serine 211. Mol. Endocrinol. 2005, 19, 1569–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nader, N.; Ng, S.S.M.; Lambrou, G.; Pervanidou, P.; Wang, Y.; Chrousos, G.P.; Kino, T. AMPK Regulates Metabolic Actions of Glucocorticoids by Phosphorylating the Glucocorticoid Receptor through p38 MAPK. Mol. Endocrinol. 2010, 24, 1748–1764. [Google Scholar] [CrossRef] [PubMed]
- Bouazza, B.; Debba-Pavard, M.; Amrani, Y.; Isaacs, L.; O’Connell, D.; Ahamed, S.; Formella, D.; Tliba, O. Basal p38 mitogen-activated protein kinase regulates unliganded glucocorticoid receptor function in airway smooth muscle cells. Am. J. Respir. Cell Mol. Biol. 2014, 50, 301–315. [Google Scholar] [PubMed]
- Itoh, M.; Adachi, M.; Yasui, H.; Takekawa, M.; Tanaka, H.; Imai, K. Nuclear Export of Glucocorticoid Receptor is Enhanced by c-Jun N-Terminal Kinase-Mediated Phosphorylation. Mol. Endocrinol. 2002, 16, 2382–2392. [Google Scholar] [CrossRef] [PubMed]
- Mercado, N.; To, Y.; Kobayashi, Y.; Adcock, I.; Barnes, P.J.; Ito, K. p38 Mitogen-Activated Protein Kinase-γ Inhibition by Long-Acting β2 Adrenergic Agonists Reversed Steroid Insensitivity in Severe Asthma. Mol. Pharmacol. 2011, 80, 1128–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercado, N.; Hakim, A.; Kobayashi, Y.; Meah, S.; Usmani, O.S.; Chung, K.F.; Barnes, P.J.; Ito, K. Restoration of Corticosteroid Sensitivity by p38 Mitogen Activated Protein Kinase Inhibition in Peripheral Blood Mononuclear Cells from Severe Asthma. PLoS ONE 2012, 7, e41582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lea, S.; Li, J.; Plumb, J.; Gaffey, K.; Mason, S.; Gaskell, R.; Harbron, C.; Singh, D. P38 MAPK and glucocorticoid receptor crosstalk in bronchial epithelial cells. Klin. Wochenschr. 2020, 98, 361–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliher-Beckley, A.J.; Williams, J.G.; Cidlowski, J. Ligand-Independent Phosphorylation of the Glucocorticoid Receptor Integrates Cellular Stress Pathways with Nuclear Receptor Signaling. Mol. Cell. Biol. 2011, 31, 4663–4675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan Anderson, T.M.; Ma, S.H.; Raj, G.V.; Cidlowski, J.A.; Helle, T.M.; Knutson, T.P.; Krutilina, R.I.; Seagroves, T.N.; Lange, C.A. Breast Tumor Kinase (Brk/PTK6) Is Induced by HIF, Glucocorticoid Receptor, and PELP1-Mediated Stress Signaling in Triple-Negative Breast Cancer. Cancer Res. 2016, 76, 1653–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerkvliet, C.P.; Dwyer, A.R.; Diep, C.H.; Oakley, R.H.; Liddle, C.; Cidlowski, J.A.; Lange, C.A. Glucocorticoid receptors are required effectors of TGFβ1-induced p38 MAPK signaling to advanced cancer phenotypes in triple-negative breast cancer. Breast Cancer Res. 2020, 22, 39. [Google Scholar] [CrossRef] [PubMed]
