
Synergistic and Detrimental Effects of Alcohol Intake on Progression of Liver Steatosis

 , , and
, , and
Abstract
:1. Introduction
2. Definition
2.1. NAFLD
2.2. ALD
3. Liver Disease in Obesity
4. Alcohol Metabolism
- (1)
- Cytosol: the enzyme ADH is responsible for most of the ethanol metabolism. The reaction involves an intermediate carrier of electrons, nicotinamide adenine dinucleotide (NAD+), which is reduced by two electrons to form NADH.
- (2)
- Endoplasmic reticulum: the enzyme cytochrome P450 2E1 (CYP2E1) is NADPH cofactor-dependent and belongs to the microsomal ethanol oxidizing system (MEOS). However, CYP2E1 is only active after a person has consumed large amounts of alcohol. This pathway is important in metabolizing ethanol to acetaldehyde at elevated ethanol concentrations.
- (3)
- Peroxisomes: the enzyme catalase uses hydrogen peroxide (H2O2) to oxidize alcohol. Catalase metabolizes only a small fraction of alcohol in the body.
5. Mechanisms of Damage
5.1. Steatosis
5.2. Steatohepatitis
6. Fibrogenesis
7. Multiple Effects of Alcohol
7.1. Alcohol and Metabolic Disorders
7.2. ALD and Obesity
7.3. Alcohol and NAFLD
8. Genetic Predisposition
9. Extrahepatic Manifestations
10. Synergistic Effects of ALD and NAFLD
11. ALD, NAFLD, and HCC
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Asrani, S.K.; Devarbhavi, H.; Eaton, J.; Kamath, P.S. Burden of liver diseases in the world. J. Hepatol. 2019, 70, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ejaz, A.; Tayal, A.; Spolverato, G.; Bridges, J.F.; Anders, R.A.; Pawlik, T.M. Temporal trends in population-based death rates associated with chronic liver disease and liver cancer in the United States over the last 30 years. Cancer 2014, 120, 3058–3065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pimpin, L.; Cortez-Pinto, H.; Negro, F.; Corbould, E.; Lazarus, J.V.; Webber, L.; Sheron, N.; Committee, E.H.S. Burden of liver disease in Europe: Epidemiology and analysis of risk factors to identify prevention policies. J. Hepatol. 2018, 69, 718–735. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.; Dang, K.; Ladhani, S.; Singal, A.K.; Wong, R.J. Prevalence of Alcoholic Fatty Liver Disease among Adults in the United States, 2001-2016. JAMA 2019, 321, 1723–1725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.F.; Qu, F.; Zheng, S.J.; Wu, H.L.; Liu, M.; Liu, S.; Ren, Y.; Ren, F.; Chen, Y.; Duan, Z.P.; et al. Elevated plasma sphingomyelin (d18:1/22:0) is closely related to hepatic steatosis in patients with chronic hepatitis C virus infection. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1725–1732. [Google Scholar] [CrossRef]
- Yasui, K.; Harano, Y.; Mitsuyoshi, H.; Tsuji, K.; Endo, M.; Nakajima, T.; Minami, M.; Itoh, Y.; Zen, Y.; Nakanuma, Y.; et al. Steatosis and hepatic expression of genes regulating lipid metabolism in Japanese patients infected with hepatitis C virus. J. Gastroenterol. 2010, 45, 95–104. [Google Scholar] [CrossRef]
- Wu, Y.J.; Chen, L.S.; Qiang, W.G. Effects of fatty liver and related factors on the efficacy of combination antiviral therapy in patients with chronic hepatitis C. Liver Int. 2006, 26, 166–172. [Google Scholar] [CrossRef]
- Hwang, S.J.; Luo, J.C.; Chu, C.W.; Lai, C.R.; Lu, C.L.; Tsay, S.H.; Wu, J.C.; Chang, F.Y.; Lee, S.D. Hepatic steatosis in chronic hepatitis C virus infection: Prevalence and clinical correlation. J. Gastroenterol. Hepatol. 2001, 16, 190–195. [Google Scholar] [CrossRef]
- Satapathy, S.K.; Kuwajima, V.; Nadelson, J.; Atiq, O.; Sanyal, A.J. Drug-induced fatty liver disease: An overview of pathogenesis and management. Ann. Hepatol. 2015, 14, 789–806. [Google Scholar] [CrossRef]
- Stattermayer, A.F.; Traussnigg, S.; Dienes, H.P.; Aigner, E.; Stauber, R.; Lackner, K.; Hofer, H.; Stift, J.; Wrba, F.; Stadlmayr, A.; et al. Hepatic steatosis in Wilson disease--Role of copper and PNPLA3 mutations. J. Hepatol. 2015, 63, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.; Popovic, P.; Rotovnik Kozjek, N. Liver steatosis in adult patients on home parenteral nutrition. Eur. J. Clin. Nutr. 2020, 74, 255–260. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver; European Association for the Study of Diabetes; European Association for the Study of Obesity. EASL-EASD-EASO Clinical Practice Guidelines for the management of non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, J.C.; Horton, J.D.; Hobbs, H.H. Human fatty liver disease: Old questions and new insights. Science 2011, 332, 1519–1523. [Google Scholar] [CrossRef] [Green Version]
- Szczepaniak, L.S.; Nurenberg, P.; Leonard, D.; Browning, J.D.; Reingold, J.S.; Grundy, S.; Hobbs, H.H.; Dobbins, R.L. Magnetic resonance spectroscopy to measure hepatic triglyceride content: Prevalence of hepatic steatosis in the general population. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E462–E468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Vernon, G.; Baranova, A.; Younossi, Z.M. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011, 34, 274–285. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The Third National Health and Nutrition Examination Survey, 1988-1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Blissett, D.; Blissett, R.; Henry, L.; Stepanova, M.; Younossi, Y.; Racila, A.; Hunt, S.; Beckerman, R. The economic and clinical burden of nonalcoholic fatty liver disease in the United States and Europe. Hepatology 2016, 64, 1577–1586. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Afendy, M.; Fang, Y.; Younossi, Y.; Mir, H.; Srishord, M. Changes in the prevalence of the most common causes of chronic liver diseases in the United States from 1988 to 2008. Clin. Gastroenterol. Hepatol. 2011, 9, 524–530.e1. [Google Scholar] [CrossRef]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Rinella, M.E.; Sanyal, A.J.; Harrison, S.A.; Brunt, E.M.; Goodman, Z.; Cohen, D.E.; Loomba, R. From NAFLD to MAFLD: Implications of a Premature Change in Terminology. Hepatology 2021, 73, 1194–1198. [Google Scholar] [CrossRef] [PubMed]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef] [Green Version]
- Molina-Molina, E.; Krawczyk, M.; Stachowska, E.; Lammert, F.; Portincasa, P. Non-Alcoholic Fatty Liver Disease in Non-Obese Individuals: Prevalence, Pathogenesis and Treatment. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 638–645. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, F.; Wang, W.; Zhang, X.J.; Ji, Y.X.; Zhang, P.; She, Z.G.; Zhu, L.; Cai, J.; Li, H. Epidemiological Features of NAFLD From 1999 to 2018 in China. Hepatology 2020, 71, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Kim, W.R. Nonobese Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2017, 15, 474–485. [Google Scholar] [CrossRef] [Green Version]
- Loomba, R.; Sanyal, A.J. The global NAFLD epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic fatty liver disease as a nexus of metabolic and hepatic diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Di Palo, D.M.; Garruti, G.; Di Ciaula, A.; Molina-Molina, E.; Shanmugam, H.; De Angelis, M.; Portincasa, P. Increased Colonic Permeability and Lifestyles as Contributing Factors to Obesity and Liver Steatosis. Nutrients 2020, 12, 564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ciaula, A.; Carbone, F.; Shanmugham, H.; Molina-Molina, E.; Bonfrate, L.; Ministrini, S.; Montecucco, F.; Portincasa, P. Adiponectin involved in portal flow hepatic extraction of 13C-methacetin in obesity and non-alcoholic fatty liver. Eur. J. Intern. Med. 2021, 89, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708. [Google Scholar] [CrossRef]
- Krawczyk, M.; Grünhage, F.; Mihalache, F.; Acalovschi, M.; Lammert, F. The common adiponutrin variant p. I148M, a common genetic risk factor for severe forms of NAFLD and ALD, in gallstone patients. Z. Für Gastroenterol. 2010, 48, P408. [Google Scholar] [CrossRef]
- Palmentieri, B.; de Sio, I.; La Mura, V.; Masarone, M.; Vecchione, R.; Bruno, S.; Torella, R.; Persico, M. The role of bright liver echo pattern on ultrasound B-mode examination in the diagnosis of liver steatosis. Dig. Liver Dis. 2006, 38, 485–489. [Google Scholar] [CrossRef]
- Powell, E.E.; Wong, V.W.; Rinella, M. Non-alcoholic fatty liver disease. Lancet 2021, 397, 2212–2224. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: A systematic review and meta-analysis of paired-biopsy studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e9. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, J.; Viggiano, T.R.; McGill, D.B.; Oh, B.J. Nonalcoholic steatohepatitis: Mayo Clinic experiences with a hitherto unnamed disease. Mayo Clin. Proc. Mayo Clin. 1980, 55, 434–438. [Google Scholar]
- Sheth, S.G.; Gordon, F.D.; Chopra, S. Nonalcoholic steatohepatitis. Ann. Intern. Med. 1997, 126, 137–145. [Google Scholar] [CrossRef]
- Caldwell, S.H.; Oelsner, D.H.; Iezzoni, J.C.; Hespenheide, E.E.; Battle, E.H.; Driscoll, C.J. Cryptogenic cirrhosis: Clinical characterization and risk factors for underlying disease. Hepatology 1999, 29, 664–669. [Google Scholar] [CrossRef] [PubMed]
- Browning, J.D.; Kumar, K.S.; Saboorian, M.H.; Thiele, D.L. Ethnic differences in the prevalence of cryptogenic cirrhosis. Am. J. Gastroenterol. 2004, 99, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Nasr, P.; Ignatova, S.; Kechagias, S.; Ekstedt, M. Natural history of nonalcoholic fatty liver disease: A prospective follow-up study with serial biopsies. Hepatol. Commun. 2018, 2, 199–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e121. [Google Scholar] [CrossRef] [Green Version]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Torbenson, M.S.; Yeh, M.M. Steatohepatitic hepatocellular carcinoma. Hepatoma Res. 2021, 7, 38. [Google Scholar] [CrossRef]
- Caussy, C.; Soni, M.; Cui, J.; Bettencourt, R.; Schork, N.; Chen, C.H.; Ikhwan, M.A.; Bassirian, S.; Cepin, S.; Gonzalez, M.P.; et al. Nonalcoholic fatty liver disease with cirrhosis increases familial risk for advanced fibrosis. J. Clin. Investig. 2017, 127, 2697–2704. [Google Scholar] [CrossRef] [Green Version]
- Stender, S.; Loomba, R. PNPLA3 Genotype and Risk of Liver and All-Cause Mortality. Hepatology 2020, 71, 777–779. [Google Scholar] [CrossRef]
- Krawczyk, M.; Portincasa, P.; Lammert, F. PNPLA3-associated steatohepatitis: Toward a gene-based classification of fatty liver disease. Semin. Liver Dis. 2013, 33, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Moschen, A.R.; Kaser, S.; Tilg, H. Non-alcoholic steatohepatitis: A microbiota-driven disease. Trends Endocrinol. Metab. TEM 2013, 24, 537–545. [Google Scholar] [CrossRef]
- Loomba, R.; Lim, J.K.; Patton, H.; El-Serag, H.B. AGA Clinical Practice Update on Screening and Surveillance for Hepatocellular Carcinoma in Patients With Nonalcoholic Fatty Liver Disease: Expert Review. Gastroenterology 2020, 158, 1822–1830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, L.A.; Lymp, J.F.; St Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease-An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Rinella, M.E.; Sanyal, A.J. Management of NAFLD: A stage-based approach. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Yoshitaka, H.; Hamaguchi, M.; Kojima, T.; Fukuda, T.; Ohbora, A.; Fukui, M. Nonoverweight nonalcoholic fatty liver disease and incident cardiovascular disease: A post hoc analysis of a cohort study. Medicine 2017, 96, e6712. [Google Scholar] [CrossRef]
- Alcohol. World Health Organization (WHO) Factsheet 2018 [20/04/2021]. 2021. Available online: http://www.who.int/mediacentre/factsheets/fs349/en/ (accessed on 24 January 2022).
