
Inhibition of PKCθ Improves Dystrophic Heart Phenotype and Function in a Novel Model of DMD Cardiomyopathy

 ,
,  ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
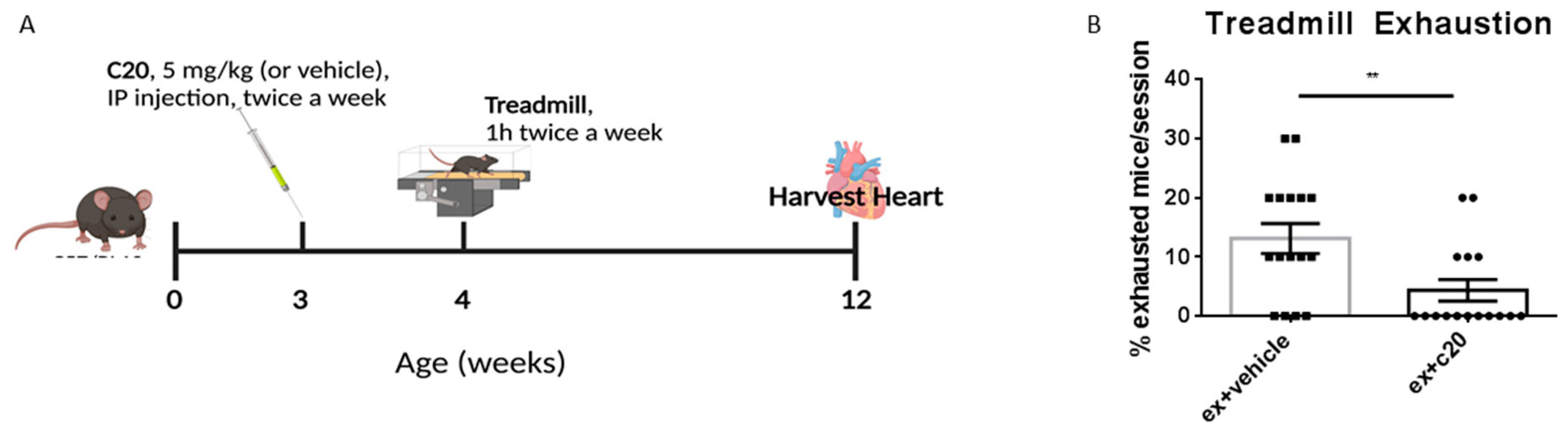
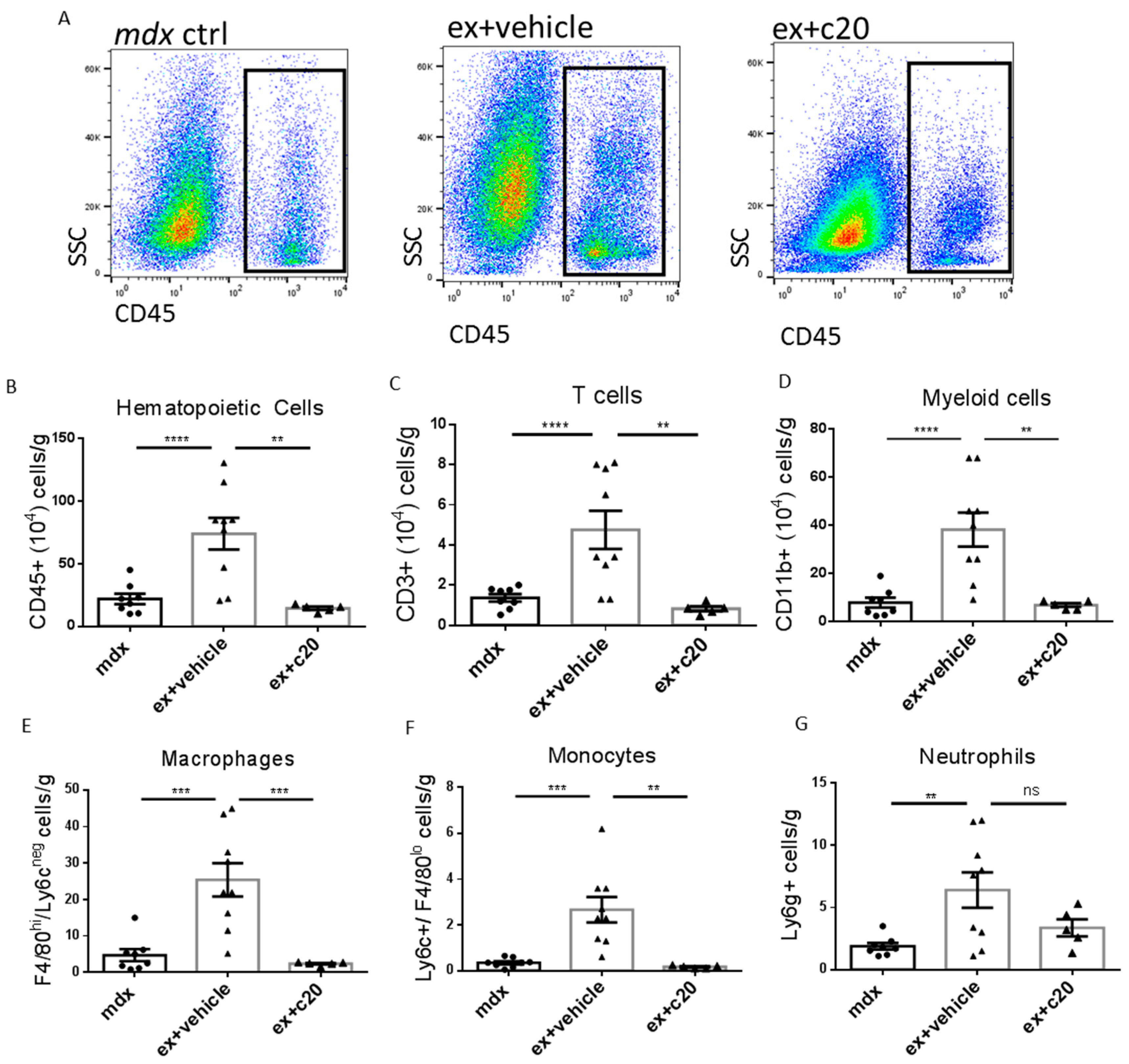
2.1. C20 Treatment Reduces Immune Cell Infiltration in Ex Mdx Heart
2.2. C20 Treatment Reduces Immune Cells Infiltration of the Heart
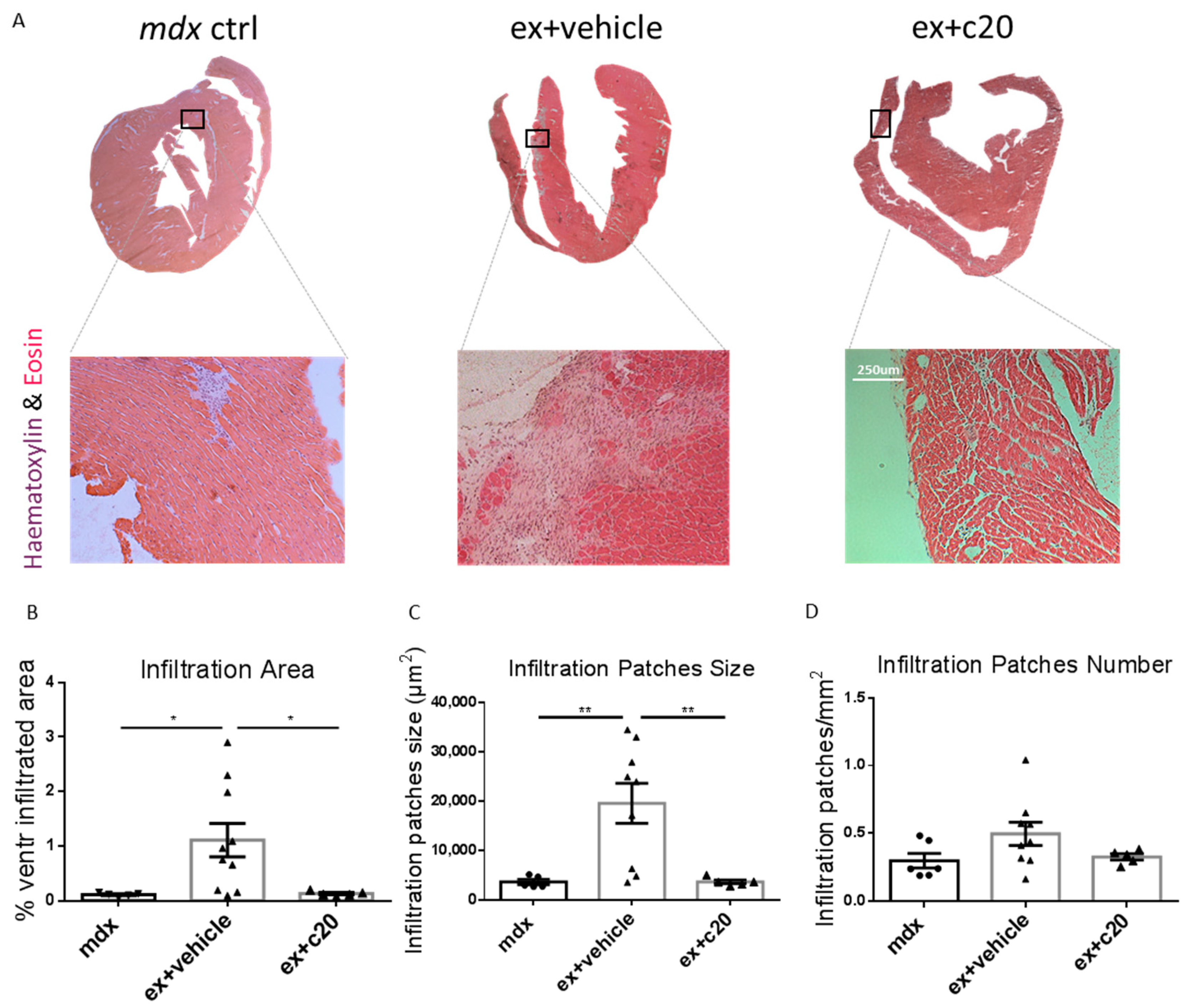
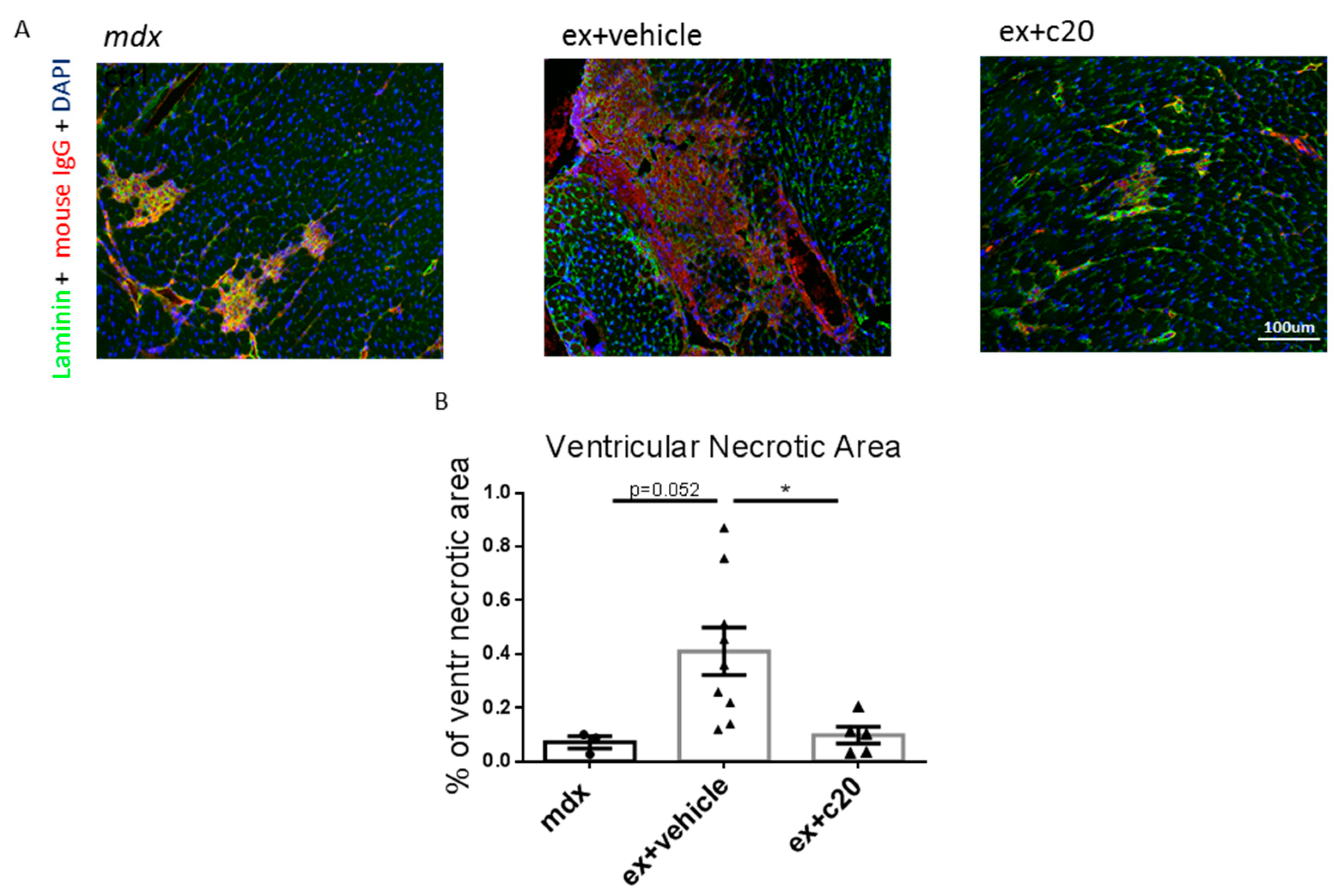
2.3. C20 Treatment Reduces the Cells Infiltration Area and Cardiomyocyte Necrosis in Mdx Ventricles
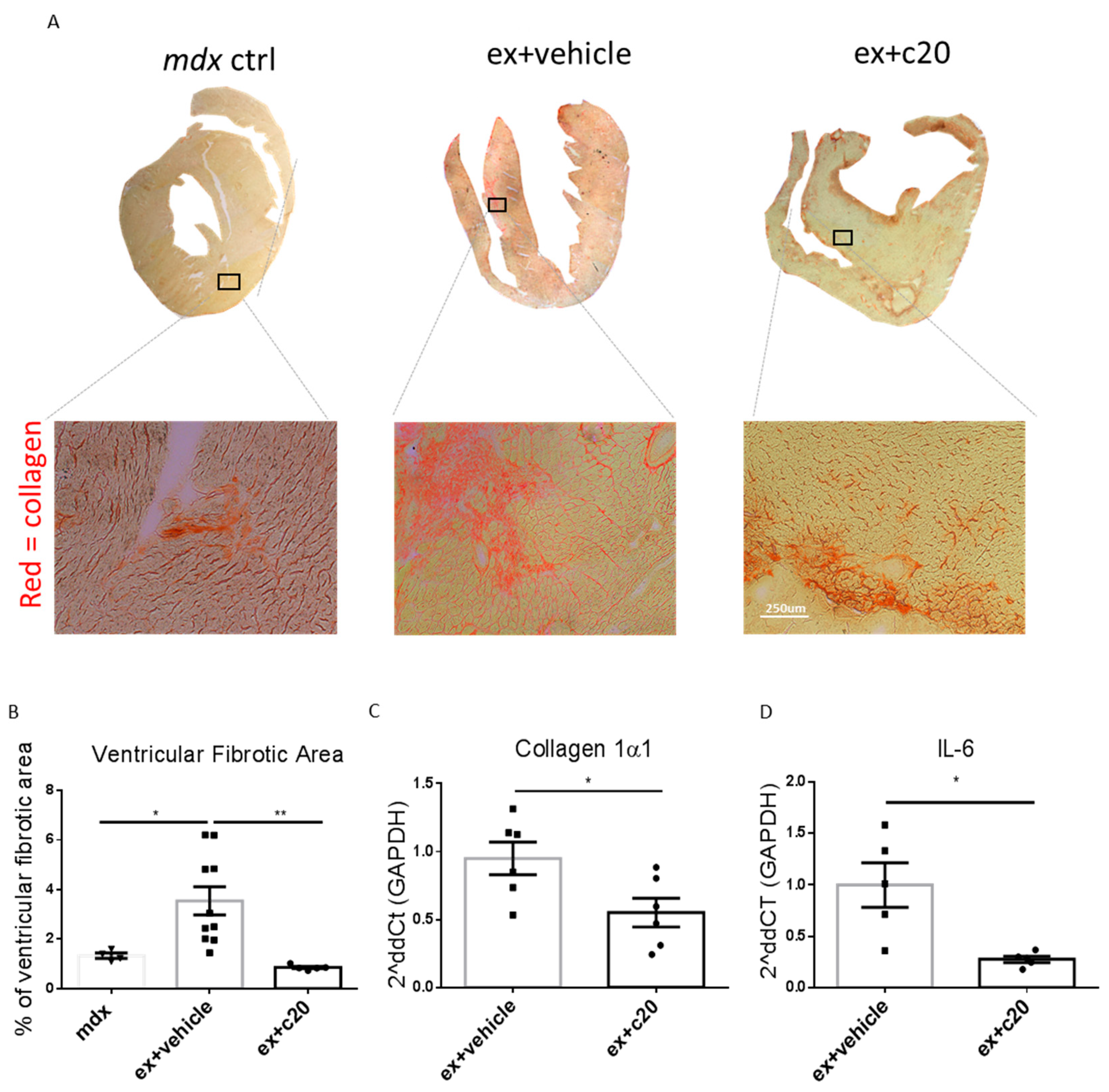
2.4. C20 Treatment Reduces Fibrotic Tissue Deposition in Ex-Mdx Ventricles
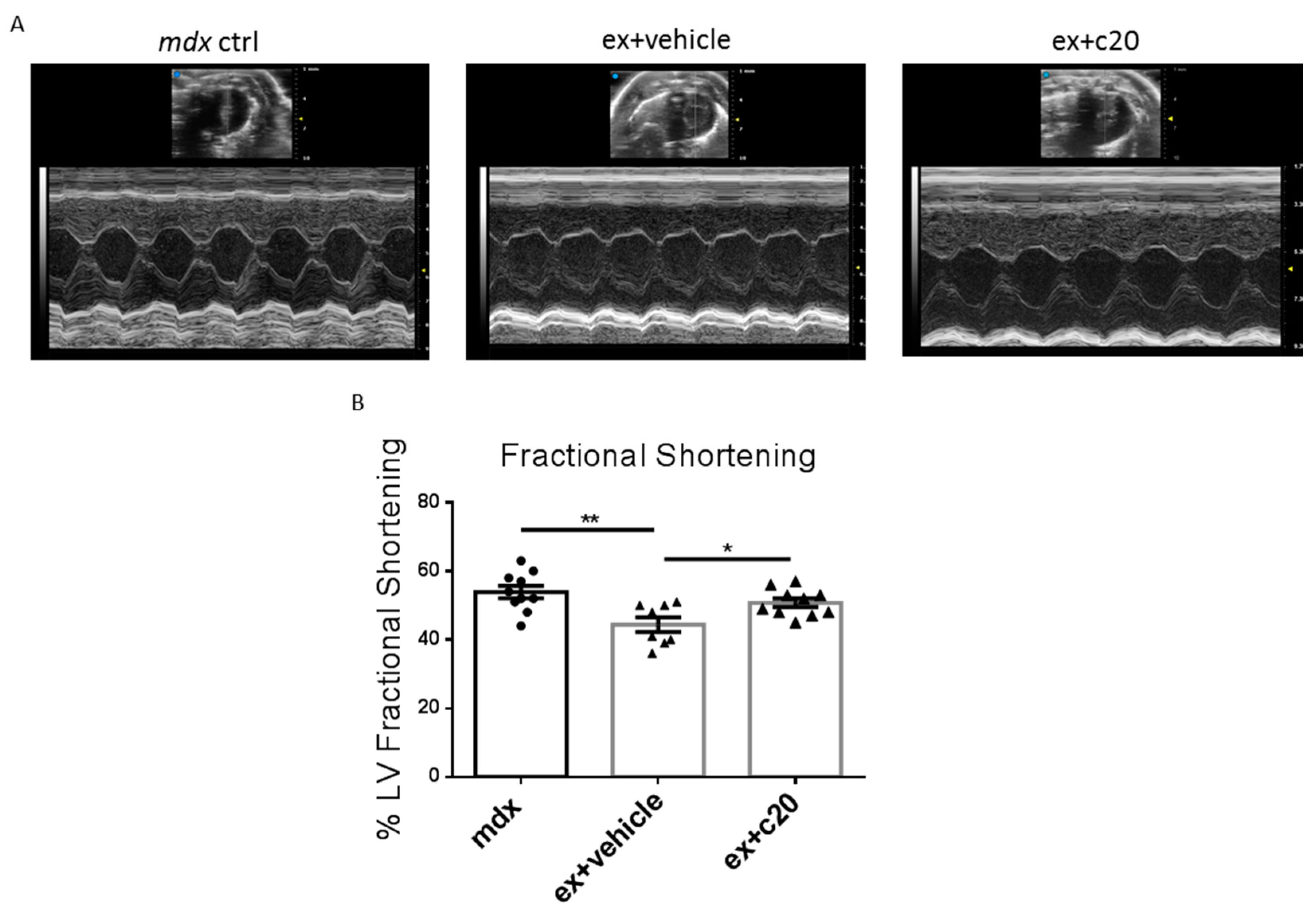
2.5. Heart Function Is Preserved in C20-Treated Exercised Mdx
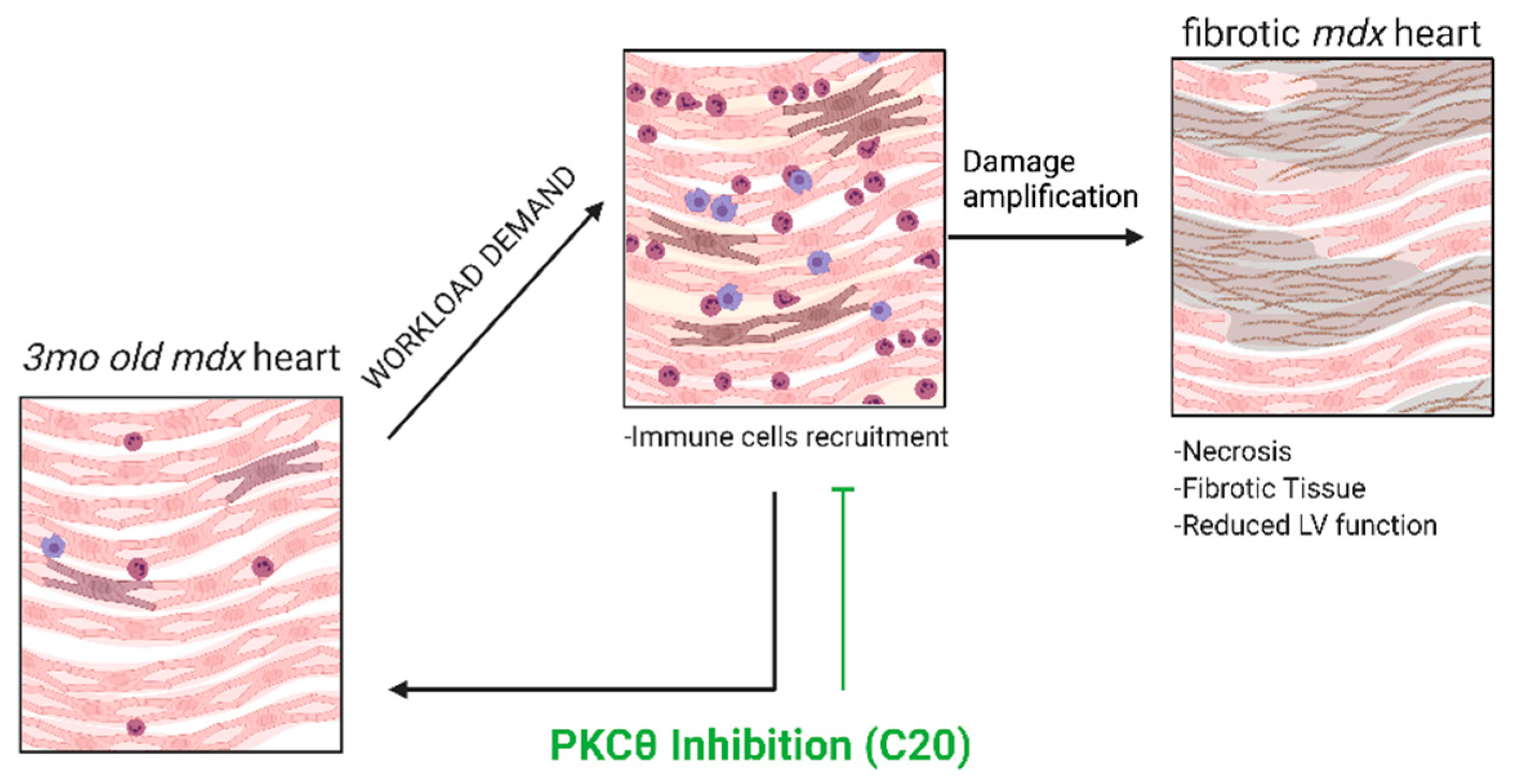
3. Discussion
4. Materials and Methods
4.1. Animal Models
4.2. Treadmill Exercise
4.3. C20 Treatment
4.4. Histology
4.5. Flow Cytometry
4.6. qRT-PCR
4.7. Echocardiography
4.8. C20 Synthesis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef]
- Duan, D.; Goemans, N.; Takeda, S.; Mercuri, E.; Aartsma-Rus, A. Duchenne muscular dystrophy. Nat. Rev. Dis. Prim. 2012, 7, 13. [Google Scholar] [CrossRef]
- Magri, F.; Govoni, A.; D’Angelo, M.G.; Del Bo, R.; Ghezzi, S.; Sandra, G.; Turconi, A.C.; Sciacco, M.; Ciscato, P.; Bordoni, A.; et al. Genotype and phenotype characterization in a large dystrophinopathic cohort with extended follow-up. J. Neurol. 2011, 258, 1610–1623. [Google Scholar] [CrossRef]
- Mercuri, E.; Muntoni, F. Muscular dystrophies. Lancet 2013, 381, 845–860. [Google Scholar] [CrossRef]
- Kamdar, F.; Garry, D.J. Dystrophin-deficient cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef]
- D’Amario, D.; Gowran, A.; Canonico, F.; Castiglioni, E.; Rovina, D.; Santoro, R.; Spinelli, P.; Adorisio, R.; Amodeo, A.; Perrucci, G.; et al. Dystrophin cardiomyopathies: Clinical management, molecular pathogenesis and evolution towards precision medicine. J. Clin. Med. 2018, 7, 291. [Google Scholar] [CrossRef] [Green Version]
- Landfeldt, E.; Thompson, R.; Sejersen, T.; McMillan, H.J.; Kirschner, J.; Lochmüller, H. Life expectancy at birth in Duchenne muscular dystrophy: A systematic review and meta-analysis. Eur. J. Epidemiol. 2020, 35, 643–653. [Google Scholar] [CrossRef] [Green Version]
- Mosqueira, M.; Zeiger, U.; Förderer, M.; Brinkmeier, H.; Fink, R.H. Cardiac and respiratory dysfunction in duchenne muscular dystrophy and the role of second messengers. Med. Res. Rev. 2013, 33, 1174–1213. [Google Scholar] [CrossRef] [PubMed]
