
Short-Term Pharmacological Induction of Arterial Stiffness and Hypertension with Angiotensin II Does Not Affect Learning and Memory and Cerebral Amyloid Load in Two Murine Models of Alzheimer’s Disease

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. AngII Treatment Affects hAPP23+/− and hAPPswe/PSEN1dE9 Mice Differently
2.2. AngII Treatment Induces Hypertrophic Cardiomyopathy
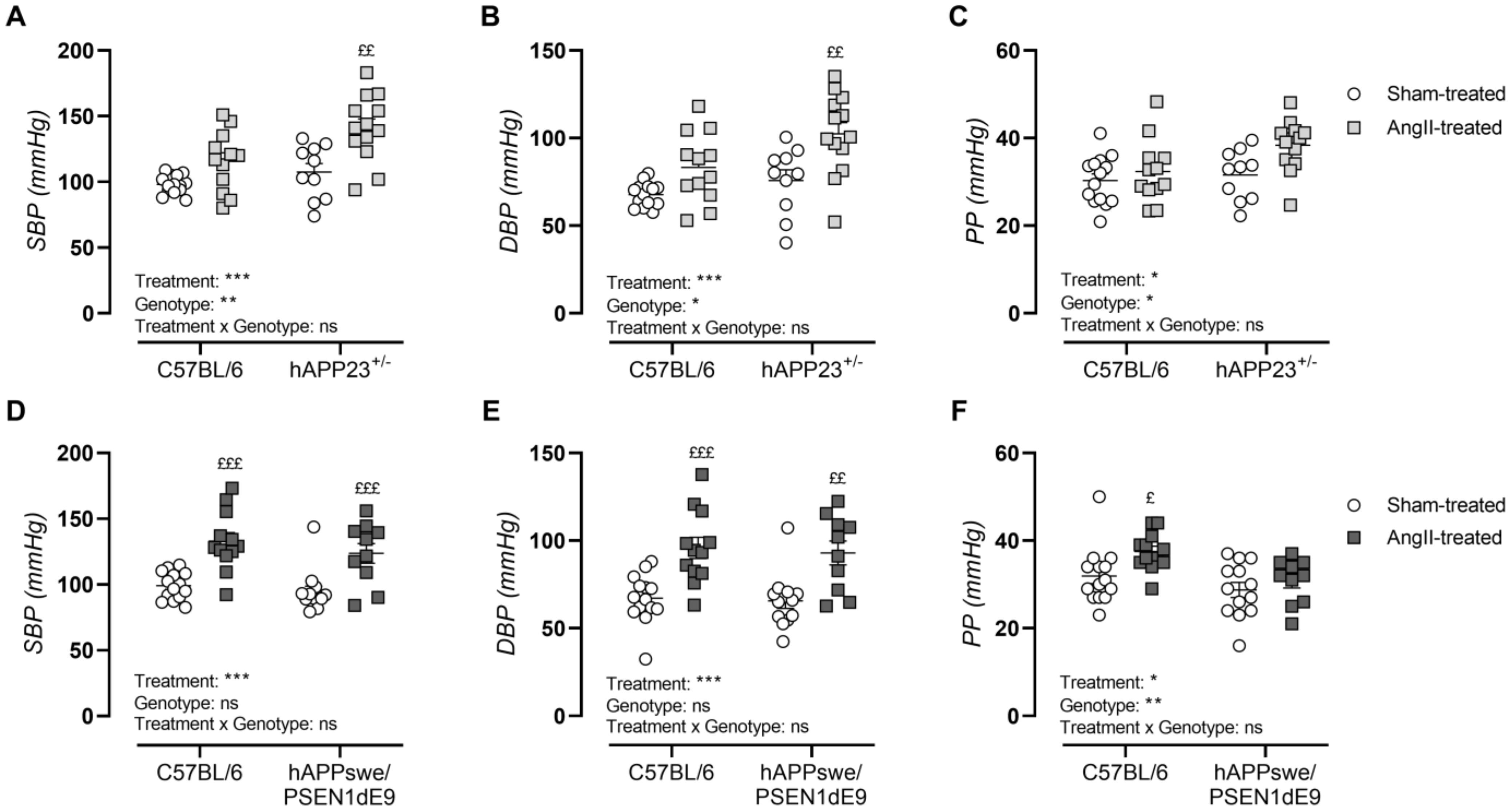