- Adult Obesity in the United States. Available online: http://stateofobesity.org/adult-obesity (accessed on 24 January 2022).
- Terris, M. Epidemiology of cirrhosis of the liver: National mortality data. Am. J. Public Health Nations Health 1967, 57, 2076–2088. [Google Scholar] [CrossRef]
- Seitz, H.K.; Neuman, M.G. The History of Alcoholic Liver Disease: From an Unrecognized Disease to One of the Most Frequent Diseases in Hepatology. J. Clin. Med. 2021, 10, 858. [Google Scholar] [CrossRef]
- Guirguis, J.; Chhatwal, J.; Dasarathy, J.; Rivas, J.; McMichael, D.; Nagy, L.E.; McCullough, A.J.; Dasarathy, S. Clinical impact of alcohol-related cirrhosis in the next decade: Estimates based on current epidemiological trends in the United States. Alcohol. Clin. Exp. Res. 2015, 39, 2085–2094. [Google Scholar] [CrossRef] [Green Version]
- Peery, A.F.; Crockett, S.D.; Barritt, A.S.; Dellon, E.S.; Eluri, S.; Gangarosa, L.M.; Jensen, E.T.; Lund, J.L.; Pasricha, S.; Runge, T.; et al. Burden of Gastrointestinal, Liver, and Pancreatic Diseases in the United States. Gastroenterology 2015, 149, 1731–1741.e1733. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization. Global Status Report on Alcohol and Health 2018. Available online: https://www.who.int/substance_abuse/ (accessed on 20 April 2021).
- Gilmore, W.; Chikritzhs, T.; Stockwell, T.; Jernigan, D.; Naimi, T.; Gilmore, I. Alcohol: Taking a population perspective. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 426–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagstrom, H.; Hemmingsson, T.; Discacciati, A.; Andreasson, A. Alcohol consumption in late adolescence is associated with an increased risk of severe liver disease later in life. J. Hepatol. 2018, 68, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Askgaard, G.; Gronbaek, M.; Kjaer, M.S.; Tjonneland, A.; Tolstrup, J.S. Alcohol drinking pattern and risk of alcoholic liver cirrhosis: A prospective cohort study. J. Hepatol. 2015, 62, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- McClain, C.J.; Song, Z.; Barve, S.S.; Hill, D.B.; Deaciuc, I. Recent advances in alcoholic liver disease. IV. Dysregulated cytokine metabolism in alcoholic liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G497–G502. [Google Scholar] [CrossRef] [PubMed]
- Namachivayam, A.; Valsala Gopalakrishnan, A. A review on molecular mechanism of alcoholic liver disease. Life Sci. 2021, 274, 119328. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Seth, D.; Day, C.P. Genetic factors that affect risk of alcoholic and nonalcoholic fatty liver disease. Gastroenterology 2016, 150, 1728–1744.e1727. [Google Scholar] [CrossRef]
- Boyle, M.; Masson, S.; Anstee, Q.M. The bidirectional impacts of alcohol consumption and the metabolic syndrome: Cofactors for progressive fatty liver disease. J. Hepatol. 2018, 68, 251–267. [Google Scholar] [CrossRef] [Green Version]
- Caballería, L.; Pera, G.; Arteaga, I.; Rodríguez, L.; Alumà, A.; Morillas, R.M.; de la Ossa, N.; Díaz, A.; Expósito, C.; Miranda, D. High prevalence of liver fibrosis among European adults with unknown liver disease: A population-based study. Clin. Gastroenterol. Hepatol. 2018, 16, 1138–1145.e1135. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.; Harman, D.J.; Card, T.R.; Aithal, G.P.; Guha, I.N. Prevalence of clinically significant liver disease within the general population, as defined by non-invasive markers of liver fibrosis: A systematic review. Lancet Gastroenterol. Hepatol. 2017, 2, 288–297. [Google Scholar] [CrossRef]
- Hagstrom, H.; Tynelius, P.; Rasmussen, F. High BMI in late adolescence predicts future severe liver disease and hepatocellular carcinoma: A national, population-based cohort study in 1.2 million men. Gut 2018, 67, 1536–1542. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Balkwill, A.; Reeves, G.; Beral, V.; Million Women Study Collaborators. Body mass index and risk of liver cirrhosis in middle aged UK women: Prospective study. BMJ 2010, 340, c912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Chiang, D.J.; McCullough, A.J. The impact of obesity and metabolic syndrome on alcoholic liver disease. Clin. Liver Dis. 2014, 18, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, D.; Visser, M.; Sepulveda, D.; Pierson, R.N.; Harris, T.; Heymsfield, S.B. How useful is body mass index for comparison of body fatness across age, sex, and ethnic groups? Am. J. Epidemiol. 1996, 143, 228–239. [Google Scholar] [CrossRef]
- Mei, Z.; Grummer-Strawn, L.M.; Pietrobelli, A.; Goulding, A.; Goran, M.I.; Dietz, W.H. Validity of body mass index compared with other body-composition screening indexes for the assessment of body fatness in children and adolescents. Am. J. Clin. Nutr. 2002, 75, 978–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obesity: Preventing and managing the global epidemic. Report of a WHO consultation. In World Health Organization Technical Report Series; World Health Organization: Geneva, Switzerland, 2000; Volume 894, pp. i–xii, 1–253.
- Consultation, W.E. Appropriate body-mass index for Asian populations and its implications for policy and intervention strategies. Lancet 2004, 363, 157–163. [Google Scholar]
- National Institute of Health. Clinical guidelines for the identification, evaluation, and treatment of overweight and obesity in adults-the evidence report. Obes. Res. 1998, 6, 51S–209S. [Google Scholar]
- Jensen, M.D.; Ryan, D.H.; Apovian, C.M.; Ard, J.D.; Comuzzie, A.G.; Donato, K.A.; Hu, F.B.; Hubbard, V.S.; Jakicic, J.M.; Kushner, R.F.; et al. 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation 2014, 129, S102–S138. [Google Scholar] [CrossRef] [Green Version]
- Gnatiuc, L.; Alegre-Diaz, J.; Wade, R.; Ramirez-Reyes, R.; Tapia-Conyer, R.; Garcilazo-Avila, A.; Chiquete, E.; Gonzales-Carballo, C.; Solano-Sanchez, M.; Clarke, R.; et al. General and Abdominal Adiposity and Mortality in Mexico City: A Prospective Study of 150,000 Adults. Ann. Intern. Med. 2019, 171, 397–405. [Google Scholar] [CrossRef]
- Janssen, I.; Katzmarzyk, P.T.; Ross, R. Waist circumference and not body mass index explains obesity-related health risk. Am. J. Clin. Nutr. 2004, 79, 379–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson, J.A.; MacInnis, R.J.; Peeters, A.; Hopper, J.L.; Giles, G.G.; English, D.R. A comparison of adiposity measures as predictors of all-cause mortality: The Melbourne Collaborative Cohort Study. Obesity 2007, 15, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Koster, A.; Leitzmann, M.F.; Schatzkin, A.; Mouw, T.; Adams, K.F.; van Eijk, J.T.; Hollenbeck, A.R.; Harris, T.B. Waist circumference and mortality. Am. J. Epidemiol. 2008, 167, 1465–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobs, E.J.; Newton, C.C.; Wang, Y.; Patel, A.V.; McCullough, M.L.; Campbell, P.T.; Thun, M.J.; Gapstur, S.M. Waist circumference and all-cause mortality in a large US cohort. Arch. Intern. Med. 2010, 170, 1293–1301. [Google Scholar] [CrossRef]
- Tsai, A.G.; Wadden, T.A. In the clinic: Obesity. Ann. Intern. Med. 2013, 159, ITC3-1–ITC3-15. [Google Scholar] [CrossRef]
- Åberg, F.; Helenius-Hietala, J.; Puukka, P.; Färkkilä, M.; Jula, A. Interaction between alcohol consumption and metabolic syndrome in predicting severe liver disease in the general population. Hepatology 2018, 67, 2141–2149. [Google Scholar] [CrossRef] [Green Version]
- Aberg, F.; Jula, A. The sagittal abdominal diameter: Role in predicting severe liver disease in the general population. Obes. Res. Clin. Pract. 2018, 12, 394–396. [Google Scholar] [CrossRef] [Green Version]
- Andreasson, A.; Carlsson, A.C.; Önnerhag, K.; Hagström, H. Waist/hip ratio better predicts development of severe liver disease within 20 years than body mass index: A population-based cohort study. Clin. Gastroenterol. Hepatol. 2017, 15, 1294–1301.e1292. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Weiss, N.S.; Boyko, E.J.; Kowdley, K.V.; Kahn, S.E.; Carithers, R.L., Jr.; Tsai, E.C.; Dominitz, J.A. Is central obesity associated with cirrhosis-related death or hospitalization? A population-based, cohort study. Clin. Gastroenterol. Hepatol. 2005, 3, 67–74. [Google Scholar] [CrossRef]
- Pang, Q.; Zhang, J.Y.; Song, S.D.; Qu, K.; Xu, X.S.; Liu, S.S.; Liu, C. Central obesity and nonalcoholic fatty liver disease risk after adjusting for body mass index. World J. Gastroenterol. WJG 2015, 21, 1650–1662. [Google Scholar] [CrossRef]
- Ampuero, J.; Aller, R.; Gallego-Duran, R.; Banales, J.M.; Crespo, J.; Garcia-Monzon, C.; Pareja, M.J.; Vilar-Gomez, E.; Caballeria, J.; Escudero-Garcia, D.; et al. The effects of metabolic status on non-alcoholic fatty liver disease-related outcomes, beyond the presence of obesity. Aliment. Pharmacol. Ther. 2018, 48, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Grobe, Y.; Juarez-Hernandez, E.; Sanchez-Jimenez, B.A.; Uribe-Ramos, M.H.; Ramos-Ostos, M.H.; Uribe, M.; Chavez-Tapia, N.C. Less liver fibrosis in metabolically healthy compared with metabolically unhealthy obese patients with non-alcoholic fatty liver disease. Diabetes Metab. 2017, 43, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Argo, C.K.; Northup, P.G.; Al-Osaimi, A.M.; Caldwell, S.H. Systematic review of risk factors for fibrosis progression in non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Schult, A.; Mehlig, K.; Björkelund, C.; Wallerstedt, S.; Kaczynski, J. Waist-to-hip ratio but not body mass index predicts liver cirrhosis in women. Scand. J. Gastroenterol. 2018, 53, 212–217. [Google Scholar] [CrossRef]
- Vecchie, A.; Dallegri, F.; Carbone, F.; Bonaventura, A.; Liberale, L.; Portincasa, P.; Fruhbeck, G.; Montecucco, F. Obesity phenotypes and their paradoxical association with cardiovascular diseases. Eur. J. Intern. Med. 2018, 48, 6–17. [Google Scholar] [CrossRef]
- Stepanova, M.; Rafiq, N.; Younossi, Z.M. Components of metabolic syndrome are independent predictors of mortality in patients with chronic liver disease: A population-based study. Gut 2010, 59, 1410–1415. [Google Scholar] [CrossRef]
- Wainwright, P.; Scorletti, E.; Byrne, C.D. Type 2 Diabetes and Hepatocellular Carcinoma: Risk Factors and Pathogenesis. Curr. Diabetes Rep. 2017, 17, 20. [Google Scholar] [CrossRef] [Green Version]
- Whitfield, J.B.; Masson, S.; Liangpunsakul, S.; Mueller, S.; Aithal, G.P.; Eyer, F.; Gleeson, D.; Thompson, A.; Stickel, F.; Soyka, M.; et al. Obesity, Diabetes, Coffee, Tea, and Cannabis Use Alter Risk for Alcohol-Related Cirrhosis in 2 Large Cohorts of High-Risk Drinkers. Am. J. Gastroenterol. 2021, 116, 106–115. [Google Scholar] [CrossRef]
- Zakhari, S. Overview: How Is Alcohol Metabolized by the Body? Available online: https://pubs.niaaa.nih.gov/publications/arh294/245-255.htm (accessed on 24 January 2022).
