
A Histone Deacetylase (HDAC) Inhibitor with Pleiotropic In Vitro Anti-Toxoplasma and Anti-Plasmodium Activities Controls Acute and Chronic Toxoplasma Infection in Mice

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results
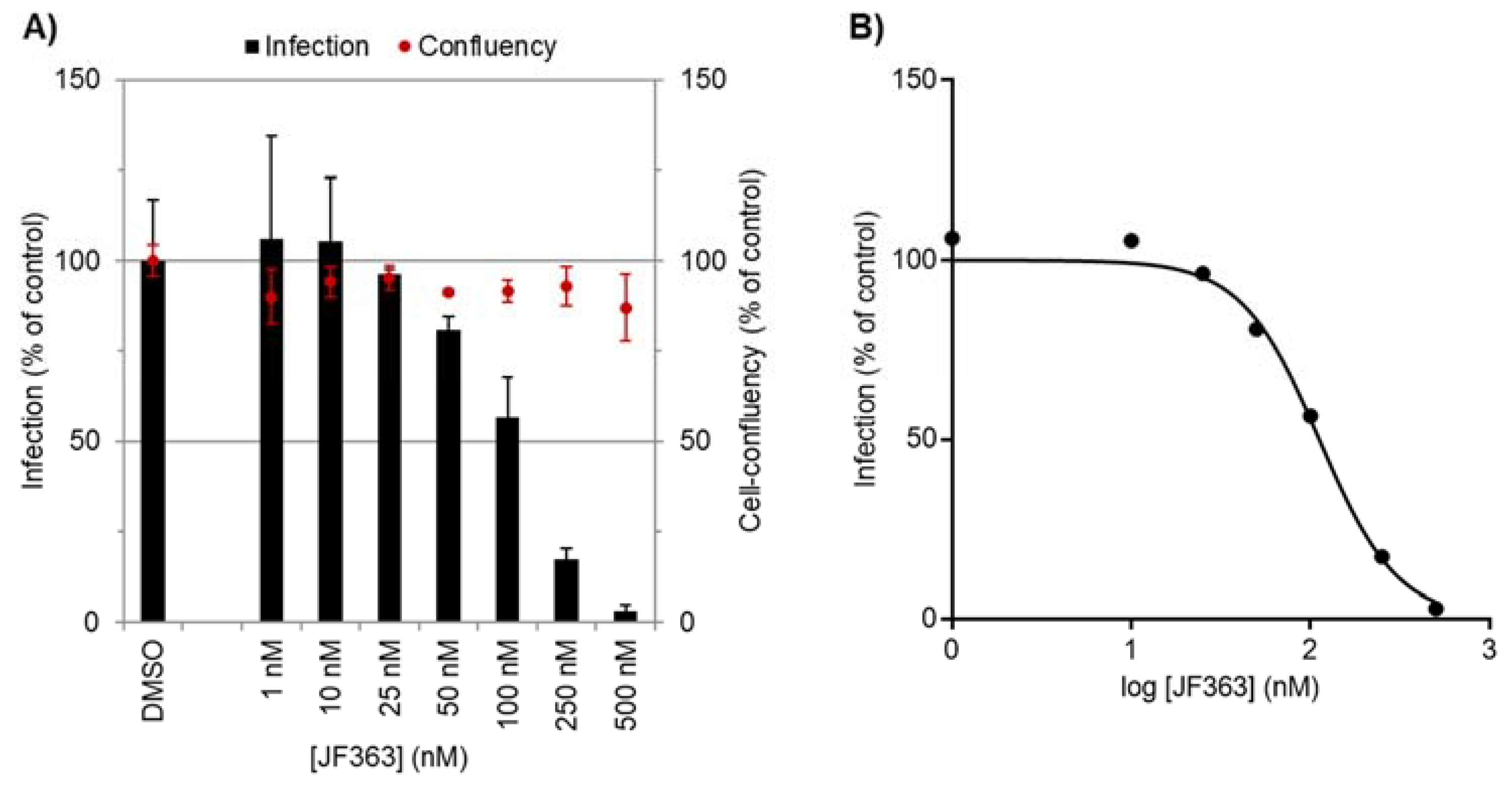
2.1. Anti-Proliferative Activity of JF363
