
Metabolic Response in Endothelial Cells to Catecholamine Stimulation Associated with Increased Vascular Permeability

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
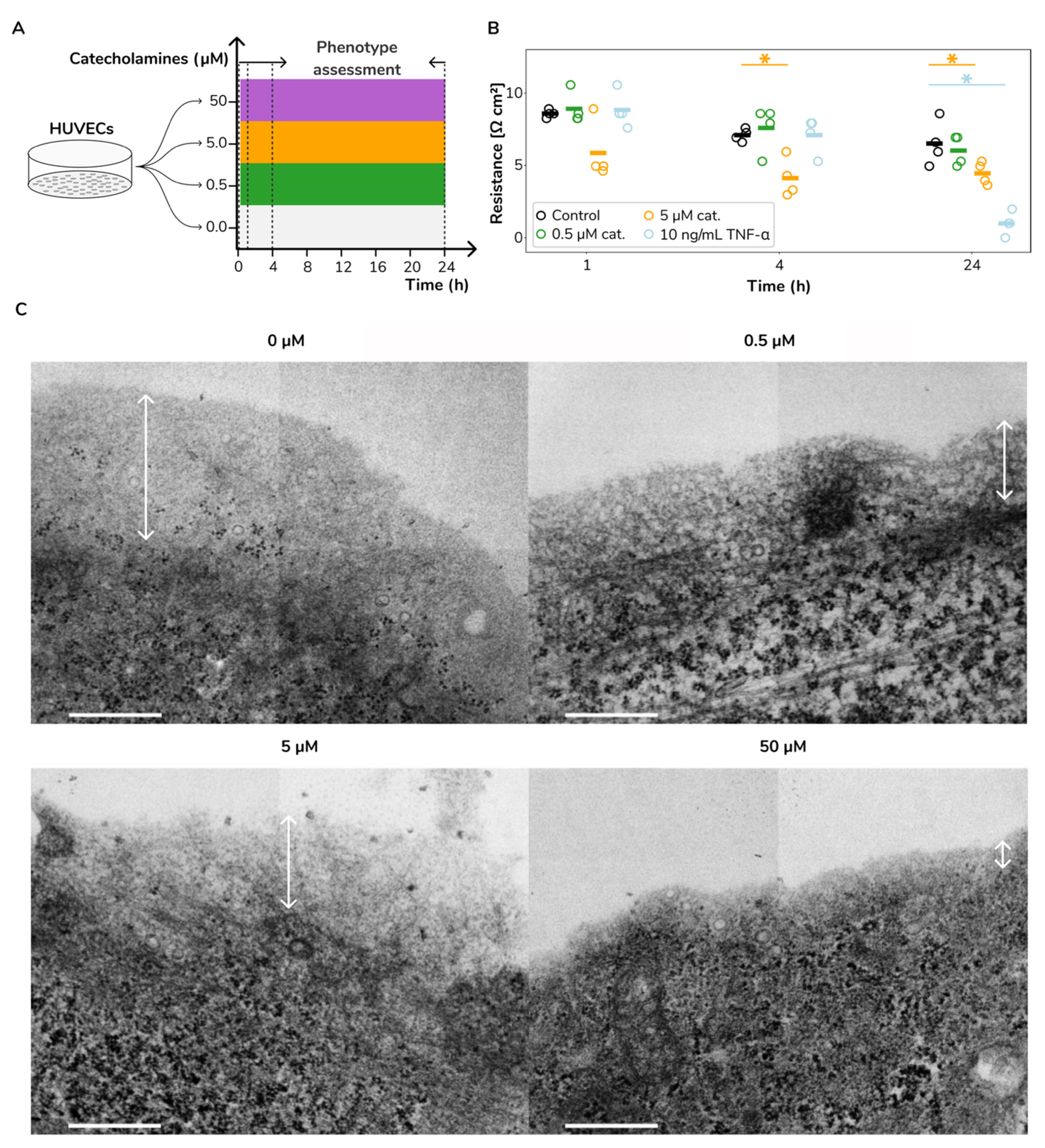
2.1. Catecholamine Stimulation Leads to Increased Vascular Permeability and Glycocalyx Loss in HUVECs
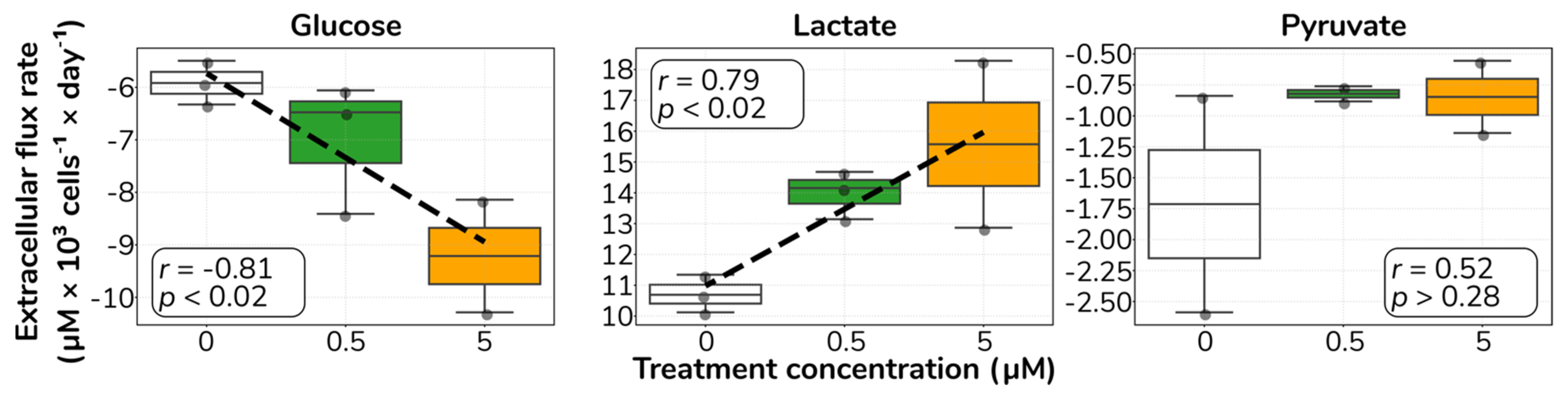