- Tan, H.K.; Yates, E.; Lilly, K.; Dhanda, A.D. Oxidative stress in alcohol-related liver disease. World J. Hepatol. 2020, 12, 332–349. [Google Scholar] [CrossRef]
- Israel, Y.; Orrego, H.; Carmichael, F.J. Acetate-mediated effects of ethanol. Alcohol. Clin. Exp. Res. 1994, 18, 144–148. [Google Scholar] [CrossRef]
- Werner, J.; Saghir, M.; Warshaw, A.L.; Lewandrowski, K.B.; Laposata, M.; Iozzo, R.V.; Carter, E.A.; Schatz, R.J.; Fernández-del Castillo, C. Alcoholic pancreatitis in rats: Injury from nonoxidative metabolites of ethanol. Am. J. Physiol.-Gastrointest. Liver Physiol. 2002, 283, G65–G73. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Cederbaum, A.I. Alcohol, oxidative stress, and free radical damage. Alcohol. Res. Health 2003, 27, 277–284. [Google Scholar] [PubMed]
- Zakim, D.; Alexande, D.; Sleiseng, M.H. Effect of ethanol on hepatic secretion of triglycerides into plasma. J. Clin. Investig. 1965, 44, 1115. [Google Scholar]
- Adachi, M.; Brenner, D.A. Clinical syndromes of alcoholic liver disease. Dig. Dis. 2005, 23, 255–263. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Fischer, M.; Deeg, M.A.; Crabb, D.W. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J. Biol. Chem. 2002, 277, 29342–29347. [Google Scholar] [CrossRef] [Green Version]
- Szabo, G.; Mandrekar, P.; Dolganiuc, A. Innate immune response and hepatic inflammation. Semin. Liver Dis. 2007, 27, 339–350. [Google Scholar] [CrossRef]
- Laso, F.J.; Vaquero, J.M.; Almeida, J.; Marcos, M.; Orfao, A. Chronic alcohol consumption is associated with changes in the distribution, immunophenotype, and the inflammatory cytokine secretion profile of circulating dendritic cells. Alcohol. Clin. Exp. Res. 2007, 31, 846–854. [Google Scholar] [CrossRef]
- Marentette, J.O.; Wang, M.; Michel, C.R.; Powell, R.; Zhang, X.; Reisdorph, N.; Fritz, K.S.; Ju, C. Multi-omics Analysis of Liver Infiltrating Macrophages Following Ethanol Consumption. Sci. Rep. 2019, 9, 7776. [Google Scholar] [CrossRef]
- Li, S.; Tan, H.Y.; Wang, N.; Feng, Y.; Wang, X.; Feng, Y. Recent Insights into the Role of Immune Cells in Alcoholic Liver Disease. Front. Immunol. 2019, 10, 1328. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Aragones, G.; Colom-Pellicer, M.; Aguilar, C.; Guiu-Jurado, E.; Martinez, S.; Sabench, F.; Antonio Porras, J.; Riesco, D.; Del Castillo, D.; Richart, C.; et al. Circulating microbiota-derived metabolites: A "liquid biopsy? Int. J. Obes. 2020, 44, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small intestinal bacterial overgrowth and toll-like receptor signaling in patients with non-alcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Xi, X.P. Incomplete compensation of enhanced hepatic oxygen consumption in rats with alcoholic centrilobular liver necrosis. Hepatology 1989, 9, 302–306. [Google Scholar] [CrossRef] [PubMed]
- Arteel, G.E.; Raleigh, J.A.; Bradford, B.U.; Thurman, R.G. Acute alcohol produces hypoxia directly in rat liver tissue in vivo: Role of Kupffer cells. Am. J. Physiol. 1996, 271, G494–G500. [Google Scholar] [CrossRef]
- Ishak, K.G.; Zimmerman, H.J.; Ray, M.B. Alcoholic liver disease: Pathologic, pathogenetic and clinical aspects. Alcohol. Clin. Exp. Res. 1991, 15, 45–66. [Google Scholar] [CrossRef]
- Sheron, N.; Bird, G.; Koskinas, J.; Portmann, B.; Ceska, M.; Lindley, I.; William, R. Circulating and tissue levels of the neutrophil chemotaxin interleukin-8 are elevated in severe acute alcoholic hepatitis, and tissue levels correlate with neutrophil infiltration. Hepatology 1993, 18, 41–46. [Google Scholar]
- Hill, D.B.; Marsano, L.S.; McClain, C.J. Increased plasma interleukin-8 concentrations in alcoholic hepatitis. Hepatology 1993, 18, 576–580. [Google Scholar] [CrossRef]
- Masumoto, T.; Onji, M.; Horiike, N.; Ohta, Y. Assay of serum interleukin 8 levels in patients with alcoholic hepatitis. Alcohol. Alcohol. Suppl. 1993, 1A, 99–102. [Google Scholar] [CrossRef]
- Roll, F.J.; Perez, H.D.; Serhan, C.N. Characterization of a novel arachidonic acid-derived neutrophil chemoattractant. Biochem. Biophys. Res. Commun. 1992, 186, 269–276. [Google Scholar] [CrossRef]
- Roll, F.J.; Alexander, M.A.; Cua, D.; Swanson, W.; Perez, H.D. Metabolism of ethanol by rat hepatocytes results in generation of a lipid chemotactic factor: Studies using a cell-free system and role of oxygen-derived free radicals. Arch. Biochem. Biophys. 1991, 287, 218–224. [Google Scholar] [CrossRef]
- Barry, R.E.; Williams, A.J.; McGivan, J.D. The detection of acetaldehyde/liver plasma membrane protein adduct formed in vivo by alcohol feeding. Liver 1987, 7, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Lumeng, L.; Shahidi, S.; Kelly, T.; Pound, D.C. Protein-acetaldehyde adducts in serum of alcoholic patients. Alcohol. Clin. Exp. Res. 1990, 14, 438–443. [Google Scholar] [CrossRef]
- Tuma, D.J. Role of malondialdehyde-acetaldehyde adducts in liver injury. Free Radic. Biol. Med. 2002, 32, 303–308. [Google Scholar] [CrossRef]
- Tuma, D.J.; Casey, C.A. Dangerous byproducts of alcohol breakdown--focus on adducts. Alcohol. Res. Health 2003, 27, 285–290. [Google Scholar]
- Bradford, B.U.; Kono, H.; Isayama, F.; Kosyk, O.; Wheeler, M.D.; Akiyama, T.E.; Bleye, L.; Krausz, K.W.; Gonzalez, F.J.; Koop, D.R.; et al. Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology 2005, 41, 336–344. [Google Scholar] [CrossRef]
- Nagy, L.E. Molecular aspects of alcohol metabolism: Transcription factors involved in early ethanol-induced liver injury. Annu. Rev. Nutr. 2004, 24, 55–78. [Google Scholar] [CrossRef]
- Crabb, D.W.; Pinairs, J.; Hasanadka, R.; Fang, M.; Leo, M.A.; Lieber, C.S.; Tsukamoto, H.; Motomura, K.; Miyahara, T.; Ohata, M.; et al. Alcohol and retinoids. Alcohol. Clin. Exp. Res. 2001, 25, 207S–217S. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Schuppan, D.; Hahn, E.G.; Seitz, H.K. Cocarcinogenic effects of alcohol in hepatocarcinogenesis. Gut 2002, 51, 132–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poschl, G.; Seitz, H.K. Alcohol and cancer. Alcohol Alcohol. 2004, 39, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niemela, O.; Parkkila, S.; Yla-Herttuala, S.; Halsted, C.; Witztum, J.L.; Lanca, A.; Israel, Y. Covalent protein adducts in the liver as a result of ethanol metabolism and lipid peroxidation. Lab. Investig. 1994, 70, 537–546. [Google Scholar] [PubMed]
- Holstege, A.; Bedossa, P.; Poynard, T.; Kollinger, M.; Chaput, J.C.; Houglum, K.; Chojkier, M. Acetaldehyde-modified epitopes in liver biopsy specimens of alcoholic and nonalcoholic patients: Localization and association with progression of liver fibrosis. Hepatology 1994, 19, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Clot, P.; Parola, M.; Bellomo, G.; Dianzani, U.; Carini, R.; Tabone, M.; Arico, S.; Ingelman-Sundberg, M.; Albano, E. Plasma membrane hydroxyethyl radical adducts cause antibody-dependent cytotoxicity in rat hepatocytes exposed to alcohol. Gastroenterology 1997, 113, 265–276. [Google Scholar] [CrossRef]
- Chen, W.Y.; Zhang, J.; Ghare, S.; Barve, S.; McClain, C.; Joshi-Barve, S. Acrolein Is a Pathogenic Mediator of Alcoholic Liver Disease and the Scavenger Hydralazine Is Protective in Mice. Cell. Mol. Gastroenterol. Hepatol. 2016, 2, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Parola, M.; Robino, G. Oxidative stress-related molecules and liver fibrosis. J. Hepatol. 2001, 35, 297–306. [Google Scholar] [CrossRef]
- Sampey, B.P.; Stewart, B.J.; Petersen, D.R. Ethanol-induced modulation of hepatocellular extracellular signal-regulated kinase-1/2 activity via 4-hydroxynonenal. J. Biol. Chem. 2007, 282, 1925–1937. [Google Scholar] [CrossRef] [Green Version]
- Sastre, J.; Serviddio, G.; Pereda, J.; Minana, J.B.; Arduini, A.; Vendemiale, G.; Poli, G.; Pallardo, F.V.; Vina, J. Mitochondrial function in liver disease. Front. Biosci. A J. Virtual Libr. 2007, 12, 1200–1209. [Google Scholar] [CrossRef] [Green Version]
- Lieber, C.S.; DeCarli, L.M. Ethanol oxidation by hepatic microsomes: Adaptive increase after ethanol feeding. Science 1968, 162, 917–918. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Lasker, J.M.; Takahashi, T.; Lieber, C.S. In vivo induction of hepatic P4502E1 by ethanol: Role of increased enzyme synthesis. Arch. Biochem. Biophys. 1993, 304, 209–218. [Google Scholar] [CrossRef]
- Zima, T.; Kalousova, M. Oxidative stress and signal transduction pathways in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2005, 29, 110S–115S. [Google Scholar] [CrossRef]
- Dryden, G.W., Jr.; Deaciuc, I.; Arteel, G.; McClain, C.J. Clinical implications of oxidative stress and antioxidant therapy. Curr. Gastroenterol. Rep. 2005, 7, 308–316. [Google Scholar] [CrossRef] [PubMed]
- Yuan, G.J.; Zhou, X.R.; Gong, Z.J.; Zhang, P.; Sun, X.M.; Zheng, S.H. Expression and activity of inducible nitric oxide synthase and endothelial nitric oxide synthase correlate with ethanol-induced liver injury. World J. Gastroenterol. WJG 2006, 12, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Huang, M.; Xin, Y.; Zhang, Y.; Zhang, X.; Wang, G.; Liu, S.; Wan, J.; Ahmadi, A.R.; Sun, Z.; et al. The epigenetic regulator SIRT6 protects the liver from alcohol-induced tissue injury by reducing oxidative stress in mice. J. Hepatol. 2019, 71, 960–969. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, T.; Li, Y.M.; Yin, S.; Xu, M.J.; Feng, D.; Zhou, Z.; Zang, M.; Mukhopadhyay, P.; Varga, Z.V.; Pacher, P.; et al. Aging aggravates alcoholic liver injury and fibrosis in mice by downregulating sirtuin 1 expression. J. Hepatol. 2017, 66, 601–609. [Google Scholar] [CrossRef] [Green Version]
- Torok, N.J. Update on Alcoholic Hepatitis. Biomolecules 2015, 5, 2978–2986. [Google Scholar] [CrossRef] [Green Version]
- Llopis, M.; Cassard, A.M.; Wrzosek, L.; Boschat, L.; Bruneau, A.; Ferrere, G.; Puchois, V.; Martin, J.C.; Lepage, P.; Le Roy, T.; et al. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut 2016, 65, 830–839. [Google Scholar] [CrossRef]
