
Hypoxia-Inducible Factors and Burn-Associated Acute Kidney Injury—A New Paradigm?

 ,
,
Abstract
:1. Introduction
2. Hypoxia-Inducible Factors: Structure, Roles, and Involvement in Pathology
3. HIFs and Acute Kidney Injury (AKI)
3.1. Renal Biology and AKI
3.2. HIFs and AKI
3.3. Severe Burns, AKI, and HIFs
3.4. HIFs and Mitochondria in Patients with Major Burns
3.5. HIFs and Reactive Oxygen Species/Reactive Nitrogen Species in the Presence of Major Burns
4. Hypoxia, Inflammation, HIFs, and Kidney Lesions in Patients with Severe Burns
5. HIFs and Acute Hypoxic Cell Death in Kidneys in Severe Burns
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kling, L.; Schreiber, A.; Eckardt, K.U.; Kettritz, R. Hypoxia-inducible factors not only regulate but also are myeloid-cell treatment targets. J. Leukoc. Biol. 2021, 110, 61–75. [Google Scholar] [CrossRef]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8(+) T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majmundar, A.J.; Wong, W.J.; Simon, M.C. Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 2010, 22, 294–309. [Google Scholar] [CrossRef] [Green Version]
- Szymczak, D.; Dybko, J.; Kuliczkowski, K. The role of hypoxia-inducible factors in leukemias. Adv. Clin. Exp. Med. 2018, 27, 271–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akanji, M.A.; Rotimi, D.; Adeyemi, O.S. Hypoxia-Inducible Factors as an Alternative Source of Treatment Strategy for Cancer. Oxid. Med. Cell. Longev. 2019, 14, 8547846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hickey, M.M.; Simon, M.C. Regulation of angiogenesis by hypoxia and hypoxia-inducible factors. Curr. Top. Dev. Biol. 2006, 76, 217–257. [Google Scholar]
- Zhang, X.; Agborbesong, E.; Li, X. The Role of mitochondria in acute kidney injury and chronic kidney disease and its therapeutic potential. Int. J. Mol. Sci. 2021, 19, 11253. [Google Scholar] [CrossRef] [PubMed]
- Nezu, M.; Suzuki, N. Roles of Nrf2 in protecting the kidney from oxidative damage. Int. J. Mol. Sci. 2020, 22, 2951. [Google Scholar] [CrossRef] [PubMed]
- Molema, G.; Aird, W.C. Vascular heterogeneity in the kidney. Semin. Nephrol. 2012, 32, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Wilcox, C.S.; Arendshorst, W.J. Renal autoregulation in health and disease. Physiol. Rev. 2015, 95, 405–511. [Google Scholar] [CrossRef] [Green Version]
- Gunton, J.E. Hypoxia-inducible factors and diabetes. J. Clin. Investig. 2020, 1, 5063–5073. [Google Scholar] [CrossRef] [PubMed]
- Dengler, V.L.; Galbraith, M.; Espinosa, J.M. Transcriptional regulation by hypoxia inducible factors. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebersole, J.L.; Novak, M.J.; Orraca, L.; Martinez-Gonzalez, J.; Kirakodu, S.; Chen, K.C.; Stromberg, A.; Gonzalez, O.A. Hypoxia-inducible transcription factors, HIF1A and HIF2A, increase in aging mucosal tissues. Immunology 2018, 154, 452–464. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Potluri, N.; Lu, J.; Kim, Y.; Rastinejad, F. Structural integration in hypoxia-inducible factors. Nature 2015, 20, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Haase, V.H. Hypoxia-inducible factors in the kidney. Am. J. Physiol. Renal Physiol. 2006, 291, F271–F281. [Google Scholar] [CrossRef]
- Tejiram, S.; Romanowski, K.S.; Palmieri, T.L. Initial management of severe burn injury. Curr. Opin. Crit. Care 2019, 25, 647–652. [Google Scholar] [CrossRef]
- Cancio, L.C.; Bohanon, F.J.; Kramer, G.C. Burn Resuscitation. In Total Burn Care, 5th ed.; Herndon, D.N., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 77–86. [Google Scholar]
- Hoste, E.A.; Bagshaw, S.M.; Bellomo, R.; Cely, C.M.; Colman, R.; Cruz, D.N.; Edipidis, K.; Forni, L.G.; Gomersall, C.D.; Govil, D.; et al. Epidemiology of acute kidney injury in critically ill patients: The multinational AKI-EPI study. Intensive Care Med. 2015, 41, 1411–1423. [Google Scholar] [CrossRef]
- Søvik, S.; Isachsen, M.S.; Nordhuus, K.M.; Tveiten, C.K.; Eken, T.; Sunde, K.; Brurberg, K.G.; Beitland, S. Acute kidney injury in trauma patients admitted to the ICU: A systematic review and meta-analysis. Intensive Care Med. 2019, 45, 407–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, A.; Neyra, J.A.; Madni, T.; Imran, J.; Phelan, H.; Arnoldo, B.; Wolf, S.E. Acute kidney injury after burn. Burns 2017, 43, 898–908. [Google Scholar] [CrossRef]
- Clark, A.T.; Li, X.; Kulangara, R.; Adams-Huet, B.; Huen, S.C.; Madni, T.D.; Imran, J.B.; Phelan, H.A.; Arnoldo, B.D.; Moe, O.W.; et al. Acute Kidney Injury After Burn: A cohort study from the parkland burn intensive care unit. J. Burn Care Res. 2019, 1, 72–78. [Google Scholar] [CrossRef]
- Ostermann, M.; Bellomo, R.; Burdmann, E.A.; Doi, K.; Endre, Z.H.; Goldstein, S.L.; Kane-Gill, S.L.; Liu, K.D.; Prowle, J.R.; Shaw, A.D. Conference Participants. Controversies in acute kidney injury: Conclusions from a kidney disease: Improving global outcomes (KDIGO) conference. Kidney Int. 2020, 98, 294–309. [Google Scholar] [CrossRef]
- Brusselaers, N.; Monstrey, S.; Colpaert, K.; Decruyenaere, J.; Blot, S.I.; Hoste, E.A. Outcome of acute kidney injury in severe burns: A systematic review and meta-analysis. Intensive Care Med. 2010, 36, 915–925. [Google Scholar] [CrossRef]
- Palmieri, T.; Lavrentieva, A.; Greenhalgh, D.G. Acute kidney injury in critically ill burn patients. Risk factors, progression and impact on mortality. Burns 2010, 36, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.P.; Evans, J.; McGonigle, R.J. The dialysis debate: Acute renal failure in burns patients. Burns 1994, 20, 71–73. [Google Scholar] [CrossRef]