- Meyers, T.A.; Townsend, D.W. Cardiac pathophysiology and the future of cardiac therapies in duchenne muscular dystrophy. Int. J. Mol. Sci. 2019, 20, 4098. [Google Scholar] [CrossRef] [Green Version]
- Buddhe, S.; Cripe, L.; Friedland-Little, J.; Kertesz, N.; Eghtesady, P.; Finder, J.; Hor, K.; Judge, D.P.; Kinnett, K.; McNally, E.M.; et al. Cardiac management of the patient with Duchenne muscular dystrophy. Pediatrics 2018, 142, S72–S81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Nigro, G.; Comi, L.I.; Politano, L.; Bain, R.J.I. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Howard, Z.M.; Dorn, L.E.; Lowe, J.; Gertzen, M.D.; Ciccone, P.; Rastogi, N.; Odom, G.L.; Accornero, F.; Chamberlain, J.S.; Rafael-Fortney, J.A. Micro-dystrophin gene therapy prevents heart failure in an improved Duchenne muscular dystrophy cardiomyopathy mouse model. JCI Insight 2021, 6, e146511. [Google Scholar] [CrossRef] [PubMed]
- Verhaart, I.E.C.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Guiraud, S.; Chen, H.; Burns, D.T.; Davies, K.E. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp. Physiol. 2015, 100, 1458–1467. [Google Scholar] [CrossRef]
- Wong, T.W.Y.; Cohn, R.D. Therapeutic applications of CRISPR/Cas for Duchenne muscular dystrophy. Curr. Gene Ther. 2017, 17, 301–308. [Google Scholar] [CrossRef]
- Fortunato, F.; Rossi, R.; Falzarano, M.S.; Ferlini, A. Innovative therapeutic approaches for Duchenne muscular dystrophy. J. Clin. Med. 2021, 10, 820. [Google Scholar] [CrossRef]
- Sun, C.; Shen, L.; Zhang, Z.; Xie, X. Therapeutic strategies for Duchenne muscular dystrophy: An update. Genes 2020, 11, 837. [Google Scholar] [CrossRef]
- Hussein, M.R.; Hamed, S.A.; Mostafa, M.G.; Abu-Dief, E.E.; Kamel, N.F.; Kandil, M.R. The effects of glucocorticoid therapy on the inflammatory and Dendritic cells in muscular dystrophies. Int. J. Exp. Pathol. 2006, 87, 451–461. [Google Scholar] [CrossRef]
- Szabo, S.M.; Salhany, R.M.; Deighton, A.; Harwood, M.; Mah, J.; Gooch, K.L. The clinical course of Duchenne muscular dystrophy in the corticosteroid treatment era: A systematic literature review. Orphanet J. Rare Dis. 2021, 16, 1–13. [Google Scholar] [CrossRef]
