
Experimental Evidence of Intrinsic Disorder and Amyloid Formation by the Henipavirus W Proteins

 , and
, and 
Abstract
:1. Introduction

2. Results
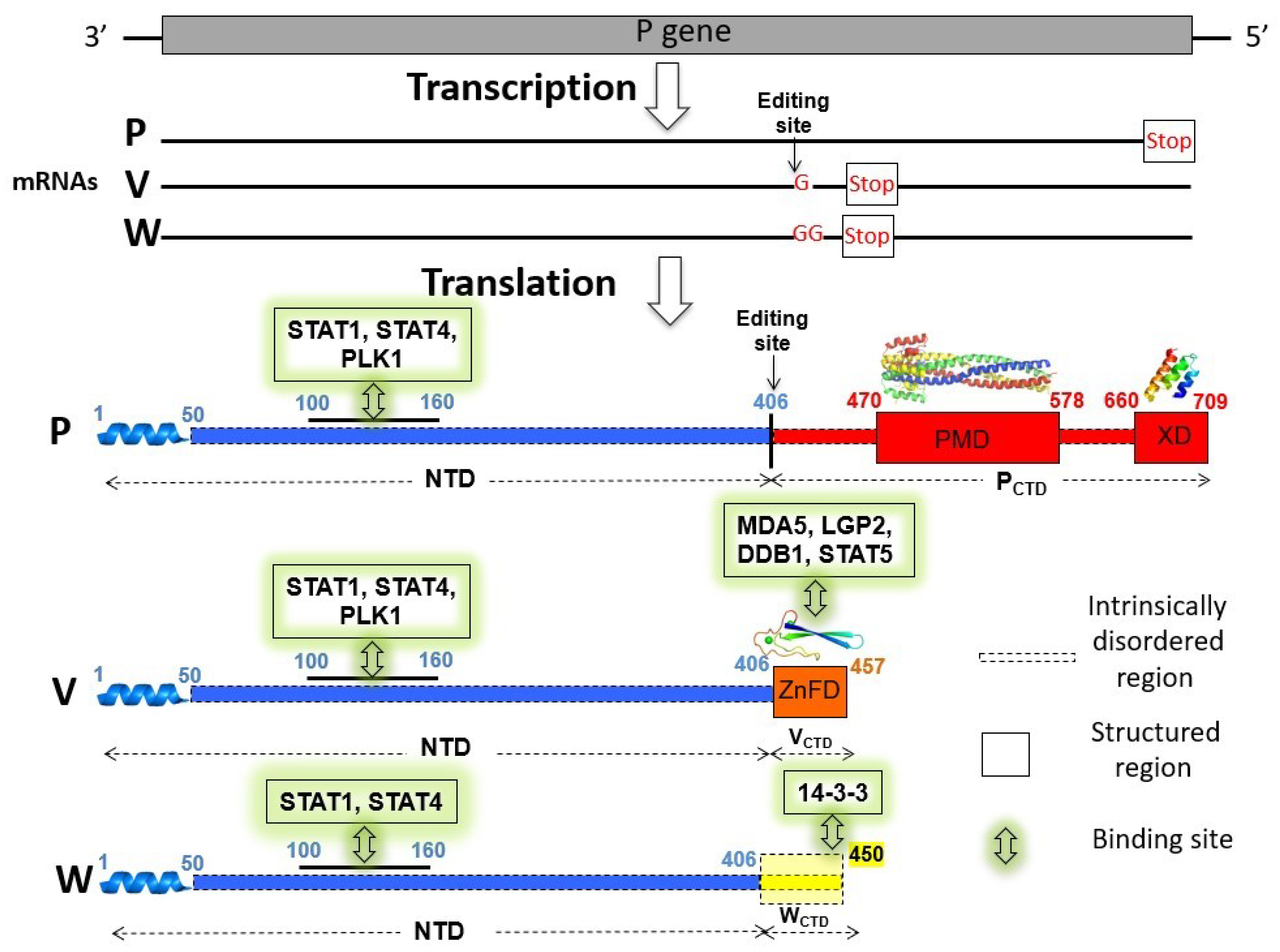
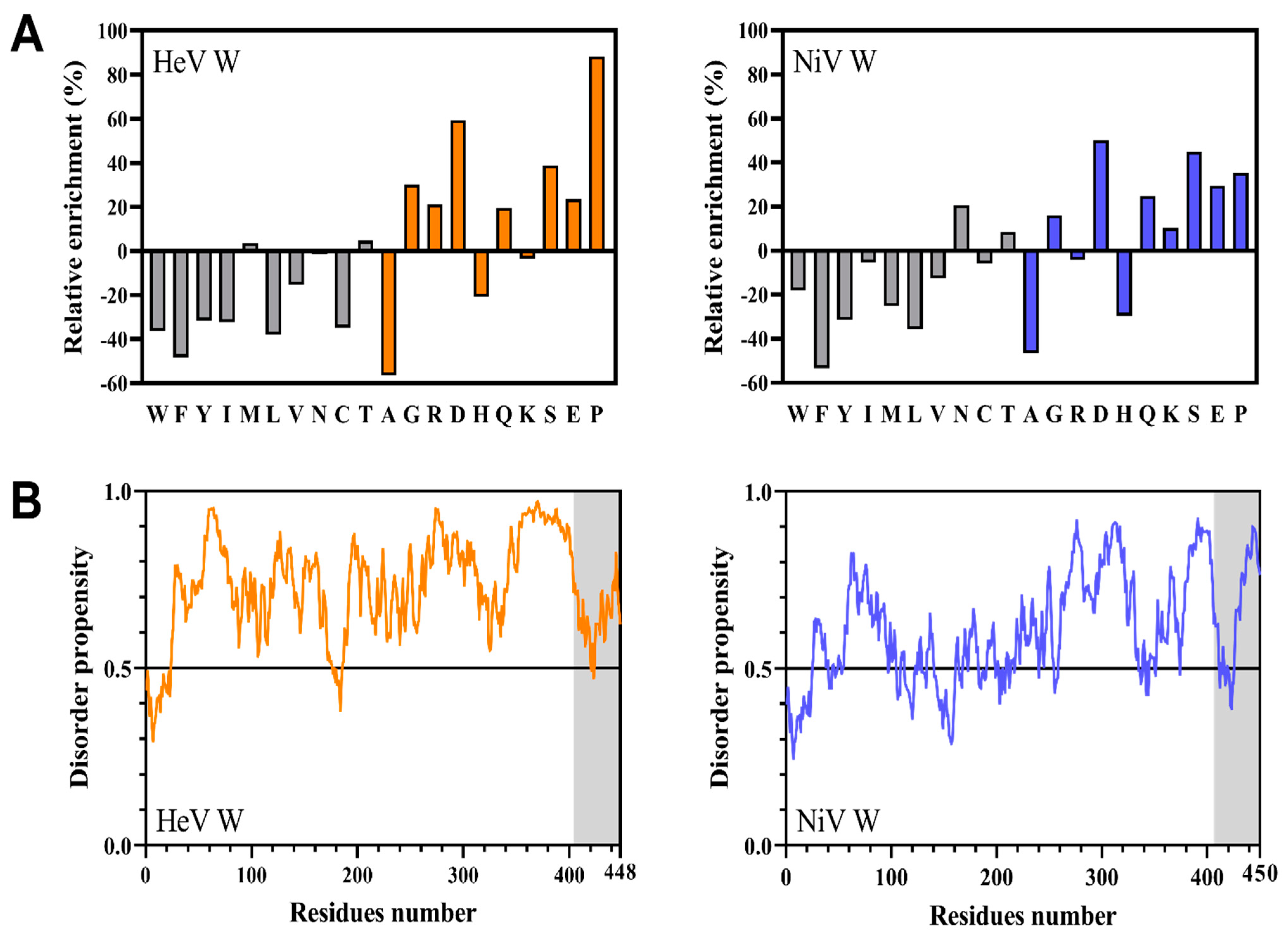
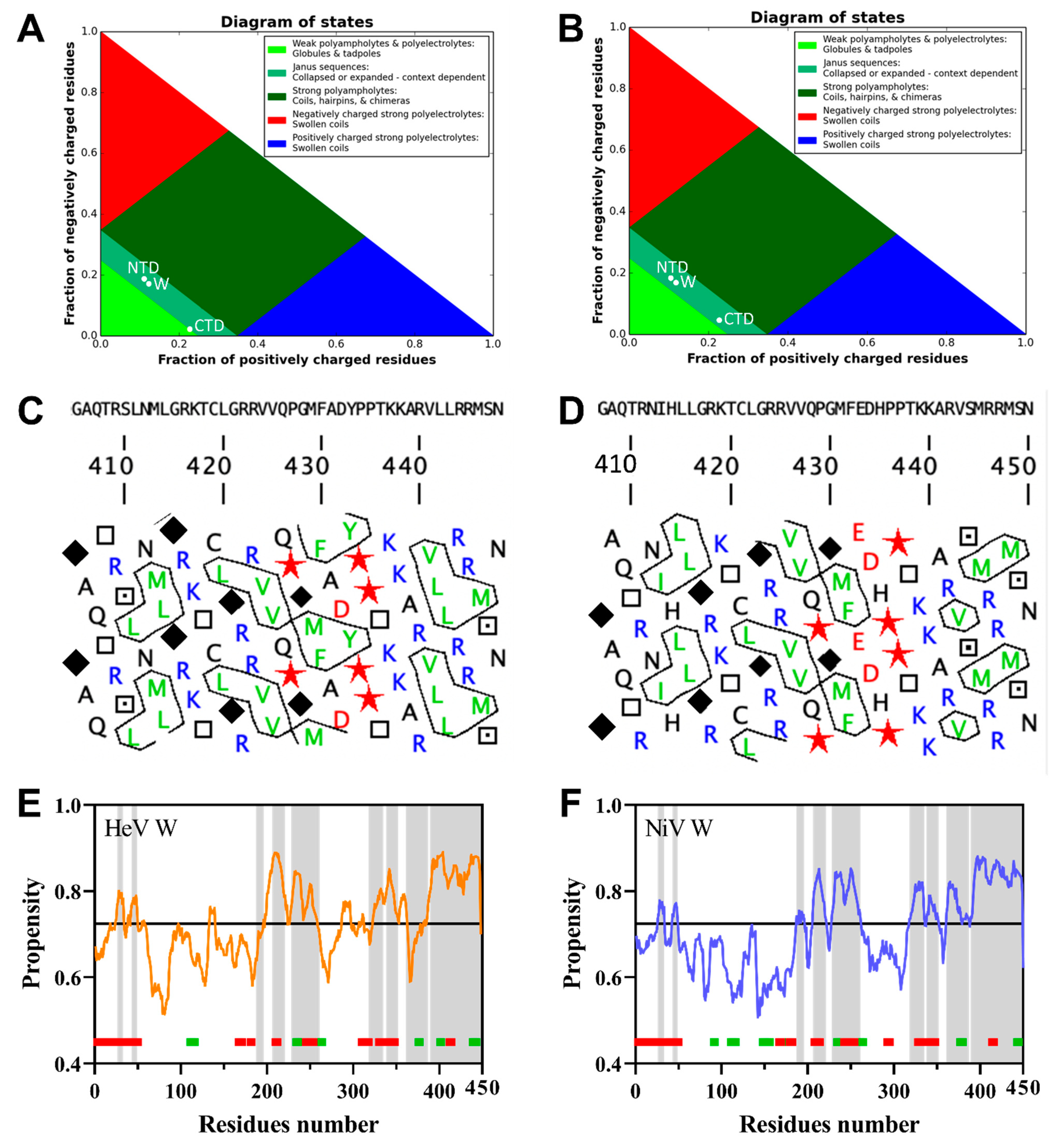
2.1. Bioinformatic Analysis of the HeV and NiV W Protein Sequences

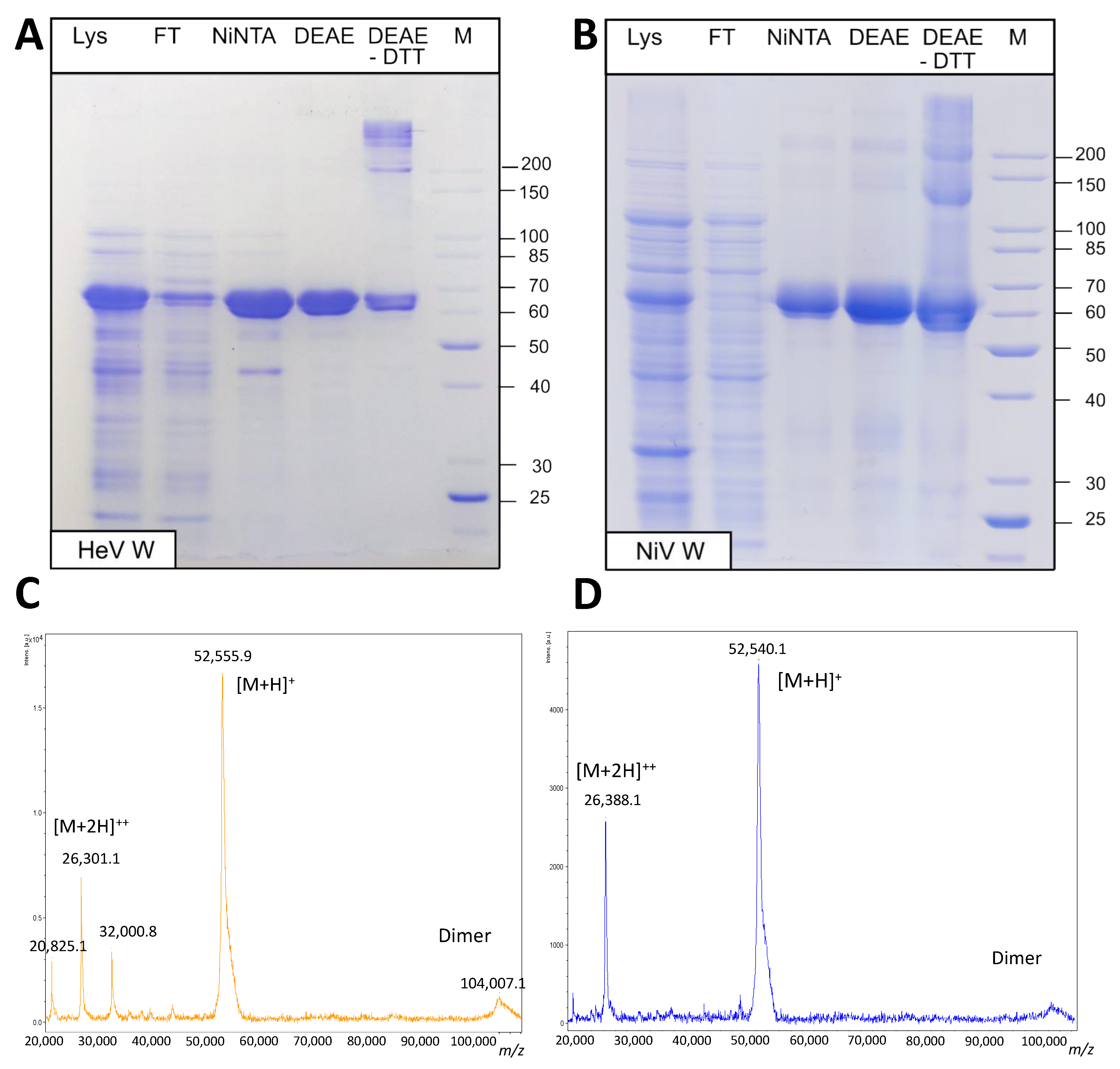
2.2. Expression and Purification of the W Proteins
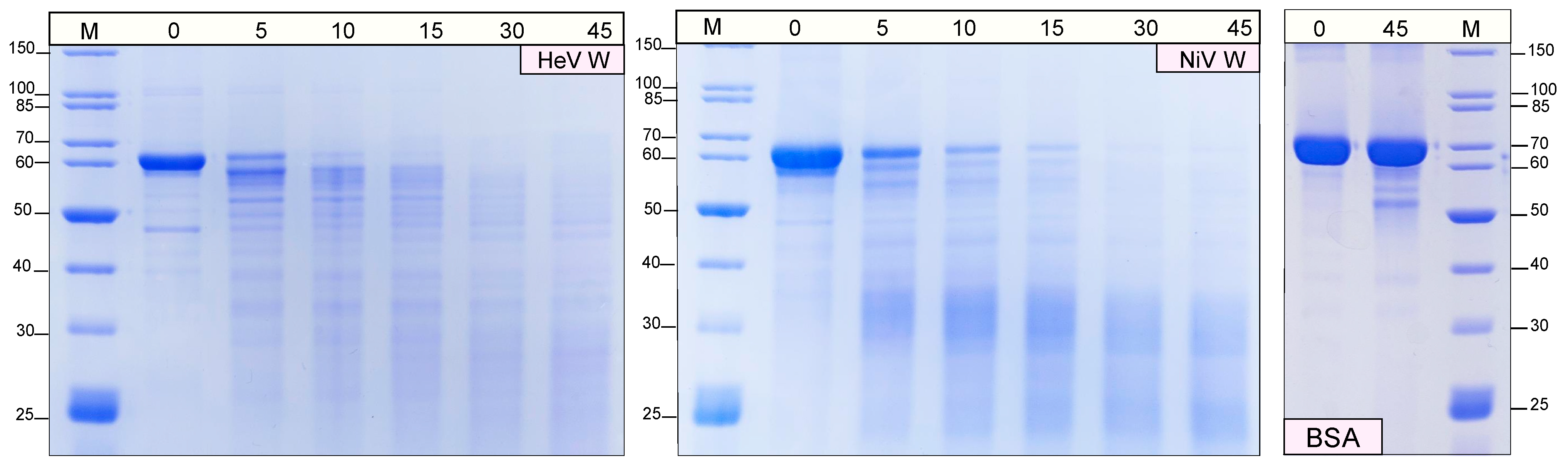
2.3. Protease Sensitivity of the W Proteins
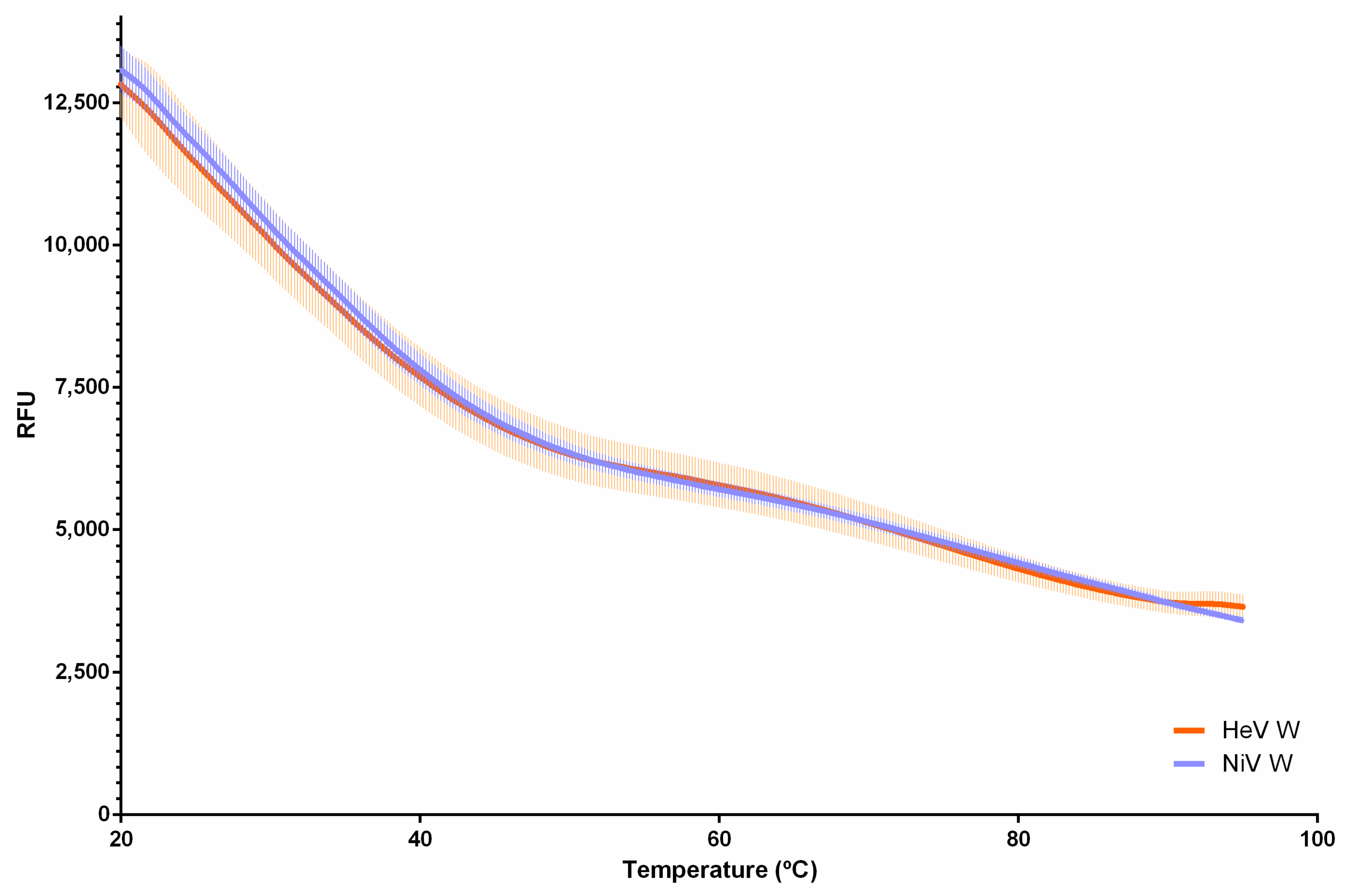
2.4. Differential Scanning Fluorimetry of the W Proteins
2.5. Hydrodynamiques Properties of the W Proteins from Size Exclusion Chromatography (SEC)
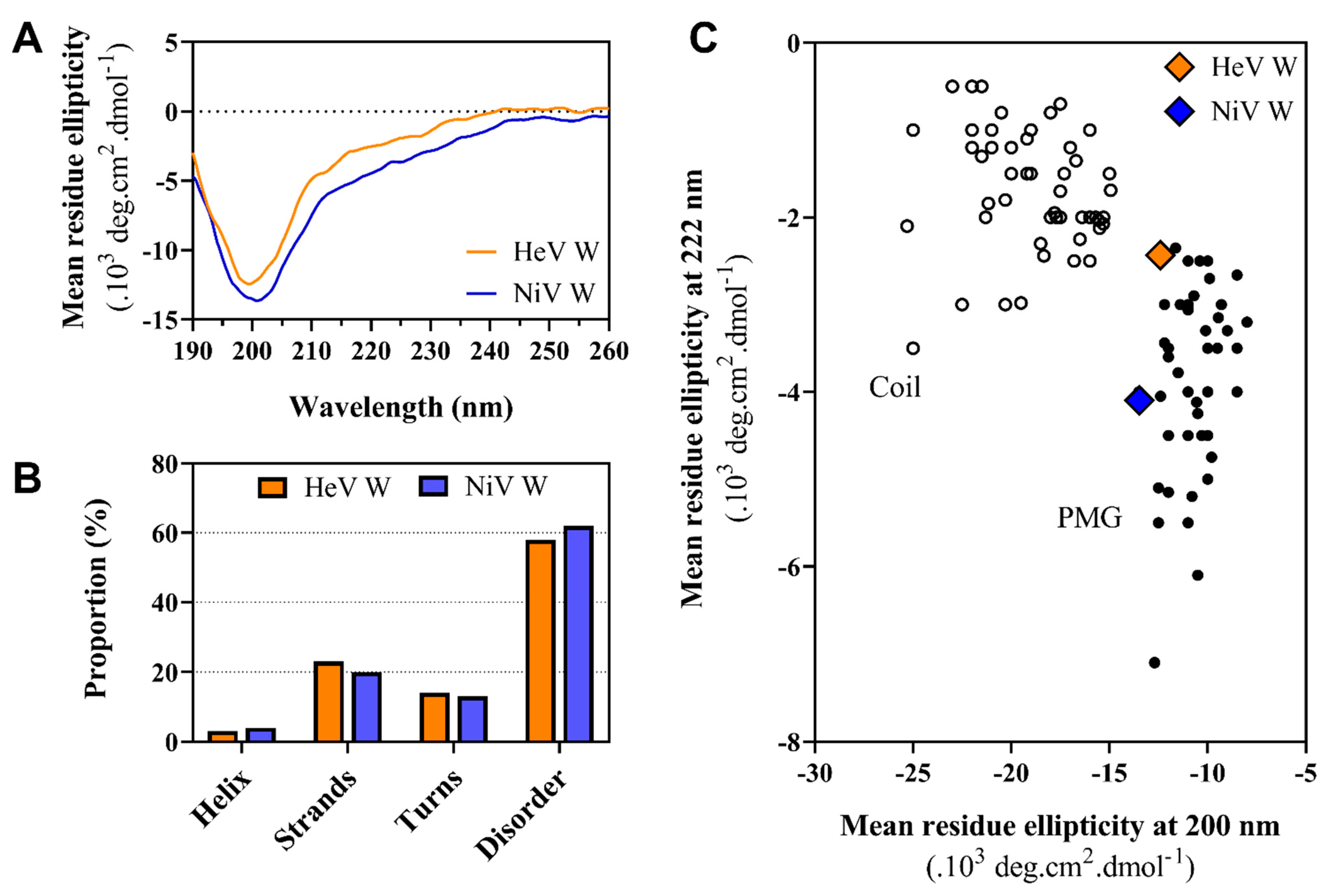
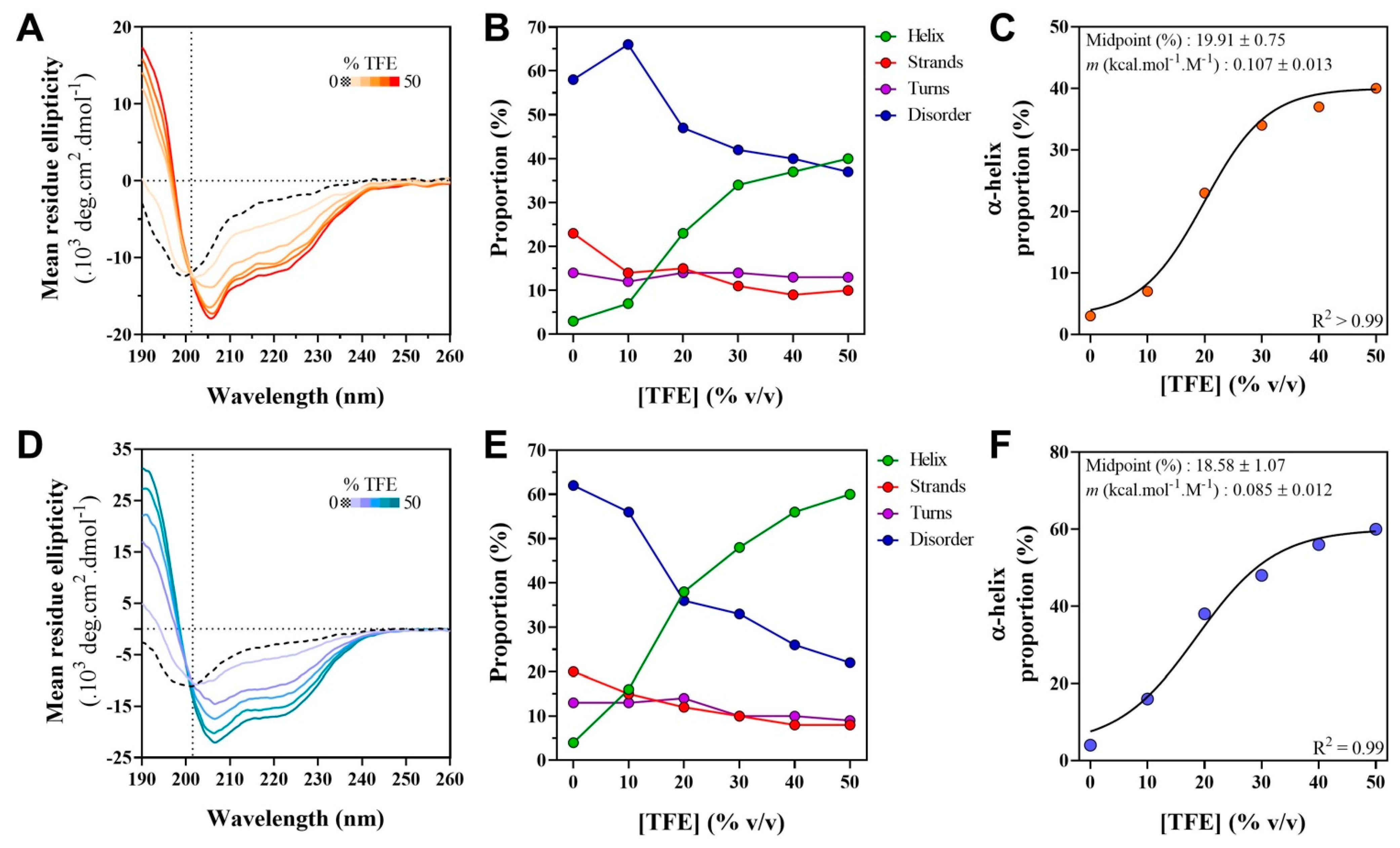
2.6. Far-UV Circular Dichroism (CD) Studies of the W Proteins
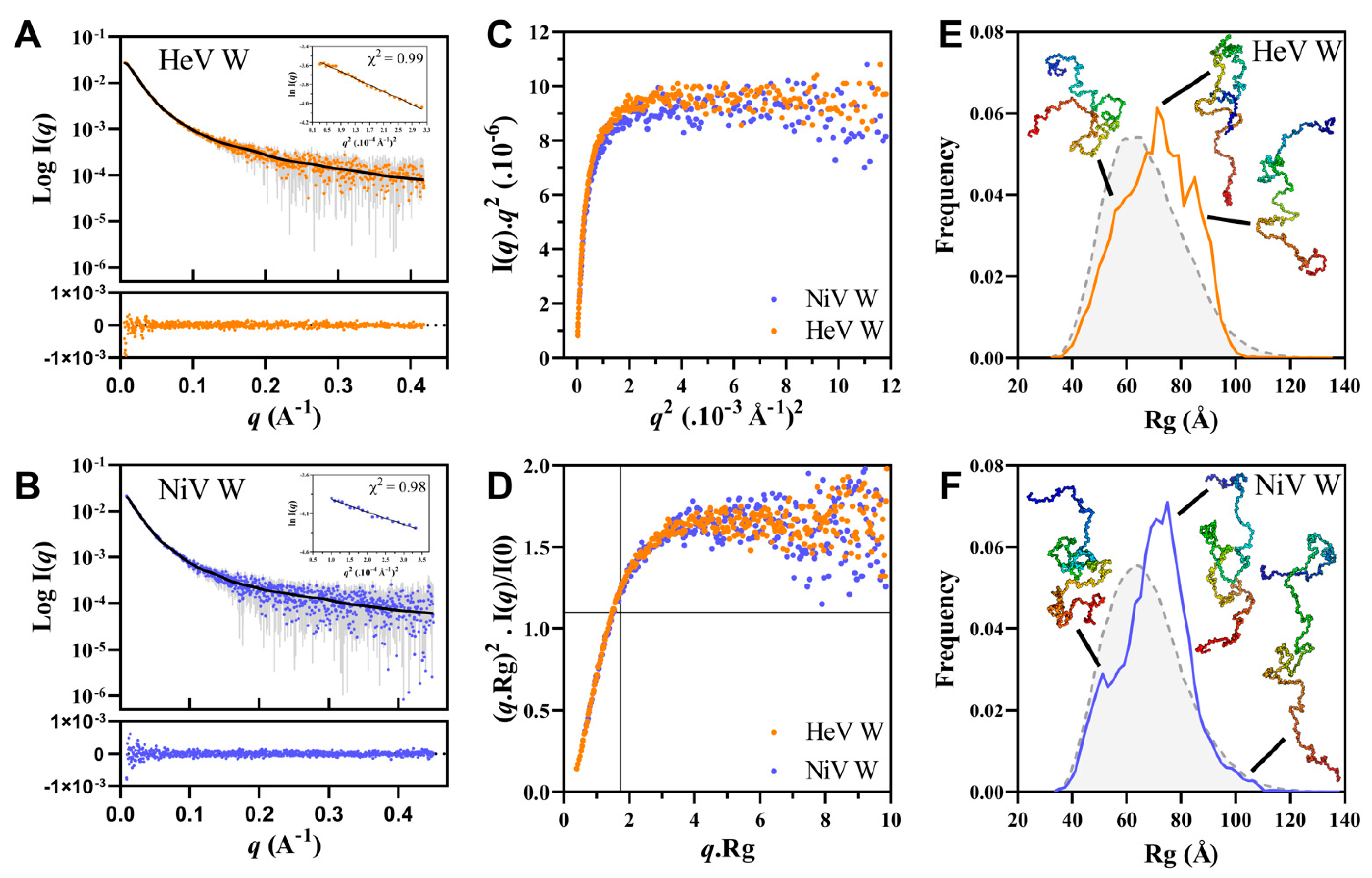
2.7. Small-Angle X-ray Scattering (SAXS) Studies of the W Proteins

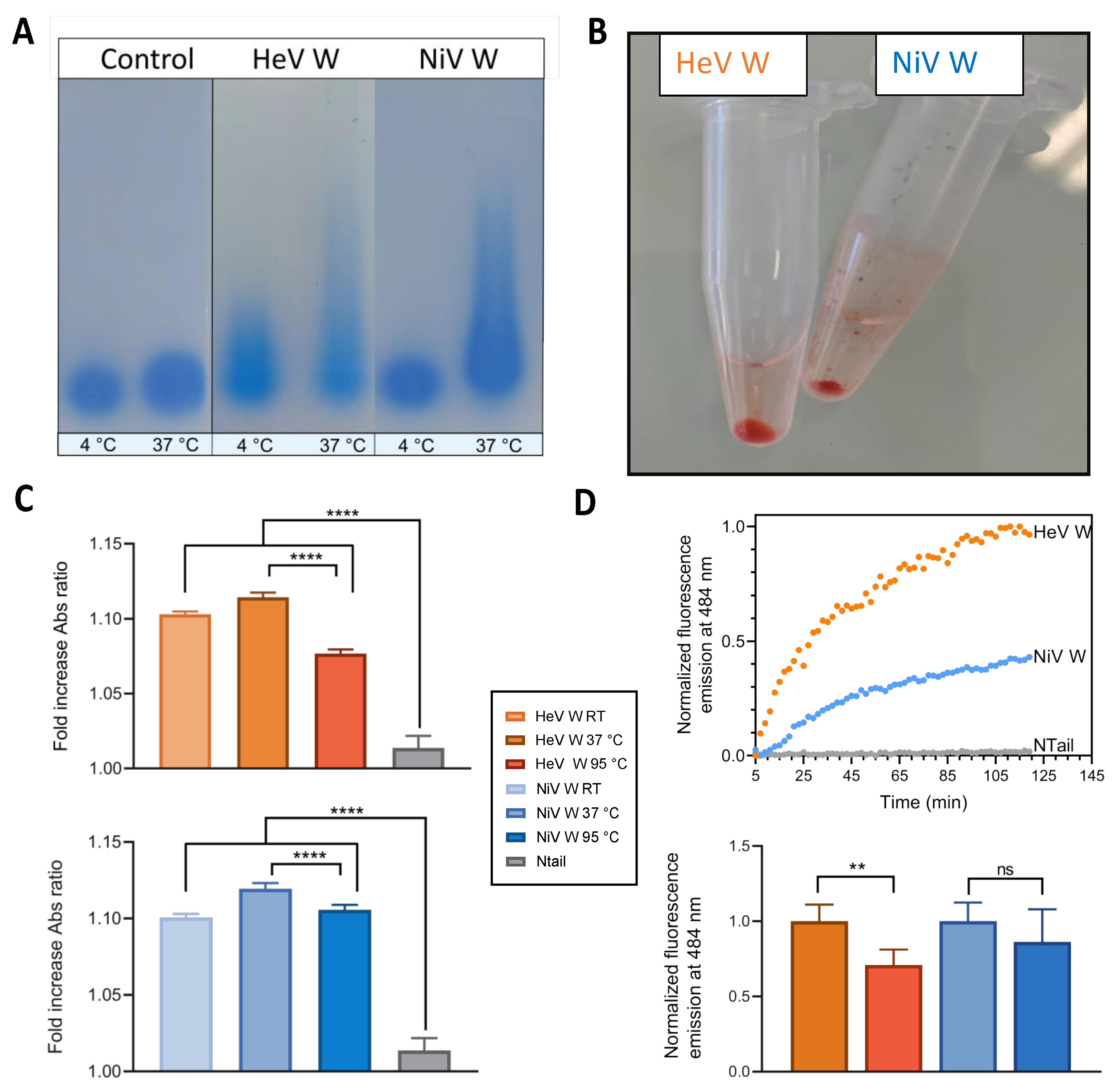
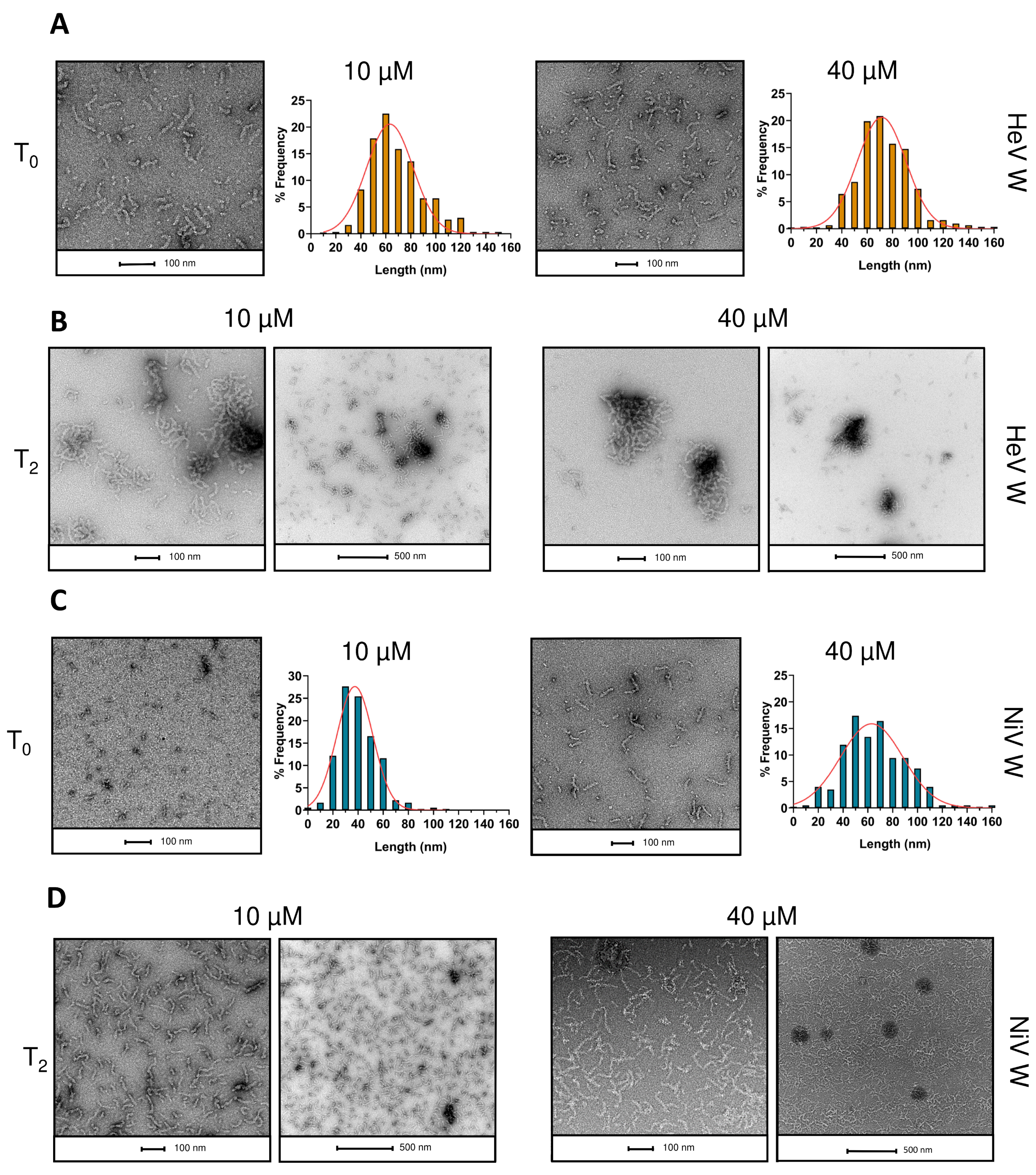
2.8. Phase Separation and Fibrillation Abilities of the W Proteins
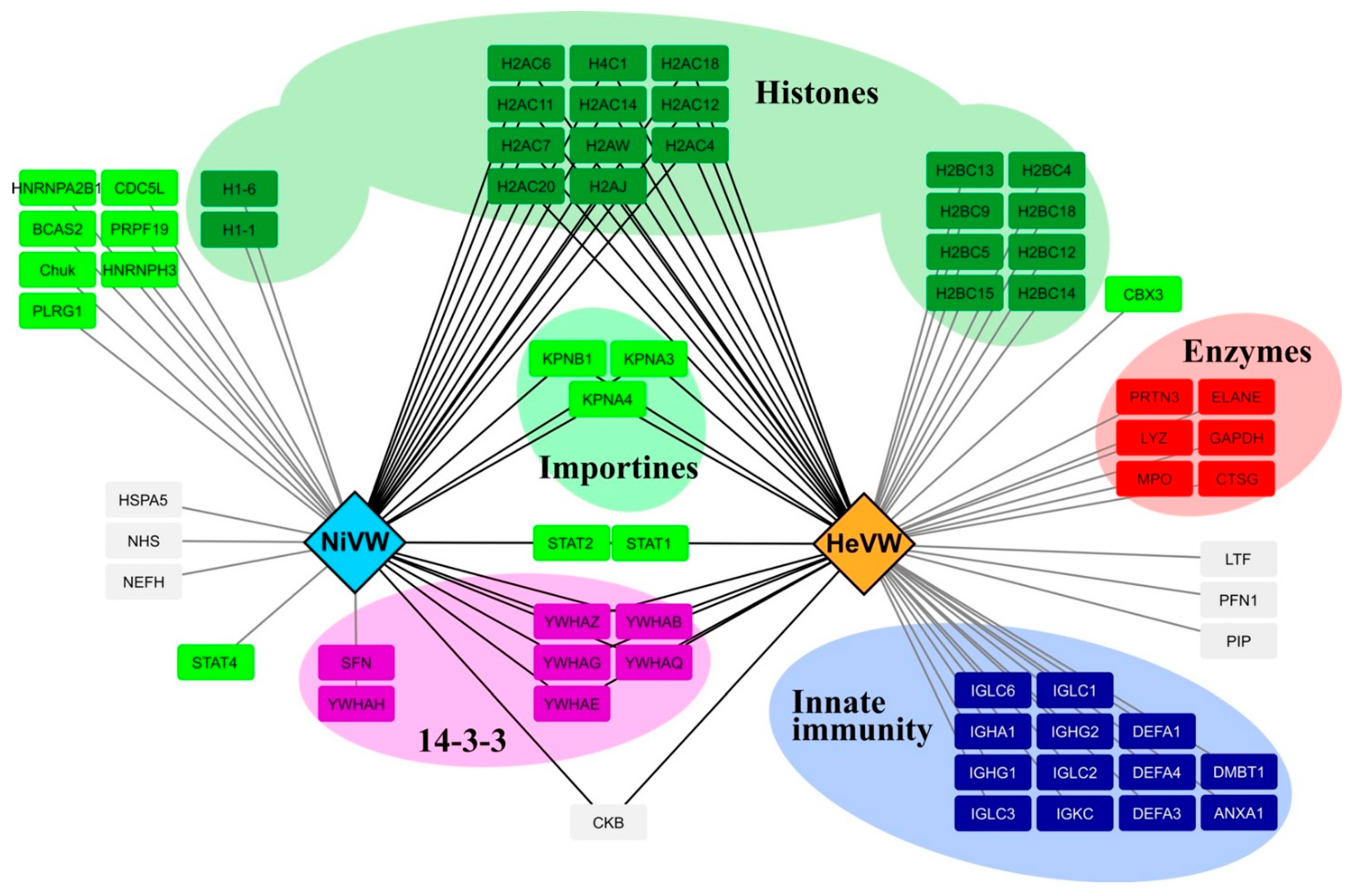
3. Discussion
4. Materials and Methods
4.1. Bioinformatic Analyses
4.2. Generation of the W Expression Constructs
4.3. Expression and Purification of the W Proteins
4.4. Mass Spectrometry
4.4.1. Intact Protein Mass Analysis