- Piovan, E.; Yu, J.; Tosello, V.; Herranz, D.; Ambesi-Impiombato, A.; Da Silva, A.C.; Sanchez-Martin, M.; Perez-Garcia, A.; Rigo, I.; Castillo, M.; et al. Direct Reversal of Glucocorticoid Resistance by AKT Inhibition in Acute Lymphoblastic Leukemia. Cancer Cell 2013, 24, 766–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habib, T.; Sadoun, A.; Nader, N.; Suzuki, S.; Liu, W.; Jithesh, P.V.; Kino, T. AKT1 has dual actions on the glucocorticoid receptor by cooperating with 14-3-3. Mol. Cell. Endocrinol. 2017, 439, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Kino, T.; Souvatzoglou, E.; De Martino, M.U.; Tsopanomihalu, M.; Wan, Y.; Chrousos, G.P. Protein 14-3-3sigma interacts with and favors cytoplasmic subcellular localization of the glucocorticoid receptor, acting as a negative regulator of the glucocorticoid signaling pathway. J. Biol. Chem. 2003, 278, 25651–25656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basta-Kaim, A.; Budziszewska, B.; Jaworska-Feil, L.; Tetich, M.; Kubera, M.; Zajicova, A.; Holan, V.; Lasoń, W. Effects of lipopolysaccharide and chlorpromazine on glucocorticoid receptor-mediated gene transcription and immunoreactivity: A possible involvement of p38-MAP kinase. Eur. Neuropsychopharmacol. 2004, 14, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wu, H.; Miller, A.H. Interleukin 1α (IL-1α) induced activation of p38 mitogen-activated protein kinase inhibits glucocorticoid receptor function. Mol. Psychiatry 2003, 9, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blind, R.D.; Garabedian, M.J. Differential recruitment of glucocorticoid receptor phospho-isoforms to glucocorticoid-induced genes. J. Steroid Biochem. Mol. Biol. 2008, 109, 150–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Dang, T.; Blind, R.D.; Wang, Z.; Cavasotto, C.N.; Hittelman, A.B.; Rogatsky, I.; Logan, S.K.; Garabedian, M.J. Glucocorticoid Receptor Phosphorylation Differentially Affects Target Gene Expression. Mol. Endocrinol. 2008, 22, 1754–1766. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.H.; McLaughlin, W.A.; Kumar, R. Site-specific phosphorylation regulates the structure and function of an intrinsically disordered domain of the glucocorticoid receptor. Sci. Rep. 2017, 7, 15440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avenant, C.; Kotitschke, A.; Hapgood, J.P. Glucocorticoid Receptor Phosphorylation Modulates Transcription Efficacy through GRIP-1 Recruitment. Biochemistry 2010, 49, 972–985. [Google Scholar] [CrossRef] [PubMed]