- Tong, W.W.; Tong, G.H.; Liu, Y. Cancer stem cells and hypoxia-inducible factors (Review). Int. J. Oncol. 2018, 53, 469–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loboda, A.; Jozkowicz, A.; Dulak, J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol. Cells 2010, 29, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Lou, T. Hypoxia inducible factors in hepatocellular carcinoma. Oncotarget 2017, 8, 46691–46703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammadzada, P.; Corredoira, P.M.; André, H. The role of hypoxia-inducible factors in neovascular age-related macular degeneration: A gene therapy perspective. Cell Mol. Life Sci. 2020, 77, 819–833. [Google Scholar] [CrossRef] [Green Version]
- Hirota, K. An intimate crosstalk between iron homeostasis and oxygen metabolism regulated by the hypoxia-inducible factors (HIFs). Free Radic. Biol. Med. 2019, 133, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Bernhardt, W.; Willam, C.; Wiesener, M. Hypoxia-inducible transcription factors and their role in renal disease. Semin. Nephrol. 2007, 27, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Pawlus, M.R.; Hu, C.J. Enhanceosomes as integrators of hypoxia inducible factor (HIF) and other transcription factors in the hypoxic transcriptional response. Cell. Signal. 2013, 25, 1895–1903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanimoto, K. Genetics of the hypoxia-inducible factors in human cancers. Exp. Cell Res. 2017, 15, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Yuen, V.W.; Wong, C.C. Hypoxia-inducible factors and innate immunity in liver cancer. J. Clin. Investig. 2020, 1, 5052–5062. [Google Scholar] [CrossRef] [PubMed]
- Kapitsinou, P.P.; Haase, V.H. Molecular mechanisms of ischemic preconditioning in the kidney. Am. J. Physiol. Renal Physiol. 2015, 15, F821–F834. [Google Scholar] [CrossRef]
- Gruber, M.; Simon, M.C. Hypoxia-inducible factors, hypoxia, and tumor angiogenesis. Curr. Opin. Hematol. 2006, 13, 169–174. [Google Scholar] [CrossRef]
- Maynard, M.A.; Ohh, M. The role of hypoxia-inducible factors in cancer. Cell Mol. Life Sci. 2007, 64, 2170–2180. [Google Scholar] [CrossRef]
- Wei, W.; Yu, X.D. Hypoxia-inducible factors: Crosstalk between their protein stability and protein degradation. Cancer Lett. 2007, 18, 145–156. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef] [Green Version]
- Heyman, S.N.; Rosen, S.; Rosenberger, C. Hypoxia-inducible factors and the prevention of acute organ injury. Crit. Care 2011, 15, 209. [Google Scholar] [CrossRef] [Green Version]
- Hammond, F.R.; Lewis, A.; Elks, P.M. If it’s not one thing, HIF’s another: Immunoregulation by hypoxia inducible factors in disease. FEBS J. 2020, 287, 3907–3916. [Google Scholar] [CrossRef] [PubMed]
- Kerber, E.L.; Padberg, C.; Koll, N.; Schuetzhold, V.; Fandrey, J.; Winning, S. The importance of hypoxia-inducible factors (HIF-1 and HIF-2) for the pathophysiology of inflammatory bowel disease. Int. J. Mol. Sci. 2020, 13, 8551. [Google Scholar] [CrossRef] [PubMed]
- Frede, S.; Berchner-Pfannschmidt, U.; Fandrey, J. Regulation of hypoxia-inducible factors during inflammation. Methods Enzymol. 2007, 435, 405–419. [Google Scholar] [PubMed]
- Zou, J.X.; Chen, K. Roles and molecular mechanisms of hypoxia-inducible factors in renal cell carcinoma. Yi Chuan 2018, 20, 341–356. [Google Scholar]
- Packer, M. Mechanisms Leading to differential hypoxia-inducible factor signaling in the diabetic kidney: Modulation by SGLT2 inhibitors and hypoxia mimetics. Am. J. Kidney Dis. 2021, 77, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Pirri, D.; Fragiadaki, M.; Evans, P.C. Diabetic atherosclerosis: Is there a role for the hypoxia-inducible factors? Biosci. Rep. 2020, 28, BSR20200026. [Google Scholar] [CrossRef]
- Evans, M.A.; Sano, S.; Walsh, K. Cardiovascular disease, aging, and clonal hematopoiesis. Annu. Rev. Pathol. 2020, 24, 419–438. [Google Scholar] [CrossRef] [Green Version]
- Danescu, I.L.; Tudosie, M.S.; Caragea, G.; Avram, R.; Ştefani, C.; Cioca, G.; Macovei, R.A. Survival following repetitive ventricular fibrillation caused by monoamine oxidase inhibitor overdosing (MAOI). RJMM 2019, CXXII, 71–76. [Google Scholar]
- Al-Mallah, M.H.; Sakr, S.; Al-Qunaibet, A. Cardiorespiratory Fitness and cardiovascular disease prevention: An update. Curr. Atheroscler. Rep. 2018, 16, 1. [Google Scholar] [CrossRef]
- Li, Z.; Rich, J.N. Hypoxia and hypoxia inducible factors in cancer stem cell maintenance. Curr. Top. Microbiol. Immunol. 2010, 345, 21–30. [Google Scholar]
- Vaupel, P.; Multhoff, G. Fatal alliance of hypoxia-/HIF-1alpha-driven microenvironmental traits promoting cancer progression. Adv. Exp. Med. Biol. 2020, 1232, 169–176. [Google Scholar]
- Schödel, J.; Grampp, S.; Maher, E.R.; Moch, H.; Ratcliffe, P.J.; Russo, P.; Mole, D.R. Hypoxia, hypoxia-inducible transcription factors, and renal cancer. Eur. Urol. 2016, 69, 646–657. [Google Scholar] [CrossRef] [Green Version]
- Balamurugan, K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int. J. Cancer 2016, 1, 1058–1066. [Google Scholar] [CrossRef]
- Gerber, P.A.; Rutter, G.A. The role of oxidative stress and hypoxia in pancreatic beta-cell dysfunction in diabetes mellitus. Antioxid. Redox Signal. 2017, 1, 501–518. [Google Scholar] [CrossRef] [Green Version]