2.1.1. Pleiotropic In Vitro Anti-Toxoplasma Activity
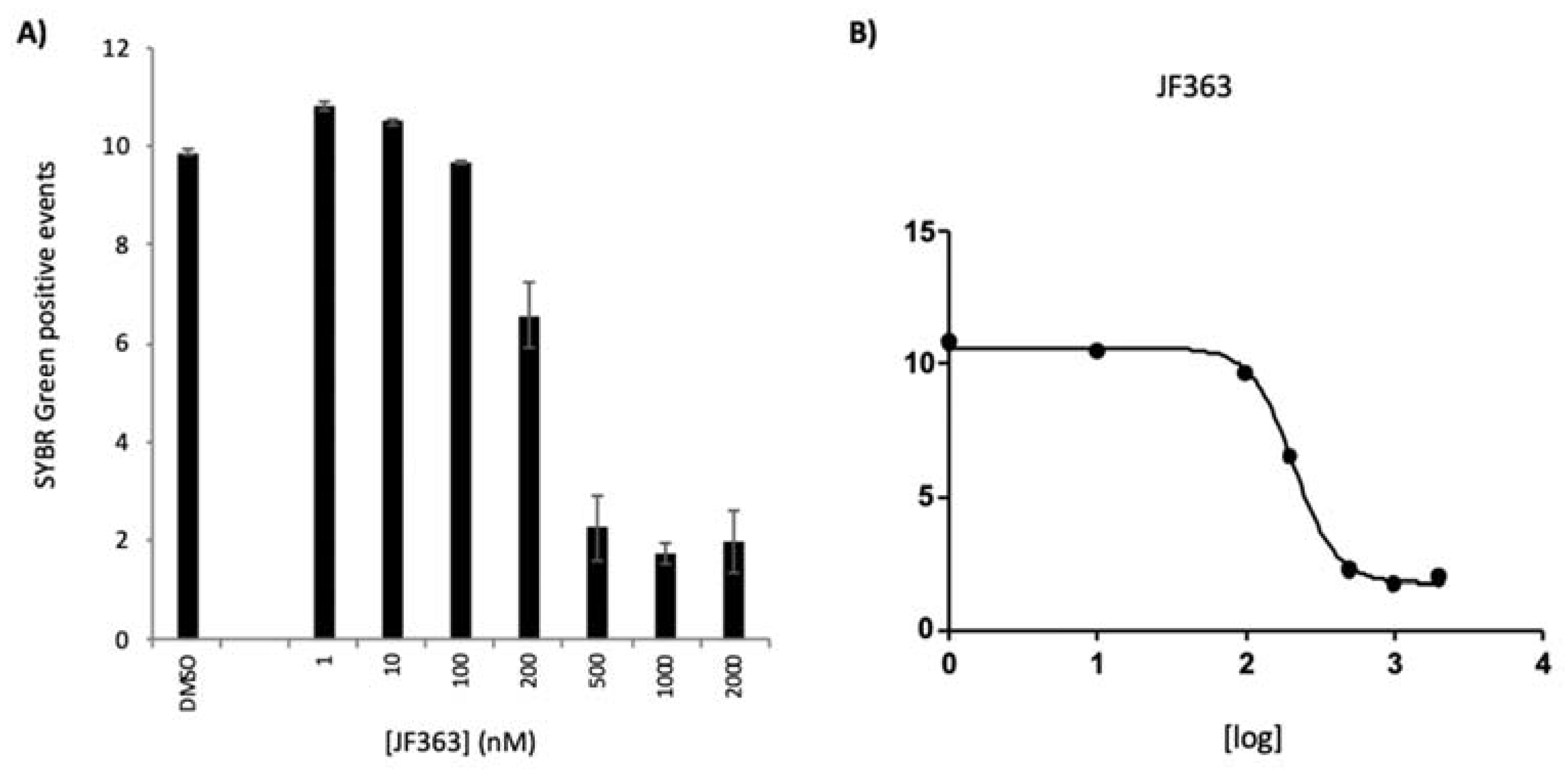
2.1.2. Anti-Plasmodium Activity
2.2. In Vivo Anti-Toxoplasma Activity
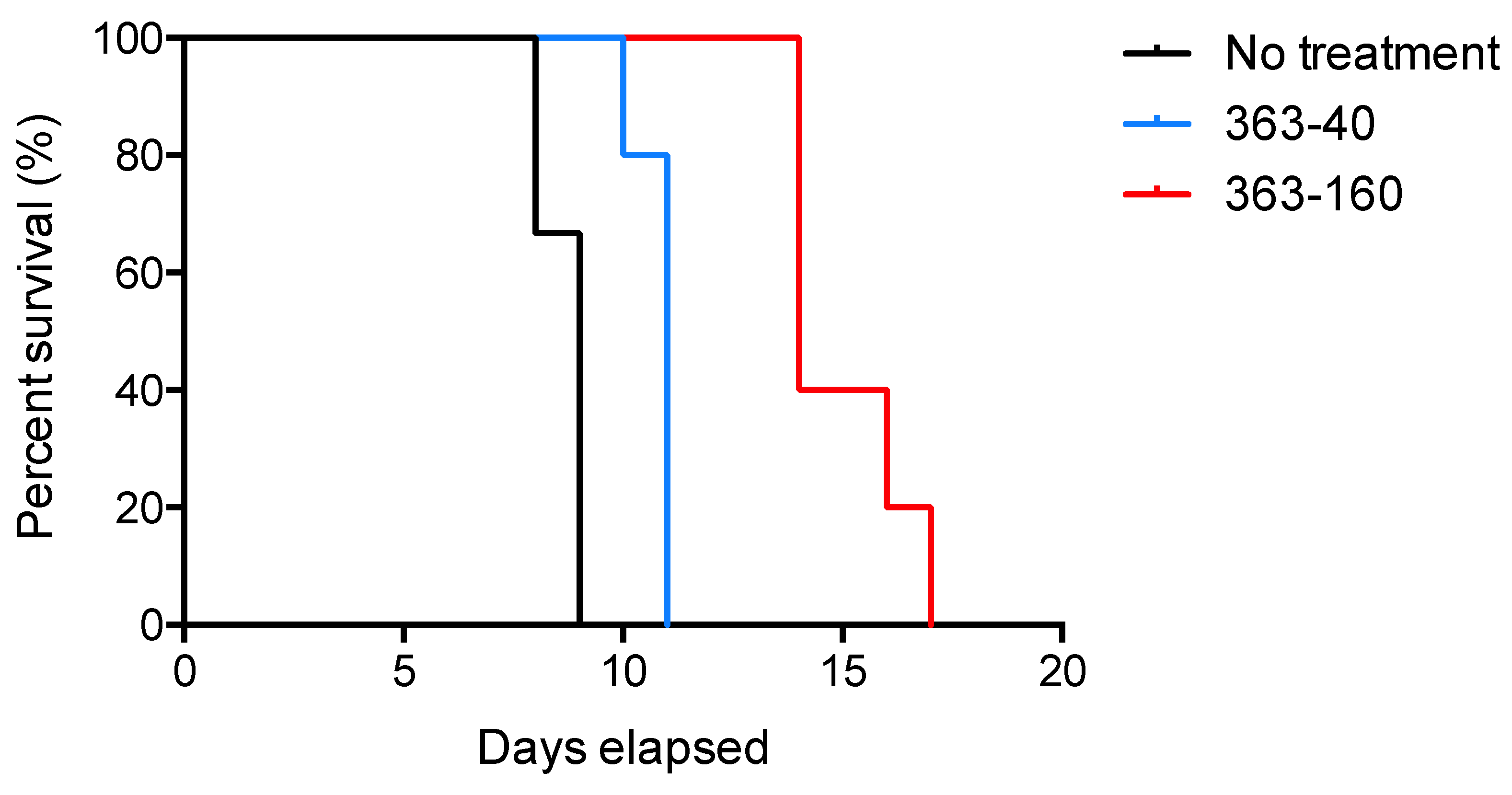
2.2.1. Effect of JF363 on Mouse Survival during the Acute Phase of T. gondii Infection
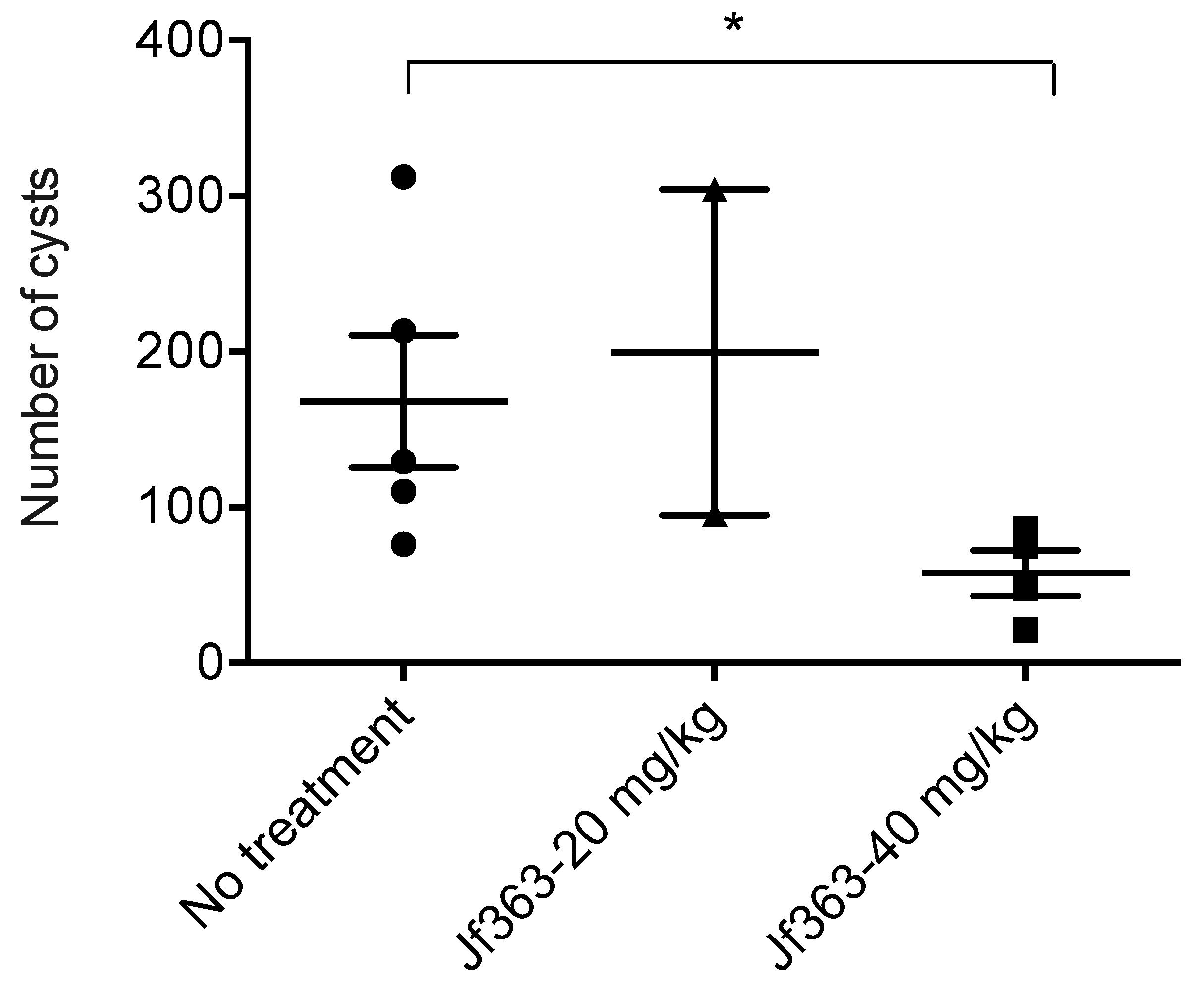