2.3. AngII Treatment Induces Hypertension
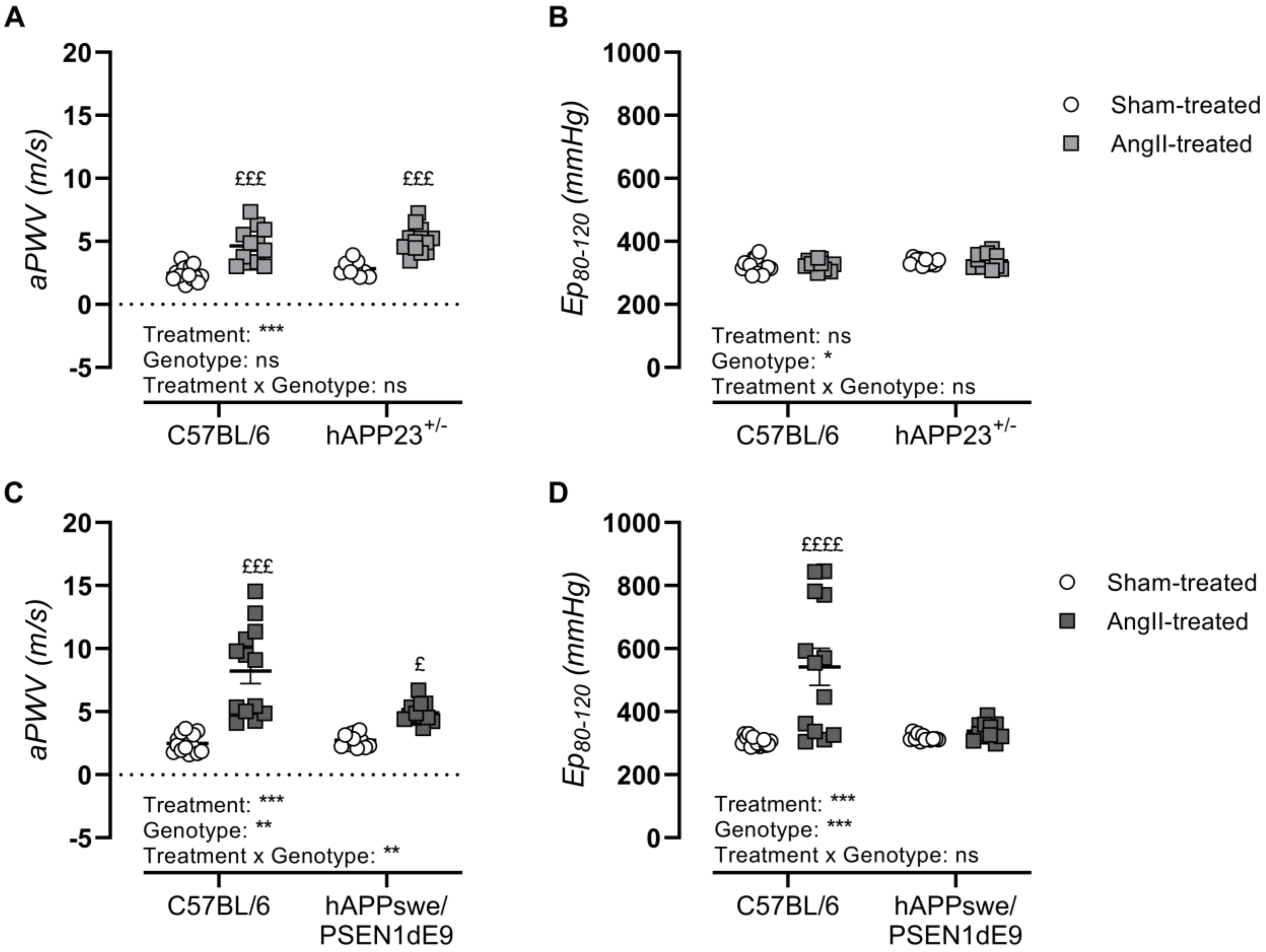
2.4. AngII Treatment Induces In Vivo Arterial Stiffness
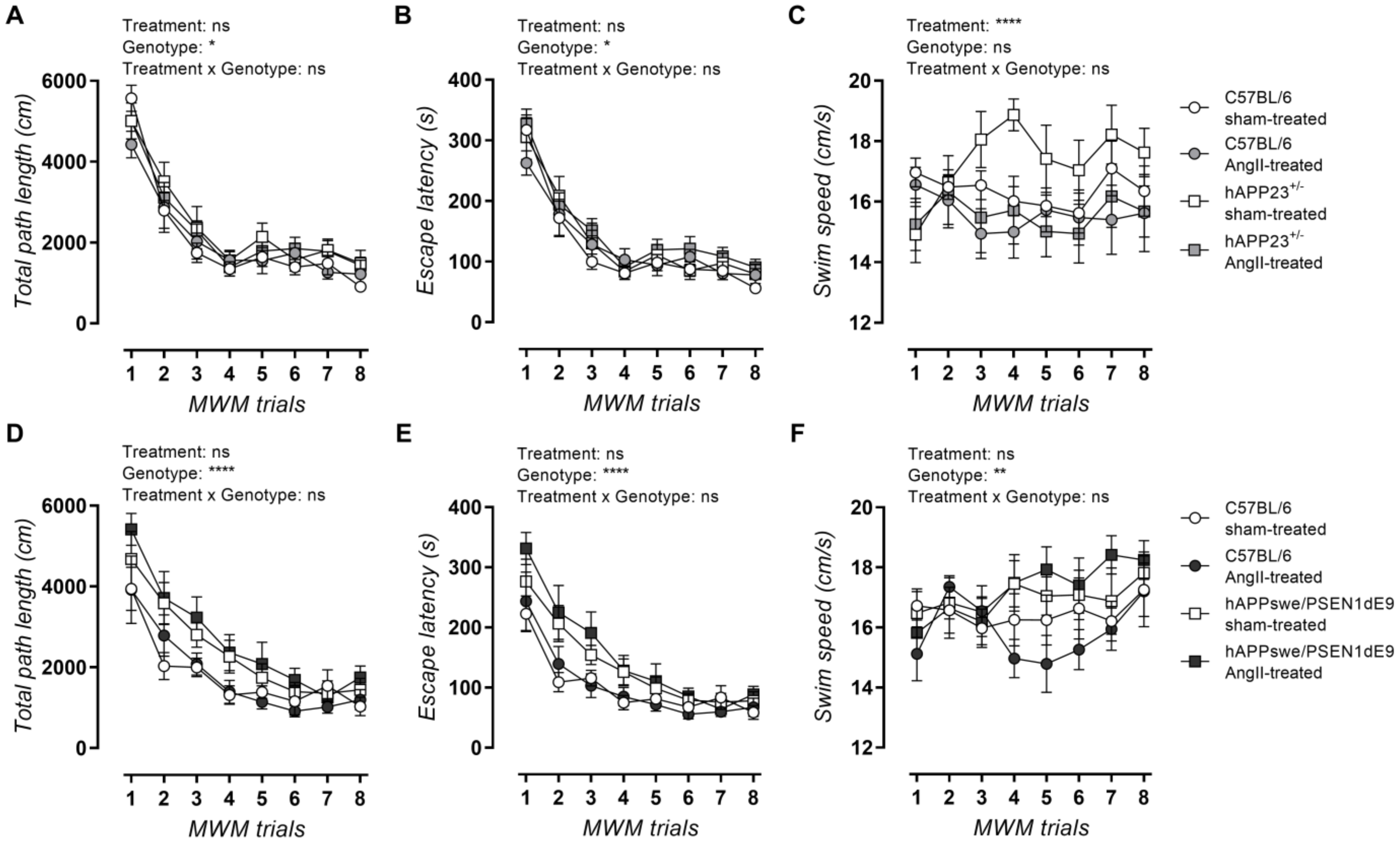
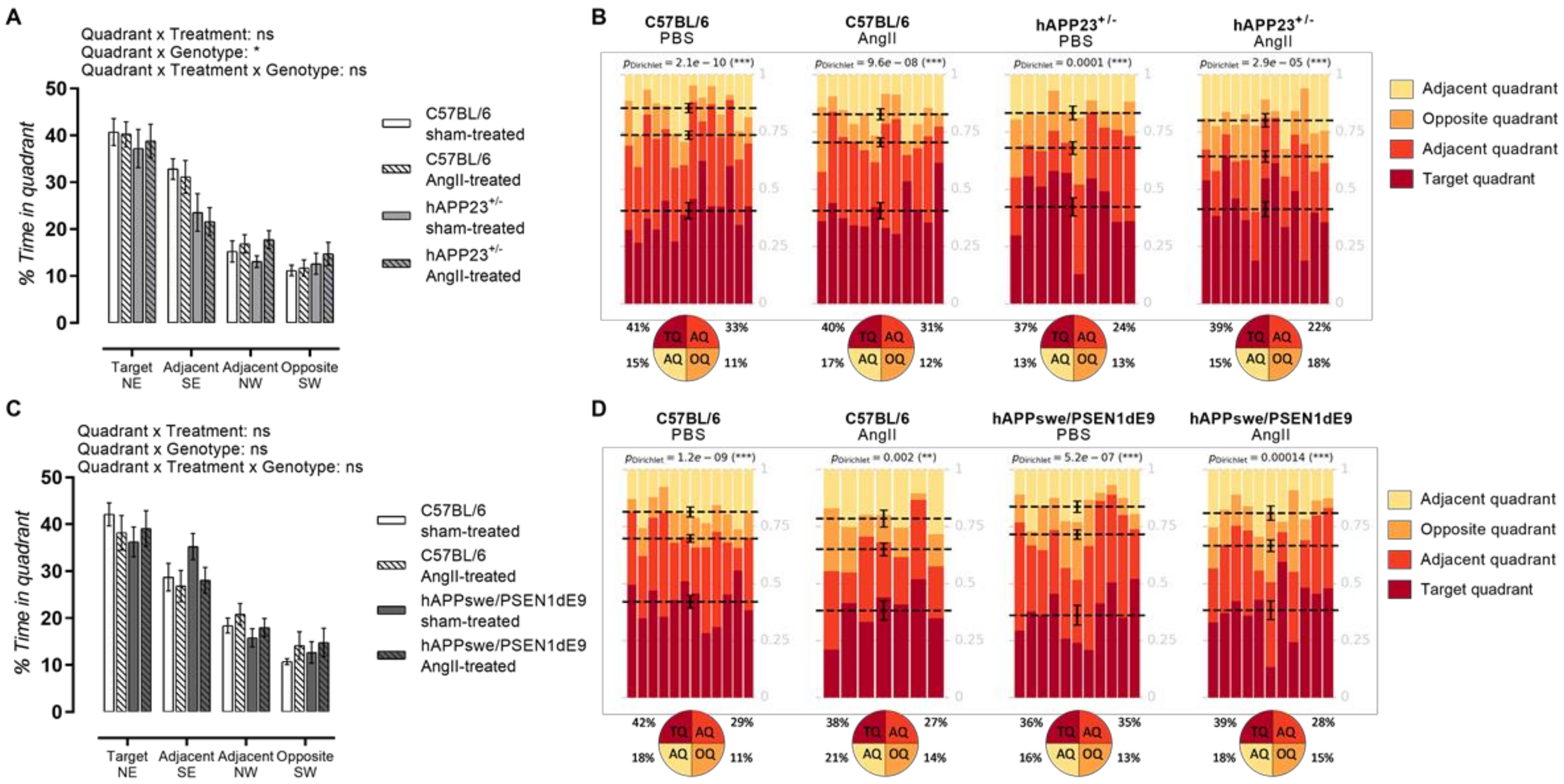
2.5. AngII Treatment Does Not Lead to Impaired MWM Acquisition Trial Performance
2.6. AngII Treatment Does Not Result in Impaired Probe Trial Performances
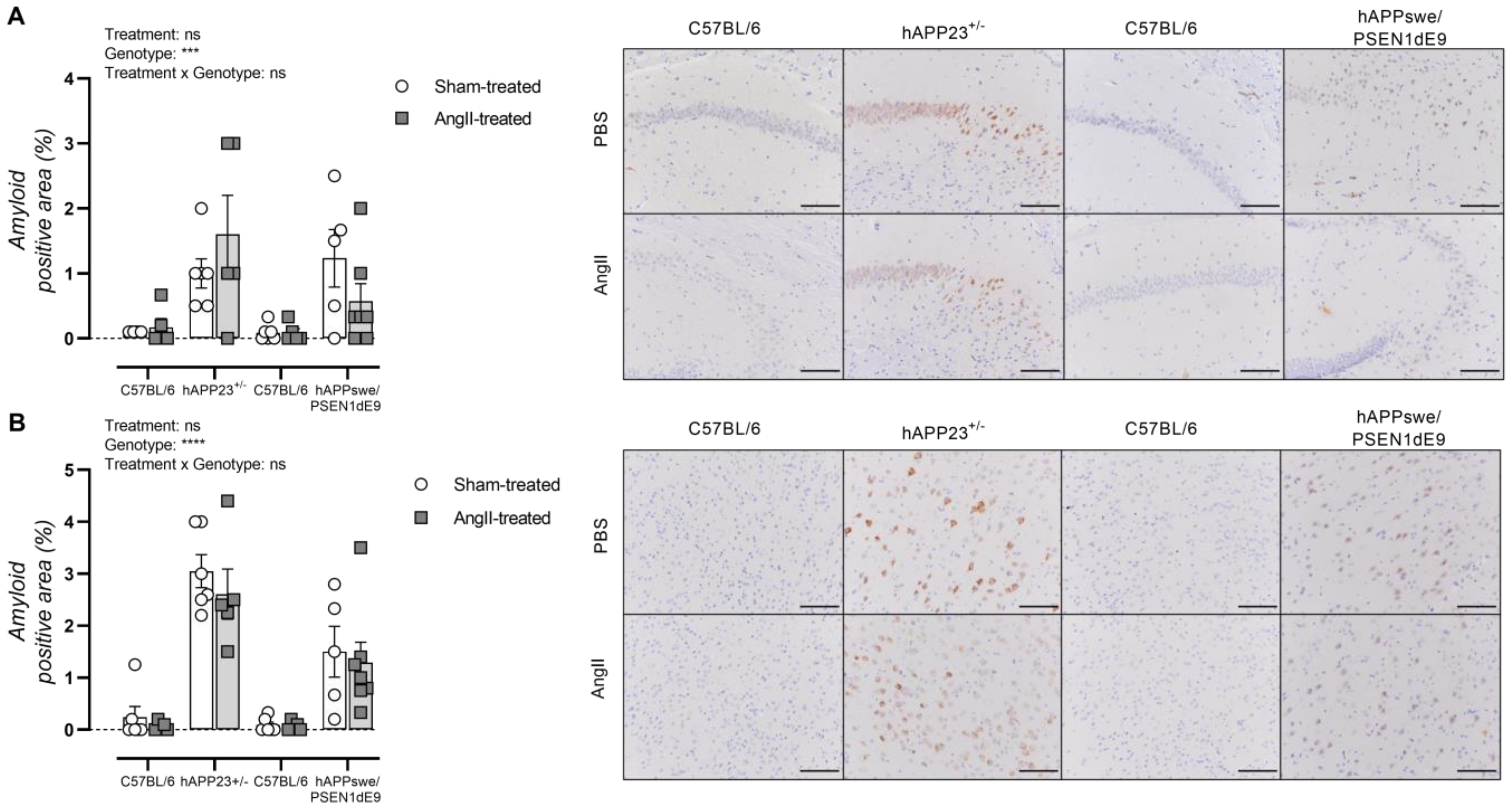