- Wehling-Henricks, M.; Lee, J.J.; Tidball, J.G. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul. Disord. 2004, 14, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Matthews, E.; Brassington, R.; Kuntzer, T.; Jichi, F.; Manzur, A.Y. Corticosteroids for the treatment of Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2016, 2016, CD003725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mojumdar, K.; Liang, F.; Giordano, C.; Lemaire, C.; Danialou, G.; Okazaki, T.; Bourdon, J.; Rafei, M.; Galipeau, J.; Divangahi, M.; et al. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med. 2014, 6, 1476–1492. [Google Scholar] [CrossRef] [PubMed]
- Farini, A.; Meregalli, M.; Belicchi, M.; Battistelli, M.; Parolini, D.; D’Antona, G.; Gavina, M.; Ottoboni, L.; Constantin, G.; Bottinelli, R.; et al. T and B lymphocyte depletion has a marked effect on the fibrosis of dystrophic skeletal muscles in the scid/mdx mouse. J. Pathol. 2007, 213, 229–238. [Google Scholar] [CrossRef]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFα function with Etanercept in mdx mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef]
- Spencer, M.J.; Montecino-Rodriguez, E.; Dorshkind, K.; Tidball, J.G. Helper (CD4+) and cytotoxic (CD8+) T cells promote the pathology of dystrophin-deficient muscle. Clin. Immunol. 2001, 98, 235–243. [Google Scholar] [CrossRef]
- Villalta, S.A.; Deng, B.; Rinaldi, C.; Wehling-Henricks, M.; Tidball, J.G. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol. 2011, 187, 5419–5428. [Google Scholar] [CrossRef] [Green Version]
- Spencer, M.J.; Tidball, J.G. Do immune cells promote the pathology of dystrophin-deficient myopathies? Neuromuscul. Disord. 2001, 11, 556–564. [Google Scholar] [CrossRef]
- Rizzo, G.; Di Maggio, R.; Benedetti, A.; Morroni, J.; Bouche, M.; Lozanoska-Ochser, B. Splenic Ly6Chi monocytes are critical players in dystrophic muscle injury and repair. JCI Insight 2020, 5, e130807. [Google Scholar] [CrossRef] [Green Version]
- Lozanoska-Ochser, B.; Benedetti, A.; Rizzo, G.; Marrocco, V.; Di Maggio, R.; Fiore, P.; Bouche, M. Targeting early PKCθ-dependent T-cell infiltration of dystrophic muscle reduces disease severity in a mouse model of muscular dystrophy. J. Pathol. 2018, 244, 323–333. [Google Scholar] [CrossRef]
- Marrocco, V.; Fiore, P.; Benedetti, A.; Pisu, S.; Rizzuto, E.; Musarò, A.; Madaro, L.; Lozanoska-Ochser, B.; Bouché, M. Pharmacological inhibition of PKCθ counteracts muscle disease in a mouse model of duchenne muscular dystrophy. EBioMedicine 2017, 16, 150–161. [Google Scholar] [CrossRef] [Green Version]
- Isakov, N.; Altman, A. Protien kinase Cθ in T cell activation. Annu. Rev. Immunol. 2002, 20, 761–794. [Google Scholar] [CrossRef]
- Chand, S.; Mehta, N.; Singh Bahia, M.; Dixit, A.; Silakari, O. Protein kinase C-theta inhibitors: A novel therapy for inflammatory disorders. Curr. Pharm. Des. 2012, 18, 4725–4746. [Google Scholar] [CrossRef] [PubMed]