- Zhu, W.J.; Li, P.; Wang, L.; Xu, Y.C. Hypoxia-inducible factor-1: A potential pharmacological target to manage psoriasis. Int. Immunopharmacol. 2020, 86, 106689. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, A.M.A.; Stefani, C.; Grajdeanu, I.V.; Serban, B.; Ciobanu, G.; Diaconu, C.C. Severity of psoriasis associated with metabolic syndrome. Rev. Chim. 2019, 70, 2072–2079. [Google Scholar] [CrossRef]
- Makrecka-Kuka, M.; Korzh, S.; Vilks, K.; Vilskersts, R.; Cirule, H.; Dambrova, M.; Liepinsh, E. Mitochondrial function in the kidney and heart, but not the brain, is mainly altered in an experimental model of endotoxaemia. Shock 2019, 52, e153–e162. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.Y.; Chiang, W.C.; Chen, Y.M. The journey from erythropoietin to 2019 Nobel Prize: Focus on hypoxia-inducible factors in the kidney. J. Formos. Med Assoc. 2021, 120, 60–67. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [Green Version]
- Andringa, K.K.; Agarwal, A. Role of hypoxia-inducible factors in acute kidney injury. Nephron Clin. Pract. 2014, 127, 70–74. [Google Scholar] [CrossRef]
- Jang, I.A.; Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Effects of resveratrol on the renin-angiotensin system in the aging kidney. Nutrients 2018, 12, 1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khwaja, A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin. Pract. 2012, 120, c179–c184. [Google Scholar] [CrossRef]
- Thomas, M.E.; Blaine, C.; Dawnay, A.; Devonald, M.A.; Ftouh, S.; Laing, C.; Latchem, S.; Lewington, A.; Milford, D.V.; Ostermann, M. The definition of acute kidney injury and its use in practice. Kidney Int. 2015, 87, 62–73. [Google Scholar] [CrossRef]
- Lopes, J.A.; Fernandes, P.; Jorge, S.; Gonçalves, S.; Alvarez, A.; Costa e Silva, Z.; França, C.; Prata, M.M. Acute kidney injury in intensive care unit patients: A comparison between the RIFLE and the acute kidney injury network classifications. Crit. Care 2008, 12, R110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izawa, J.; Uchino, S.; Takinami, M. A detailed evaluation of the new acute kidney injury criteria by KDIGO in critically ill patients. J. Anesth. 2016, 30, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.K.; Stewart, I.J.; Gisler, C.; Simmons, J.W.; Aden, J.K.; Tilley, M.A.; Cotant, C.L.; White, C.E.; Wolf, S.E.; Renz, E.M. The acute kidney injury network (AKIN) criteria applied in burns. J. Burn Care Res. 2012, 33, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.S.; Goverman, J.; Fagan, S.P. Acute Renal Failure in Association with Thermal Injury. In Total Burn Care, 5th ed.; Herndon, D.N., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 318–326. [Google Scholar]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R.H.; Stiehl, D.P.; Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci STKE 2005, 18, re12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandau, K.B.; Zhou, J.; Kietzmann, T.; Brüne, B. Regulation of the hypoxia-inducible factor 1alpha by the inflammatory mediators nitric oxide and tumor necrosis factor-alpha in contrast to desferroxamine and phenylarsine oxide. J. Biol Chem. 2001, 26, 39805–39811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellwig-Bürgel, T.; Rutkowski, K.; Metzen, E.; Fandrey, J.; Jelkmann, W. Interleukin-1beta and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood 1999, 1, 1561–1567. [Google Scholar] [CrossRef]
- Stiehl, D.P.; Jelkmann, W.; Wenger, R.H.; Hellwig-Bürgel, T. Normoxic induction of the hypoxia-inducible factor 1alpha by insulin and interleukin-1beta involves the phosphatidylinositol 3-kinase pathway. FEBS Lett. 2002, 13, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Treins, C.; Giorgetti-Peraldi, S.; Murdaca, J.; Semenza, G.L.; Van Obberghen, E. Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 2002, 2, 27975–27981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori-Cintra, A.R.; Mara, C.S.; Argolo, D.L.; Coimbra, I.B. Regulation of hypoxia-inducible factor-1α (HIF-1α) expression by interleukin-1β (IL-1 β), insulin-like growth factors I (IGF-I) and II (IGF-II) in human osteoarthritic chondrocytes. Clinics (Sao Paulo) 2012, 67, 35–40. [Google Scholar] [CrossRef]
- Feldser, D.; Agani, F.; Iyer, N.V.; Pak, B.; Ferreira, G.; Semenza, G.L. Reciprocal positive regulation of hypoxia-inducible factor 1alpha and insulin-like growth factor 2. Cancer Res. 1999, 15, 3915–3918. [Google Scholar]
- Richard, D.E.; Berra, E.; Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1alpha in vascular smooth muscle cells. J. Biol. Chem. 2000, 1, 26765–26771. [Google Scholar] [CrossRef]
- Brüne, B.; von Knethen, A.; Sandau, K.B. Transcription factors p53 and HIF-1alpha as targets of nitric oxide. Cell. Signal. 2001, 13, 525–533. [Google Scholar] [CrossRef]
- Metzen, E.; Zhou, J.; Jelkmann, W.; Fandrey, J.; Brüne, B. Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol. Biol. Cell 2003, 14, 3470–3481. [Google Scholar] [CrossRef] [Green Version]
- Braverman, J.; Stanley, S.A. Nitric Oxide Modulates Macrophage Responses to Mycobacterium tuberculosis Infection through Activation of HIF-1α and Repression of NF-κB. J. Immunol. 2017, 1, 1805–1816. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.M.; Zhang, W.H.; Han, X.X.; Li, Y.Y.; Lu, Y.; Pan, J.; Mao, J.Q.; Zhu, L.Y.; Deng, J.J.; Huang, W.; et al. Hypoxia-induced ROS contribute to myoblast pyroptosis during obstructive sleep apnea via the NF-κB/HIF-1α signaling pathway. Oxid. Med. Cell. Longev. 2019, 11, 4596368. [Google Scholar] [CrossRef] [Green Version]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative stress in cancer. Cancer Cell 2020, 10, 167–197. [Google Scholar] [CrossRef]