- Szatmáry, Z.; Garabedian, M.J.; Vilček, J. Inhibition of Glucocorticoid Receptor-mediated Transcriptional Activation by p38 Mitogen-activated Protein (MAP) Kinase. J. Biol. Chem. 2004, 279, 43708–43715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, I.; Berghe, W.V.; Vermeulen, L.; Yamamoto, K.R.; Haegeman, G.; De Bosscher, K. Crosstalk in Inflammation: The Interplay of Glucocorticoid Receptor-Based Mechanisms and Kinases and Phosphatases. Endocr. Rev. 2009, 30, 830–882. [Google Scholar] [CrossRef] [PubMed]
- Ayroldi, E.; Cannarile, L.; Migliorati, G.; Nocentini, G.; Delfino, D.V.; Riccardi, C. Mechanisms of the anti-inflammatory effects of glucocorticoids: Genomic and nongenomic interference with MAPK signaling pathways. FASEB J. 2012, 26, 4805–4820. [Google Scholar] [CrossRef] [PubMed]
- Kassel, O.; Sancono, A.; Krätzschmar, J.; Kreft, B.; Stassen, M.; Cato, A.C. Glucocorticoids inhibit MAP kinase via increased expression and decreased degradation of MKP-1. EMBO J. 2001, 20, 7108–7116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seternes, O.-M.; Kidger, A.M.; Keyse, S.M. Dual-specificity MAP kinase phosphatases in health and disease. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 124–143. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Brown, D.E.; Brewer, J.A.; Vogt, S.K.; Muglia, L.J. Macrophage glucocorticoid receptors regulate Toll-like receptor 4–mediated inflammatory responses by selective inhibition of p38 MAP kinase. Blood 2007, 109, 4313–4319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, S.M.; Lawrence, T.; Kleiman, A.; Warden, P.; Medghalchi, M.; Tuckermann, J.P.; Saklatvala, J.; Clark, A.R. Antiinflammatory effects of dexamethasone are partly dependent on induction of dual specificity phosphatase 1. J. Exp. Med. 2006, 203, 1883–1889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Qiu, J.; Wang, J.; Zhong, Y.; Zhu, J.; Chen, Y. Corticosterone-induced rapid phosphorylation of p38 and JNK mitogen-activated protein kinases in PC12 cells. FEBS Lett. 2001, 492, 210–214. [Google Scholar] [CrossRef] [Green Version]
- Qi, A.-Q.; Qiu, J.; Xiao, L.; Chen, Y.-Z. Rapid activation of JNK and p38 by glucocorticoids in primary cultured hippocampal cells. J. Neurosci. Res. 2005, 80, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Arthurton, L.; Akujuru, E.; Pearson, T.; Steverding, D.; Protasi, F.; Mutungi, G. Membrane glucocorticoid receptors are localised in the extracellular matrix and signal through the MAPK pathway in mammalian skeletal muscle fibres. J. Physiol. 2015, 593, 2679–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Cai, G.Q.; Peng, J.P.; Shen, C. Glucocorticoids induce apoptosis and matrix metalloproteinase-13 expression in chondrocytes through the NOX4/ROS/p38 MAPK pathway. J. Steroid Biochem. Mol. Biol. 2018, 181, 52–62. [Google Scholar] [CrossRef]