4.4.2. Peptide Mass Fingerprintings
4.5. Estimation of the Hydrodynamic Radius by SEC
4.6. Protease Sensitivity Assay
4.7. Differential Scanning Fluorimetry (DSF)
4.8. Circular Dichroism
4.9. Small-Angle X-ray Scattering (SAXS)
4.10. SDD-AGE Analysis, and Congo Red and Thioflavin T Binding Assays
4.11. Negative-Staining Transmission Electron Microscopy (TEM)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Eaton, B.T.; Broder, C.C.; Middleton, D.; Wang, L.F. Hendra and Nipah viruses: Different and dangerous. Nat. Rev. Microbiol. 2006, 4, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.F.; Yu, M.; Hansson, E.; Pritchard, L.I.; Shiell, B.; Michalski, W.P.; Eaton, B.T. The exceptionally large genome of Hendra virus: Support for creation of a new genus within the family Paramyxoviridae. J. Virol. 2000, 74, 9972–9979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurley, E.S.; Montgomery, J.M.; Hossain, M.J.; Bell, M.; Azad, A.K.; Islam, M.R.; Molla, M.A.; Carroll, D.S.; Ksiazek, T.G.; Rota, P.A.; et al. Person-to-person transmission of Nipah virus in a Bangladeshi community. Emerg. Infect. Dis. 2007, 13, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Homaira, N.; Rahman, M.; Hossain, M.J.; Epstein, J.H.; Sultana, R.; Khan, M.S.; Podder, G.; Nahar, K.; Ahmed, B.; Gurley, E.S.; et al. Nipah virus outbreak with person-to-person transmission in a district of Bangladesh, 2007. Epidemiol. Infect. 