- He, M.; Zhou, C.; Lu, Y.; Mao, L.; Xi, Y.; Mei, X.; Wang, X.; Zhang, L.; Yu, Z.; Zhou, Z. Melatonin antagonizes nickel-induced aerobic glycolysis by blocking ROS-mediated HIF-1α/miR210/ISCU axis activation. Oxid. Med. Cell. Longev. 2020, 28, 5406284. [Google Scholar] [CrossRef] [PubMed]
- Del Vecchio, L.; Locatelli, F. Hypoxia response and acute lung and kidney injury: Possible implications for therapy of COVID-19. Clin. Kidney. J. 2020, 2, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Townley-Tilson, W.H.; Pi, X.; Xie, L. The role of oxygen sensors, hydroxylases, and HIF in cardiac function and disease. Oxid. Med. Cell Longev. 2015, 2015, 676893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benita, Y.; Kikuchi, H.; Smith, A.D.; Zhang, M.Q.; Chung, D.C.; Xavier, R.J. An integrative genomics approach identifies hypoxia inducible factor-1 (HIF-1)-target genes that form the core response to hypoxia. Nucleic Acids Res. 2009, 37, 4587–4602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debangshu, S.; Gregg, L.S. Maintenance of redox homeostasis by hypoxia-inducible factors. Redox. Biol. 2017, 13, 331–335. [Google Scholar]
- Semenza, G.L. Hypoxia-inducible factors: Mediators of cancer progression and targets for cancer therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, J.T.; Wang, Z.; Nuyten, D.S.; Rodriguez, E.H.; Schaner, M.E.; Salim, A.; Wang, Y.; Kristensen, G.B.; Helland, A.; Børresen-Dale, A.L.; et al. Gene expression programs in response to hypoxia: Cell type specificity and prognostic significance in human cancers. PLoS Med. 2006, 3, e47. [Google Scholar] [CrossRef] [PubMed]
- Mole, D.R.; Blancher, C.; Copley, R.R.; Pollard, P.J.; Gleadle, J.M.; Ragoussis, J.; Ratcliffe, P.J. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 2009, 19, 16767–16775. [Google Scholar] [CrossRef] [Green Version]
- Shu, S.; Wang, Y.; Zheng, M.; Liu, Z.; Cai, J.; Tang, C.; Dong, Z. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells 2019, 28, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villar, D.; Ortiz-Barahona, A.; Gómez-Maldonado, L.; Pescador, N.; Sánchez-Cabo, F.; Hackl, H.; Rodriguez, B.A.; Trajanoski, Z.; Dopazo, A.; Huang, T.H.; et al. Cooperativity of stress-responsive transcription factors in core hypoxia-inducible factor binding regions. PLoS ONE 2012, 7, e45708. [Google Scholar] [CrossRef] [Green Version]
- Firth, J.D.; Ebert, B.L.; Ratcliffe, P.J. Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 1995, 8, 21021–21027. [Google Scholar] [CrossRef] [Green Version]
- Schödel, J.; Oikonomopoulos, S.; Ragoussis, J.; Pugh, C.W.; Ratcliffe, P.J.; Mole, D.R. High-resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 2011, 9, e207–e217. [Google Scholar] [CrossRef] [Green Version]
- Xia, X.; Kung, A.L. Preferential binding of HIF-1 to transcriptionally active loci determines cell-type specific response to hypoxia. Genome Biol. 2009, 10, R113. [Google Scholar] [CrossRef]
- Bell, O.; Tiwari, V.K.; Thomä, N.H.; Schübeler, D. Determinants and dynamics of genome accessibility. Nat. Rev. Genet. 2011, 12, 554–564. [Google Scholar] [CrossRef]
- Lai, W.K.M.; Pugh, B.F. Understanding nucleosome dynamics and their links to gene expression and DNA replication. Nat. Rev. Mol. Cell Biol. 2017, 18, 548–562. [Google Scholar] [CrossRef]
- Jeschke, M.G. Postburn hypermetabolism: Past, present, and future. J. Burn Care Res. 2016, 37, 86–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emami, A.; Javanmardi, F.; Rajaee, M.; Pirbonyeh, N.; Keshavarzi, A.; Fotouhi, M.; Hosseini, S.M. Predictive biomarkers for acute kidney injury in burn patients. J. Burn Care Res. 2019, 14, 601–605. [Google Scholar] [CrossRef]
- Bagshaw, S.M.; Wald, R.; Adhikari, N.K.J.; Bellomo, R.; da Costa, B.R.; Dreyfuss, D.; Du, B.; Gallagher, M.P.; Gaudry, S.; Hoste, E.A.; et al. Timing of Initiation of renal-replacement therapy in acute kidney injury. N. Engl. J. Med. 2020, 16, 240–251. [Google Scholar]
- Auger, C.; Samadi, O.; Jeschke, M.G. The biochemical alterations underlying post-burn hypermetabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 2633–2644. [Google Scholar] [CrossRef]
- Wolfe, R.R. Review: Acute versus chronic response to burn injury. Circ. Shock 1981, 8, 105–115. [Google Scholar] [PubMed]
- Bastin, A.J.; Ostermann, M.; Slack, A.J.; Diller, G.P.; Finney, S.J.; Evans, T.W. Acute kidney injury after cardiac surgery according to risk/injury/failure/loss/end-stage, acute kidney injury network, and kidney disease: Improving global outcomes classifications. J. Crit. Care 2013, 28, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Schödel, J.; Ratcliffe, P.J. Mechanisms of hypoxia signalling: New implications for nephrology. Nat. Rev. Nephrol. 2019, 15, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Soilleux, E.J.; Turley, H.; Tian, Y.M.; Pugh, C.W.; Gatter, K.C.; Harris, A.L. Use of novel monoclonal antibodies to determine the expression and distribution of the hypoxia regulatory factors PHD-1, PHD-2, PHD-3 and FIH in normal and neoplastic human tissues. Histopathology 2005, 47, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 10, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Schödel, J.; Klanke, B.; Weidemann, A.; Buchholz, B.; Bernhardt, W.; Bertog, M.; Amann, K.; Korbmacher, C.; Wiesener, M.; Warnecke, C.; et al. HIF-prolyl hydroxylases in the rat kidney: Physiologic expression patterns and regulation in acute kidney injury. Am. J. Pathol. 2009, 174, 1663–1674. [Google Scholar] [CrossRef] [Green Version]