- Schnabl, B. Liver capsule: Mechanisms of alcoholic hepatitis. Hepatology 2016, 64, 276. [Google Scholar] [CrossRef] [Green Version]
- Bode, C.; Kugler, V.; Bode, J.C. Endotoxemia in patients with alcoholic and non-alcoholic cirrhosis and in subjects with no evidence of chronic liver disease following acute alcohol excess. J. Hepatol. 1987, 4, 8–14. [Google Scholar] [CrossRef]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. 1), 38–42. [Google Scholar] [CrossRef] [Green Version]
- Petrasek, J.; Iracheta-Vellve, A.; Csak, T.; Satishchandran, A.; Kodys, K.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Szabo, G. STING-IRF3 pathway links endoplasmic reticulum stress with hepatocyte apoptosis in early alcoholic liver disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16544–16549. [Google Scholar] [CrossRef] [Green Version]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.; Bradford, B.U.; Wheeler, M.D.; Uesugi, T.; Froh, M.; Goyert, S.M.; Thurman, R.G. Reduced early alcohol-induced liver injury in CD14-deficient mice. J. Immunol. 2001, 166, 4737–4742. [Google Scholar] [CrossRef] [Green Version]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Wheeler, M.D.; Gabele, E.; Isayama, F.; Thurman, R.G. Role of lipopolysaccharide-binding protein in early alcohol-induced liver injury in mice. J. Immunol. 2002, 168, 2963–2969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, B.M.; Ma, D.W.; Simons, B.; Noureldin, S.A.; Therapondos, G.; Guindi, M.; Sherman, M.; Allard, J.P. Nonalcoholic fatty liver disease is associated with lower hepatic and erythrocyte ratios of phosphatidylcholine to phosphatidylethanolamine. Appl. Physiol. Nutr. Metab. 2013, 38, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarvelainen, H.A.; Fang, C.; Ingelman-Sundberg, M.; Lindros, K.O. Effect of chronic coadministration of endotoxin and ethanol on rat liver pathology and proinflammatory and anti-inflammatory cytokines. Hepatology 1999, 29, 1503–1510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McClain, C.; Hill, D.; Schmidt, J.; Diehl, A.M. Cytokines and alcoholic liver disease. Semin. Liver Dis. 1993, 13, 170–182. [Google Scholar] [CrossRef]
- Morales-Ibanez, O.; Affo, S.; Rodrigo-Torres, D.; Blaya, D.; Millan, C.; Coll, M.; Perea, L.; Odena, G.; Knorpp, T.; Templin, M.F.; et al. Kinase analysis in alcoholic hepatitis identifies p90RSK as a potential mediator of liver fibrogenesis. Gut 2016, 65, 840–851. [Google Scholar] [CrossRef] [Green Version]
- Iimuro, Y.; Gallucci, R.M.; Luster, M.I.; Kono, H.; Thurman, R.G. Antibodies to tumor necrosis factor alfa attenuate hepatic necrosis and inflammation caused by chronic exposure to ethanol in the rat. Hepatology 1997, 26, 1530–1537. [Google Scholar] [CrossRef]
- Yin, M.; Wheeler, M.D.; Kono, H.; Bradford, B.U.; Gallucci, R.M.; Luster, M.I.; Thurman, R.G. Essential role of tumor necrosis factor alpha in alcohol-induced liver injury in mice. Gastroenterology 1999, 117, 942–952. [Google Scholar] [CrossRef]
- Mundt, B.; Wirth, T.; Zender, L.; Waltemathe, M.; Trautwein, C.; Manns, M.P.; Kuhnel, F.; Kubicka, S. Tumour necrosis factor related apoptosis inducing ligand (TRAIL) induces hepatic steatosis in viral hepatitis and after alcohol intake. Gut 2005, 54, 1590–1596. [Google Scholar] [CrossRef] [Green Version]
- Ma, H.Y.; Yamamoto, G.; Xu, J.; Liu, X.; Karin, D.; Kim, J.Y.; Alexandrov, L.B.; Koyama, Y.; Nishio, T.; Benner, C.; et al. IL-17 signaling in steatotic hepatocytes and macrophages promotes hepatocellular carcinoma in alcohol-related liver disease. J. Hepatol. 2020, 72, 946–959. [Google Scholar] [CrossRef]
- Nanji, A.A.; Zakim, D.; Rahemtulla, A.; Daly, T.; Miao, L.; Zhao, S.; Khwaja, S.; Tahan, S.R.; Dannenberg, A.J. Dietary saturated fatty acids down-regulate cyclooxygenase-2 and tumor necrosis factor alfa and reverse fibrosis in alcohol-induced liver disease in the rat. Hepatology 1997, 26, 1538–1545. [Google Scholar] [CrossRef]
- Nanji, A.A.; Khwaja, S.; Rahemtulla, A.; Miao, L.; Zhao, S.; Tahan, S.R. Thromboxane inhibitors attenuate pathological changes in alcoholic liver disease in the rat. Gastroenterology 1997, 112, 200–207. [Google Scholar] [CrossRef]
- Nanji, A.A.; Miao, L.; Thomas, P.; Rahemtulla, A.; Khwaja, S.; Zhao, S.; Peters, D.; Tahan, S.R.; Dannenberg, A.J. Enhanced cyclooxygenase-2 gene expression in alcoholic liver disease in the rat. Gastroenterology 1997, 112, 943–951. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet. 2010, 42, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Blaya, D.; Coll, M.; Rodrigo-Torres, D.; Vila-Casadesus, M.; Altamirano, J.; Llopis, M.; Graupera, I.; Perea, L.; Aguilar-Bravo, B.; Diaz, A.; et al. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut 2016, 65, 1535–1545. [Google Scholar] [CrossRef] [PubMed]
- Argemi, J.; Latasa, M.U.; Atkinson, S.R.; Blokhin, I.O.; Massey, V.; Gue, J.P.; Cabezas, J.; Lozano, J.J.; Van Booven, D.; Bell, A.; et al. Defective HNF4alpha-dependent gene expression as a driver of hepatocellular failure in alcoholic hepatitis. Nat. Commun. 2019, 10, 3126. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, T.; Bell, A.; Brooks, J.M.; Setoyama, K.; Melis, M.; Han, B.; Fukumitsu, K.; Handa, K.; Tian, J.; Kaestner, K.H.; et al. Resetting the transcription factor network reverses terminal chronic hepatic failure. J. Clin. Investig. 2015, 125, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Ferrere, G.; Wrzosek, L.; Cailleux, F.; Turpin, W.; Puchois, V.; Spatz, M.; Ciocan, D.; Rainteau, D.; Humbert, L.; Hugot, C.; et al. Fecal microbiota manipulation prevents dysbiosis and alcohol-induced liver injury in mice. J. Hepatol. 2017, 66, 806–815. [Google Scholar] [CrossRef]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. WJG 2014, 20, 17756–17772. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Gaal, K.; French, S.W. Insights into the pathogenesis of alcoholic liver necrosis and fibrosis: Status report. Hepatology 1990, 12, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Okanoue, T.; Burbige, E.J.; French, S.W. The role of the Ito cell in perivenular and intralobular fibrosis in alcoholic hepatitis. Arch. Pathol. Lab. Med. 1983, 107, 459–463. [Google Scholar]
- Casini, A.; Cunningham, M.; Rojkind, M.; Lieber, C.S. Acetaldehyde increases procollagen type I and fibronectin gene transcription in cultured rat fat-storing cells through a protein synthesis-dependent mechanism. Hepatology 1991, 13, 758–765. [Google Scholar]
- Kawase, T.; Kato, S.; Lieber, C.S. Lipid peroxidation and antioxidant defense systems in rat liver after chronic ethanol feeding. Hepatology 1989, 10, 815–821. [Google Scholar] [CrossRef]
- Kamimura, S.; Gaal, K.; Britton, R.S.; Bacon, B.R.; Triadafilopoulos, G.; Tsukamoto, H. Increased 4-hydroxynonenal levels in experimental alcoholic liver disease: Association of lipid peroxidation with liver fibrogenesis. Hepatology 1992, 16, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Brenner, D.A. Mechanisms of alcohol-induced hepatic fibrosis: A summary of the Ron Thurman Symposium. Hepatology 2006, 43, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Albano, E. Alcohol, oxidative stress and free radical damage. Proc. Nutr. Soc. 2006, 65, 278–290. [Google Scholar] [CrossRef] [Green Version]
- Crabb, D.W.; Im, G.Y.; Szabo, G.; Mellinger, J.L.; Lucey, M.R. Diagnosis and Treatment of Alcohol-Associated Liver Diseases: 2019 Practice Guidance From the American Association for the Study of Liver Diseases. Hepatology 2020, 71, 306–333. [Google Scholar] [CrossRef] [Green Version]
- Dam, M.K.; Flensborg-Madsen, T.; Eliasen, M.; Becker, U.; Tolstrup, J.S. Smoking and risk of liver cirrhosis: A population-based cohort study. Scand. J. Gastroenterol. 2013, 48, 585–591. [Google Scholar] [CrossRef]
- Rinella, M.; Charlton, M. The Globalization of Nonalcoholic Fatty Liver Disease: Prevalence and Impact on World Health. Hepatology 2015, 64, 19–22. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Taylor, B.; Mohapatra, S.; Irving, H.; Baliunas, D.; Patra, J.; Roerecke, M. Alcohol as a risk factor for liver cirrhosis: A systematic review and meta-analysis. Drug Alcohol Rev. 2010, 29, 437–445. [Google Scholar] [CrossRef]
- Teli, M.R.; Day, C.P.; Burt, A.D.; Bennett, M.K.; James, O.F. Determinants of progression to cirrhosis or fibrosis in pure alcoholic fatty liver. Lancet 1995, 346, 987–990. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Costa, G.; Tiribelli, C.; Manenti, F.; Sodde, M.; Croce, L.S.; Sasso, F.; Pozzato, G.; Cristianini, G. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997, 41, 845–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OECD. Obesity Update. Available online: http://www.oecd.org/health/obesity-update.htm (accessed on 24 February 2021).
- Sahlman, P.; Nissinen, M.; Puukka, P.; Jula, A.; Salomaa, V.; Männistö, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Färkkilä, M. Genetic and lifestyle risk factors for advanced liver disease among men and women. J. Gastroenterol. Hepatol. 2020, 35, 291–298. [Google Scholar] [CrossRef]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Davey Smith, G. Effect of body mass index and alcohol consumption on liver disease: Analysis of data from two prospective cohort studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [Green Version]
- Åberg, F.; Färkkilä, M. Drinking and obesity: Alcoholic liver disease/nonalcoholic fatty liver disease interactions. Semin. Liver Dis. 2020, 40, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Baumeister, S.E.; Lieb, W.; Meffert, P.J.; Lerch, M.M.; Mayerle, J.; Volzke, H. The combined effects of alcohol consumption and body mass index on hepatic steatosis in a general population sample of European men and women. Aliment. Pharmacol. Ther. 2015, 41, 467–476. [Google Scholar] [CrossRef]
- Rethinking Drinking. Alcohol & Your Health. Available online: https://www.rethinkingdrinking.niaaa.nih.gov/ (accessed on 20 April 2021).