2.2.2. Effect of the JF363 on Cyst Formation in Chronically Infected Mice
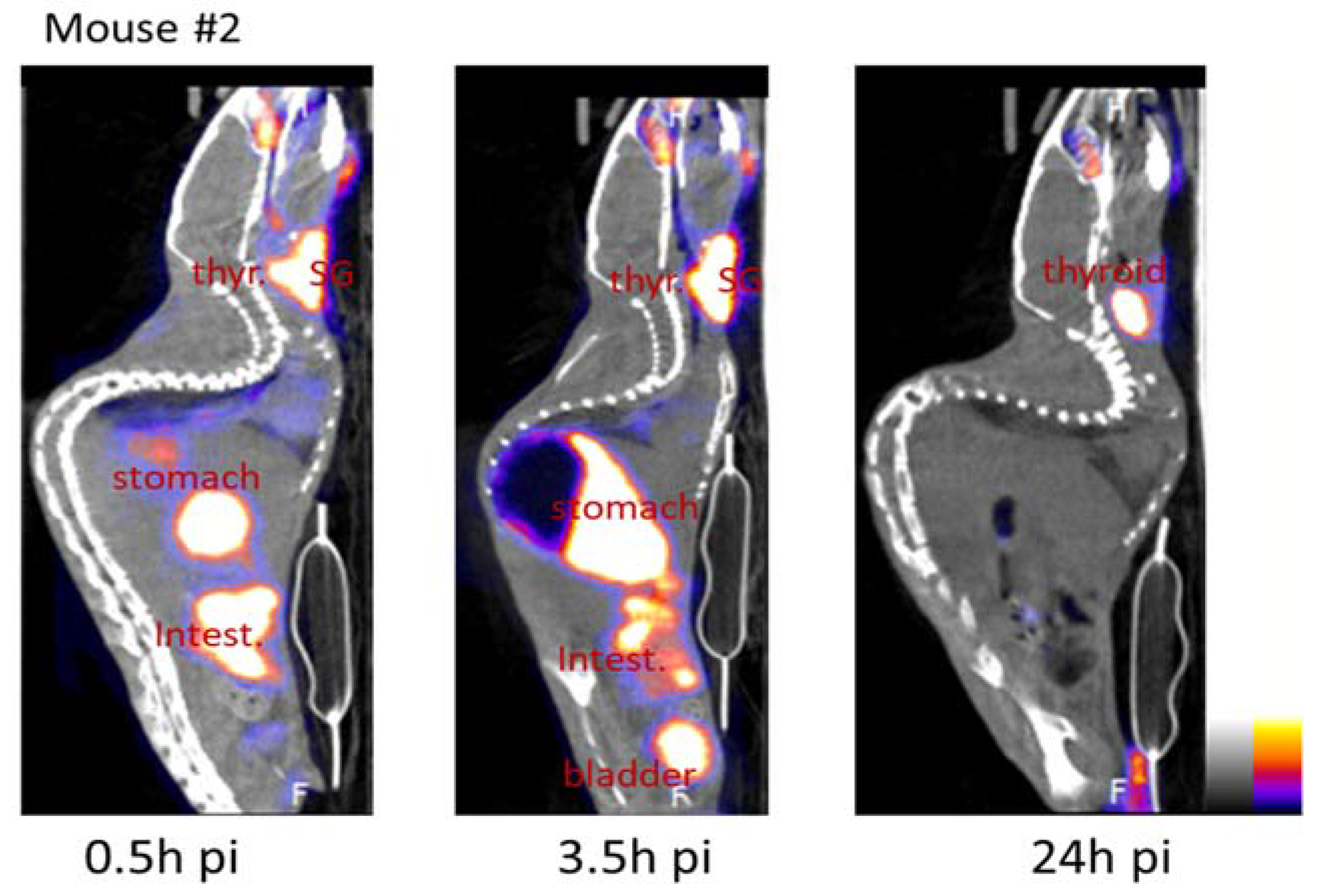
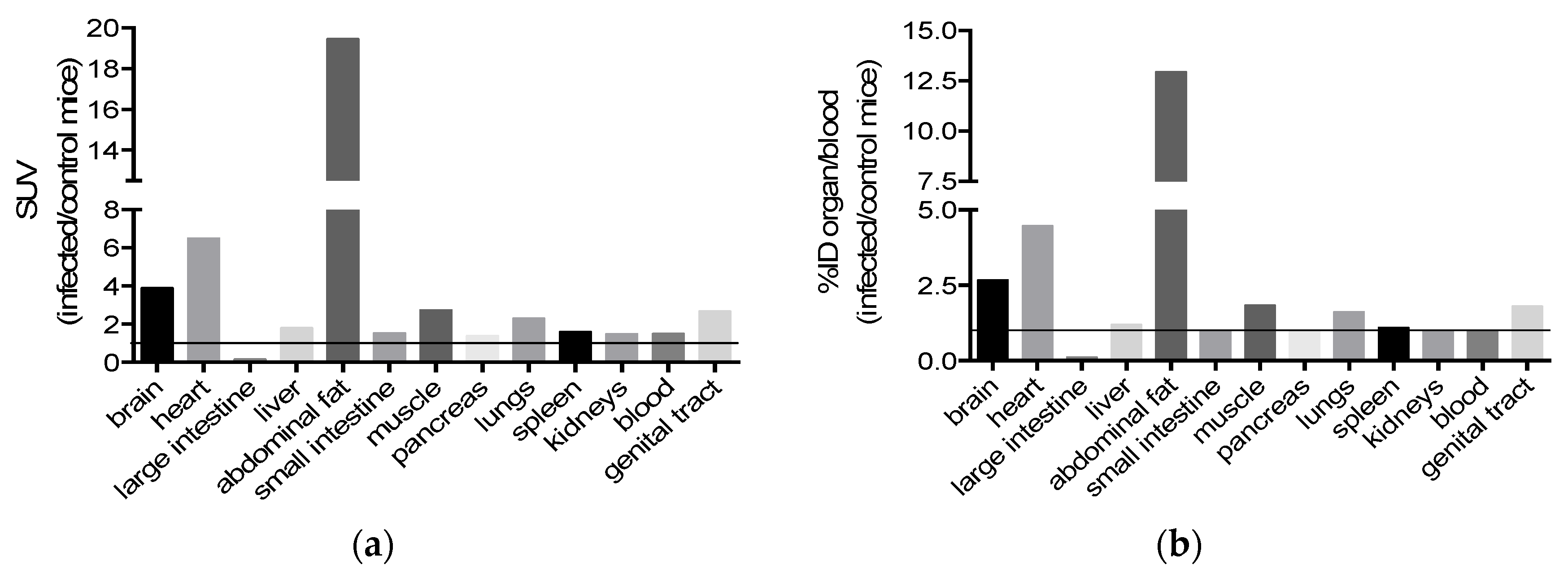
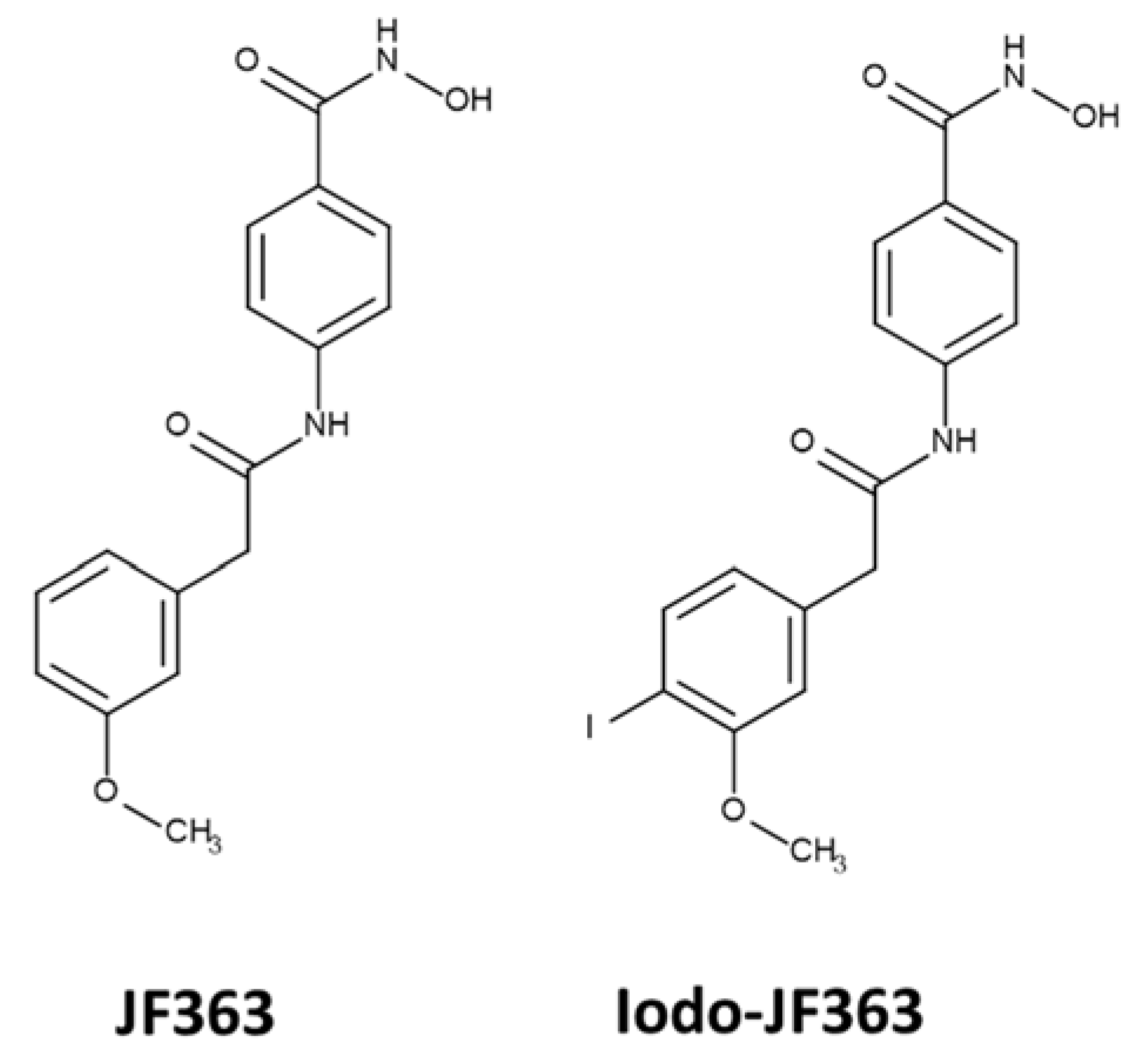
2.3. Bioavailability
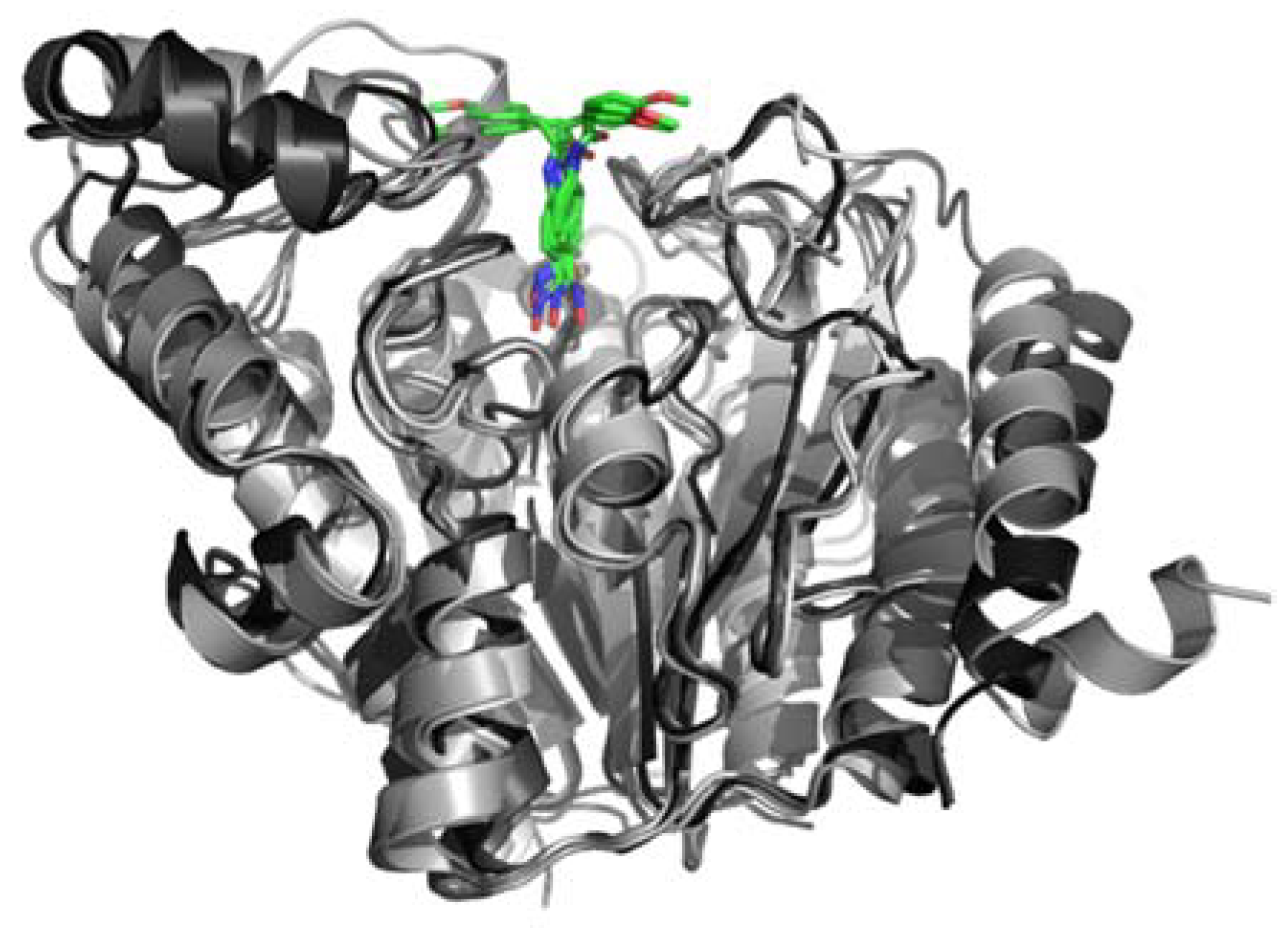
2.4. In Silico Modeling and Docking
3. Discussion
4. Materials and Methods
4.1. Parasites
4.2. Cell Culture and In Vitro Antimicrobial Test
4.2.1. In Vitro Anti-Toxoplasma Activity
4.2.2. In Vitro Anti-Plasmodium Activity
4.3. Acute Phase Study
4.4. Chronic Phase Study
4.5. In Vivo Biodisponibility
4.5.1. Synthesis of the Iodo-JF363 Compound
4.5.2. Radiolabeling with Iodine-125
4.5.3. SPECT/CT Imaging and Ex Vivo Biodistribution in Mice
4.6. Structure Modeling and Ligand Docking
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. World Malaria Report 2021. Available online: www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2021 (accessed on 20 December 2021).