- Vandewalle, J.; Luypaert, A.; De Bosscher, K.; Libert, C. Therapeutic Mechanisms of Glucocorticoids. Trends Endocrinol. Metab. 2018, 29, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Bhavsar, P.K.; Khorasani, N.; Baker, J.; Johnson, M.; Chung, K.F. Reversal of corticosteroid insensitivity by p38 MAPK inhibition in peripheral blood mononuclear cells from COPD. Int. J. Chronic Obstr. Pulm. Dis. 2015, 10, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higham, A.; Singh, D. Dexamethasone and p38 MAPK inhibition of cytokine production from human lung fibroblasts. Fundam. Clin. Pharmacol. 2021, 35, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Rebeyrol, C.; Saint-Criq, V.; Guillot, L.; Riffault, L.; Corvol, H.; Chadelat, K.; Ray, D.; Clement, A.; Tabary, O.; Le Rouzic, P. Glucocorticoids reduce inflammation in cystic fibrosis bronchial epithelial cells. Cell. Signal. 2012, 24, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Xu, R.; Li, M.; Zhao, Q.; Ren, X.; Li, Z.; Cao, J.; Zang, W. Glucocorticoid receptor inhibit the activity of NF-κB through p38 signaling pathway in spinal cord in the spared nerve injury rats. Life Sci. 2018, 208, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Yubero, S.; Ramudo, L.; Manso, M.; De Dios, I. Mechanisms of dexamethasone-mediated chemokine down-regulation in mild and severe acute pancreatitis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2009, 1792, 1205–1211. [Google Scholar] [CrossRef] [PubMed]
- Bläuer, M.; Sand, J.; Laukkarinen, J. Regulation of p38 MAPK and glucocorticoid receptor activation by hydrocortisone in mono-and co-cultured pancreatic acinar and stellate cells. Pancreatology 2021, 21, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Roila, F.; Fatigoni, S. New antiemetic drugs. Ann. Oncol. 2006, 17, ii96–ii100. [Google Scholar] [CrossRef] [PubMed]
- Hesketh, P.J.; Kris, M.G.; Basch, E.; Bohlke, K.; Barbour, S.Y.; Clark-Snow, R.A.; Danso, M.A.; Dennis, K.; Dupuis, L.L.; Dusetzina, S.B.; et al. Antiemetics: ASCO Guideline Update. J. Clin. Oncol. 2020, 38, 2782–2797. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ge, J.; Li, Q.; Guo, X.; Gu, L.; Ma, Z.-G.; Li, X.-H.; Zhu, Y.-P. Low-dose anisomycin sensitizes glucocorticoid-resistant T-acute lymphoblastic leukemia CEM-C1 cells to dexamethasone-induced apoptosis through activation of glucocorticoid receptor and p38-MAPK/JNK. Leuk. Lymphoma 2014, 55, 2179–2188. [Google Scholar] [CrossRef] [PubMed]