- Sayon-Orea, C.; Martinez-Gonzalez, M.A.; Bes-Rastrollo, M. Alcohol consumption and body weight: A systematic review. Nutr. Rev. 2011, 69, 419–431. [Google Scholar] [CrossRef]
- Traversy, G.; Chaput, J.P. Alcohol Consumption and Obesity: An Update. Curr. Obes. Rep. 2015, 4, 122–130. [Google Scholar] [CrossRef] [Green Version]
- Bendsen, N.T.; Christensen, R.; Bartels, E.M.; Kok, F.J.; Sierksma, A.; Raben, A.; Astrup, A. Is beer consumption related to measures of abdominal and general obesity? A systematic review and meta-analysis. Nutr. Rev. 2013, 71, 67–87. [Google Scholar] [CrossRef]
- Parker, R.; Kim, S.J.; Gao, B. Alcohol, adipose tissue and liver disease: Mechanistic links and clinical considerations. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Kechagias, S.; Zanjani, S.; Gjellan, S.; Leinhard, O.D.; Kihlberg, J.; Smedby, O.; Johansson, L.; Kullberg, J.; Ahlstrom, H.; Lindstrom, T.; et al. Effects of moderate red wine consumption on liver fat and blood lipids: A prospective randomized study. Ann. Med. 2011, 43, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, J.K.; Olafsson, S.; Bergmann, O.M.; Runarsdottir, V.; Hansdottir, I.; Sigurdardottir, R.; Björnsson, E.S. Lifetime drinking history in patients with alcoholic liver disease and patients with alcohol use disorder without liver disease. Scand. J. Gastroenterol. 2017, 52, 762–767. [Google Scholar] [CrossRef]
- Stokkeland, K.; Hilm, G.; Spak, F.; Franck, J.; Hultcrantz, R. Different drinking patterns for women and men with alcohol dependence with and without alcoholic cirrhosis. Alcohol Alcohol. 2008, 43, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatton, J.; Burton, A.; Nash, H.; Munn, E.; Burgoyne, L.; Sheron, N. Drinking patterns, dependency and life-time drinking history in alcohol-related liver disease. Addiction 2009, 104, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J.C. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.F.; Hermon, C.; Liu, B.; Green, J.; Reeves, G.K.; Beral, V.; Floud, S.; Million Women Study Collaborators. Alcohol drinking patterns and liver cirrhosis risk: Analysis of the prospective UK Million Women Study. Lancet Public Health 2019, 4, e41–e48. [Google Scholar] [CrossRef] [Green Version]
- Aberg, F.; Helenius-Hietala, J.; Puukka, P.; Jula, A. Binge drinking and the risk of liver events: A population-based cohort study. Liver Int. 2017, 37, 1373–1381. [Google Scholar] [CrossRef]
- Betrapally, N.S.; Gillevet, P.M.; Bajaj, J.S. Changes in the Intestinal Microbiome and Alcoholic and Nonalcoholic Liver Diseases: Causes or Effects? Gastroenterology 2016, 150, 1745–1755.e1743. [Google Scholar] [CrossRef] [Green Version]
- Åberg, F.; Puukka, P.; Sahlman, P.; Nissinen, M.; Salomaa, V.; Männistö, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Jula, A. Metabolic risk factors for advanced liver disease among alcohol risk users in the general population. J. Hepatol. 2019, 70, E273. [Google Scholar] [CrossRef]
- Hejlova, I.; Honsova, E.; Sticova, E.; Lanska, V.; Hucl, T.; Spicak, J.; Jirsa, M.; Trunecka, P. Prevalence and risk factors of steatosis after liver transplantation and patient outcomes. Liver Transplant. 2016, 22, 644–655. [Google Scholar] [CrossRef] [Green Version]
- Losurdo, G.; Castellaneta, A.; Rendina, M.; Carparelli, S.; Leandro, G.; Di Leo, A. Systematic review with meta-analysis: De novo non-alcoholic fatty liver disease in liver-transplanted patients. Aliment. Pharmacol. Ther. 2018, 47, 704–714. [Google Scholar] [CrossRef]
- Sookoian, S.; Pirola, C.J. Systems biology elucidates common pathogenic mechanisms between nonalcoholic and alcoholic-fatty liver disease. PLoS ONE 2013, 8, e58895. [Google Scholar] [CrossRef] [Green Version]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, L.E.; Ding, W.X.; Cresci, G.; Saikia, P.; Shah, V.H. Linking Pathogenic Mechanisms of Alcoholic Liver Disease With Clinical Phenotypes. Gastroenterology 2016, 150, 1756–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakaguchi, S.; Takahashi, S.; Sasaki, T.; Kumagai, T.; Nagata, K. Progression of alcoholic and non-alcoholic steatohepatitis: Common metabolic aspects of innate immune system and oxidative stress. Drug Metab. Pharm. 2011, 26, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Kwong, E.K.; Puri, P. Gut microbiome changes in Nonalcoholic fatty liver disease & alcoholic liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 3. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Suzuki, H.; Sakaguchi, S. Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J. Toxicol. Sci. 2007, 32, 453–468. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246. [Google Scholar] [CrossRef]
- Liu, Y.M.; Moldes, M.; Bastard, J.P.; Bruckert, E.; Viguerie, N.; Hainque, B.; Basdevant, A.; Langin, D.; Pairault, J.; Clement, K. Adiponutrin: A new gene regulated by energy balance in human adipose tissue. J. Clin. Endocrinol. Metab. 2004, 89, 2684–2689. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, N.S.; Duseja, A. Genetic and epigenetic disease modifiers: Non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD). Transl. Gastroenterol. Hepatol. 2021, 6, 2. [Google Scholar] [CrossRef]
- Trepo, E.; Gustot, T.; Degre, D.; Lemmers, A.; Verset, L.; Demetter, P.; Ouziel, R.; Quertinmont, E.; Vercruysse, V.; Amininejad, L.; et al. Common polymorphism in the PNPLA3/adiponutrin gene confers higher risk of cirrhosis and liver damage in alcoholic liver disease. J. Hepatol. 2011, 55, 906–912. [Google Scholar] [CrossRef]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Grimaudo, S.; Pipitone, R.M.; Pennisi, G.; Celsa, C.; Cammà, C.; Di Marco, V.; Barcellona, M.R.; Boemi, R.; Enea, M.; Giannetti, A. Association between PNPLA3 rs738409 C> G variant and liver-related outcomes in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2020, 18, 935–944.e933. [Google Scholar] [CrossRef] [PubMed]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 2016, 48, 69–75. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Donati, B.; Fares, R.; Lombardi, R.; Mancina, R.M.; Romeo, S.; Valenti, L. PNPLA3 I148M polymorphism and progressive liver disease. World J. Gastroenterol. WJG 2013, 19, 6969–6978. [Google Scholar] [CrossRef]
- Stickel, F.; Moreno, C.; Hampe, J.; Morgan, M.Y. The genetics of alcohol dependence and alcohol-related liver disease. J. Hepatol. 2017, 66, 195–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Vecchione, G.; Grasselli, E.; Compalati, A.D.; Ragazzoni, M.; Cortese, K.; Gallo, G.; Voci, A.; Vergani, L. Ethanol and fatty acids impair lipid homeostasis in an in vitro model of hepatic steatosis. Food Chem. Toxicol. 2016, 90, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Xu, J.; Lai, K.K.Y.; Verlinsky, A.; Lugea, A.; French, S.W.; Cooper, M.P.; Ji, C.; Tsukamoto, H. Synergistic steatohepatitis by moderate obesity and alcohol in mice despite increased adiponectin and p-AMPK. J. Hepatol. 2011, 55, 673–682. [Google Scholar] [CrossRef] [Green Version]
- Grasselli, E.; Voci, A.; Demori, I.; De Matteis, R.; Compalati, A.D.; Gallo, G.; Vergani, L. Effects of binge ethanol on lipid homeostasis and oxidative stress in a rat model of nonalcoholic fatty liver disease. J. Physiol. Biochem. 2014, 70, 341–353. [Google Scholar] [CrossRef]
- de Medeiros, I.C.; de Lima, J.G. Is nonalcoholic fatty liver disease an endogenous alcoholic fatty liver disease?–A mechanistic hypothesis. Med. Hypotheses 2015, 85, 148–152. [Google Scholar] [CrossRef]
- Guo, F.; Zheng, K.; Benede-Ubieto, R.; Cubero, F.J.; Nevzorova, Y.A. The Lieber-DeCarli Diet-A Flagship Model for Experimental Alcoholic Liver Disease. Alcohol. Clin. Exp. Res. 2018, 42, 1828–1840. [Google Scholar] [CrossRef] [PubMed]
- Minato, T.; Tsutsumi, M.; Tsuchishima, M.; Hayashi, N.; Saito, T.; Matsue, Y.; Toshikuni, N.; Arisawa, T.; George, J. Binge alcohol consumption aggravates oxidative stress and promotes pathogenesis of NASH from obesity-induced simple steatosis. Mol. Med. 2014, 20, 490–502. [Google Scholar] [CrossRef]
- Duly, A.M.; Alani, B.; Huang, E.Y.; Yee, C.; Haber, P.S.; McLennan, S.V.; Seth, D. Effect of multiple binge alcohol on diet-induced liver injury in a mouse model of obesity. Nutr. Diabetes 2015, 5, e154. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.S.; Baker, R.D.; Liu, W.; Nowak, N.J.; Zhu, L. Role of alcohol metabolism in non-alcoholic steatohepatitis. PLoS ONE 2010, 5, e9570. [Google Scholar] [CrossRef] [Green Version]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Croce, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef]