2010, 138, 1630–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ching, P.K.; de los Reyes, V.C.; Sucaldito, M.N.; Tayag, E.; Columna-Vingno, A.B.; Malbas, F.F., Jr.; Bolo, G.C., Jr.; Sejvar, J.J.; Eagles, D.; Playford, G.; et al. Outbreak of henipavirus infection, Philippines, 2014. Emerg. Infect. Dis. 2015, 21, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Plowright, R.K.; Becker, D.J.; Crowley, D.E.; Washburne, A.D.; Huang, T.; Nameer, P.O.; Gurley, E.S.; Han, B.A. Prioritizing surveillance of Nipah virus in India. PLoS Negl. Trop. Dis. 2019, 13, e0007393. [Google Scholar] [CrossRef] [Green Version]
- Ker, D.; Jenkins, H.; Greive, S.; Antson, A. CryoEM structure of the Nipah virus nucleocapsid assembly. PLoS Pathog. 2021, 17, e1009740. [Google Scholar] [CrossRef] [PubMed]
- Bloyet, L.M.; Welsch, J.; Enchery, F.; Mathieu, C.; de Breyne, S.; Horvat, B.; Grigorov, B.; Gerlier, D. HSP90 Chaperoning in Addition to Phosphoprotein Required for Folding but Not for Supporting Enzymatic Activities of Measles and Nipah Virus L Polymerases. J. Virol. 2016, 90, 6642–6656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdella, R.; Aggarwal, M.; Okura, T.; Lamb, R.A.; He, Y. Structure of a paramyxovirus polymerase complex reveals a unique methyltransferase-CTD conformation. Proc. Natl. Acad. Sci. USA 2020, 117, 4931–4941. [Google Scholar] [CrossRef]