- Keith, B.D. Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 2008, 8, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tonsing-Carter, E.; Hernandez, K.M.; Kim, C.; Harkless, R.V.; Oh, A.; Bowie, K.; West-Szymanski, D.C.; Betancourt-Ponce, M.A.; Green, B.D.; Lastra, R.R.; et al. Glucocorticoid receptor modulation decreases ER-positive breast cancer cell proliferation and suppresses wild-type and mutant ER chromatin association. Breast Cancer Res. BCR 2019, 21, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.-X.; Mu, D.-L.; Jin, K.-M.; Li, X.-Y.; Wang, D.-X. Perioperative Glucocorticoids are Associated with Improved Recurrence-Free Survival After Pancreatic Cancer Surgery: A Retrospective Cohort Study with Propensity Score-Matching. Ther. Clin. Risk Manag. 2021, 17, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, H.T.; Mellemkjaer, L.; Nielsen, G.L.; Baron, J.A.; Olsen, J.H.; Karagas, M.R. Skin Cancers and Non-Hodgkin Lymphoma Among Users of Systemic Glucocorticoids: A Population-Based Cohort Study. JNCI J. Natl. Cancer Inst. 2004, 96, 709–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obradovic, M.; Hamelin, B.; Manevski, N.; Couto, J.P.; Sethi, A.; Coissieux, M.-M.; Muenst, S.; Okamoto, R.; Kohler, H.; Schmidt, A.; et al. Glucocorticoids promote breast cancer metastasis. Nature 2019, 567, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, G.; Zhang, H.; Xiong, Q.; Cheng, F.; Wang, H.; Luo, J.; Zhang, Y.; Shi, P.; Xu, J.; et al. Dexamethasone enhances the lung metastasis of breast cancer via a PI3K-SGK1-CTGF pathway. Oncogene 2021, 40, 5367–5378. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aleksandrowicz, E.; Schönsiegel, F.; Gröner, D.; Bauer, N.; Nwaeburu, C.C.; Zhao, Z.; Gladkich, J.; Hoppe-Tichy, T.; Yefenof, E.; et al. Dexamethasone mediates pancreatic cancer progression by glucocorticoid receptor, TGFβ and JNK/AP-1. Cell Death Dis. 2017, 8, e3064. [Google Scholar] [CrossRef] [PubMed]
- Regan Anderson, T.M.; Ma, S.; Perez Kerkvliet, C.; Peng, Y.; Helle, T.M.; Krutilina, R.I.; Raj, G.V.; Cidlowski, J.A.; Ostrander, J.H.; Schwertfeger, K.L.; et al. Taxol Induces Brk-dependent Prosurvival Phenotypes in TNBC Cells through an AhR/GR/HIF-driven Signaling Axis. Mol. Cancer Res. MCR 2018, 16, 1761–1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mattern, J.; Büchler, M.W.; Herr, I. Cell Cycle Arrest by Glucocorticoids May Protect Normal Tissue and Solid Tumors from Cancer Therapy. Cancer Biol. Ther. 2007, 6, 1341–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- de Olano, N.; Koo, C.Y.; Monteiro, L.J.; Pinto, P.H.; Gomes, A.R.; Aligue, R.; Lam, E.W.-F. The p38 MAPK–MK2 Axis Regulates E2F1 and FOXM1 Expression after Epirubicin Treatment. Mol. Cancer Res. 2012, 10, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
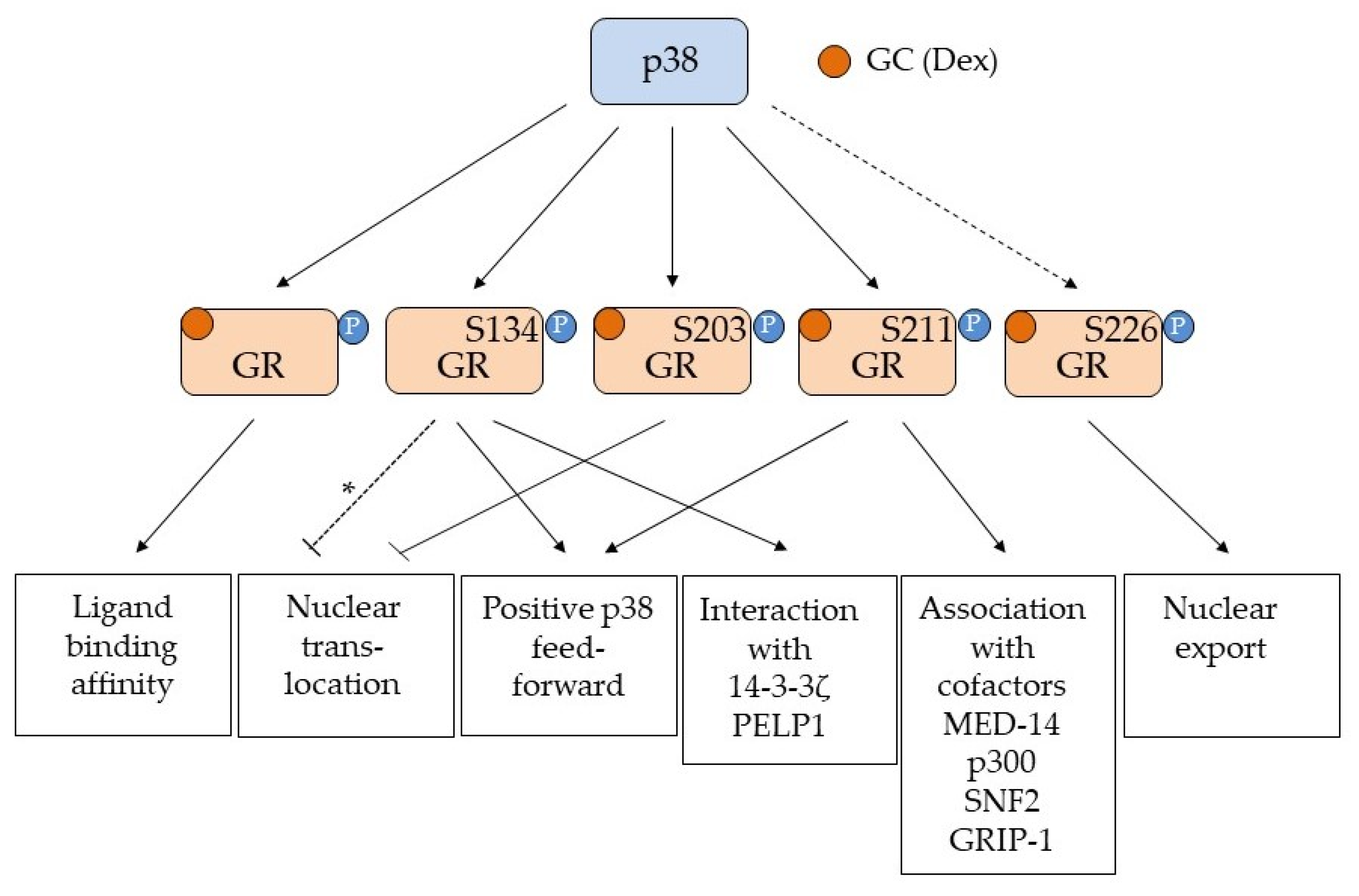
Figure 1.