- Zekar, L.; Sharman, T. Plasmodium falciparum malaria. In StatPearls; StatPearls PublishingCopyright © 2022; StatPearls Publishing LLC.: Treasure Island, FL, USA, 2021. [Google Scholar]
- Menard, D.; Dondorp, A. Antimalarial drug resistance: A threat to malaria elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molan, A.; Nosaka, K.; Hunter, M.; Wang, W. Global status of Toxoplasma gondii infection: Systematic review and prevalence snapshots. Trop. Biomed. 2019, 36, 898–925. [Google Scholar] [PubMed]
- Galal, L.; Hamidović, A.; Dardé, M.L.; Mercier, M. Diversity of Toxoplasma gondii strains at the global level and its determinants. Food Waterborne Parasitol. 2019, 15, e00052. [Google Scholar] [CrossRef]
- Dubey, J.P.; Jones, J.L. Toxoplasma gondii infection in humans and animals in the United States. Int. J. Parasitol. 2008, 38, 1257–1278. [Google Scholar] [CrossRef]
- Carme, B.; Demar, M.; Ajzenberg, D.; Dardé, M.L. Severe acquired toxoplasmosis caused by wild cycle of Toxoplasma gondii, French Guiana. Emerg. Infect. Dis. 2009, 15, 656–658. [Google Scholar] [CrossRef]
- Carme, B.; Aznar, C.; Motard, A.; Demar, M.; de Thoisy, B. Serologic survey of Toxoplasma gondii in noncarnivorous free-ranging neotropical mammals in French Guiana. Vector Borne Zoonotic Dis. 2002, 2, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Demar, M.; Ajzenberg, D.; Serrurier, B.; Dardé, M.L.; Carme, B. Atypical Toxoplasma gondii strain from a free-living jaguar (Panthera onca) in French Guiana. Am. J. Trop. Med. Hyg. 2008, 78, 195–197. [Google Scholar] [CrossRef]
- Carme, B.; Bissuel, F.; Ajzenberg, D.; Bouyne, R.; Aznar, C.; Demar, M.; Bichat, S.; Louvel, D.; Bourbigot, A.M.; Peneau, C.; et al. Severe acquired toxoplasmosis in immunocompetent adult patients in French Guiana. J. Clin. Microbiol. 2002, 40, 4037–4044. [Google Scholar] [CrossRef] [Green Version]
- Vyas, A. Mechanisms of Host Behavioral change in Toxoplasma gondii rodent association. PLoS Pathog. 2015, 11, e1004935. [Google Scholar] [CrossRef] [Green Version]
- Abdulai-Saiku, S.; Tong, W.H.; Vyas, A. Behavioral manipulation by Toxoplasma gondii: Does brain residence matter? Trends Parasitol. 2021, 37, 381–390. [Google Scholar] [CrossRef]
- Sutterland, A.L.; Fond, G.; Kuin, A.; Koeter, M.W.; Lutter, R.; van Gool, T.; Yolken, R.; Szoke, A.; Leboyer, M.; de Haan, L. Beyond the association. Toxoplasma gondii in schizophrenia, bipolar disorder, and addiction: Systematic review and meta-analysis. Acta Psychiatr. Scand. 2015, 132, 161–179. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.; Ramana, B.V. Schizophrenia and bipolar disorders: The Toxoplasma connection. Trop. Parasitol. 2019, 9, 71–76. [Google Scholar] [CrossRef]
- Ben-Harari, R.R.; Goodwin, E.; Casoy, J. Adverse event profile of pyrimethamine-based therapy in toxoplasmosis: A Systematic Review. Drugs R D 2017, 17, 523–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montazeri, M.; Mehrzadi, S.; Sharif, M.; Sarvi, S.; Tanzifi, A.; Aghayan, S.A.; Daryani, A. Drug resistance in Toxoplasma gondii. Front. Microbiol. 2018, 9, 2587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunay, I.R.; Gajurel, K.; Dhakal, R.; Liesenfeld, O.; Montoya, J.G. Treatment of toxoplasmosis: Historical perspective, animal models, and current clinical practice. Clin. Microbiol. Rev. 2018, 31, e00057-17. [Google Scholar] [CrossRef] [Green Version]
- Porter, S.B.; Sande, M.A. Toxoplasmosis of the central nervous system in the acquired immunodeficiency syndrome. N. Engl. J. Med. 1992, 327, 1643–1648. [Google Scholar] [CrossRef]
- Luft, B.J.; Remington, J.S. Toxoplasmic encephalitis in AIDS. Clin. Infect. Dis. 1992, 15, 211–222. [Google Scholar] [CrossRef]
- Montoya, J.G.; Liesenfeld, O. Toxoplasmosis. Lancet 2004, 363, 1965–1976. [Google Scholar] [CrossRef]
- Connolly, M.P.; Goodwin, E.; Schey, C.; Zummo, J. Toxoplasmic encephalitis relapse rates with pyrimethamine-based therapy: Systematic review and meta-analysis. Pathog. Glob. Health 2017, 111, 31–44. [Google Scholar] [CrossRef]
- Konstantinovic, N.; Guegan, H.; Stäjner, T.; Belaz, S.; Robert-Gangneux, F. Treatment of toxoplasmosis: Current options and future perspectives. Food Waterborne Parasitol. 2019, 15, e00036. [Google Scholar] [CrossRef]