2.7. AngII Treatment of AD Mice Does Not Result in Increased Amyloid Load
3. Discussion
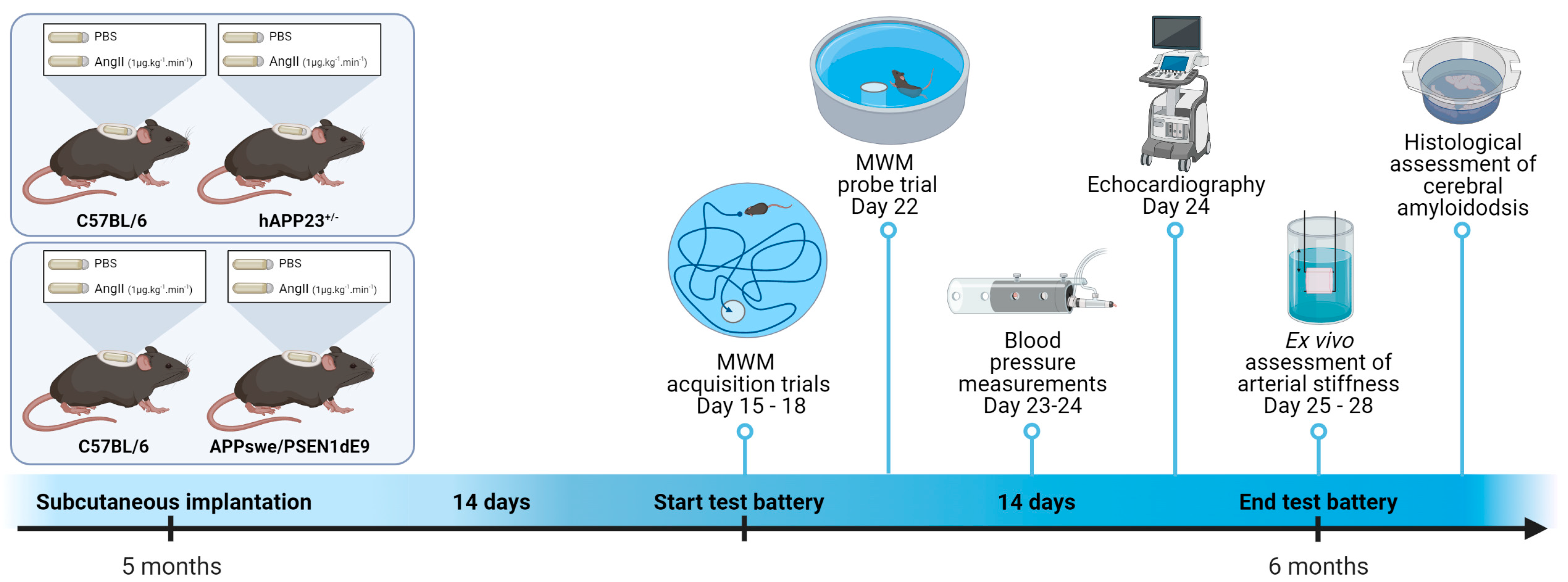
4. Materials and Methods
4.1. Experimental Animals and Osmotic Minipump Implantation
4.2. Visuospatial Learning and Memory
4.3. Blood Pressure Measurements
4.4. Echocardiography
4.5. Non-Invasive Pulse Wave Velocity (PWV) Measurements of the Aortic Abdominal Aorta (aPWV)
4.6. Rodent Oscillatory Tension Set-Up for Arterial Compliance (ROTSAC)
4.7. Histology
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Report on Ageing and Health. Available online: https://www.who.int/news-room/fact-sheets/detail/ageing-and-health (accessed on 30 January 2022).
- WHO Cardiovascular Diseases (CVDs). Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds)#:~:text=Cardiovascular%20diseases%20(CVDs)%20are%20the,%2D%20and%20middle%2Dincome%20countries (accessed on 30 January 2022).