- Berra, E.; Benizri, E.; Ginouvès, A.; Volmat, V.; Roux, D.; Pouysségur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 15, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Schödel, J.; Bohr, D.; Klanke, B.; Schley, G.; Schlötzer-Schrehardt, U.; Warnecke, C.; Kurtz, A.; Amann, K.; Eckardt, K.U.; Willam, C. Factor inhibiting HIF limits the expression of hypoxia-inducible genes in podocytes and distal tubular cells. Kidney Int. 2010, 78, 857–867. [Google Scholar] [CrossRef] [Green Version]
- Dayan, F.; Roux, D.; Brahimi-Horn, M.C.; Pouyssegur, J.; Mazure, N.M. The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res. 2006, 1, 3688–3698. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr.; Ratcliffe, P.J. Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 2008, 23, 393–402. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 1, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, S.; Perez, M.E.; Kanakubo, E.; Yasuhara, Y.; Shin, Y.S.; Kaneki, M.; Fujita, T.; Martyn, J.A. Skeletal muscle apoptosis after burns is associated with activation of proapoptotic signals. Am. J. Physiol. Endocrinol. Metab. 2000, 279, E1114–E1121. [Google Scholar] [CrossRef] [PubMed]
- Szczesny, B.; Brunyánszki, A.; Ahmad, A.; Oláh, G.; Porter, C.; Toliver-Kinsky, T.; Sidossis, L.; Herndon, D.N.; Szabo, C. Time-dependent and organ-specific changes in mitochondrial function, mitochondrial DNA integrity, oxidative stress and mononuclear cell infiltration in a mouse model of burn injury. PLoS ONE 2015, 2, 0143730. [Google Scholar] [CrossRef] [Green Version]
- Ogawa, Y.; Matsumoto, K.; Ofuji, S. Changes in adenine nucleotide and mitochondrial metabolism of the kidney of burned rats and their relation to insulin. J. Lab. Clin. Med. 1977, 90, 457–465. [Google Scholar] [PubMed]
- Chandel, N.S.; Maltepe, E.; Goldwasser, E.; Mathieu, C.E.; Simon, M.C.; Schumacker, P.T. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 29, 11715–11720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 18, 25130–25138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, E.L.; Klimova, T.A.; Eisenbart, J.; Moraes, C.T.; Murphy, M.P.; Budinger, G.R.; Chandel, N.S. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J. Cell Biol. 2007, 18, 1029–1036. [Google Scholar] [CrossRef] [Green Version]
- Gerald, D.; Berra, E.; Frapart, Y.M.; Chan, D.A.; Giaccia, A.J.; Mansuy, D.; Pouysségur, J.; Yaniv, M.; Mechta-Grigoriou, F. Jun D reduces tumor angiogenesis by protecting cells from oxidative stress. Cell 2004, 17, 781–794. [Google Scholar] [CrossRef]
- Outten, F.W.; Theil, E.C. Iron-based redox switches in biology. Antioxid. Redox Signal. 2009, 11, 1029–1046. [Google Scholar] [CrossRef] [Green Version]
- Golinelli-Cohen, M.P.; Bouton, C. Fe-S proteins acting as redox switch: New key actors of cellular adaptive responses. Curr. Chem. Biol. Bentham Sci. 2017, 11, 70–88. [Google Scholar] [CrossRef]
- Tahara, E.B.; Navarete, F.D.; Kowaltowski, A.J. Tissue-, substrate-, and site-specific characteristics of mitochondrial reactive oxygen species generation. Free Radic. Biol. Med. 2009, 1, 1283–1297. [Google Scholar] [CrossRef]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxid. Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Lukyanova, L.D.; Kirova, Y.I.; Germanova, E.L. The role of succinate in regulation of immediate HIF-1α expression in hypoxia. Bull. Exp. Biol. Med. 2018, 164, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Waypa, G.B.; Schumacker, P.T. O(2) sensing in hypoxic pulmonary vasoconstriction: The mitochondrial door re-opens. Respir. Physiol. Neurobiol. 2002, 22, 81–91. [Google Scholar] [CrossRef]
- Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, G.; Khan, S.A.; Luo, W.; Nanduri, J.; Semenza, G.L.; Prabhakar, N.R. Hypoxia-inducible factor 1 mediates increased expression of NADPH oxidase-2 in response to intermittent hypoxia. J. Cell. Physiol. 2011, 226, 2925–2933. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.W.; Lin, S.C.; Chien, C.W.; Lin, S.C.; Lee, C.T.; Lin, B.W.; Lee, J.C.; Tsai, S.J. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 2011, 179, 1405–1414. [Google Scholar] [CrossRef]
- Chan, S.Y.; Zhang, Y.Y.; Hemann, C.; Mahoney, C.E.; Zweier, J.L.; Loscalzo, J. MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 2009, 10, 273–284. [Google Scholar] [CrossRef] [Green Version]
- Li, H.S.; Zhou, Y.N.; Li, L.; Li, S.F.; Long, D.; Chen, X.L.; Zhang, J.B.; Feng, L.; Li, Y.P. HIF-1α protects against oxidative stress by directly targeting mitochondria. Redox Biol. 2019, 25, 101109. [Google Scholar] [CrossRef]
- Diebold, I.; Petry, A.; Hess, J.; Görlach, A. The NADPH oxidase subunit NOX4 is a new target gene of the hypoxia-inducible factor-1. Mol. Biol. Cell 2010, 15, 2087–2096. [Google Scholar] [CrossRef] [Green Version]
- Diebold, I.; Petry, A.; Sabrane, K.; Djordjevic, T.; Hess, J.; Görlach, A. The HIF1 target gene NOX2 promotes angiogenesis through urotensin-II. J. Cell Sci. 2012, 15, 956–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movafagh, S.; Crook, S.; Vo, K. Regulation of hypoxia-inducible factor-1a by reactive oxygen species: New developments in an old debate. J. Cell. Biochem. 2015, 116, 696–703. [Google Scholar] [CrossRef] [PubMed]