- Altman, A.; Villalba, M. Protein kinase Cθ (PKCθ): A key enzyme in T cell life and death. J. Biochem. 2002, 132, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Brezar, V.; Tu, W.J.; Seddiki, N. PKC-theta in regulatory and effector T-cell functions. Front. Immunol. 2015, 6, 530. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z. Intervention of PKC-θ as an immunosuppressive regimen. Front. Immunol. 2012, 3, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, J.; Han, X.; Zhao, C.; Canonigo-Balancio, A.J.; Yates, J.R.; Li, Y.; Lillemeier, B.F.; Altman, A. Phosphotyrosine-dependent interaction between the kinases PKC and Zap70 promotes proximal TCR signaling. Sci. Signal. 2019, 12, eaar3349. [Google Scholar] [CrossRef]
- Zhang, E.Y.; Kong, K.F.; Altman, A. The yin and yang of protein kinase C-theta (PKCθ): A novel drug target for selective immunosuppression. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 66, pp. 267–312. ISBN 9780124047174. [Google Scholar]
- Hage-sleiman, R.; Hamze, A.B.; Reslan, L.; Kobeissy, H.; Dbaibo, G. The Novel PKC ? From Benchtop to Clinic. J. Immunol. Res. 2015, 2015, 348798. [Google Scholar] [CrossRef] [Green Version]
- Cywin, C.L.; Dahmann, G.; Prokopowicz, A.S.; Young, E.R.R.; Magolda, R.L.; Cardozo, M.G.; Cogan, D.A.; DiSalvo, D.; Ginn, J.D.; Kashem, M.A.; et al. Discovery of potent and selective PKC-θ inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 225–230. [Google Scholar] [CrossRef]
- Benedetti, A.; Fiore, P.F.; Madaro, L.; Lozanoska-Ochser, B.; Bouché, M. Targeting pkcθ promotes satellite cell self-renewal. Int. J. Mol. Sci. 2020, 21, 2419. [Google Scholar] [CrossRef] [Green Version]
- Fiore, P.F.; Benedetti, A.; Sandonà, M.; Madaro, L.; De Bardi, M.; Saccone, V.; Puri, P.L.; Gargioli, C.; Lozanoska-Ochser, B.; Bouché, M. Lack of PKCθ promotes regenerative ability of muscle stem cells in chronic muscle injury. Int. J. Mol. Sci. 2020, 21, 932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGreevy, J.W.; Hakim, C.H.; McIntosh, M.A.; Duan, D. Animal models of Duchenne muscular dystrophy: From basic mechanisms to gene therapy. DMM Dis. Model. Mech. 2015, 8, 195–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, K.; Faelan, C.; Patterson-Kane, J.C.; Rudmann, D.G.; Moore, S.A.; Frank, D.; Charleston, J.; Tinsley, J.; Young, G.D.; Milici, A.J. Duchenne and becker muscular dystrophies: A review of animal models, clinical end points, and biomarker quantification. Toxicol. Pathol. 2017, 45, 961–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [Green Version]