- GBD 2019 Collaborators. Global mortality from dementia: Application of a new method and results from the Global Burden of Disease Study 2019. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2021, 7, e12200. [Google Scholar] [CrossRef]
- Tanaka, M.; Toldi, J.; Vécsei, L. Exploring the etiological links behind neurodegenerative diseases: Inflammatory cytokines and bioactive kynurenines. Int. J. Mol. Sci. 2020, 21, 2431. [Google Scholar] [CrossRef] [Green Version]
- 2020 Alzheimer’s disease facts and figures. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2020, 16, 391–460. [CrossRef] [PubMed]
- Hendrickx, J.O.; Martinet, W.; Van Dam, D.; De Meyer, G.R. Inflammation, Nitro-Oxidative Stress, Impaired Autophagy, and Insulin Resistance as a Mechanistic Convergence Between Arterial Stiffness and Alzheimer’s Disease. Front. Mol. Biosci. 2021, 8, 185. [Google Scholar] [CrossRef] [PubMed]
- Eglit, G.M.; Weigand, A.J.; Nation, D.A.; Bondi, M.W.; Bangen, K.J. Hypertension and Alzheimer’s disease: Indirect effects through circle of Willis atherosclerosis. Brain Commun. 2020, 2, fcaa114. [Google Scholar] [CrossRef]
- Mills, K.T.; Bundy, J.D.; Kelly, T.N.; Reed, J.E.; Kearney, P.M.; Reynolds, K.; Chen, J.; He, J. Global disparities of hypertension prevalence and control: A systematic analysis of population-based studies from 90 countries. Circulation 2016, 134, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Walker, K.A.; Power, M.C.; Gottesman, R.F. Defining the relationship between hypertension, cognitive decline, and dementia: A review. Curr. Hypertens. Rep. 2017, 19, 24. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; Braskie, M.N.; Tosun, D.; Thompson, P.M.; Weiner, M.; DeCarli, C.; Carmichael, O.T.; Initiative, A.S.D.N. Cerebral amyloid and hypertension are independently associated with white matter lesions in elderly. Front. Aging Neurosci. 2015, 7, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashby, E.L.; Miners, J.S.; Kehoe, P.G.; Love, S. Effects of hypertension and anti-hypertensive treatment on amyloid-β (Aβ) plaque load and Aβ-synthesizing and Aβ-degrading enzymes in frontal cortex. J. Alzheimer’s Dis. 2016, 50, 1191–1203. [Google Scholar] [CrossRef] [Green Version]
- Charidimou, A.; Boulouis, G.; Pasi, M.; Auriel, E.; van Etten, E.S.; Haley, K.; Ayres, A.; Schwab, K.M.; Martinez-Ramirez, S.; Goldstein, J.N. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 2017, 88, 1157–1164. [Google Scholar] [CrossRef] [Green Version]
- Bierer, L.M.; Hof, P.R.; Purohit, D.P.; Carlin, L.; Schmeidler, J.; Davis, K.L.; Perl, D.P. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch. Neurol. 1995, 52, 81–88. [Google Scholar] [CrossRef]
- Loewenstein, D.; Rabbat, M. Neurological complications of systemic hypertension. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2021; Volume 177, pp. 253–259. [Google Scholar] [CrossRef]
- Arvanitakis, Z.; Capuano, A.W.; Lamar, M.; Shah, R.C.; Barnes, L.L.; Bennett, D.A.; Schneider, J.A. Late-life blood pressure association with cerebrovascular and Alzheimer disease pathology. Neurology 2018, 91, e517–e525. [Google Scholar] [CrossRef]
- Kern, K.C.; Wright, C.B.; Bergfield, K.L.; Fitzhugh, M.C.; Chen, K.; Moeller, J.R.; Nabizadeh, N.; Elkind, M.S.; Sacco, R.L.; Stern, Y. Blood pressure control in aging predicts cerebral atrophy related to small-vessel white matter lesions. Front. Aging Neurosci. 2017, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Hendrickx, J.O.; De Moudt, S.; Van Dam, D.; De Deyn, P.P.; Fransen, P.; De Meyer, G.R. Altered stress hormone levels affect in vivo vascular function in the hAPP23+/− overexpressing mouse model of Alzheimer’s disease. Am. J. Physiol. Heart Circ. Physiol. 2021, 321, H905–H919. [Google Scholar] [CrossRef]
- Roth, L.; Rombouts, M.; Schrijvers, D.M.; Lemmens, K.; De Keulenaer, G.W.; Martinet, W.; De Meyer, G.R. Chronic intermittent mental stress promotes atherosclerotic plaque vulnerability, myocardial infarction and sudden death in mice. Atherosclerosis 2015, 242, 288–294. [Google Scholar] [CrossRef]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G. Context-dependent extinction of threat memories: Influences of healthy aging. Sci. Rep. 2018, 8, 12592. [Google Scholar] [CrossRef]
- Battaglia, S.; Garofalo, S.; di Pellegrino, G.; Starita, F. Revaluing the role of vmPFC in the acquisition of Pavlovian threat conditioning in humans. J. Neurosci. 2020, 40, 8491–8500. [Google Scholar] [CrossRef]
- Battaglia, S.; Harrison, B.J.; Fullana, M.A. Does the human ventromedial prefrontal cortex support fear learning, fear extinction or both? A commentary on subregional contributions. Mol. Psychiatry 2021, 1–3. [Google Scholar] [CrossRef]