- McFarland, M.M.; Zach, S.J.; Wang, X.; Potluri, L.P.; Neville, A.J.; Vennerstrom, J.L.; Davis, P.H. Review of experimental compounds demonstrating anti-Toxoplasma activity. Antimicrob. Agents Chemother. 2016, 60, 7017–7034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bougdour, A.; Braun, L.; Cannella, D.; Hakimi, M.A. Chromatin modifications: Implications in the regulation of gene expression in Toxoplasma gondii. Cell. Microbiol. 2010, 12, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Touquet, B.; Oury, B.; Eddaikra, N.; Pons, J.L.; Guichou, J.F.; Labesse, G.; Sereno, D. Synthesis of aminophenylhydroxamate and aminobenzylhydroxamate derivatives and in vitro screening for antiparasitic and histone deacetylase inhibitory activity. Int. J. Parasitol. Drugs Drug Resist. 2018, 8, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Loeuillet, C.; Touquet, B.; Guichou, J.F.; Labesse, G.; Sereno, D. A tiny change makes a big difference in the anti-Parasitic activities of an HDAC inhibitor. Int. J. Mol. Sci. 2019, 20, 2973. [Google Scholar] [CrossRef] [Green Version]
- Strobl, J.S.; Cassell, M.; Mitchell, S.M.; Reilly, C.M.; Lindsay, D.S. Scriptaid and suberoylanilide hydroxamic acid are histone deacetylase inhibitors with potent anti-Toxoplasma gondii activity in vitro. J. Parasitol. 2007, 93, 694–700. [Google Scholar] [CrossRef]
- Darkin-Rattray, S.J.; Gurnett, A.M.; Myers, R.W.; Dulski, P.M.; Crumley, T.M.; Allocco, J.J.; Cannova, C.; Meinke, P.T.; Colletti, S.L.; Bednarek, M.A.; et al. Apicidin: A novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc. Natl. Acad. Sci. USA 1996, 93, 13143–13147. [Google Scholar] [CrossRef] [Green Version]
- Maubon, D.; Bougdour, A.; Wong, Y.S.; Brenier-Pinchart, M.P.; Curt, A.; Hakimi, M.A.; Pelloux, H. Activity of the histone deacetylase inhibitor FR235222 on Toxoplasma gondii: Inhibition of stage conversion of the parasite cyst form and study of new derivative compounds. Antimicrob. Agents Chemother. 2010, 54, 4843–4850. [Google Scholar] [CrossRef] [Green Version]
- Araujo-Silva, C.A.; De Souza, W.; Martins-Duarte, E.S.; Vommaro, R.C. HDAC inhibitors Tubastatin A and SAHA affect parasite cell division and are potential anti-Toxoplasma gondii chemotherapeutics. Int. J. Parasitol. Drugs Drug Resist. 2021, 15, 25–35. [Google Scholar] [CrossRef]
- Mouveaux, T.; Rotili, D.; Boissavy, T.; Roger, E.; Pierrot, C.; Mai, A.; Gissot, M. A potent HDAC inhibitor blocks Toxoplasma gondii tachyzoite growth and profoundly disrupts parasite gene expression. Int. J. Antimicrob. Agents 2022, 3, 106526. [Google Scholar] [CrossRef]
- Bridges, J.F.; Critchlow, M.; Irving, M.P.; Purkiss, S.C.; Taylor, D.C.; Lloyd, J.B. Radiolabeling, stability, and body distribution in rats, of low molecular weight polylactide homopolymer and polylactide-polyethyleneglycol copolymer. Biomaterials 2000, 21, 199–209. [Google Scholar] [CrossRef]
- Butler, N.J.; Furtado, J.M.; Winthrop, K.L.; Smith, J.R. Ocular toxoplasmosis II: Clinical features, pathology and management. Clin. Exp. Ophthalmol. 2013, 41, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercier, A.; Ajzenberg, D.; Devillard, S.; Demar, M.P.; de Thoisy, B.; Bonnabau, H.; Collinet, F.; Boukhari, R.; Blanchet, D.; Simon, S.; et al. Human impact on genetic diversity of Toxoplasma gondii: Example of the anthropized environment from French Guiana. Infect. Genet. Evol. 2011, 11, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.C.; Andrade, G.M.; Costa, J.G.; Pinheiro, B.V.; Vasconcelos-Santos, D.V.; Ferreira, A.M.; Su, C.; Januário, J.N.; Vitor, R.W. Genetic characterization of Toxoplasma gondii revealed highly diverse genotypes for isolates from newborns with congenital toxoplasmosis in southeastern Brazil. J. Clin. Microbiol. 2013, 51, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.A.; Reis-Cunha, J.L.; Bartholomeu, D.C.; Vítor, R.W. Genetic polymorphisms and phenotypic profiles of sulfadiazine-resistant and sensitive Toxoplasma gondii isolates obtained from newborns with congenital toxoplasmosis in minas gerais, Brazil. PLoS ONE 2017, 12, e0170689. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.A.; Fernandes, M.D.; Machado, A.S.; Reis-Cunha, J.L.; Bartholomeu, D.C.; Almeida Vitor, R.W. Efficacy of sulfadiazine and pyrimetamine for treatment of experimental toxoplasmosis with strains obtained from human cases of congenital disease in Brazil. Exp. Parasitol. 2019, 202, 7–14. [Google Scholar] [CrossRef]
- Oliveira, C.B.; Meurer, Y.S.; Andrade, J.M.; Costa, M.E.; Andrade, M.M.; Silva, L.A.; Lanza, D.C.; Vítor, R.W.; Andrade-Neto, V.F. Pathogenicity and phenotypic sulfadiazine resistance of Toxoplasma gondii isolates obtained from livestock in northeastern Brazil. Mem. Inst. Oswaldo Cruz 2016, 111, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Derouin, F.; Lacroix, C.; Sumyuen, M.H.; Romand, S.; Garin, Y.J. Experimental models of toxoplasmosis. Pharmacological applications. Parasite 1995, 2, 243–256. [Google Scholar] [CrossRef] [Green Version]