- Morroni, J.; Schirone, L.; Vecchio, D.; Nicoletti, C.; D’Ambrosio, L.; Valenti, V.; Sciarretta, S.; Lozanoska-Ochser, B.; Bouchè, M. Accelerating the Mdx heart histo-pathology through physical exercise. Life 2021, 11, 706. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The biological basis for cardiac repair after myocardial infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef] [Green Version]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Perrino, C.; Gargiulo, G.; Pironti, G.; Franzone, A.; Scudiero, L.; de Laurentis, M.; Magliulo, F.; Ilardi, F.; Carotenuto, G.; Schiattarella, G.G.; et al. Cardiovascular effects of treadmill exercise in physiological and pathological preclinical settings. Am. J. Physiol.-Hear. Circ. Physiol. 2011, 300, 1983–1989. [Google Scholar] [CrossRef] [Green Version]
- Feng, R.; Wang, L.; Li, Z.; Yang, R.; Liang, Y.; Sun, Y.; Yu, Q.; Ghartey-Kwansah, G.; Sun, Y.; Wu, Y.; et al. A systematic comparison of exercise training protocols on animal models of cardiovascular capacity. Life Sci. 2019, 217, 128–140. [Google Scholar] [CrossRef]
- Kemi, O.J.; Loennechen, J.P.; Wisløff, U.; Ellingsen, Y. Intensity-controlled treadmill running in mice: Cardiac and skeletal muscle hypertrophy. J. Appl. Physiol. 2002, 93, 1301–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibb, A.A.; Epstein, P.N.; Uchida, S.; Zheng, Y.; McNally, L.A.; Obal, D.; Katragadda, K.; Trainor, P.; Conklin, D.J.; Brittian, K.R.; et al. Exercise-induced changes in glucose metabolism promote physiological cardiac growth. Circulation 2017, 136, 2144–2157. [Google Scholar] [CrossRef] [PubMed]
- Ellison, G.M.; Waring, C.D.; Vicinanza, C.; Torella, D. Physiological cardiac remodelling in response to endurance exercise training: Cellular and molecular mechanisms. Heart 2012, 98, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Musman, J.; Pons, S.; Barau, C.; Caccia, C.; Leoni, V.; Berdeaux, A.; Ghaleh, B.; Morin, D. Regular treadmill exercise inhibits mitochondrial accumulation of cholesterol and oxysterols during myocardial ischemia-reperfusion in wild-type and ob/ob mice. Free Radic. Biol. Med. 2016, 101, 317–324. [Google Scholar] [CrossRef]