- Bloyet, L.M.; Schramm, A.; Lazert, C.; Raynal, B.; Hologne, M.; Walker, O.; Longhi, S.; Gerlier, D. Regulation of measles virus gene expression by P protein coiled-coil properties. Sci. Adv. 2019, 5, eaaw3702. [Google Scholar] [CrossRef] [Green Version]
- Yabukarski, F.; Lawrence, P.; Tarbouriech, N.; Bourhis, J.M.; Delaforge, E.; Jensen, M.R.; Ruigrok, R.W.; Blackledge, M.; Volchkov, V.; Jamin, M. Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat. Struct. Mol. Biol. 2014, 21, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Karlin, D.; Ferron, F.; Canard, B.; Longhi, S. Structural disorder and modular organization in Paramyxovirinae N and P. J. Gen. Virol. 2003, 84, 3239–3252. [Google Scholar] [CrossRef]
- Habchi, J.; Mamelli, L.; Darbon, H.; Longhi, S. Structural Disorder within Henipavirus Nucleoprotein and Phosphoprotein: From Predictions to Experimental Assessment. PLoS ONE 2010, 5, e11684. [Google Scholar] [CrossRef] [PubMed]
- Schiavina, M.; Salladini, E.; Murrali, M.G.; Tria, G.; Felli, I.C.; Pierattelli, R.; Longhi, S. Ensemble description of the intrinsically disordered N-terminal domain of the Nipah virus P/V protein from combined NMR and SAXS. Sci. Rep. 2020, 10, 19574. [Google Scholar] [CrossRef]