Overview of the GR phosphorylation sites affected by p38 MAPK. Serine (S) 134, 203 and 211 are all phosphorylation sites shown to be affected by p38 MAPK activation. Serine (S) 226 is a JNK phosphorylation site known to enhance nuclear export [36] and is indicated (but not convincingly proven) to be a p38 MAPK phospho site [37,38,39] (stippled line). The various responses to the individual p38 phosphorylation events are shown in the boxes below. Arrows indicate activation/positive regulation, while a blunted line indicates inhibition. The GR ligand is indicated as GC (glucocorticoid) and Dex (Dexamethasone). For S134, a phosphorylation by AKT1 is reported to prevent nuclear translocation [43], but since this is not proven to be the effect of p38 phosphorylation of this site, it is marked with a stippled line with an asterisk (*). p38-dependent phosphorylation of unspecified residues in GR affects ligand binding [31], while pS134 is shown to be ligand independent and to generate a positive feed forward loop with p38 [33] as well as promoting interaction with 14-3-3ζ [40,42] and PELP1 [41]. p38 dependent phosphorylation of S203 prevents nuclear translocation [35], while pS211 is associated with ligand bound and activated GR and are important for GR’s interaction with transcriptional co-factors such as MED-14 [49], p300 [34], SNF2 [34] and GRIP-1 [51]. GR pS211 will also contribute to a positive feed forward loop with p38 [42]. The sum of all these phosphorylation events will affect the transcriptional regulation exerted by GR. Importantly the phosphorylation of S203, S211 and S226 seem to influence each other, and the relationship between the phosphorylation events for pS203 and pS211 [32,35,51] and pS211 and pS226 [48,49] will contribute to the selection of genes transcribed by GR in a cell and tissue specific manner.
Figure 1.
Overview of the GR phosphorylation sites affected by p38 MAPK. Serine (S) 134, 203 and 211 are all phosphorylation sites shown to be affected by p38 MAPK activation. Serine (S) 226 is a JNK phosphorylation site known to enhance nuclear export [36] and is indicated (but not convincingly proven) to be a p38 MAPK phospho site [37,38,39] (stippled line). The various responses to the individual p38 phosphorylation events are shown in the boxes below. Arrows indicate activation/positive regulation, while a blunted line indicates inhibition. The GR ligand is indicated as GC (glucocorticoid) and Dex (Dexamethasone). For S134, a phosphorylation by AKT1 is reported to prevent nuclear translocation [43], but since this is not proven to be the effect of p38 phosphorylation of this site, it is marked with a stippled line with an asterisk (*). p38-dependent phosphorylation of unspecified residues in GR affects ligand binding [31], while pS134 is shown to be ligand independent and to generate a positive feed forward loop with p38 [33] as well as promoting interaction with 14-3-3ζ [40,42] and PELP1 [41]. p38 dependent phosphorylation of S203 prevents nuclear translocation [35], while pS211 is associated with ligand bound and activated GR and are important for GR’s interaction with transcriptional co-factors such as MED-14 [49], p300 [34], SNF2 [34] and GRIP-1 [51]. GR pS211 will also contribute to a positive feed forward loop with p38 [42]. The sum of all these phosphorylation events will affect the transcriptional regulation exerted by GR. Importantly the phosphorylation of S203, S211 and S226 seem to influence each other, and the relationship between the phosphorylation events for pS203 and pS211 [32,35,51] and pS211 and pS226 [48,49] will contribute to the selection of genes transcribed by GR in a cell and tissue specific manner.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zeyen, L.; Seternes, O.M.; Mikkola, I. Crosstalk between p38 MAPK and GR Signaling. Int. J. Mol. Sci. 2022, 23, 3322. https://doi.org/10.3390/ijms23063322
AMA Style
Zeyen L, Seternes OM, Mikkola I. Crosstalk between p38 MAPK and GR Signaling. International Journal of Molecular Sciences. 2022; 23(6):3322. https://doi.org/10.3390/ijms23063322
Chicago/Turabian StyleZeyen, Lisa, Ole Morten Seternes, and Ingvild Mikkola. 2022. "Crosstalk between p38 MAPK and GR Signaling" International Journal of Molecular Sciences 23, no. 6: 3322. https://doi.org/10.3390/ijms23063322
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.