2.2. Catecholamine Stimulation Increases Glucose Uptake and Lactate Secretion in HUVECs
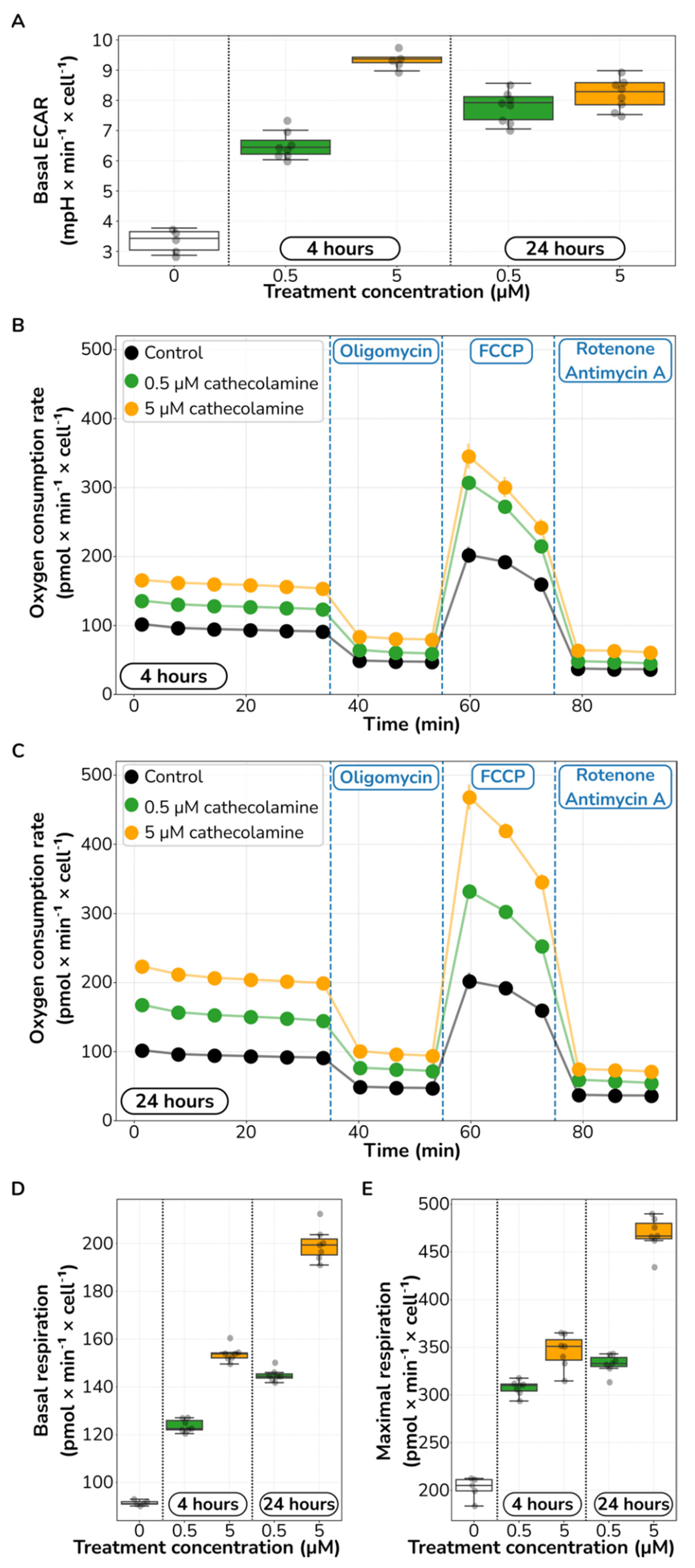
2.3. Catecholamine Stimulation Increases Glycolysis and Aerobic Respiration in HUVECs
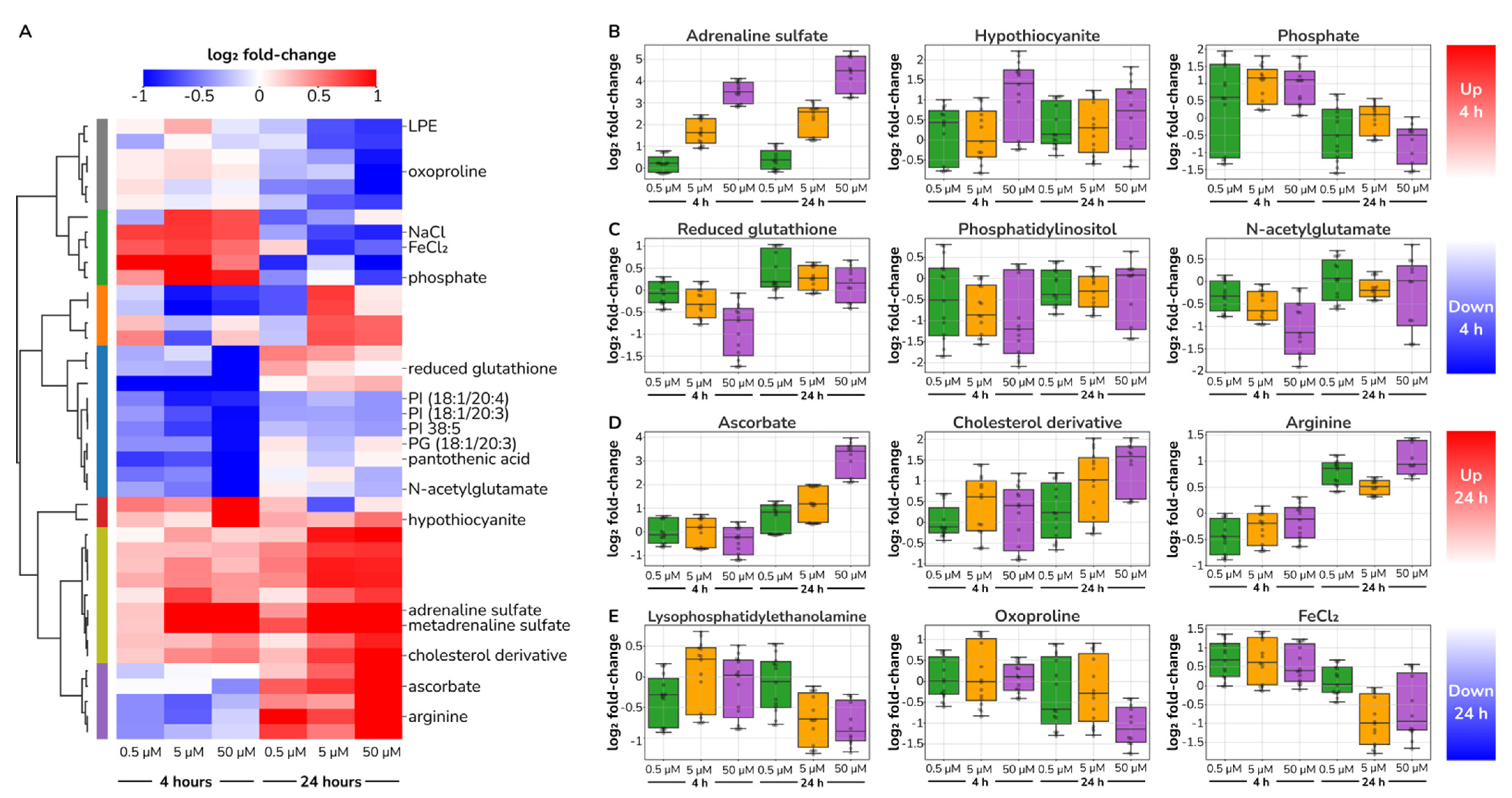
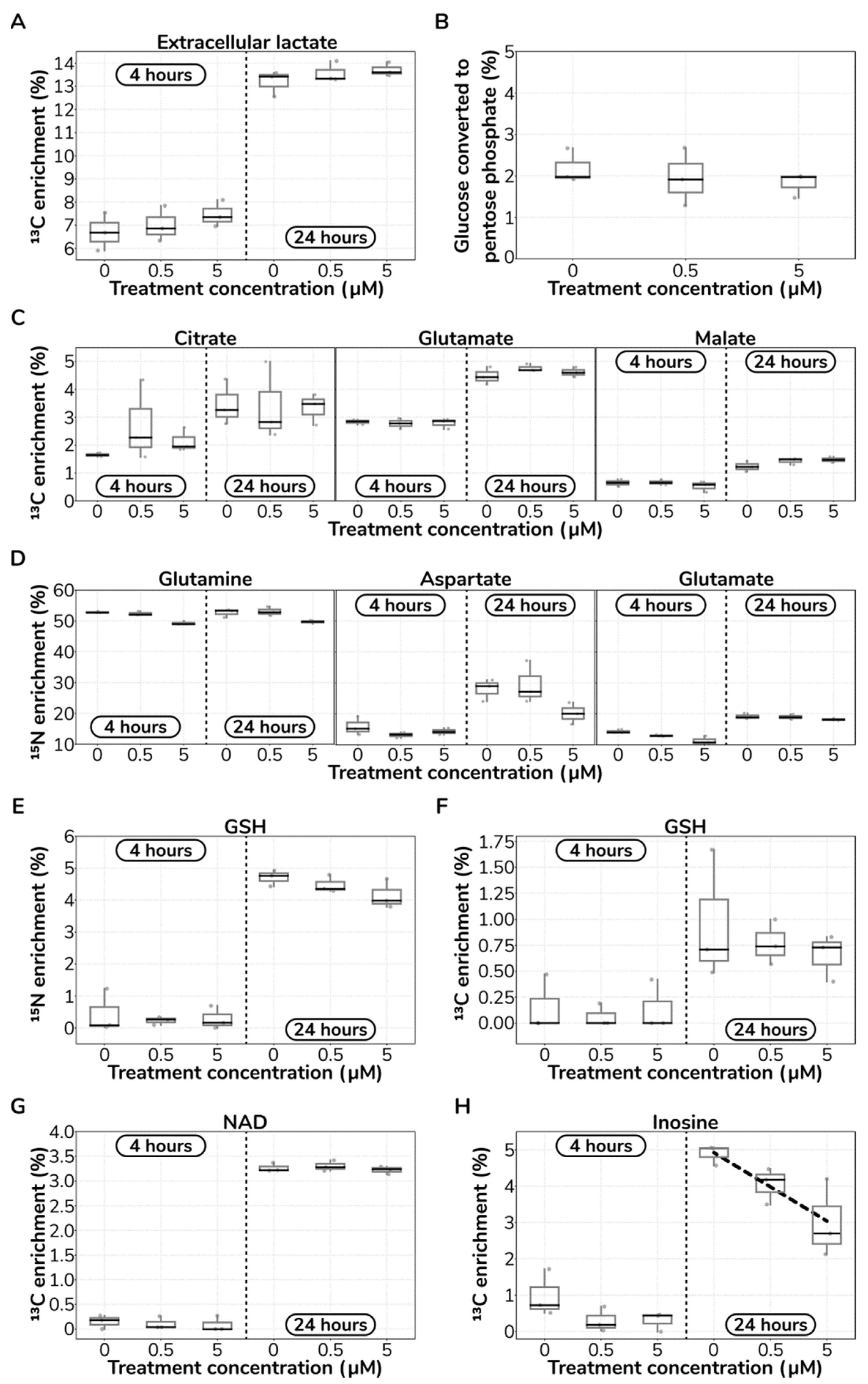
2.4. Catecholamine Dose-Responding Metabolites in HUVECs
2.5. Catecholamine Stimulation Does Not Reroute Core Metabolism in HUVECs
3. Discussion
4. Methods
4.1. Cell Culture
4.2. Endothelial Cell Culture Permeability Assay
4.3. Seahorse Assay
4.4. Biochemical Assays for Extracellular Transport Rates Measurements
4.5. Electron Microscope Imaging
4.6. Metabolomics
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aird, W.C. 3-Endothelium. In Consultative Hemostasis and Thrombosis, 3rd ed.; Kitchens, C.S., Kessler, C.M., Konkle, B.A., Eds.; W.B. Saunders: Philadelphia, PA, USA, 2013; pp. 33–41. [Google Scholar] [CrossRef]
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Blood Vessels and Endothelial Cells; Garland Science: New York, NY, USA, 2002. Available online: https://www.ncbi.nlm.nih.gov/books/NBK26848/ (accessed on 17 November 2020).