- Loomba, R.; Bettencourt, R.; Barrett-Connor, E. Synergistic association between alcohol intake and body mass index with serum alanine and aspartate aminotransferase levels in older adults: The Rancho Bernardo Study. Aliment. Pharmacol. Ther. 2009, 30, 1137–1149. [Google Scholar] [CrossRef] [Green Version]
- Ruhl, C.E.; Everhart, J.E. Joint effects of body weight and alcohol on elevated serum alanine aminotransferase in the United States population. Clin. Gastroenterol. Hepatol. 2005, 3, 1260–1268. [Google Scholar] [CrossRef]
- Alatalo, P.I.; Koivisto, H.M.; Hietala, J.P.; Puukka, K.S.; Bloigu, R.; Niemela, O.J. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am. J. Clin. Nutr. 2008, 88, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Castano, G.O.; Pirola, C.J. Modest alcohol consumption decreases the risk of non-alcoholic fatty liver disease: A meta-analysis of 43 175 individuals. Gut 2014, 63, 530–532. [Google Scholar] [CrossRef]
- Kwon, H.K.; Greenson, J.K.; Conjeevaram, H.S. Effect of lifetime alcohol consumption on the histological severity of non-alcoholic fatty liver disease. Liver Int. 2014, 34, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Hagström, H.; Nasr, P.; Ekstedt, M.; Kechagias, S.; Önnerhag, K.; Nilsson, E.; Rorsman, F.; Sheikhi, R.; Marschall, H.-U.; Hultcrantz, R. Low to moderate lifetime alcohol consumption is associated with less advanced stages of fibrosis in non-alcoholic fatty liver disease. Scand. J. Gastroenterol. 2017, 52, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Dunn, W.; Sanyal, A.J.; Brunt, E.M.; Unalp-Arida, A.; Donohue, M.; McCullough, A.J.; Schwimmer, J.B. Modest alcohol consumption is associated with decreased prevalence of steatohepatitis in patients with non-alcoholic fatty liver disease (NAFLD). J. Hepatol. 2012, 57, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Moriya, A.; Iwasaki, Y.; Ohguchi, S.; Kayashima, E.; Mitsumune, T.; Taniguchi, H.; Ikeda, F.; Shiratori, Y.; Yamamoto, K. Alcohol consumption appears to protect against non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2011, 33, 378–388. [Google Scholar] [CrossRef]
- Pietraszek, A.; Gregersen, S.; Hermansen, K. Alcohol and type 2 diabetes. A review. Nutr. Metab. Cardiovasc. Dis. NMCD 2010, 20, 366–375. [Google Scholar] [CrossRef]
- Roerecke, M.; Rehm, J. Alcohol consumption, drinking patterns, and ischemic heart disease: A narrative review of meta-analyses and a systematic review and meta-analysis of the impact of heavy drinking occasions on risk for moderate drinkers. BMC Med. 2014, 12, 182. [Google Scholar] [CrossRef] [Green Version]
- Ajmera, V.H.; Terrault, N.A.; Harrison, S.A. Is moderate alcohol use in nonalcoholic fatty liver disease good or bad? A critical review. Hepatology 2017, 65, 2090–2099. [Google Scholar] [CrossRef] [Green Version]
- Salameh, H.; Raff, E.; Erwin, A.; Seth, D.; Nischalke, H.D.; Falleti, E.; Burza, M.A.; Leathert, J.; Romeo, S.; Molinaro, A.; et al. PNPLA3 Gene Polymorphism Is Associated With Predisposition to and Severity of Alcoholic Liver Disease. Am. J. Gastroenterol. 2015, 110, 846–856. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, M.; Liebe, R.; Lammert, F. Toward Genetic Prediction of Nonalcoholic Fatty Liver Disease Trajectories: PNPLA3 and Beyond. Gastroenterology 2020, 158, 1865–1880.e1861. [Google Scholar] [CrossRef]
- Israelsen, M.; Juel, H.B.; Detlefsen, S.; Madsen, B.S.; Rasmussen, D.N.; Larsen, T.R.; Kjaergaard, M.; Fernandes Jensen, M.J.; Stender, S.; Hansen, T.; et al. Metabolic and Genetic Risk Factors Are the Strongest Predictors of Severity of Alcohol-Related Liver Fibrosis. Clin. Gastroenterol. Hepatol. 2020. [Google Scholar] [CrossRef]
- Dong, X.C. PNPLA3-A Potential Therapeutic Target for Personalized Treatment of Chronic Liver Disease. Front. Med. 2019, 6, 304. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjaerg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, B.; Linden, D.; Brolen, G.; Liljeblad, M.; Bjursell, M.; Romeo, S.; Loomba, R. Review article: The emerging role of genetics in precision medicine for patients with non-alcoholic steatohepatitis. Aliment. Pharmacol. Ther. 2020, 51, 1305–1320. [Google Scholar] [CrossRef] [PubMed]
- Johansen, A.; Rosti, R.O.; Musaev, D.; Sticca, E.; Harripaul, R.; Zaki, M.; Caglayan, A.O.; Azam, M.; Sultan, T.; Froukh, T.; et al. Mutations in MBOAT7, Encoding Lysophosphatidylinositol Acyltransferase I, Lead to Intellectual Disability Accompanied by Epilepsy and Autistic Features. Am. J. Hum. Genet. 2016, 99, 912–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stickel, F.; Lutz, P.; Buch, S.; Nischalke, H.D.; Silva, I.; Rausch, V.; Fischer, J.; Weiss, K.H.; Gotthardt, D.; Rosendahl, J.; et al. Genetic Variation in HSD17B13 Reduces the Risk of Developing Cirrhosis and Hepatocellular Carcinoma in Alcohol Misusers. Hepatology 2020, 72, 88–102. [Google Scholar] [CrossRef] [PubMed]
- Innes, H.; Buch, S.; Hutchinson, S.; Guha, I.N.; Morling, J.R.; Barnes, E.; Irving, W.; Forrest, E.; Pedergnana, V.; Goldberg, D.; et al. Genome-Wide Association Study for Alcohol-Related Cirrhosis Identifies Risk Loci in MARC1 and HNRNPUL1. Gastroenterology 2020, 159, 1276–1289.e1277. [Google Scholar] [CrossRef] [PubMed]
- Luukkonen, P.K.; Juuti, A.; Sammalkorpi, H.; Penttila, A.K.; Oresic, M.; Hyotylainen, T.; Arola, J.; Orho-Melander, M.; Yki-Jarvinen, H. MARC1 variant rs2642438 increases hepatic phosphatidylcholines and decreases severity of non-alcoholic fatty liver disease in humans. J. Hepatol. 2020, 73, 725–726. [Google Scholar] [CrossRef]
- De Vincentis, A.; Tavaglione, F.; Jamialahmadi, O.; Picardi, A.; Antonelli-Incalzi, R.; Valenti, L.; Romeo, S.; Vespasiani-Gentilucci, U. A Polygenic Risk Score to Refine Risk Stratification and Prediction for Severe Liver Disease by Clinical Fibrosis Scores. Clin. Gastroenterol. Hepatol. 2022, 20, 658–673. [Google Scholar] [CrossRef]
- Cao, Y.; Willett, W.C.; Rimm, E.B.; Stampfer, M.J.; Giovannucci, E.L. Light to moderate intake of alcohol, drinking patterns, and risk of cancer: Results from two prospective US cohort studies. BMJ 2015, 351, h4238. [Google Scholar] [CrossRef] [Green Version]
- Di Castelnuovo, A.; Costanzo, S.; Bagnardi, V.; Donati, M.B.; Iacoviello, L.; de Gaetano, G. Alcohol dosing and total mortality in men and women: An updated meta-analysis of 34 prospective studies. Arch. Intern. Med. 2006, 166, 2437–2445. [Google Scholar] [CrossRef]
- Åberg, F.; Puukka, P.; Sahlman, P.; Nissinen, M.; Salomaa, V.; Männistö, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Jula, A. In NAFLD, alcohol drinking habits and genetics predict progression to advanced liver disease: Follow-up of population surveys. J. Hepatol. 2019, 70, E141. [Google Scholar] [CrossRef]
- VanWagner, L.B.; Ning, H.; Allen, N.B.; Ajmera, V.; Lewis, C.E.; Carr, J.J.; Lloyd-Jones, D.M.; Terrault, N.A.; Siddique, J. Alcohol Use and Cardiovascular Disease Risk in Patients With Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 153, 1260–1272.e1263. [Google Scholar] [CrossRef] [PubMed]
- Wood, A.M.; Kaptoge, S.; Butterworth, A.S.; Willeit, P.; Warnakula, S.; Bolton, T.; Paige, E.; Paul, D.S.; Sweeting, M.; Burgess, S.; et al. Risk thresholds for alcohol consumption: Combined analysis of individual-participant data for 599,912 current drinkers in 83 prospective studies. Lancet 2018, 391, 1513–1523. [Google Scholar] [CrossRef] [Green Version]
- Staufer, K.; Strebinger, G.; Huber-Schoenauer, U.; Suesse, S.; Teufelhart, M.; Prager, G.; Paulweber, B.; Ferenci, P.; Stimpfl, T.; Yegles, M. Ethyl glucuronide in hair uncovers a high rate of harmful alcohol consumption in patients with presumed NAFLD. J. Hepatol. 2019, 70, E784. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Ong, J.; Yilmaz, Y.; Duseja, A.; Eguchi, Y.; El Kassas, M.; Castellanos-Fernandez, M.; George, J.; Jacobson, I.M.; et al. Effects of Alcohol Consumption and Metabolic Syndrome on Mortality in Patients With Nonalcoholic and Alcohol-Related Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 1625–1633.e1621. [Google Scholar] [CrossRef]
- Hajifathalian, K.; Torabi Sagvand, B.; McCullough, A.J. Effect of Alcohol Consumption on Survival in Nonalcoholic Fatty Liver Disease: A National Prospective Cohort Study. Hepatology 2019, 70, 511–521. [Google Scholar] [CrossRef]
- Vilar-Gomez, E.; Calzadilla-Bertot, L.; Wong, V.W.-S.; Castellanos, M.; Aller-de la Fuente, R.; Metwally, M.; Eslam, M.; Gonzalez-Fabian, L.; Sanz, M.A.-Q.; Conde-Martin, A.F. Fibrosis severity as a determinant of cause-specific mortality in patients with advanced nonalcoholic fatty liver disease: A multi-national cohort study. Gastroenterology 2018, 155, 443–457.e417. [Google Scholar] [CrossRef]