- Wright, P.E.; Dyson, H.J. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999, 293, 321–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Tompa, P. Intrinsically disordered proteins: A 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Dunker, A.K.; Babu, M.M.; Barbar, E.; Blackledge, M.; Bondos, S.E.; Dosztányi, Z.; Dyson, H.J.; Forman-Kay, J.; Fuxreiter, M.; Gsponer, J.; et al. What’s in a name? Why these proteins are intrinsically disordered: Why these proteins are intrinsically disordered. Intrinsically Disord. Proteins 2013, 1, e24157. [Google Scholar] [CrossRef] [Green Version]
- Habchi, J.; Tompa, P.; Longhi, S.; Uversky, V.N. Introducing Protein Intrinsic Disorder. Chem. Rev. 2014, 114, 6561–6588. [Google Scholar] [CrossRef] [Green Version]
- Jensen, M.R.; Yabukarski, F.; Communie, G.; Condamine, E.; Mas, C.; Volchkova, V.; Tarbouriech, N.; Bourhis, J.M.; Volchkov, V.; Blackledge, M.; et al. Structural Description of the Nipah Virus Phosphoprotein and Its Interaction with STAT1. Biophys. J. 2020, 118, 2470–2488. [Google Scholar] [CrossRef] [PubMed]
- Bruhn-Johannsen, J.F.; Barnett, K.; Bibby, J.; Thomas, J.; Keegan, R.; Rigden, D.; Bornholdt, Z.A.; Saphire, E.O. Crystal structure of the Nipah virus phosphoprotein tetramerization domain. J. Virol. 