- Jaffe, E.A. Cell biology of endothelial cells. Hum. Pathol. 1987, 18, 234–239. [Google Scholar] [CrossRef]
- Verhamme, P.; Hoylaerts, M.F. The pivotal role of the endothelium in haemostasis and thrombosis. Acta Clin. Belg. 2006, 61, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Dejana, E.; McDonald, D.M. Permeability of the Endothelial Barrier: Identifying and Reconciling Controversies. Trends Mol. Med. 2021, 27, 314–331. [Google Scholar] [CrossRef]
- Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 2015, 15, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Trepels, T.; Zeiher, A.M.; Fichtlscherer, S. The endothelium and inflammation. Endothelium 2006, 13, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Tavora, B.; Mederer, T.; Wessel, K.J.; Ruffing, S.; Sadjadi, M.; Missmahl, M.; Ostendorf, B.N.; Liu, X.; Kim, J.-Y.; Olsen, O.; et al. Tumoural activation of TLR3-SLIT2 axis in endothelium drives metastasis. Nature 2020, 586, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef]
- Li, X.; Sun, X.; Carmeliet, P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metab. 2019, 30, 414–433. [Google Scholar] [CrossRef]
- Leung, S.W.S.; Shi, Y. The glycolytic process in endothelial cells and its implications. Acta Pharmacol. Sin. 2021, 43, 251–259. [Google Scholar] [CrossRef]
- Du, W.; Ren, L.; Hamblin, M.H.; Fan, Y. Endothelial Cell Glucose Metabolism and Angiogenesis. Biomedicines 2021, 9, 147. [Google Scholar] [CrossRef] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, B.; Li, J.; Jang, C.; Arany, Z. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J. 2017, 36, 2321–2333. [Google Scholar] [CrossRef] [PubMed]
- Kalucka, J.; Bierhansl, L.; Conchinha, N.V.; Missiaen, R.; Elia, I.; Brüning, U.; Scheinok, S.; Treps, L.; Cantelmo, A.R.; Dubois, C.; et al. Quiescent Endothelial Cells Upregulate Fatty Acid β-Oxidation for Vasculoprotection via Redox Homeostasis. Cell Metab. 2018, 28, 881–894.e13. [Google Scholar] [CrossRef] [Green Version]
- Patella, F.; Schug, Z.T.; Persi, E.; Neilson, L.J.; Erami, Z.; Avanzato, D.; Maione, F.; Hernandez-Fernaud, J.R.; Mackay, G.; Zheng, L.; et al. Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol. Cell Proteom. 2015, 14, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris, C.; Papageorgiou, N.; Stefanadis, C. The role of nitric oxide on endothelial function. Curr. Vasc. Pharmacol. 2012, 10, 4–18. [Google Scholar] [CrossRef]
- Franses, J.W.; Drosu, N.C.; Gibson, W.J.; Chitalia, V.C.; Edelman, E.R. Dysfunctional endothelial cells directly stimulate cancer inflammation and metastasis. Int. J. Cancer 2013, 133, 1334–1344. [Google Scholar] [CrossRef] [Green Version]
- Kaur, R.; Kaur, M.; Singh, J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: Molecular insights and therapeutic strategies. Cardiovasc. Diabetol. 2018, 17, 121. [Google Scholar] [CrossRef]
- Davignon, J.; Ganz, P. Role of endothelial dysfunction in atherosclerosis. Circulation 2004, 109, III27–III32. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Ostrowski, S.R. Shock induced endotheliopathy (SHINE) in acute critical illness—A unifying pathophysiologic mechanism. Crit. Care 2017, 21, 25. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, N.K.J.; Fowler, R.A.; Bhagwanjee, S.; Rubenfeld, G.D. Critical care and the global burden of critical illness in adults. Lancet 2010, 376, 1339–1346. [Google Scholar] [CrossRef]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. High circulating adrenaline levels at admission predict increased mortality after trauma. J. Trauma Acute Care Surg. 2012, 72, 428–436. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.V.; Liberati, D.M.; Diebel, L.N. Disparate effects of catecholamines under stress conditions on endothelial glycocalyx injury: An in vitro model. Am. J. Surg. 2017, 214, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Leite, A.R.; Borges-Canha, M.; Cardoso, R.; Neves, J.S.; Castro-Ferreira, R.; Leite-Moreira, A. Novel Biomarkers for Evaluation of Endothelial Dysfunction. Angiology 2020, 71, 397–410. [Google Scholar] [CrossRef]