- Aberg, F.; Puukka, P.; Salomaa, V.; Mannisto, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Jula, A.; Farkkila, M. Combined Effects of Alcohol and Metabolic Disorders in Patients With Chronic Liver Disease. Clin. Gastroenterol. Hepatol. 2020, 18, 995–997.e992. [Google Scholar] [CrossRef]
- Irvine, L.; Crombie, I.K.; Cunningham, K.B.; Williams, B.; Sniehotta, F.F.; Norrie, J.; Melson, A.J.; Jones, C.; Rice, P.; Slane, P.W.; et al. Modifying Alcohol Consumption to Reduce Obesity: A Randomized Controlled Feasibility Study of a Complex Community-based Intervention for Men. Alcohol Alcohol. 2017, 52, 677–684. [Google Scholar] [CrossRef] [Green Version]
- Eslam, M.; Sanyal, A.J.; George, J. Toward More Accurate Nomenclature for Fatty Liver Diseases. Gastroenterology 2019, 157, 590–593. [Google Scholar] [CrossRef] [Green Version]
- Bacon, B.R.; Farahvash, M.J.; Janney, C.G.; Neuschwander-Tetri, B.A. Nonalcoholic steatohepatitis: An expanded clinical entity. Gastroenterology 1994, 107, 1103–1109. [Google Scholar] [CrossRef]
- Powell, E.E.; Cooksley, W.G.; Hanson, R.; Searle, J.; Halliday, J.W.; Powell, L.W. The natural history of nonalcoholic steatohepatitis: A follow-up study of forty-two patients for up to 21 years. Hepatology 1990, 11, 74–80. [Google Scholar] [CrossRef]
- Yeh, M.M.; Brunt, E.M. Pathology of nonalcoholic fatty liver disease. Am. J. Clin. Pathol. 2007, 128, 837–847. [Google Scholar] [CrossRef]
- Rubin, E.; Lieber, C.S. Alcohol-induced hepatic injury in nonalcoholic volunteers. N. Engl. J. Med. 1968, 278, 869–876. [Google Scholar] [CrossRef]
- Lefkowitch, J.H. Morphology of alcoholic liver disease. Clin. Liver Dis. 2005, 9, 37–53. [Google Scholar] [CrossRef]
- Christoffersen, P.; Braendstrup, O.; Juhl, E.; Poulsen, H. Lipogranulomas in human liver biopsies with fatty change. A morphological, biochemical and clinical investigation. Acta Pathol. Microbiol. Scand. A 1971, 79, 150–158. [Google Scholar] [CrossRef]
- Denk, H.; Lackinger, E. Cytoskeleton in liver diseases. Semin. Liver Dis. 1986, 6, 199–211. [Google Scholar] [CrossRef]
- French, S.W.; Nash, J.; Shitabata, P.; Kachi, K.; Hara, C.; Chedid, A.; Mendenhall, C.L. Pathology of alcoholic liver disease. VA Cooperative Study Group 119. Semin. Liver Dis. 1993, 13, 154–169. [Google Scholar] [CrossRef]
- Nakano, M.; Worner, T.M.; Lieber, C.S. Perivenular fibrosis in alcoholic liver injury: Ultrastructure and histologic progression. Gastroenterology 1982, 83, 777–785. [Google Scholar] [CrossRef]
- Worner, T.M.; Lieber, C.S. Perivenular fibrosis as precursor lesion of cirrhosis. JAMA 1985, 254, 627–630. [Google Scholar] [CrossRef]
- Fauerholdt, L.; Schlichting, P.; Christensen, E.; Poulsen, H.; Tygstrup, N.; Juhl, E. Conversion of micronodular cirrhosis into macronodular cirrhosis. Hepatology 1983, 3, 928–931. [Google Scholar] [CrossRef]
- Pomier-Layrargues, G.; Kusielewicz, D.; Willems, B.; Villeneuve, J.P.; Marleau, D.; Cote, J.; Huet, P.M. Presinusoidal portal hypertension in non-alcoholic cirrhosis. Hepatology 1985, 5, 415–418. [Google Scholar] [CrossRef]
- Farrokhi, F.; Chandra, P.; Smiley, D.; Pasquel, F.; Peng, L.; Newton, C.; Umpierrez, G. Glucose variability is an independent predictor of mortality in hospitalized patients treated with total parenteral nutrition. Endocr. Pract. 2013, 20, 41–45. [Google Scholar] [CrossRef]
- Dunn, W.; Angulo, P.; Sanderson, S.; Jamil, L.H.; Stadheim, L.; Rosen, C.; Malinchoc, M.; Kamath, P.S.; Shah, V.H. Utility of a new model to diagnose an alcohol basis for steatohepatitis. Gastroenterology 2006, 131, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, K.A.; Petrick, J.L.; El-Serag, H.B. Epidemiology of Hepatocellular Carcinoma. Hepatology 2021, 73 (Suppl. 1), 4–13. [Google Scholar] [CrossRef]
- Nahon, P.; Sutton, A.; Rufat, P.; Ziol, M.; Akouche, H.; Laguillier, C.; Charnaux, N.; Ganne-Carrie, N.; Grando-Lemaire, V.; N’Kontchou, G.; et al. Myeloperoxidase and superoxide dismutase 2 polymorphisms comodulate the risk of hepatocellular carcinoma and death in alcoholic cirrhosis. Hepatology 2009, 50, 1484–1493. [Google Scholar] [CrossRef]
- McKillop, I.H.; Schrum, L.W. Role of alcohol in liver carcinogenesis. Semin. Liver Dis. 2009, 29, 222–232. [Google Scholar] [CrossRef]
- Mancebo, A.; Gonzalez-Dieguez, M.L.; Cadahia, V.; Varela, M.; Perez, R.; Navascues, C.A.; Sotorrios, N.G.; Martinez, M.; Rodrigo, L.; Rodriguez, M. Annual incidence of hepatocellular carcinoma among patients with alcoholic cirrhosis and identification of risk groups. Clin. Gastroenterol. Hepatol. 2013, 11, 95–101. [Google Scholar] [CrossRef]
- Ganne-Carrie, N.; Chaffaut, C.; Bourcier, V.; Archambeaud, I.; Perarnau, J.M.; Oberti, F.; Roulot, D.; Moreno, C.; Louvet, A.; Dao, T.; et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J. Hepatol. 2018, 69, 1274–1283. [Google Scholar] [CrossRef]
- Tsukuma, H.; Hiyama, T.; Tanaka, S.; Nakao, M.; Yabuuchi, T.; Kitamura, T.; Nakanishi, K.; Fujimoto, I.; Inoue, A.; Yamazaki, H.; et al. Risk factors for hepatocellular carcinoma among patients with chronic liver disease. N. Engl. J. Med. 1993, 328, 1797–1801. [Google Scholar] [CrossRef]
- Donato, F.; Tagger, A.; Gelatti, U.; Parrinello, G.; Boffetta, P.; Albertini, A.; Decarli, A.; Trevisi, P.; Ribero, M.L.; Martelli, C.; et al. Alcohol and hepatocellular carcinoma: The effect of lifetime intake and hepatitis virus infections in men and women. Am. J. Epidemiol. 2002, 155, 323–331. [Google Scholar] [CrossRef] [Green Version]
- Hassan, M.M.; Hwang, L.Y.; Hatten, C.J.; Swaim, M.; Li, D.; Abbruzzese, J.L.; Beasley, P.; Patt, Y.Z. Risk factors for hepatocellular carcinoma: Synergism of alcohol with viral hepatitis and diabetes mellitus. Hepatology 2002, 36, 1206–1213. [Google Scholar] [CrossRef]
- Loomba, R.; Yang, H.-I.; Su, J.; Brenner, D.; Barrett-Connor, E.; Iloeje, U.; Chen, C.-J. Synergism between obesity and alcohol in increasing the risk of hepatocellular carcinoma: A prospective cohort study. Am. J. Epidemiol. 2013, 177, 333–342. [Google Scholar] [CrossRef] [Green Version]
- Ohki, T.; Tateishi, R.; Sato, T.; Masuzaki, R.; Imamura, J.; Goto, T.; Yamashiki, N.; Yoshida, H.; Kanai, F.; Kato, N.; et al. Obesity is an independent risk factor for hepatocellular carcinoma development in chronic hepatitis C patients. Clin. Gastroenterol. Hepatol. 2008, 6, 459–464. [Google Scholar] [CrossRef]
- Saunders, D.; Seidel, D.; Allison, M.; Lyratzopoulos, G. Systematic review: The association between obesity and hepatocellular carcinoma—Epidemiological evidence. Aliment. Pharmacol. Ther. 2010, 31, 1051–1063. [Google Scholar] [CrossRef]
- El-Serag, H.B.; Tran, T.; Everhart, J.E. Diabetes increases the risk of chronic liver disease and hepatocellular carcinoma. Gastroenterology 2004, 126, 460–468. [Google Scholar] [CrossRef]
- Jee, S.H.; Ohrr, H.; Sull, J.W.; Yun, J.E.; Ji, M.; Samet, J.M. Fasting serum glucose level and cancer risk in Korean men and women. JAMA 2005, 293, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Inoue, M.; Iwasaki, M.; Otani, T.; Sasazuki, S.; Noda, M.; Tsugane, S. Diabetes mellitus and the risk of cancer: Results from a large-scale population-based cohort study in Japan. Arch. Intern. Med. 2006, 166, 1871–1877. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Li, W.W.; Yang, X.; Fan, W.B.; Yu, J.H.; Xie, S.S.; Liu, L.; Ma, L.X.; Chen, S.J.; Kato, N. Type 2 diabetes and hepatocellular carcinoma: A case-control study in patients with chronic hepatitis B. Int J Cancer 2012, 131, 1197–1202. [Google Scholar] [CrossRef]
- Lai, S.W.; Chen, P.C.; Liao, K.F.; Muo, C.H.; Lin, C.C.; Sung, F.C. Risk of hepatocellular carcinoma in diabetic patients and risk reduction associated with anti-diabetic therapy: A population-based cohort study. Am. J. Gastroenterol. 2012, 107, 46–52. [Google Scholar] [CrossRef]
- Salmon, D.; Bani-Sadr, F.; Loko, M.A.; Stitou, H.; Gervais, A.; Durant, J.; Rosenthal, E.; Quertainmont, Y.; Barange, K.; Vittecoq, D.; et al. Insulin resistance is associated with a higher risk of hepatocellular carcinoma in cirrhotic HIV/HCV-co-infected patients: Results from ANRS CO13 HEPAVIH. J. Hepatol. 2012, 56, 862–868. [Google Scholar] [CrossRef]
- Chen, H.P.; Shieh, J.J.; Chang, C.C.; Chen, T.T.; Lin, J.T.; Wu, M.S.; Lin, J.H.; Wu, C.Y. Metformin decreases hepatocellular carcinoma risk in a dose-dependent manner: Population-based and in vitro studies. Gut 2013, 62, 606–615. [Google Scholar] [CrossRef] [PubMed]
- Arase, Y.; Kobayashi, M.; Suzuki, F.; Suzuki, Y.; Kawamura, Y.; Akuta, N.; Kobayashi, M.; Sezaki, H.; Saito, S.; Hosaka, T.; et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology 2013, 57, 964–973. [Google Scholar] [CrossRef]