2014, 88, 758–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blocquel, D.; Beltrandi, M.; Erales, J.; Barbier, P.; Longhi, S. Biochemical and structural studies of the oligomerization domain of the Nipah virus phosphoprotein: Evidence for an elongated coiled-coil homotrimer. Virology 2013, 446, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Beltrandi, M.; Blocquel, D.; Erales, J.; Barbier, P.; Cavalli, A.; Longhi, S. Insights into the coiled-coil organization of the Hendra virus phosphoprotein from combined biochemical and SAXS studies. Virology 2015, 477, 42–55. [Google Scholar] [CrossRef] [PubMed]
- Habchi, J.; Blangy, S.; Mamelli, L.; Ringkjobing Jensen, M.; Blackledge, M.; Darbon, H.; Oglesbee, M.; Shu, Y.; Longhi, S. Characterization of the interactions between the nucleoprotein and the phosphoprotein of Henipaviruses. J. Biol. Chem. 2011, 286, 13583–13602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Communie, G.; Habchi, J.; Yabukarski, F.; Blocquel, D.; Schneider, R.; Tarbouriech, N.; Papageorgiou, N.; Ruigrok, R.W.; Jamin, M.; Ringkjøbing-Jensen, M.; et al. Atomic resolution description of the interaction between the nucleoprotein and phosphoprotein of Hendra virus. PLoS Pathog. 2013, 9, e1003631. [Google Scholar] [CrossRef]
- Douglas, J.; Drummond, A.J.; Kingston, R.L. Evolutionary history of cotranscriptional editing in the paramyxoviral phosphoprotein gene. Virus Evol. 2021, 7, veab028. [Google Scholar] [CrossRef]