- Battaglia, S. Neurobiological advances of learned fear in humans. Adv. Clin. Exp. Med. 2022. [Google Scholar] [CrossRef]
- Gabin, J.M.; Tambs, K.; Saltvedt, I.; Sund, E.; Holmen, J. Association between blood pressure and Alzheimer disease measured up to 27 years prior to diagnosis: The HUNT Study. Alzheimer’s Res. Ther. 2017, 9, 37. [Google Scholar] [CrossRef]
- Glodzik, L.; Rusinek, H.; Pirraglia, E.; McHugh, P.; Tsui, W.; Williams, S.; Cummings, M.; Li, Y.; Rich, K.; Randall, C. Blood pressure decrease correlates with tau pathology and memory decline in hypertensive elderly. Neurobiol. Aging 2014, 35, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebouvier, T.; Chen, Y.; Duriez, P.; Pasquier, F.; Bordet, R. Antihypertensive agents in Alzheimer’s disease: Beyond vascular protection. Expert Rev. Neurother. 2020, 20, 175–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoenes, M.; Neuberger, H.; Volpe, M.; Khan, B.; Kirch, W.; Böhm, M. Antihypertensive drug therapy and blood pressure control in men and women: An international perspective. J. Hum. Hypertens. 2010, 24, 336–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Protogerou, A.; Blacher, J.; Stergiou, G.S.; Achimastos, A.; Safar, M.E. Blood pressure response under chronic antihypertensive drug therapy: The role of aortic stiffness in the REASON (Preterax in Regression of Arterial Stiffness in a Controlled Double-Blind) study. J. Am. Coll. Cardiol. 2009, 53, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Iulita, M.F.; Noriega de la Colina, A.; Girouard, H. Arterial stiffness, cognitive impairment and dementia: Confounding factor or real risk? J. Neurochem. 2018, 144, 527–548. [Google Scholar] [CrossRef] [Green Version]
- Pasha, E.P.; Rutjes, E.; Tomoto, T.; Tarumi, T.; Stowe, A.; Claassen, J.A.; Munro Cullum, C.; Zhu, D.C.; Zhang, R. Carotid Stiffness is Associated with Brain Amyloid-β Burden in Amnestic Mild Cognitive Impairment. J. Alzheimer’s Dis. 2020, 74, 925–935. [Google Scholar] [CrossRef]
- Suri, S.; Chiesa, S.T.; Zsoldos, E.; Mackay, C.E.; Filippini, N.; Griffanti, L.; Mahmood, A.; Singh-Manoux, A.; Shipley, M.J.; Brunner, E.J. Associations between arterial stiffening and brain structure, perfusion, and cognition in the Whitehall II Imaging Sub-study: A retrospective cohort study. PLoS Med. 2020, 17, e1003467. [Google Scholar] [CrossRef]
- Pase, M.P.; Himali, J.J.; Mitchell, G.F.; Beiser, A.; Maillard, P.; Tsao, C.; Larson, M.G.; DeCarli, C.; Vasan, R.S.; Seshadri, S. Association of aortic stiffness with cognition and brain aging in young and middle-aged adults: The Framingham Third Generation Cohort Study. Hypertension 2016, 67, 513–519. [Google Scholar] [CrossRef] [Green Version]
- Hughes, T.M.; Kuller, L.H.; Barinas-Mitchell, E.J.; McDade, E.M.; Klunk, W.E.; Cohen, A.D.; Mathis, C.A.; DeKosky, S.T.; Price, J.C.; Lopez, O.L. Arterial stiffness and β-amyloid progression in nondemented elderly adults. JAMA Neurol. 2014, 71, 562–568. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; Luo, R.; Tang, X.; Tang, L.; Huang, H.X.; Wen, X.; Hu, S.; Peng, B. Age-related progression of arterial stiffness and its elevated positive association with blood pressure in healthy people. Atherosclerosis 2015, 238, 147–152. [Google Scholar] [CrossRef]
- Villemagne, V.L.; Burnham, S.; Bourgeat, P.; Brown, B.; Ellis, K.A.; Salvado, O.; Szoeke, C.; Macaulay, S.L.; Martins, R.; Maruff, P. Amyloid β deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer’s disease: A prospective cohort study. Lancet Neurol. 2013, 12, 357–367. [Google Scholar] [CrossRef]
- Suvila, K.; Lima, J.A.; Yano, Y.; Tan, Z.S.; Cheng, S.; Niiranen, T.J. Early-but Not Late-Onset Hypertension Is Related to Midlife Cognitive Function. Hypertension 2021, 77, 972–979. [Google Scholar] [CrossRef]
- Maugard, M.; Doux, C.; Bonvento, G. A new statistical method to analyze Morris Water Maze data using Dirichlet distribution. F1000Research 2019, 8, 1601. [Google Scholar] [CrossRef]
- Takagi, H.; Umemoto, T.; Group, A. Association of hypertension with abdominal aortic aneurysm expansion. Ann. Vasc. Surg. 2017, 39, 74–89. [Google Scholar] [CrossRef]
- Åström Malm, I.; De Basso, R.; Blomstrand, P.; Bjarnegård, N. Increased arterial stiffness in males with abdominal aortic aneurysm. Clin. Physiol. Funct. Imaging 2021, 41, 68–75. [Google Scholar] [CrossRef]
- Takagi, H.; Furukawa, M.; Umeda, Y.; Fukumoto, Y.; Yoshida, N.; Yamada, T.; Mori, Y. Aortic arch aneurysm with dissection in Cushing syndrome. J. Thorac. Cardiovasc. Surg. 2004, 127, 278–279. [Google Scholar] [CrossRef] [Green Version]