- Wang, H.; Bei, Y.; Lu, Y.; Sun, W.; Liu, Q.; Wang, Y.; Cao, Y.; Chen, P.; Xiao, J.; Kong, X. Exercise prevents cardiac injury and improves mitochondrial biogenesis in advanced diabetic cardiomyopathy with PGC-1α and Akt activation. Cell. Physiol. Biochem. 2015, 35, 2159–2168. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, M.; Bryant, J.L.; Ma, J.; Xu, X. Exercise-induced myokine FNDC5/irisin functions in cardiovascular protection and intracerebral retrieval of synaptic plasticity. Cell Biosci. 2019, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Marrocco, V.; Fiore, P.; Madaro, L.; Crupi, A.; Lozanoska-Ochser, B.; Bouché, M. Targeting PKCθ in skeletal muscle and muscle diseases: Good or bad? Biochem. Soc. Trans. 2014, 42, 1550–1555. [Google Scholar] [CrossRef]
- Berg-Brown, N.N.; Gronski, M.A.; Jones, R.G.; Elford, A.R.; Deenick, E.K.; Odermatt, B.; Littman, D.R.; Ohashi, P.S. PKCθ Signals activation versus tolerance in vivo. J. Exp. Med. 2004, 199, 743–752. [Google Scholar] [CrossRef]
- Curnock, A.; Bolton, C.; Chiu, P.; Doyle, E.; Fraysse, D.; Hesse, M.; Jones, J.; Weber, P.; Jimenez, J.M. Selective protein kinase Cθ (PKCθ) inhibitors for the treatment of autoimmune diseases. Biochem. Soc. Trans. 2014, 42, 1524–1528. [Google Scholar] [CrossRef]
- Villalta, S.A.; Rosenberg, A.S.; Bluestone, J.A. The immune system in Duchenne muscular dystrophy: Friend or foe. Rare Dis. 2015, 3, e1010966. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, A.S.; Puig, M.; Nagaraju, K.; Hoffman, E.P.; Villalta, S.A.; Rao, V.A.; Wakefield, L.M.; Woodcock, J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015, 7, 299rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’Amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; De Benedetti, F.; et al. Functional and morphological improvement of dystrophic muscle by interleukin 6 receptor blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Erp, C.; Irwin, N.G.; Hoey, A.J. Long-term administration of pirfenidone improves cardiac function in mdx mice. Muscle Nerve 2006, 34, 327–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, M.J.; Oggiam, D.S.; Cristina, I.; Barbin, C. Long-term therapy with deflazacort decreases myocardial fibrosis in mdx mice. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med. 2009, 40, 466–468. [Google Scholar] [CrossRef]
- Laurila, P.-P.; Wohlwend, M.; Luan, P.; Zanou, N.; Crisol, B.; Imamura de Lima, T.; Goeminne, L.J.E.; Gallart-Ayala, H.; Shong, M.; Ivanisevic, J.; et al. Inhibition of sphingolipid de novo synthesis counteracts muscular dystrophy. Sci. Adv. 2021, 8, eabh4423. [Google Scholar] [CrossRef]