- Fontana, J.M.; Bankamp, B.; Rota, P.A. Inhibition of interferon induction and signaling by paramyxoviruses. Immunol. Rev. 2008, 225, 46–67. [Google Scholar] [CrossRef]
- Audsley, M.D.; Moseley, G.W. Paramyxovirus evasion of innate immunity: Diverse strategies for common targets. World J. Virol. 2013, 2, 57–70. [Google Scholar] [CrossRef]
- Tsimbalyuk, S.; Cross, E.M.; Hoad, M.; Donnelly, C.M.; Roby, J.A.; Forwood, J.K. The Intrinsically Disordered W Protein Is Multifunctional during Henipavirus Infection, Disrupting Host Signalling Pathways and Nuclear Import. Cells 2020, 9, 1913. [Google Scholar] [CrossRef]
- Childs, K.; Randall, R.; Goodbourn, S. Paramyxovirus V proteins interact with the RNA Helicase LGP2 to inhibit RIG-I-dependent interferon induction. J. Virol. 2012, 86, 3411–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludlow, L.E.; Lo, M.K.; Rodriguez, J.J.; Rota, P.A.; Horvath, C.M. Henipavirus V protein association with Polo-like kinase reveals functional overlap with STAT1 binding and interferon evasion. J. Virol. 2008, 82, 6259–6271. [Google Scholar] [CrossRef] [Green Version]
- Ulane, C.M.; Horvath, C.M. Paramyxoviruses SV5 and HPIV2 assemble STAT protein ubiquitin ligase complexes from cellular components. Virology 2002, 304, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salladini, E.; Delauzun, V.; Longhi, S. The Henipavirus V protein is a prevalently unfolded protein with a zinc-finger domain involved in binding to DDB1. Mol. Biosyst. 2017, 13, 2254–2267. [Google Scholar] [CrossRef]