- Callapina, M.; Zhou, J.; Schnitzer, S.; Metzen, E.; Lohr, C.; Deitmer, J.W.; Brüne, B. Nitric oxide reverses desferrioxamine- and hypoxia-evoked HIF-1alpha accumulation--implications for prolyl hydroxylase activity and iron. Exp. Cell. Res. 2005, 15, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; García-Lecea, M.; Cadenas, S.; Hernández, C.; Moncada, S. Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem. J. 2003, 1, 537–544. [Google Scholar] [CrossRef] [Green Version]
- Jeschke, M.G.; Gauglitz, G.G.; Kulp, G.A.; Finnerty, C.C.; Williams, F.N.; Kraft, R.; Suman, O.E.; Mlcak, R.P.; Herndon, D.N. Long-term persistance of the pathophysiologic response to severe burn injury. PLoS ONE 2011, 6, e21245. [Google Scholar] [CrossRef] [Green Version]
- Greenhalgh, D.G. Management of burns. N. Engl. J. Med. 2019, 380, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Barayan, D.; Abdullahi, A.; Vinaik, R.; Knuth, C.M.; Auger, C.; Jeschke, M.G. Interleukin-6 blockade, a potential adjunct therapy for post-burn hypermetabolism. FASEB J. 2021, 35, 21596. [Google Scholar] [CrossRef]
- Westra, J.; Brouwer, E.; Bos, R.; Posthumus, M.D.; Doornbos-van der Meer, B.; Kallenberg, C.G.; Limburg, P.C. Regulation of cytokine-induced HIF-1alpha expression in rheumatoid synovial fibroblasts. Ann. N. Y. Acad. Sci. 2007, 1108, 340–348. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, X.; Zhao, T.; Qiao, M.; Zhao, X.; Zhao, M.; Xu, L.; Zhao, Y.; Wu, L.; Wu, K.; et al. Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav. Immun. 2017, 64, 266–275. [Google Scholar] [CrossRef] [Green Version]
- Finnerty, C.C.; Herndon, D.N.; Przkora, R.; Pereira, C.T.; Oliveira, H.M.; Queiroz, D.M.; Rocha, A.M.; Jeschke, M.G. Cytokine expression profile over time in severely burned pediatric patients. Shock 2006, 26, 13–19. [Google Scholar] [CrossRef]
- Gauglitz, G.G.; Herndon, D.N.; Kulp, G.A.; Meyer III, W.J.; Jeschke, M.G. Abnormal insulin sensitivity persists up to three years in pediatric patients post-burn. J. Clin. Endocrinol. Metab. 2009, 94, 1656–1664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabryelska, A.; Karuga, F.F.; Szmyd, B.; Białasiewicz, P. HIF-1α as a mediator of insulin resistance, T2DM, and its complications: Potential links with obstructive sleep apnea. Front. Physiol. 2020, 9, 1035. [Google Scholar] [CrossRef] [PubMed]
- Chobanyan-Jürgens, K.; Scheibe, R.J.; Potthast, A.B.; Hein, M.; Smith, A.; Freund, R.; Tegtbur, U.; Das, A.M.; Engeli, S.; Jordan, J.; et al. Influences of hypoxia exercise on whole-body insulin sensitivity and oxidative metabolism in older individuals. J. Clin. Endocrinol. Metab. 2019, 104, 5238–5248. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Mazure, N.M.; Cooper, J.A.; Giaccia, A.J. Hypoxia activates a platelet-derived growth factor receptor/phosphatidylinositol 3-kinase/Akt pathway that results in glycogen synthase kinase-3 inactivation. Cancer Res. 2001, 15, 2429–2433. [Google Scholar]
- Zhong, H.; Chiles, K.; Feldser, D.; Laughner, E.; Hanrahan, C.; Georgescu, M.M.; Simons, J.W.; Semenza, G.L. Modulation of hypoxia-inducible factor 1alpha expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: Implications for tumor angiogenesis and therapeutics. Cancer Res. 2000, 15, 1541–1545. [Google Scholar]
- Ota, H.; Fujita, Y.; Yamauchi, M.; Muro, S.; Kimura, H.; Takasawa, S. Relationship between intermittent hypoxia and type 2 diabetes in sleep apnea syndrome. Int. J. Mol. Sci. 2019, 25, 4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, T.; Eltzschig, H.K.; Karhausen, J.; Colgan, S.P.; Shelley, C.S. Leukocyte adhesion during hypoxia is mediated by HIF-1-dependent induction of beta2 integrin gene expression. Proc. Natl. Acad. Sci. USA 2004, 13, 10440–10445. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, A.; Johnson, R.S. Biology of HIF-1alpha. Cell Death Differ. 2008, 15, 621–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 2003, 7, 645–657. [Google Scholar] [CrossRef] [Green Version]
- Caldwell, C.C.; Kojima, H.; Lukashev, D.; Armstrong, J.; Farber, M.; Apasov, S.G.; Sitkovsky, M.V. Differential effects of physiologically relevant hypoxic conditions on T lymphocyte development and effector functions. J. Immunol. 2001, 1, 6140–6149. [Google Scholar] [CrossRef]
- Kojima, H.; Gu, H.; Nomura, S.; Caldwell, C.C.; Kobata, T.; Carmeliet, P.; Semenza, G.L.; Sitkovsky, M.V. Abnormal B lymphocyte development and autoimmunity in hypoxia-inducible factor 1alpha -deficient chimeric mice. Proc. Natl. Acad. Sci. USA 2002, 19, 2170–2174. [Google Scholar] [CrossRef] [Green Version]
- Neumann, A.K.; Yang, J.; Biju, M.P.; Joseph, S.K.; Johnson, R.S.; Haase, V.H.; Freedman, B.D.; Turka, L.A. Hypoxia inducible factor 1 alpha regulates T cell receptor signal transduction. Proc. Natl. Acad. Sci. USA 2005, 22, 17071–17076. [Google Scholar] [CrossRef] [Green Version]
- Biju, M.P.; Akai, Y.; Shrimanker, N.; Haase, V.H. Protection of HIF-1-deficient primary renal tubular epithelial cells from hypoxia-induced cell death is glucose dependent. Am. J. Physiol. Renal Physiol. 2005, 289, F1217–F1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greijer, A.E.; van der Wall, E. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J. Clin. Pathol. 2004, 57, 1009–1014. [Google Scholar] [CrossRef]