- Zschüntzsch, J.; Jouvenal, P.V.; Zhang, Y.; Klinker, F.; Tiburcy, M.; Liebetanz, D.; Malzahn, D.; Brinkmeier, H.; Schmidt, J. Long-term human IgG treatment improves heart and muscle function in a mouse model of Duchenne muscular dystrophy. J. Cachexia. Sarcopenia Muscle 2020, 11, 1018–1031. [Google Scholar] [CrossRef]
- Kumar, B.V.; Connors, T.; Farber, D.L. Human T cell development, localization, and function throughout life. Immunity 2018, 48, 202–213. [Google Scholar] [CrossRef] [Green Version]
- Cronkite, D.A.; Strutt, T.M. The regulation of inflammation by innate and adaptive lymphocytes. J. Immunol. Res. 2018, 2018, 1467538. [Google Scholar] [CrossRef]
- Madaro, L.; Pelle, A.; Nicoletti, C.; Crupi, A.; Marrocco, V.; Bossi, G.; Soddu, S.; Bouché, M. PKC theta ablation improves healing in a mouse model of muscular dystrophy. PLoS ONE 2012, 7, e31515. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, J.H.; Zhang, Y.Y.; Wang, Y.Z.; Zhao, Y.; Jin, X.X.; Xue, G.L.; Li, P.H.; Sun, Y.L.; Huang, Q.H.; et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFβ1 and miR-29 pathways. Sci. Rep. 2016, 6, 23010. [Google Scholar] [CrossRef]
- González, G.E.; Rhaleb, N.E.; D’Ambrosio, M.A.; Nakagawa, P.; Liu, Y.; Leung, P.; Dai, X.; Yang, X.P.; Peterson, E.L.; Carretero, O.A. Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunctionwithout affecting blood pressure in angiotensin II-high salt-induced hypertension. J. Hypertens. 2015, 33, 144–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, D.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.K.; Fillmore, J.J.; Sunshine, M.J.; Albrecht, B.; Higashimori, T.; Kim, D.-W.; Liu, Z.-X.; Soos, T.J.; Cline, G.W.; O’Brien, W.R.; et al. PKC-θ knockout mice are protected from fat-induced insulin resistance. J. Clin. Investig. 2004, 114, 823–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicolle, A.; Zhang, Y.; Belguise, K. The emerging function of pkctheta in cancer. Biomolecules 2021, 11, 221. [Google Scholar] [CrossRef]
- Cohen, S.; Braiman, A.; Shubinsky, G.; Isakov, N. Protein kinase C-theta in platelet activation. FEBS Lett. 2011, 585, 3208–3215. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, Y.; He, J.; Deng, W.; Cheng, L.; Jiang, Z.; Wang, D. Protein Kinase C Theta inhibition attenuates lipopolysaccharide-induced acute lung injury through notch signaling pathway via suppressing Th17 cell response in mice. Inflammation 2019, 42, 1980–1989. [Google Scholar] [CrossRef]
- Paoletti, R.; Maffei, A.; Madaro, L.; Notte, A.; Stanganello, E.; Cifelli, G.; Carullo, P.; Molinaro, M.; Lembo, G.; Bouché, M. Protein kinase Cθ is required for cardiomyocyte survival and cardiac remodeling. Cell Death Dis. 2010, 1, e45. [Google Scholar] [CrossRef]