- Nare, B.; Allocco, J.J.; Liberator, P.A.; Donald, R.G. Evaluation of a cyclic GMP-dependent protein kinase inhibitor in treatment of murine toxoplasmosis: Gamma interferon is required for efficacy. Antimicrob. Agents Chemother. 2002, 46, 300–307. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.L.; Si, H.F.; Shang, X.F.; Zhang, X.K.; Li, B.; Zhou, X.Z.; Zhang, J.Y. New life for an old drug: In vitro and in vivo effects of the anthelmintic drug niclosamide against Toxoplasma gondii RH strain. Int. J. Parasitol. Drugs Drug Resist. 2019, 9, 27–34. [Google Scholar] [CrossRef]
- Montazeri, M.; Sharif, M.; Sarvi, S.; Mehrzadi, S.; Ahmadpour, E.; Daryani, A. A Systematic Review of in vitro and in vivo activities of anti-Toxoplasma drugs and compounds (2006–2016). Front. Microbiol. 2017, 8, 25. [Google Scholar] [CrossRef] [Green Version]
- Takeda, M.; Imaizumi, M.; Sawano, S.; Manabe, Y.; Fushiki, T. Long-term optional ingestion of corn oil induces excessive caloric intake and obesity in mice. Nutrition 2001, 17, 117–120. [Google Scholar] [CrossRef]
- Ploemen, I.H.; Prudêncio, M.; Douradinha, B.G.; Ramesar, J.; Fonager, J.; van Gemert, G.J.; Luty, A.J.; Hermsen, C.C.; Sauerwein, R.W.; Baptista, F.G.; et al. Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS ONE 2009, 4, e7881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponnudurai, T.; Leeuwenberg, A.D.; Meuwissen, J.H. Chloroquine sensitivity of isolates of Plasmodium falciparum adapted to in vitro culture. Trop. Geogr. Med. 1981, 33, 50–54. [Google Scholar] [PubMed]
- Azevedo, R.; Markovic, M.; Machado, M.; Franke-Fayard, B.; Mendes, A.M.; Prudêncio, M. Bioluminescence method for in vitro screening of Plasmodium transmission-blocking compounds. Antimicrob. Agents Chemother. 2017, 61, e02699-16. [Google Scholar] [CrossRef] [Green Version]
- Sergent, V.; Cautain, B.; Khalife, J.; Deslée, D.; Bastien, P.; Dao, A.; Dubremetz, J.F.; Fournié, G.J.; Saoudi, A.; Cesbron-Delauw, M.F. Innate refractoriness of the Lewis rat to toxoplasmosis is a dominant trait that is intrinsic to bone marrow-derived cells. Infect. Immun. 2005, 73, 6990–6997. [Google Scholar] [CrossRef] [Green Version]
- Aldebert, D.; Hypolite, M.; Cavailles, P.; Touquet, B.; Flori, P.; Loeuillet, C.; Cesbron-Delauw, M.F. Development of high-throughput methods to quantify cysts of Toxoplasma gondii. Cytometry A 2011, 79, 952–958. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parasite Strain | Haplogroup | Clade | IC50 (μM) |
|---|---|---|---|
| RH-YFP | 1 | A | 0.56 ± 0.05 |
| Prugniaud | 2 | D | 0.27 ± 0.02 |
| VEG | 3 | C | 0.18 ± 0.07 |
| MAS | 4 | B | 0.21 ± 0.01 |
| GUY008-ABE | 5 | F | 0.30 ± 0.21 |
| GUY009-AKO | 10 | F | 0.43 ± 0.12 |
| GUY021-TOJ | 10 | F | 0.27 ± 0.03 |
| GUY-JAG1 | 11 | / | 0.17 ± 0.34 |
| Mouse #2 | Mouse #3 | ||||||
|---|---|---|---|---|---|---|---|
| Time Pi | 0.5 h | 3.5 h | 24 h | 0.5 h | 3.5 h | 24 h | |
| Organs | |||||||
| Stomach | 12.1 | 17.1 | 4.2 | 45.0 | 42.7 | 8.9 | |
| Intestine | 16.7 | 13.8 | 0.5 | 3.8 | 4.9 | 1.5 | |
| Salivary glands | 16.7 | 37.6 | 3.0 | 8.1 | 20.3 | 3.3 | |
| Thyroid | 28.9 | 18.8 | 26.6 | 4.1 | 9.9 | 29.5 | |
| Bladder | 5.5 | 19.1 | 0.6 | 6.4 | 8.2 | 3.9 | |
| Heart | 3.1 | 2.1 | 0.2 | 1.0 | 1.0 | 0.2 | |
| Lungs | 1.8 | 1.5 | 0.1 | 0.6 | 0.5 | 0.2 | |
| Mouth | 10.0 | 8.4 | 4.7 | 10.5 | 6.8 | 3.4 | |
| Neck ganglia | 8.5 | 9.5 | 1.3 | 4.0 | 5.0 | 1.1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jublot, D.; Cavaillès, P.; Kamche, S.; Francisco, D.; Fontinha, D.; Prudêncio, M.; Guichou, J.-F.; Labesse, G.; Sereno, D.; Loeuillet, C. A Histone Deacetylase (HDAC) Inhibitor with Pleiotropic In Vitro Anti-Toxoplasma and Anti-Plasmodium Activities Controls Acute and Chronic Toxoplasma Infection in Mice. Int. J. Mol. Sci. 2022, 23, 3254. https://doi.org/10.3390/ijms23063254
Jublot D, Cavaillès P, Kamche S, Francisco D, Fontinha D, Prudêncio M, Guichou J-F, Labesse G, Sereno D, Loeuillet C. A Histone Deacetylase (HDAC) Inhibitor with Pleiotropic In Vitro Anti-Toxoplasma and Anti-Plasmodium Activities Controls Acute and Chronic Toxoplasma Infection in Mice. International Journal of Molecular Sciences. 2022; 23(6):3254. https://doi.org/10.3390/ijms23063254
Chicago/Turabian StyleJublot, Delphine, Pierre Cavaillès, Salima Kamche, Denise Francisco, Diana Fontinha, Miguel Prudêncio, Jean-Francois Guichou, Gilles Labesse, Denis Sereno, and Corinne Loeuillet. 2022. "A Histone Deacetylase (HDAC) Inhibitor with Pleiotropic In Vitro Anti-Toxoplasma and Anti-Plasmodium Activities Controls Acute and Chronic Toxoplasma Infection in Mice" International Journal of Molecular Sciences 23, no. 6: 3254. https://doi.org/10.3390/ijms23063254