- Gonzalez Rodriguez, E.; Ostrowski, S.R.; Cardenas, J.C.; Baer, L.A.; Tomasek, J.S.; Henriksen, H.H.; Stensballe, J.; Cotton, B.A.; Holcomb, J.B.; Johansson, P.I.; et al. Syndecan-1: A Quantitative Marker for the Endotheliopathy of Trauma. J. Am. Coll. Surg. 2017, 225, 419–427. [Google Scholar] [CrossRef] [Green Version]
- Jackson, N.C.; Carroll, P.V.; Russell-Jones, D.L.; Sönksen, P.H.; Treacher, D.F.; Umpleby, A.M. The metabolic consequences of critical illness: Acute effects on glutamine and protein metabolism. Am. J. Physiol. 1999, 276, E163–E170. [Google Scholar] [CrossRef]
- Mizock, B.A. Alterations in fuel metabolism in critical illness: Hyperglycaemia. Best Pract. Res. Clin. Endocrinol. Metab. 2001, 15, 533–551. [Google Scholar] [CrossRef]
- Kuehn, B.M. Guideline promotes early, aggressive sepsis treatment to boost survival. JAMA 2013, 309, 969–970. [Google Scholar] [CrossRef]
- Bruning, R.; Dykes, H.; Jones, T.W.; Wayne, N.B.; Sikora Newsome, A. Beta-Adrenergic Blockade in Critical Illness. Front. Pharmacol. 2021, 12, 735841. [Google Scholar] [CrossRef]
- Hu, Z.; Cano, I.; D’amore, P.A. Update on the role of the endothelial glycocalyx in angiogenesis and vascular inflammation. Front. Cell Dev. Biol. 2021, 9, 734276. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Jacob, M.; Paul, O.; Rehm, M.; Welsch, U.; Stoeckelhuber, M.; Conzen, P.; Becker, B.F. The Glycocalyx of the Human Umbilical Vein Endothelial Cell: An Impressive Structure Ex Vivo but Not in Culture. Circ. Res. 2009, 104, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.I.; Stensballe, J.; Rasmussen, L.S.; Ostrowski, S.R. A High Admission Syndecan-1 Level, a Marker of Endothelial Glycocalyx Degradation, Is Associated with Inflammation, Protein C Depletion, Fibrinolysis, and Increased Mortality in Trauma Patients. Ann. Surg. 2011, 254, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.R.; Pedersen, S.H.; Jensen, J.S.; Mogelvang, R.; Johansson, P.I. Acute myocardial infarction is associated with endothelial glycocalyx and cell damage and a parallel increase in circulating catecholamines. Crit. Care 2013, 17, R32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strott, C.A. Sulfonation and molecular action. Endocr. Rev. 2002, 23, 703–732. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ramírez, A.; Ortiz-Balderas, E.; Cardozo-Saldaña, G.; Diaz-Diaz, E.; El-Hafidi, M. Glycine restores glutathione and protects against oxidative stress in vascular tissue from sucrose-fed rats. Clin. Sci. 2014, 126, 19–29. [Google Scholar] [CrossRef]
- Espinosa-Díez, C.; Miguel, V.; Vallejo, S.; Sánchez, F.J.; Sandoval, E.; Blanco, E.; Cannata, P.; Peiró, C.; Sánchez-Ferrer, C.F.; Lamas, S. Role of glutathione biosynthesis in endothelial dysfunction and fibrosis. Redox Biol. 2018, 14, 88–99. [Google Scholar] [CrossRef]
- Bozonet, S.M.; Scott-Thomas, A.P.; Nagy, P.; Vissers, M.C.M. Hypothiocyanous acid is a potent inhibitor of apoptosis and caspase 3 activation in endothelial cells. Free Radic. Biol. Med. 2010, 49, 1054–1063. [Google Scholar] [CrossRef]
- Montecinos, V.; Guzmán, P.; Barra, V.; Villagrán, M.; Muñoz-Montesino, C.; Sotomayor, K.; Escobar, E.; Godoy, A.; Mardones, L.; Sotomayor, P.; et al. Vitamin C is an essential antioxidant that enhances survival of oxidatively stressed human vascular endothelial cells in the presence of a vast molar excess of glutathione. J. Biol. Chem. 2007, 282, 15506–15515. [Google Scholar] [CrossRef] [Green Version]
- Van Der Pol, A.; Gil, A.; Silljé, H.H.W.; Tromp, J.; Ovchinnikova, E.S.; Vreeswijk-Baudoin, I.; Hoes, M.; Domian, I.J.; Van De Sluis, B.; van Deursen, J.M.; et al. Accumulation of 5-Oxoproline in Myocardial Dysfunction and the Protective Effects of OPLAH. Sci. Transl. Med. 2017, 9, eaam8574. [Google Scholar] [CrossRef] [Green Version]
- Gambardella, J.; Khondkar, W.; Morelli, M.B.; Wang, X.; Santulli, G.; Trimarco, V. Arginine and Endothelial Function. Biomedicines 2020, 8, 277. [Google Scholar] [CrossRef] [PubMed]