- Wang, C.; Wang, X.; Gong, G.; Ben, Q.; Qiu, W.; Chen, Y.; Li, G.; Wang, L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: A systematic review and meta-analysis of cohort studies. Int. J. Cancer 2012, 130, 1639–1648. [Google Scholar] [CrossRef]
- Wang, P.; Kang, D.; Cao, W.; Wang, Y.; Liu, Z. Diabetes mellitus and risk of hepatocellular carcinoma: A systematic review and meta-analysis. Diabetes Metab. Res. Rev. 2012, 28, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Va, P.; Bray, F.; Gao, S.; Gao, J.; Li, H.L.; Xiang, Y.B. The role of pre-existing diabetes mellitus on hepatocellular carcinoma occurrence and prognosis: A meta-analysis of prospective cohort studies. PLoS ONE 2011, 6, e27326. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Lin, M.C.; Muo, C.H.; Yeh, S.Y.; Sung, F.C.; Kao, C.H. Combination Therapy of Metformin and Statin May Decrease Hepatocellular Carcinoma Among Diabetic Patients in Asia. Medicine 2015, 94, e1013. [Google Scholar] [CrossRef]
- Donadon, V.; Balbi, M.; Mas, M.D.; Casarin, P.; Zanette, G. Metformin and reduced risk of hepatocellular carcinoma in diabetic patients with chronic liver disease. Liver Int. 2010, 30, 750–758. [Google Scholar] [CrossRef]
- Hassan, M.M.; Curley, S.A.; Li, D.; Kaseb, A.; Davila, M.; Abdalla, E.K.; Javle, M.; Moghazy, D.M.; Lozano, R.D.; Abbruzzese, J.L.; et al. Association of diabetes duration and diabetes treatment with the risk of hepatocellular carcinoma. Cancer 2010, 116, 1938–1946. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. Metformin and risk of hepatocellular carcinoma in patients with type 2 diabetes. Liver Int. 2018, 38, 2018–2027. [Google Scholar] [CrossRef]
- Singh, S.; Singh, P.P.; Singh, A.G.; Murad, M.H.; Sanchez, W. Anti-diabetic medications and the risk of hepatocellular cancer: A systematic review and meta-analysis. Am. J. Gastroenterol. 2013, 108, 881–891. [Google Scholar] [CrossRef]
- Welzel, T.M.; Graubard, B.I.; Zeuzem, S.; El-Serag, H.B.; Davila, J.A.; McGlynn, K.A. Metabolic syndrome increases the risk of primary liver cancer in the United States: A study in the SEER-Medicare database. Hepatology 2011, 54, 463–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyda Seydel, G.; Kucukoglu, O.; Altinbasv, A.; Demir, O.O.; Yilmaz, S.; Akkiz, H.; Otan, E.; Sowa, J.P.; Canbay, A. Economic growth leads to increase of obesity and associated hepatocellular carcinoma in developing countries. Ann. Hepatol. 2016, 15, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.C.; Loo, W.M.; Goh, K.L.; Sugano, K.; Chan, W.K.; Chiu, W.Y.; Choi, M.G.; Gonlachanvit, S.; Lee, W.J.; Lee, W.J.; et al. Asian consensus on the relationship between obesity and gastrointestinal and liver diseases. J. Gastroenterol. Hepatol. 2016, 31, 1405–1413. [Google Scholar] [CrossRef] [Green Version]
- Larsson, S.C.; Wolk, A. Overweight, obesity and risk of liver cancer: A meta-analysis of cohort studies. Br. J. Cancer 2007, 97, 1005–1008. [Google Scholar] [CrossRef] [PubMed]
- Ascha, M.S.; Hanouneh, I.A.; Lopez, R.; Tamimi, T.A.; Feldstein, A.F.; Zein, N.N. The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 2010, 51, 1972–1978. [Google Scholar] [CrossRef]
- Yasui, K.; Hashimoto, E.; Komorizono, Y.; Koike, K.; Arii, S.; Imai, Y.; Shima, T.; Kanbara, Y.; Saibara, T.; Mori, T.; et al. Characteristics of patients with nonalcoholic steatohepatitis who develop hepatocellular carcinoma. Clin. Gastroenterol. Hepatol. 2011, 9, 428–433. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e743. [Google Scholar] [CrossRef] [Green Version]
- Ioannou, G.N.; Green, P.; Kerr, K.F.; Berry, K. Models estimating risk of hepatocellular carcinoma in patients with alcohol or NAFLD-related cirrhosis for risk stratification. J. Hepatol. 2019, 71, 523–533. [Google Scholar] [CrossRef]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [Green Version]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients With Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e1822. [Google Scholar] [CrossRef] [Green Version]
- Dyson, J.; Jaques, B.; Chattopadyhay, D.; Lochan, R.; Graham, J.; Das, D.; Aslam, T.; Patanwala, I.; Gaggar, S.; Cole, M.; et al. Hepatocellular cancer: The impact of obesity, type 2 diabetes and a multidisciplinary team. J. Hepatol. 2014, 60, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Åberg, F.; Puukka, P.; Salomaa, V.; Männistö, S.; Lundqvist, A.; Valsta, L.; Perola, M.; Färkkilä, M.; Jula, A. Risks of light and moderate alcohol use in fatty liver disease: Follow-up of population cohorts. Hepatology 2020, 71, 835–848. [Google Scholar] [CrossRef] [PubMed]



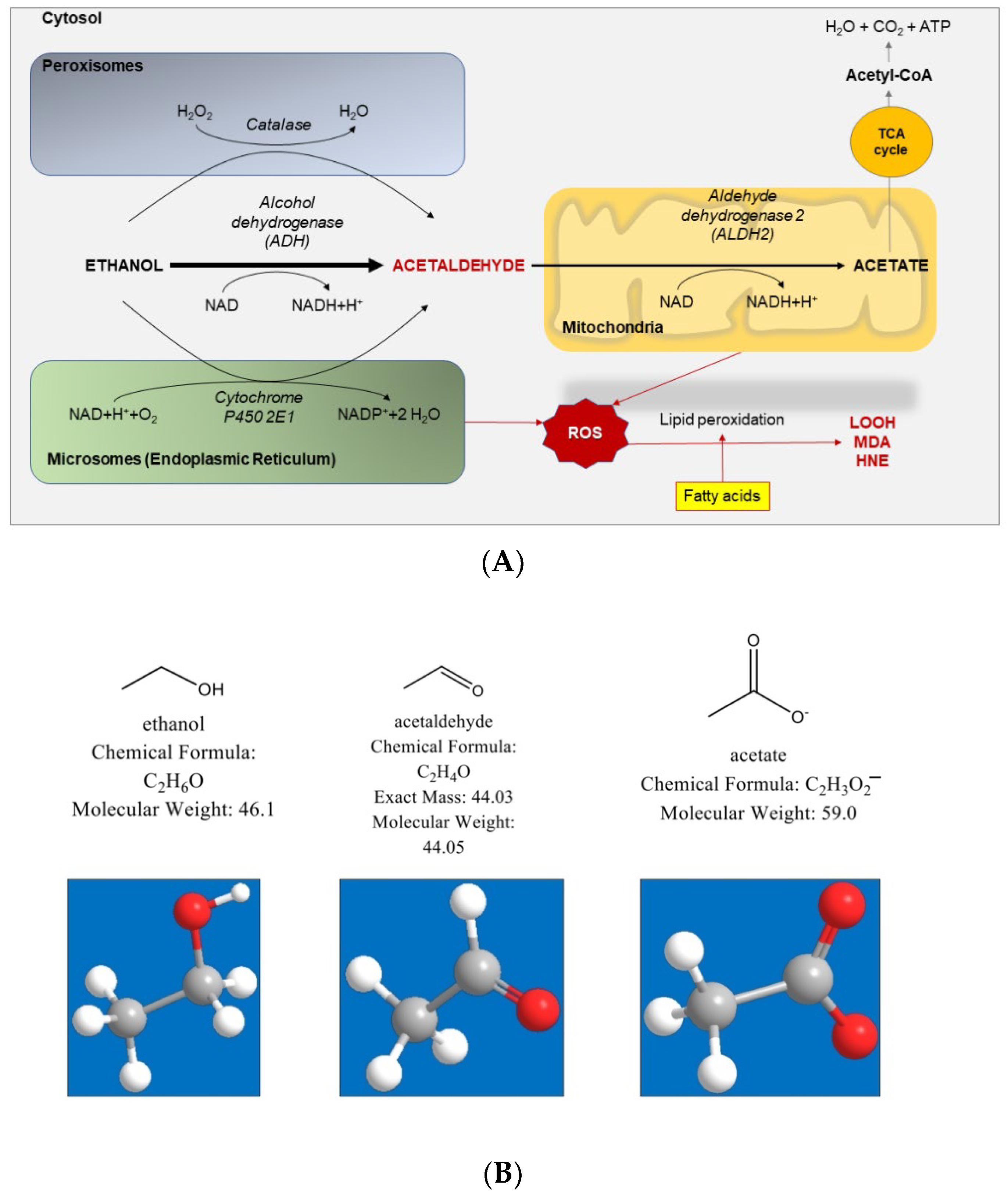{kind=link}
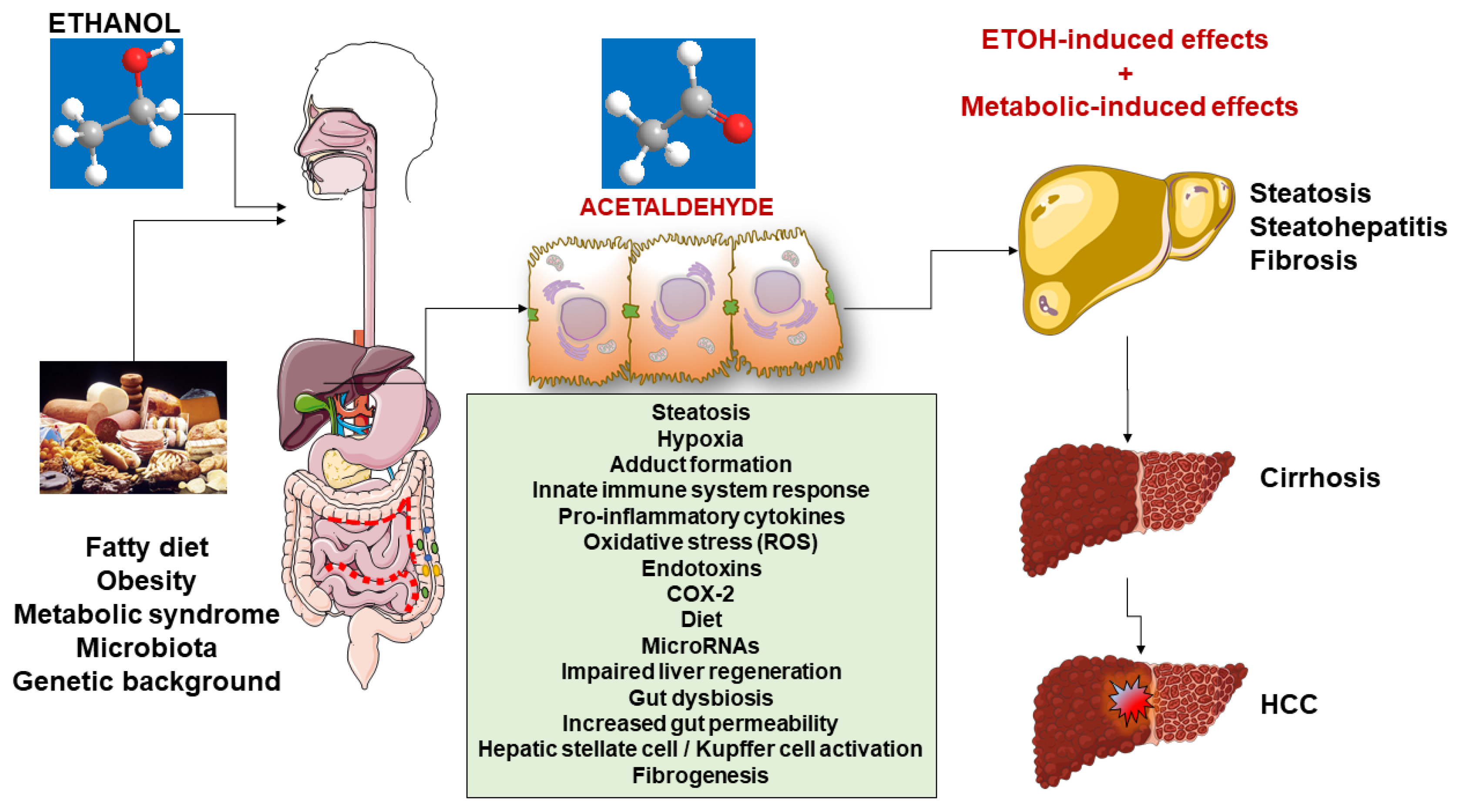{kind=link}
| Author | Setting | Experiment | Notes |
|---|---|---|---|
| Vecchione et al. [233] | FaO hepatoma cell culture | Incubation of cells with 0.35 mM free fatty acids oleate/palmitate alone, in combination with 100 mM ethanol, or ethanol alone. |
|
| Cope et al. [234] | Mice model |
To determine if the intestinal production of ethanol is increased in obesity. Breath collected from genetically obese, ob/ob male C57BL/6 mice and lean male littermates at different ages (14, 20, and 24 weeks) and times of the day (9 a.m., 3 p.m., and 9 p.m.). Obese mice (24 weeks old) were then treated with neomycin (1 mg/mL) for 5 days, and sampling was repeated. |
|
| Nagata et al. [222] | Animal models | Review of mechanisms of damage in ALD/NAFLD |
|
| Xu et al. [235] | Mice | 170% overnutrition in calories (intragastric overfeeding of high fat diet). Alcohol (low or high dose) was then co-administrated. |
|
| Grasselli et al. [236] | Rats | Effects produced by binge ethanol consumption in the liver of male Wistar rats fed a standard (Ctrl) or a high-fat diet HFD. |
|
| de Medeiros et al. [237] | Animal models/human context | Review of mechanisms leading to increased production of endogenous ethanol in NAFLD. |
|
| Guo et al. [238] | Rodents | Lieber–De Carli liquid diet. Induction of alcoholic liver injury with addition of fat components |
|
| Minato et al. [239] | Rat model |
|
|
| Duly et al. [240] | Mice model |
|
|
| Baker et al. [241] | Human model | To test the expression of inflammation, fibrosis, and alcohol metabolism-related genes in the liver tissues of NASH patients and normal controls. Microarray and quantitative real-time PCR. |
|
| Zhu et al. [116] | Human model | Composition of gut bacterial communities of NASH, obese, and healthy children (16S ribosomal RNA pyrosequencing). Peripheral blood ethanol levels assessed as marker of endogenous ethanol production of patients and healthy controls. |
|
| Parker et al. [205] | Human model | Review on the effect of alcohol on adiposity and adipose tissue and the relationship between alcohol, adipose tissue, and the liver. |
|
| Aragonès et al. [117] | Human model | Measurement of circulating microbiota-derived metabolites from women with normal weight, morbid obesity, with or without NAFLD. Liver biopsy performed to differentiate between simple steatosis and steatohepatitis. Measurement of choline and its derivatives, betaine, endogenous ethanol, bile acids, short-chain fatty acids, and soluble Toll-like receptors. |
|
| Incomplete study design |
| Unclear endpoints |
| No adjustment for dietary factors, physical exercise, smoking, coffee consumption, or economic and social aspects |
| Lack of proper stratification for ethnicity |
| Poor information about comorbidities |
| Insufficient information about pattern and type of alcohol use, lifetime alcohol intake (more than average alcohol intake), distinction between lifetime abstainers vs. current abstainers (which might have included former heavy drinkers) |
| Underreporting alcohol use |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Ciaula, A.; Bonfrate, L.; Krawczyk, M.; Frühbeck, G.; Portincasa, P. Synergistic and Detrimental Effects of Alcohol Intake on Progression of Liver Steatosis. Int. J. Mol. Sci. 2022, 23, 2636. https://doi.org/10.3390/ijms23052636
Di Ciaula A, Bonfrate L, Krawczyk M, Frühbeck G, Portincasa P. Synergistic and Detrimental Effects of Alcohol Intake on Progression of Liver Steatosis. International Journal of Molecular Sciences. 2022; 23(5):2636. https://doi.org/10.3390/ijms23052636
Chicago/Turabian StyleDi Ciaula, Agostino, Leonilde Bonfrate, Marcin Krawczyk, Gema Frühbeck, and Piero Portincasa. 2022. "Synergistic and Detrimental Effects of Alcohol Intake on Progression of Liver Steatosis" International Journal of Molecular Sciences 23, no. 5: 2636. https://doi.org/10.3390/ijms23052636