- Wiesmann, M.; Roelofs, M.; van der Lugt, R.; Heerschap, A.; Kiliaan, A.J.; Claassen, J.A. Angiotensin II, hypertension and angiotensin II receptor antagonism: Roles in the behavioural and brain pathology of a mouse model of Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2017, 37, 2396–2413. [Google Scholar] [CrossRef]
- Wiesmann, M.; Capone, C.; Zerbi, V.; Mellendijk, L.; Heerschap, A.; AHR Claassen, J.; Kiliaan, A.J. Hypertension impairs cerebral blood flow in a mouse model for Alzheimer’s disease. Curr. Alzheimer Res. 2015, 12, 914–922. [Google Scholar] [CrossRef]
- Quitterer, U.; AbdAlla, S. Improvements of symptoms of Alzheimers disease by inhibition of the angiotensin system. Pharmacol. Res. 2020, 154, 104230. [Google Scholar] [CrossRef]
- Ongali, B.; Nicolakakis, N.; Tong, X.-K.; Aboulkassim, T.; Papadopoulos, P.; Rosa-Neto, P.; Lecrux, C.; Imboden, H.; Hamel, E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol. Dis. 2014, 68, 126–136. [Google Scholar] [CrossRef]
- Royea, J.; Lacalle-Aurioles, M.; Trigiani, L.J.; Fermigier, A.; Hamel, E. AT2R’s (Angiotensin II Type 2 Receptor’s) role in cognitive and cerebrovascular deficits in a mouse model of Alzheimer disease. Hypertension 2020, 75, 1464–1474. [Google Scholar] [CrossRef]
- Royea, J.; Zhang, L.; Tong, X.-K.; Hamel, E. Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 5562–5573. [Google Scholar] [CrossRef]
- Ems, R.; Garg, A.; Ostergard, T.A.; Miller, J.P. Potential deep brain stimulation targets for the management of refractory hypertension. Front. Neurosci. 2019, 13, 93. [Google Scholar] [CrossRef]
- Chang, C.-H.; Lane, H.-Y.; Lin, C.-H. Brain stimulation in Alzheimer’s disease. Front. Psychiatry 2018, 9, 201. [Google Scholar] [CrossRef] [Green Version]
- Sanches, C.; Stengel, C.; Godard, J.; Mertz, J.; Teichmann, M.; Migliaccio, R.; Valero-Cabré, A. Past, present, and future of non-invasive brain stimulation approaches to treat cognitive impairment in neurodegenerative diseases: Time for a comprehensive critical review. Front. Aging Neurosci. 2021, 12, 578339. [Google Scholar] [CrossRef]
- Borgomaneri, S.; Battaglia, S.; Sciamanna, G.; Tortora, F.; Laricchiuta, D. Memories are not written in stone: Re-writing fear memories by means of non-invasive brain stimulation and optogenetic manipulations. Neurosci. Biobehav. Rev. 2021, 127, 334–352. [Google Scholar] [CrossRef]
- Chung, C.-M.; Cheng, H.-W.; Chang, J.-J.; Lin, Y.-S.; Hsiao, J.-F.; Chang, S.-T.; Hsu, J.-T. Relationship between resistant hypertension and arterial stiffness assessed by brachial-ankle pulse wave velocity in the older patient. Clin. Interv. Aging 2014, 9, 1495. [Google Scholar] [CrossRef] [Green Version]
- Alzheimer’s Disease International. Dementia Statistics. Available online: https://www.alzint.org/about/dementia-facts-figures/dementia-statistics/ (accessed on 30 January 2022).
- Kilkenny, C.; Browne, W.; Cuthill, I.; Emerson, M.; Altman, D.G. The ARRIVE guidelines. ReqartoCom 2010, 8, e1000412. [Google Scholar] [CrossRef] [Green Version]
- Underwood, W.; Anthony, R. AVMA Guidelines for the Euthanasia of Animals: 2020 Edition; AVMA: Schaumburg, IL, USA, 2013; Volume 30. [Google Scholar]
- Van Dam, D.; d’Hooge, R.; Staufenbiel, M.; Van Ginneken, C.; Van Meir, F.; De Deyn, P.P. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur. J. Neurosci. 2003, 17, 388–396. [Google Scholar] [CrossRef]
- Van Dam, D.; Lenders, G.; De Deyn, P.P. Effect of Morris water maze diameter on visual-spatial learning in different mouse strains. Neurobiol. Learn. Mem. 2006, 85, 164–172. [Google Scholar] [CrossRef]
- Feng, M.; Whitesall, S.; Zhang, Y.; Beibel, M.; Alecy, L.D.; DiPetrillo, K. Validation of volume–pressure recording tail-cuff blood pressure measurements. Am. J. Hypertens. 2008, 21, 1288–1291. [Google Scholar] [CrossRef] [PubMed]
- Di Lascio, N.; Stea, F.; Kusmic, C.; Sicari, R.; Faita, F. Non-invasive assessment of pulse wave velocity in mice by means of ultrasound images. Atherosclerosis 2014, 237, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Leloup, A.J.; Hove, C.E.; Kurdi, A.; Moudt, S.; Martinet, W.; Meyer, G.R.; Schrijvers, D.M.; Keulenaer, G.W.; Fransen, P. A novel set-up for the ex vivo analysis of mechanical properties of mouse aortic segments stretched at physiological pressure and frequency. J. Physiol. 2016, 594, 6105–6115. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C57BL/6 | hAPP23+/− | ||||||
|---|---|---|---|---|---|---|---|