- Kou, R.; Michel, T. Epinephrine regulation of the endothelial nitric-oxide synthase: Roles of RAC1 and β3-adrenergic receptors in endothelial NO signaling. J. Biol. Chem. 2007, 282, 32719–32729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seya, Y.; Fukuda, T.; Isobe, K.; Kawakami, Y.; Takekoshi, K. Effect of norepinephrine on RhoA, MAP kinase, proliferation and VEGF expression in human umbilical vein endothelial cells. Eur. J. Pharmacol. 2006, 553, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Queen, L.R.; Ji, Y.; Xu, B.; Young, L.; Yao, K.; Wyatt, A.W.; Rowlands, D.J.; Siow, R.C.M.; Mann, G.E.; Ferro, A. Mechanisms underlying beta2-adrenoceptor-mediated nitric oxide generation by human umbilical vein endothelial cells. J. Physiol. 2006, 576, 585–594. [Google Scholar] [CrossRef]
- Schena, M.; Mulatero, P.; Schiavone, D.; Mengozzi, G.; Tesio, L.; Chiandussi, L.; Veglio, F. Vasoactive hormones induce nitric oxide synthase mRNA expression and nitric oxide production in human endothelial cells and monocytes. Am. J. Hypertens. 1999, 12, 388–397. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.N.; Boros, L.G.; Puigjaner, J.; Bassilian, S.; Lim, S.; Cascante, M. Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am. J. Physiol. 1998, 274, E843–E851. [Google Scholar] [CrossRef]
- Mateuszuk, Ł.; Campagna, R.; Kutryb-Zając, B.; Kuś, K.; Słominska, E.M.; Smolenski, R.T.; Chlopicki, S. Reversal of endothelial dysfunction by nicotinamide mononucleotide via extracellular conversion to nicotinamide riboside. Biochem. Pharmacol. 2020, 178, 114019. [Google Scholar] [CrossRef]
- Beurskens, D.M.; Bol, M.E.; Delhaas, T.; van de Poll, M.C.; Reutelingsperger, C.P.; Nicolaes, G.A.; Sels, J.-W.E. Decreased endothelial glycocalyx thickness is an early predictor of mortality in sepsis. Anaesth. Intensive Care 2020, 48, 221–228. [Google Scholar] [CrossRef]
- Rahbar, E.; Cardenas, J.C.; Baimukanova, G.; Usadi, B.; Bruhn, R.; Pati, S.; Ostrowski, S.R.; Johansson, P.I.; Holcomb, J.B.; Wade, C.E. Endothelial glycocalyx shedding and vascular permeability in severely injured trauma patients. J. Transl. Med. 2015, 13, 117. [Google Scholar] [CrossRef] [Green Version]
- Wik, J.A.; Phung, D.; Kolan, S.; Haraldsen, G.; Skålhegg, B.S.; Hol Fosse, J. Inflammatory activation of endothelial cells increases glycolysis and oxygen consumption despite inhibiting cell proliferation. FEBS Open Bio 2021, 11, 1719–1730. [Google Scholar] [CrossRef]
- Ziogas, A.; Sajib, M.S.; Lim, J.-H.; Alves, T.C.; Das, A.; Witt, A.; Hagag, E.; Androulaki, N.; Grossklaus, S.; Gerlach, M.; et al. Glycolysis is integral to histamine-induced endothelial hyperpermeability. FASEB J. 2021, 35, e21425. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, Z.S.; Gokce, N.; Loscalzo, J. Mechanisms of oxidative stress and vascular dysfunction. Postgrad. Med. J. 2003, 79, 195–199, quiz 198–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Preiser, J.-C.; Ichai, C.; Orban, J.-C.; Groeneveld, A.B.J. Metabolic response to the stress of critical illness. Br. J. Anaesth. 2014, 113, 945–954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravi, S.; Gunawan, R. ΔFBA-Predicting metabolic flux alterations using genome-scale metabolic models and differential transcriptomic data. PLoS Comput. Biol. 2021, 17, e1009589. [Google Scholar] [CrossRef]
- Johansson, P.I.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Ostrowski, S.R. Traumatic Endotheliopathy: A Prospective Observational Study of 424 Severely Injured Patients. Ann. Surg. 2017, 265, 597–603. [Google Scholar] [CrossRef] [Green Version]
- Ostrowski, S.R.; Henriksen, H.H.; Stensballe, J.; Gybel-Brask, M.; Cardenas, J.C.; Baer, L.A.; Cotton, B.A.; Holcomb, J.B.; Wade, C.E.; Johansson, P.I. Sympathoadrenal activation and endotheliopathy are drivers of hypocoagulability and hyperfibrinolysis in trauma: A prospective observational study of 404 severely injured patients. J. Trauma Acute Care Surg. 2017, 82, 293–301. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Y.; Gorshkov, B.; Zhou, Y.; Yang, Q.; Xu, J.; Ma, Q.; Zhang, X.; Wang, J.; Mao, X.; et al. Ablation of endothelial Pfkfb3 protects mice from acute lung injury in LPS-induced endotoxemia. Pharmacol. Res. 2019, 146, 104292. [Google Scholar] [CrossRef]