- Simonis, G.; Briem, S.K.; Schoen, S.P.; Bock, M.; Marquetant, R.; Strasser, R.H. Protein kinase C in the human heart: Differential regulation of the isoforms in aortic stenosis or dilated cardiomyopathy. Mol. Cell. Biochem. 2007, 305, 103–111. [Google Scholar] [CrossRef]
- Sciarretta, S.; Yee, D.; Nagarajan, N.; Bianchi, F.; Saito, T.; Valenti, V.; Tong, M.; Del Re, D.P.; Vecchione, C.; Schirone, L.; et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J. Am. Coll. Cardiol. 2018, 71, 1999–2010. [Google Scholar] [CrossRef]
- Sciarretta, S.; Zhai, P.; Maejima, Y.; DelRe, D.P.; Nagarajan, N.; Yee, D.; Liu, T.; Magnuson, M.A.; Volpe, M.; Frati, G.; et al. MTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep. 2015, 11, 125–136. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| End-Diastolic Diameter [mm] | End-Systolic Diameter [mm] | Anterior Wall Thickness [mm] | Posterior Wall Thickness [mm] | Fractional Shortening [%] | Heart Rate (bpm) | Heart Weight/Body Weight [mg/g] | |
|---|---|---|---|---|---|---|---|
| mdx | 2.78 ± 0.044 | 1.41 ± 0.09 | 1.11 ± 0.06 | 1.08 ± 0.03 | 53.9 ± 1.81 | 417 ± 16.4 | 5.95 ± 0.16 |
| ex+vehicle | 3.04 ± 0.11 | 1.5 ± 0.1 | 1.06 ± 0.04 | 1.16 ± 0.02 | 44.4 ± 2.12 ** | 441 ± 32.7 | 5.53 ± 0.28 |
| ex+c20 | 3.1 ± 0.12 | 1.53 ± 0.1 | 1.11 ± 0.07 | 1.11 ± 0.03 | 50.8 ± 1.26 # | 443 ± 43.8 | 5.55 ± 0.34 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morroni, J.; Schirone, L.; Valenti, V.; Zwergel, C.; Riera, C.S.; Valente, S.; Vecchio, D.; Schiavon, S.; Ragno, R.; Mai, A.; et al. Inhibition of PKCθ Improves Dystrophic Heart Phenotype and Function in a Novel Model of DMD Cardiomyopathy. Int. J. Mol. Sci. 2022, 23, 2256. https://doi.org/10.3390/ijms23042256
Morroni J, Schirone L, Valenti V, Zwergel C, Riera CS, Valente S, Vecchio D, Schiavon S, Ragno R, Mai A, et al. Inhibition of PKCθ Improves Dystrophic Heart Phenotype and Function in a Novel Model of DMD Cardiomyopathy. International Journal of Molecular Sciences. 2022; 23(4):2256. https://doi.org/10.3390/ijms23042256
Chicago/Turabian StyleMorroni, Jacopo, Leonardo Schirone, Valentina Valenti, Clemens Zwergel, Carles Sánchez Riera, Sergio Valente, Daniele Vecchio, Sonia Schiavon, Rino Ragno, Antonello Mai, and et al. 2022. "Inhibition of PKCθ Improves Dystrophic Heart Phenotype and Function in a Novel Model of DMD Cardiomyopathy" International Journal of Molecular Sciences 23, no. 4: 2256. https://doi.org/10.3390/ijms23042256