| PBS (n = 14) | AngII (n = 12) | PBS (n = 11) | AngII (n = 11) | ptreatment | pgenotype | ptreatment × genotype | |
| Body weight (g) | 27 ± 1 | 27 ± 1 | 27 ± 1 | 24 ± 1 $ | ns | ** | ns |
| Survival, genotype (%) | 74 | - | 80 | - | - | ns | - |
| Survival, treatment (%) | 100 | 57 | 76 | 80 | - | ns | - |
| AAA incidence (%) | - | 25 | - | 31 | ns | - | - |
| C57BL/6 | hAPPswe/ PSEN1dE9 | ||||||
| PBS (n = 14) | AngII (n = 13) | PBS (n = 12) | AngII (n = 11) | ptreatment | pgenotype | ptreatment × genotype | |
| Body weight (g) | 34 ± 1 | 32 ± 1 £ | 33 ± 1 | 32 ± 1 | ** | ns | ns |
| Survival, genotype (%) | 90 | - | 75 | - | - | ns | - |
| Survival, treatment (%) | 100 | 58 | 95 | 72 | - | 0.09 | - |
| AAA incidence (%) | - | 69 | - | 31 | *** | - | - |
| C57BL/6 | hAPP23+/− | ||||||
|---|---|---|---|---|---|---|---|
| PBS (n = 14) | AngII (n = 12) | PBS (n = 11) | AngII (n = 11) | ptreatment | pgenotype | ptreatment × genotype | |
| Heart weight (mg) | 165 ± 5 | 188 ± 5 £ | 141 ± 3 | 183 ± 8 $$$ | *** | * | ns |
| Heart weight (%) | 0.53 ± 0.01 | 0.58 ± 0.01 | 0.51 ± 0.01 | 0.69 ± 0.04 $$$ | *** | * | ** |
| IVS,d (mm) | 1.2 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.1 | ns | ns | ns |
| LVID,d (mm) | 3.7 ± 0.1 | 3.7 ± 0.1 | 3.6 ± 0.1 | 3.4 ± 0.2 | ns | 0.06 | ns |
| LVPW,d (mm) | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.1 ± 0.1 | 1.4 ± 0.1 | 0.08 | 0.07 | ns |
| LV mass (mg) | 172 ± 11 | 167 ± 10 | 157 ± 11 | 187 ± 18 | ns | ns | ns |
| LV mass/BW (10−3) | 5.5 ± 0.3 | 5.2 ± 0.3 | 5.7 ± 0.5 | 7.1 ± 0.7 | ns | * | ns |
| LV volume,d (µL) | 60 ± 4 | 61 ± 5 | 54 ± 4 | 49 ± 6 | ns | 0.08 | ns |
| Stroke volume (µL) | 42 ± 3 | 42 ± 3 | 39 ± 2 | 33 ± 3 | ns | 0.08 | ns |
| EF (%) | 71 ± 2 | 70 ± 3 | 73 ± 2 | 71 ± 3 | ns | ns | ns |
| FS (%) | 40 ± 2 | 39 ± 2 | 42 ± 2 | 40 ± 2 | ns | ns | ns |
| E/A (none) | 1.4 ± 0.1 | 1.4 ± 0.1 | 1.5 ± 0.3 | 1.6 ± 0.1 | ns | ns | ns |
| E/E’ (none) | 32 ± 3 | 29 ± 4 | 35 ± 4 | 51 ± 7 | ns | * | ns |
| IVRT (ms) | 19 ± 1 | 21 ± 1 | 23 ± 2 | 24 ± 3 | ns | 0.07 | ns |
| Deceleration (ms) | 17 ± 1 | 15 ± 1 | 16 ± 2 | 21 ± 2 | ns | ns | ns |
| C57BL/6 | hAPPswe/ PSEN1dE9 | ||||||
|---|---|---|---|---|---|---|---|
| PBS (n = 14) | AngII (n = 13) | PBS (n = 12) | AngII (n = 11) | ptreatment | pgenotype | ptreatment × genotype | |
| Heart weight (mg) | 182 ± 5 | 218 ± 7 £££ | 158 ± 3 | 204 ± 9 $$$ | *** | ** | ns |
| Heart weight (%) | 0.53 ± 0.01 | 0.68 ± 0.02 £££ | 0.48 ± 0.01 | 0.65 ± 0.02 $$$ | *** | * | ns |
| IVS,d (mm) | 1.2 ± 0.1 | 1.4 ± 0.1 | 1.1 ± 0.1 | 1.3 ± 0.1 | * | ns | ns |
| LVID,d (mm) | 3.6 ± 0.1 | 3.8 ± 0.1 | 3.6 ± 0.1 | 3.6 ± 0.2 | ns | ns | ns |
| LVPW,d (mm) | 1.0 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | 1.1 ± 0.1 | * | ns | ns |
| LV mass (mg) | 170 ± 21 | 224 ± 17 | 148 ± 15 | 186 ± 18 | * | ns | ns |
| LV mass/BW (10−3) | 4.9 ± 0.6 | 7.0 ± 0.5 £ | 4.5 ± 0.5 | 5.9 ± 0.5 | ** | ns | ns |
| LV volume,d (µL) | 57 ± 4 | 63 ± 4 | 55 ± 4 | 54 ± 6 | ns | ns | ns |
| Stroke volume (µL) | 42 ± 3 | 42 ± 2 | 38 ± 3 | 39 ± 3 | ns | ns | ns |
| EF (%) | 73 ± 2 | 68 ± 3 | 70 ± 2 | 74 ± 4 | ns | ns | ns |
| FS (%) | 42 ± 2 | 38 ± 3 | 39 ± 2 | 43 ± 4 | ns | ns | ns |
| E/A (none) | 1.6 ± 0.2 | 1.7 ± 0.1 | 1.4 ± 0.3 | 1.5 ± 0.1 | ns | ns | ns |
| E/E’ (none) | 32 ± 5 | 23 ± 4 | 31 ± 5 | 22 ± 4 | ns | ns | ns |
| IVRT (ms) | 21 ± 2 | 22 ± 1 | 22 ± 1 | 24 ± 3 | ns | ns | ns |
| Deceleration (ms) | 16 ± 1 | 18 ± 2 | 17 ± 1 | 17 ± 1 | ns | ns | ns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrickx, J.O.; Calus, E.; De Deyn, P.P.; Van Dam, D.; De Meyer, G.R.Y. Short-Term Pharmacological Induction of Arterial Stiffness and Hypertension with Angiotensin II Does Not Affect Learning and Memory and Cerebral Amyloid Load in Two Murine Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 2738. https://doi.org/10.3390/ijms23052738
Hendrickx JO, Calus E, De Deyn PP, Van Dam D, De Meyer GRY. Short-Term Pharmacological Induction of Arterial Stiffness and Hypertension with Angiotensin II Does Not Affect Learning and Memory and Cerebral Amyloid Load in Two Murine Models of Alzheimer’s Disease. International Journal of Molecular Sciences. 2022; 23(5):2738. https://doi.org/10.3390/ijms23052738
Chicago/Turabian StyleHendrickx, Jhana O., Elke Calus, Peter Paul De Deyn, Debby Van Dam, and Guido R. Y. De Meyer. 2022. "Short-Term Pharmacological Induction of Arterial Stiffness and Hypertension with Angiotensin II Does Not Affect Learning and Memory and Cerebral Amyloid Load in Two Murine Models of Alzheimer’s Disease" International Journal of Molecular Sciences 23, no. 5: 2738. https://doi.org/10.3390/ijms23052738