- Sigmarsdottir, T.B.; McGarrity, S.; de Lomana, A.L.G.; Kotronoulas, A.; Sigurdsson, S.; Yurkovich, J.T.; Rolfsson, O.; Sigurjonsson, O.E. Metabolic and Transcriptional Changes across Osteogenic Differentiation of Mesenchymal Stromal Cells. Bioengineering 2021, 8, 208. [Google Scholar] [CrossRef]
- Liu, Z.; Ruter, D.L.; Quigley, K.; Tanke, N.T.; Jiang, Y.; Bautch, V.L. Single-Cell RNA Sequencing Reveals Endothelial Cell Transcriptome Heterogeneity Under Homeostatic Laminar Flow. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 2575–2584. [Google Scholar] [CrossRef]
- Burns, M.P.; DePaola, N. Flow-conditioned HUVECs support clustered leukocyte adhesion by coexpressing ICAM-1 and E-selectin. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H194–H204. [Google Scholar] [CrossRef]
- Noris, M.; Morigi, M.; Donadelli, R.; Aiello, S.; Foppolo, M.; Todeschini, M.; Orisio, S.; Remuzzi, G.; Remuzzi, A. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ. Res. 1995, 76, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Halldórsson, H.; Thors, B.; Thorgeirsson, G. Thrombin or Ca(++)-ionophore-mediated fall in endothelial ATP levels independent of poly(ADP-Ribose) polymerase activity and NAD levels--comparison with the effects of hydrogen peroxide. Nucleosides Nucleotides Nucleic Acids 2015, 34, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.A.; Nachman, R.L.; Becker, C.G.; Richard Minick, C. Culture of Human Endothelial Cells Derived from Umbilical Veins. identification by morphologic and immunologic criteria. J. Clin. Investig. 1973, 52, 2745–2756. [Google Scholar] [CrossRef] [PubMed]
- Paglia, G.; Hrafnsdóttir, S.; Magnúsdóttir, M.; Fleming, R.M.T.; Thorlacius, S.; Palsson, B.Ø.; Thiele, I. Monitoring metabolites consumption and secretion in cultured cells using ultra-performance liquid chromatography quadrupole-time of flight mass spectrometry (UPLC-Q-ToF-MS). Anal. Bioanal. Chem. 2012, 402, 1183–1198. [Google Scholar] [CrossRef] [Green Version]
- Paglia, G.; Williams, J.P.; Menikarachchi, L.; Thompson, J.W.; Tyldesley-Worster, R.; Halldórsson, S.; Rolfsson, O.; Moseley, A.; Grant, D.; Langridge, J.; et al. Ion mobility derived collision cross sections to support metabolomics applications. Anal. Chem. 2014, 86, 3985–3993. [Google Scholar] [CrossRef] [Green Version]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, G.; Simpson, J.A.; Creek, D.J. NormalizeMets: Assessing, selecting and implementing statistical methods for normalizing metabolomics data. Metabolomics 2018, 14, 54. [Google Scholar] [CrossRef]
- Smith, C.A.; O’Maille, G.; Want, E.J.; Qin, C.; Trauger, S.A.; Brandon, T.R.; Custodio, D.E.; Abagyan, R.; Siuzdak, G. METLIN: A metabolite mass spectral database. Ther. Drug Monit. 2005, 27, 747–751. [Google Scholar] [CrossRef]
- Millard, P.; Letisse, F.; Sokol, S.; Portais, J.-C. IsoCor: Correcting MS data in isotope labeling experiments. Bioinformatics 2012, 28, 1294–1296. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
López García de Lomana, A.; Vilhjálmsson, A.I.; McGarrity, S.; Sigurðardóttir, R.; Anuforo, Ó.; Viktorsdóttir, A.R.; Kotronoulas, A.; Bergmann, A.; Franzson, L.; Halldórsson, H.; et al. Metabolic Response in Endothelial Cells to Catecholamine Stimulation Associated with Increased Vascular Permeability. Int. J. Mol. Sci. 2022, 23, 3162. https://doi.org/10.3390/ijms23063162
López García de Lomana A, Vilhjálmsson AI, McGarrity S, Sigurðardóttir R, Anuforo Ó, Viktorsdóttir AR, Kotronoulas A, Bergmann A, Franzson L, Halldórsson H, et al. Metabolic Response in Endothelial Cells to Catecholamine Stimulation Associated with Increased Vascular Permeability. International Journal of Molecular Sciences. 2022; 23(6):3162. https://doi.org/10.3390/ijms23063162
Chicago/Turabian StyleLópez García de Lomana, Adrián, Arnar Ingi Vilhjálmsson, Sarah McGarrity, Rósa Sigurðardóttir, Ósk Anuforo, Alexía Rós Viktorsdóttir, Aris Kotronoulas, Andreas Bergmann, Leifur Franzson, Haraldur Halldórsson, and et al. 2022. "Metabolic Response in Endothelial Cells to Catecholamine Stimulation Associated with Increased Vascular Permeability" International Journal of Molecular Sciences 23, no. 6: 3162. https://doi.org/10.3390/ijms23063162