- Rosenberger, C.; Mandriota, S.; Jürgensen, J.S.; Wiesener, M.S.; Hörstrup, J.H.; Frei, U.; Ratcliffe, P.J.; Maxwell, P.H.; Bachmann, S.; Eckardt, K.U. Expression of hypoxia-inducible factor-1alpha and -2alpha in hypoxic and ischemic rat kidneys. J. Am. Soc. Nephrol. 2002, 13, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Wiesener, M.S.; Jürgensen, J.S.; Rosenberger, C.; Scholze, C.K.; Hörstrup, J.H.; Warnecke, C.; Mandriota, S.; Bechmann, I.; Frei, U.A.; Pugh, C.W.; et al. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003, 17, 271–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.T.; Li, X.Z.; Pitera, J.E.; Long, D.A.; Woolf, A.S. Peritubular capillary loss after mouse acute nephrotoxicity correlates with down-regulation of vascular endothelial growth factor-A and hypoxia-inducible factor-1 alpha. Am. J. Pathol. 2003, 163, 2289–2301. [Google Scholar] [CrossRef]
- Helton, R.; Cui, J.; Scheel, J.R.; Ellison, J.A.; Ames, C.; Gibson, C.; Blouw, B.; Ouyang, L.; Dragatsis, I.; Zeitlin, S.; et al. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxic-ischemic damage. J. Neurosci. 2005, 20, 4099–4107, Erratum in J. Neurosci. 2005, 11, 25. [Google Scholar] [CrossRef]
- Ow, Y.P.; Green, D.R.; Hao, Z.; Mak, T.W. Cytochrome c: Functions beyond respiration. Nat. Rev. Mol. Cell Biol. 2008, 9, 532–542. [Google Scholar] [CrossRef]
- Carmeliet, P.; Dor, Y.; Herbert, J.M.; Fukumura, D.; Brusselmans, K.; Dewerchin, M.; Neeman, M.; Bono, F.; Abramovitch, R.; Maxwell, P.; et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature 1998, 30, 485–490, Erratum in Nature 1998, 1, 525. [Google Scholar] [CrossRef]
- Bao, X.; Zhang, J.; Huang, G.; Yan, J.; Xu, C.; Dou, Z.; Sun, C.; Zhang, H. The crosstalk between HIFs and mitochondrial dysfunctions in cancer development. Cell Death Dis. 2021, 26, 215. [Google Scholar] [CrossRef]
- Qiu, Y.; Huang, X.; He, W. The regulatory role of HIF-1 in tubular epithelial cells in response to kidney injury. Histol. Histopathol. 2020, 35, 321–330. [Google Scholar]
- Chen, N.; Chen, X.; Huang, R.; Zeng, H.; Gong, J.; Meng, W.; Lu, Y.; Zhao, F.; Wang, L.; Zhou, Q. BCL-xL is a target gene regulated by hypoxia-inducible factor-1{alpha}. J. Biol. Chem. 2009, 10, 10004–10012. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, D.; He, J.; Chen, L.; Li, H. Bcl-XL: A multifunctional anti-apoptotic protein. Pharmacol. Res. 2020, 151, 104547. [Google Scholar] [CrossRef]
- Liu, X.H.; Yu, E.Z.; Li, Y.Y.; Kagan, E. HIF-1alpha has an anti-apoptotic effect in human airway epithelium that is mediated via Mcl-1 gene expression. J. Cell. Biochem. 2006, 97, 755–765. [Google Scholar] [CrossRef]
- Palladino, M.A.; Shah, A.; Tyson, R.; Horvath, J.; Dugan, C.; Karpodinis, M. Myeloid cell leukemia-1 (Mc1–1) is a candidate target gene of hypoxia-inducible factor-1 (HIF-1) in the testis. Reprod. Biol. Endocrinol. 2012, 10, 2012. [Google Scholar] [CrossRef] [Green Version]
- Adams, J.M.; Cory, S. The BCL-2 arbiters of apoptosis and their growing role as cancer targets. Cell Death Differ. 2018, 25, 27–36. [Google Scholar] [CrossRef] [Green Version]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef]
- Fogarty, L.C.; Flemmer, R.T.; Geizer, B.A.; Licursi, M.; Karunanithy, A.; Opferman, J.T.; Hirasawa, K.; Vanderluit, J.L. Mcl-1 and Bcl-xL are essential for survival of the developing nervous system. Cell Death Differ. 2019, 26, 1501–1515. [Google Scholar] [CrossRef]
- Wang, H.; Guo, M.; Wei, H.; Chen, Y. Targeting MCL-1 in cancer: Current status and perspectives. J. Hematol. Oncol. 2021, 14, 67. [Google Scholar] [CrossRef]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1alpha-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005, 96, 394–402. [Google Scholar] [CrossRef] [PubMed]
- Erler, J.T.; Cawthorne, C.J.; Williams, K.J.; Koritzinsky, M.; Wouters, B.G.; Wilson, C.; Miller, C.; Demonacos, C.; Stratford, I.J.; Dive, C. Hypoxia-mediated down- regulation of Bid and Bax in tumors occurs via hypoxia-inducible factor 1-dependent and -inde- pendent mechanisms and contributes to drug resistance. Mol. Cell Biol. 2004, 24, 2875–2889. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Moldoveanu, T.; Llambi, F.; Parsons, M.J.; Green, D.R. The BCL-2 family reunion. Mol. Cell 2010, 12, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Tait, S.W.; Green, D.R. Mitochondria and cell death: Outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010, 11, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Lovell, J.F.; Billen, L.P.; Bindner, S.; Shamas-Din, A.; Fradin, C.; Leber, B.; Andrews, D.W. Membrane binding by tBid initiates an ordered series of events culminating in membrane permeabilization by Bax. Cell 2008, 12, 1074–1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Blanco, A.; García-Sáez, A.J. Bax, Bak and beyond-mitochondrial performance in apoptosis. FEBS J. 2018, 285, 416–431. [Google Scholar] [CrossRef] [Green Version]
- Esposti, M.D. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Wen, Y.; Zhang, S.; Zhang, W.; Chen, Y.; Shen, Y.; Lemieux, M.J.; Campbell, R.E. Photocleavable proteins that undergo fast and efficient dissociation. Chem. Sci. 2021, 31, 9658–9672. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Norberg, E.; Zhivotovsky, B.; Orrenius, S. Mitochondrial targeting of tBid/Bax: A role for the TOM complex? Cell Death Differ. 2009, 16, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.Y.; Ahn, H.J.; Ryu, J.H.; Suk, K.; Park, J.H. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J. Exp. Med. 2004, 199, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 12, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Karim, C.B.; Espinoza-Fonseca, L.M.; James, Z.M.; Hanse, E.A.; Gaynes, J.S.; Thomas, D.D.; Kelekar, A. Structural mechanism for regulation of Bcl-2 protein Noxa by phosphorylation. Sci Rep. 2015, 5, 14557, Erratum in Sci Rep. 2015, 5, 15872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Zhou, L.; Zhao, T.; Liu, X.; Zhang, P.; Liu, Y.; Zheng, X.; Li, Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget 2017, 4, 23996–24008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apop- totic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowter, H.M.; Ratcliffe, P.J.; Watson, P.; Greenberg, A.H.; Harris, A.L. HIF-1-dependent regulation of hypoxic induction of the cell death factors BNIP3 and NIX in human tumors. Cancer Res. 2001, 61, 6669–6673. [Google Scholar]
- Zhang, Y.; Liu, D.; Hu, H.; Zhang, P.; Xie, R.; Cui, W. HIF-1α/BNIP3 signaling pathway-induced-autophagy plays protective role during myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2019, 120, 109464. [Google Scholar] [CrossRef]
- Liu, K.E.; Frazier, W.A. Phosphorylation of the BNIP3 C-Terminus inhibits mitochondrial damage and cell death without blocking autophagy. PLoS ONE 2015, 23, e0129667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Wang, K. Autophagy and apoptosis in liver injury. Cell Cycle 2015, 14, 1631–1642. [Google Scholar] [CrossRef] [Green Version]
- Sendoel, A.; Hengartner, M.O. Apoptotic cell death under hypoxia. Physiology (Bethesda) 2014, 29, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Gobé, G.; Zhang, X.J.; Cuttle, L.; Pat, B.; Willgoss, D.; Hancock, J.; Barnard, R.; Endre, R.B. Bcl-2 genes and growth factors in the pathology of ischaemic acute renal failure. Immunol. Cell Biol. 1999, 77, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Borkan, S.C. The Role of BCL-2 family members in acute kidney injury. Semin. Nephrol. 2016, 36, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Aouacheria, A.; Baghdiguian, S.; Lamb, H.M.; Huska, J.D.; Pineda, F.J.; Hardwick, J.M. Connecting mitochondrial dynamics and life-or-death events via Bcl-2 family proteins. Neurochem. Int. 2017, 109, 141–161. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Research Subject | References | |
|---|---|---|
| 1. | Acute kidney injury—activator of HIFs | [69,70] |
| 2. | HIF signaling pathway might be activated by:
| |
| [71] | ||
| [72,73] | ||
| [73,74] | ||
| [74,75,76] | ||
| [77] | ||
| [78,79,80] | ||
| [81,82,83] | ||
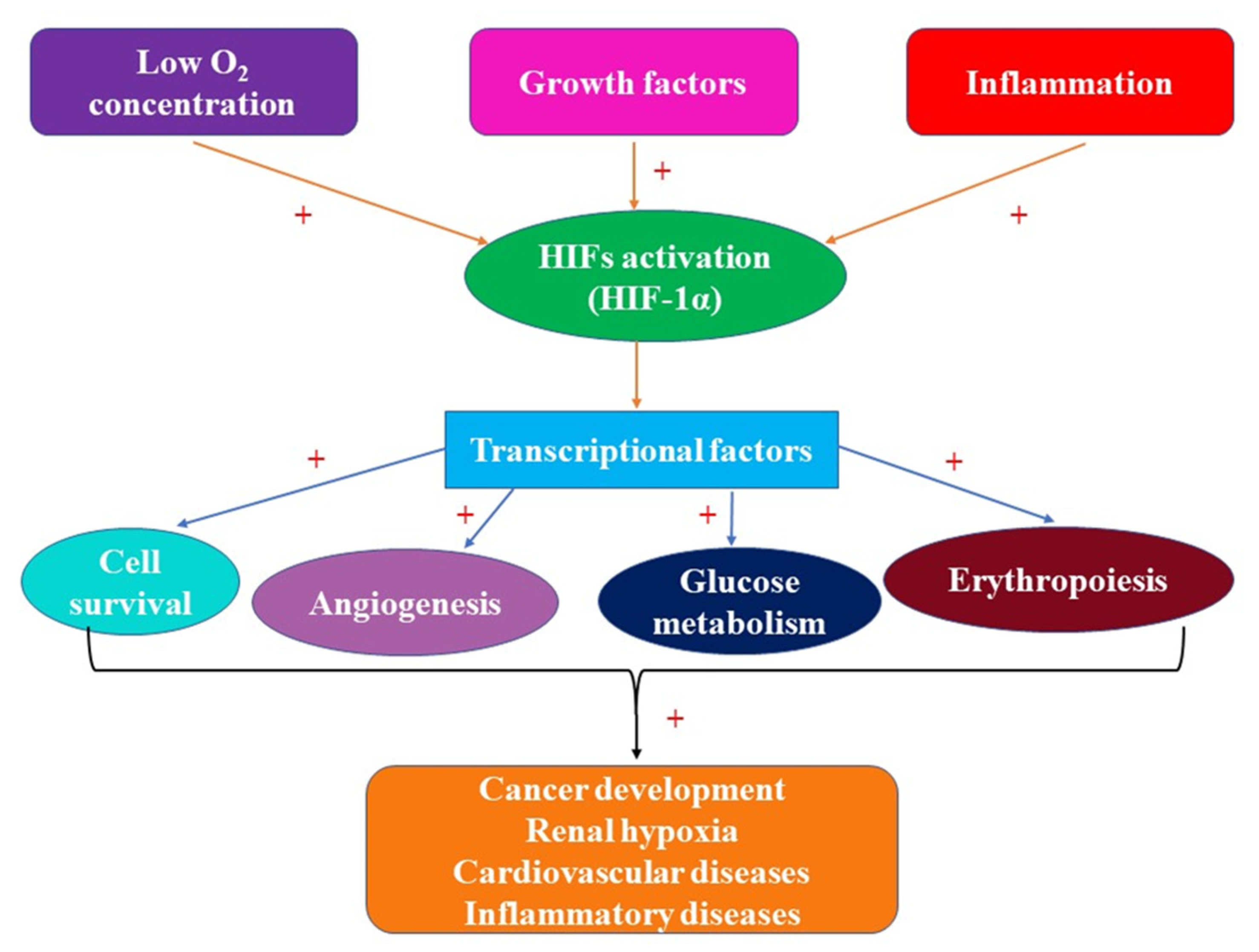
| 3. | As transcriptional factors, HIFs regulate the expression of genes involved in oxygen delivery to the renal tissues, triggering adaptation to hypoxia in the kidney | [69,70] |
| 4. | HIFs are upregulators of the genes encoding most of the glycolytic enzymes | [86,88] |
| 5. | HIF target genes (hypoxia-sensitive genes) induce the synthesis of EPO, VEGF, PGK-1, GLUT-1, transferrin and transferrin receptor, enolase 1, LDH-A (lactate dehydrogenase A), CTGF (connective tissue growth factor), vital for kidney functionality in normal conditions and for kidney adaptation to hypoxia | [15,70,91] |
| 6. | Hypoxic stabilization of HIF-α | [104] |
| 7. | In normoxia, HIF-α is hydroxylated, especially by PHD2In the process of reperfusion and reoxygenation, in the post-burn Flow Phase, HIF-α is preferentially hydroxylated by PHD3 | [106,108,109] |
| 8. | FIH (factor inhibiting HIF)—another oxygen-sensitive hydroxylase that regulates HIF transcription activity | [105,110,111,112,113] |
| 9. | Mitochondria and HIF signaling pathway complex relationship | [117] |
| 10. | HIFs stabilization in hypoxia interferes with ROS generation in two ways:
| [127,128,129,131,132,133] |
| 11. | HIF-1α stabilization increases the expression of miR-210 (microRNA-210) | [130] |
| 12 | NO (nitric oxide) and HIFs relationship during hypoxia | [134,135,136] |
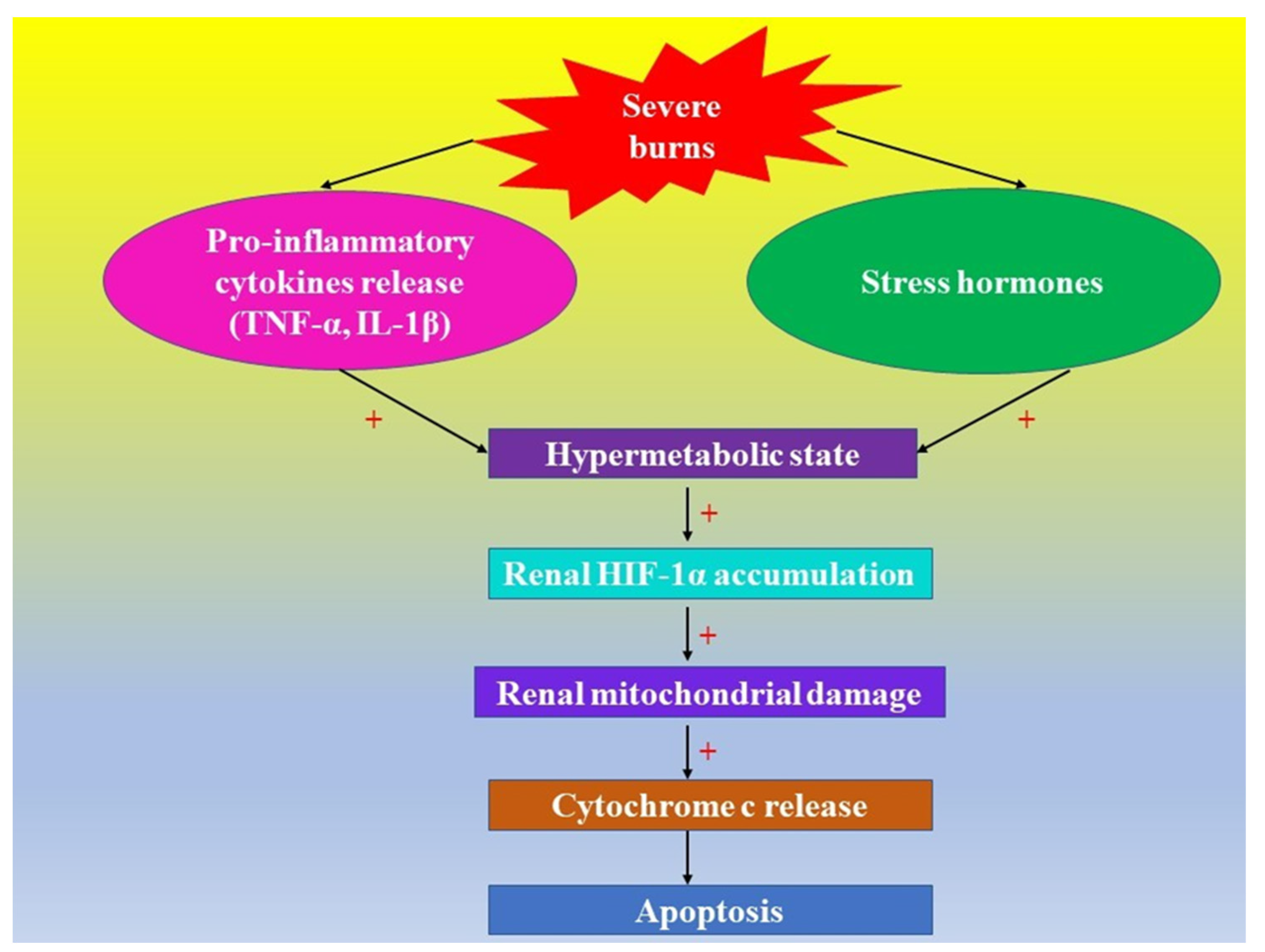
| 13. | In severe burns, the pro-inflammatory cytokines TNF-α and IL-1β increase ROS formation, triggering HIF-1α stabilization | [139,140,141] |
| 14. | HIFs and insulin resistance | [139,144,145,146,148] |
| 15. | In hypoxic renal tissue:
| [157,158,159] |
| 16. | HIF-α and apoptosis in burns | [101,161,162,163,164] |
| 17. | Anti-apoptotic effects of HIFs through:
| |
| [165,166] | ||
| [167,168,169,170,171,172] | ||
| [173,174,175,176,177,178,179,180,181] | ||
| [173] | ||
| 18. | Pro-apoptotic effects of HIFs through:
| |
| [182,183,184,185] | ||
| [186,187,188,189,190] | ||
| [186,187,188,191,192] | ||
| [173,193,194,195] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Enescu, D.M.; Parasca, S.V.; Badoiu, S.C.; Miricescu, D.; Ripszky Totan, A.; Stanescu-Spinu, I.-I.; Greabu, M.; Jinga, V. Hypoxia-Inducible Factors and Burn-Associated Acute Kidney Injury—A New Paradigm? Int. J. Mol. Sci. 2022, 23, 2470. https://doi.org/10.3390/ijms23052470
Enescu DM, Parasca SV, Badoiu SC, Miricescu D, Ripszky Totan A, Stanescu-Spinu I-I, Greabu M, Jinga V. Hypoxia-Inducible Factors and Burn-Associated Acute Kidney Injury—A New Paradigm? International Journal of Molecular Sciences. 2022; 23(5):2470. https://doi.org/10.3390/ijms23052470
Chicago/Turabian StyleEnescu, Dan Mircea, Sorin Viorel Parasca, Silviu Constantin Badoiu, Daniela Miricescu, Alexandra Ripszky Totan, Iulia-Ioana Stanescu-Spinu, Maria Greabu, and Viorel Jinga. 2022. "Hypoxia-Inducible Factors and Burn-Associated Acute Kidney Injury—A New Paradigm?" International Journal of Molecular Sciences 23, no. 5: 2470. https://doi.org/10.3390/ijms23052470