- Shaw, M.L.; Garcia-Sastre, A.; Palese, P.; Basler, C.F. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol. 2004, 78, 5633–5641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, M.L.; Cardenas, W.B.; Zamarin, D.; Palese, P.; Basler, C.F. Nuclear localization of the Nipah virus W protein allows for inhibition of both virus- and toll-like receptor 3-triggered signaling pathways. J. Virol. 2005, 79, 6078–6088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.M.; Tsimbalyuk, S.; Edwards, M.R.; Cross, E.M.; Batra, J.; Soares da Costa, T.P.; Aragao, D.; Basler, C.F.; Forwood, J.K. Structural basis for importin alpha 3 specificity of W proteins in Hendra and Nipah viruses. Nat. Commun. 2018, 9, 3703. [Google Scholar] [CrossRef]
- Audsley, M.D.; Jans, D.A.; Moseley, G.W. Nucleocytoplasmic trafficking of Nipah virus W protein involves multiple discrete interactions with the nuclear import and export machinery. Biochem. Biophys. Res. Commun. 2016, 479, 429–433. [Google Scholar] [CrossRef]
- Keiffer, T.R.; Ciancanelli, M.J.; Edwards, M.R.; Basler, C.F. Interactions of the Nipah Virus P, V, and W Proteins across the STAT Family of Transcription Factors. mSphere 2020, 5, e00449-20. [Google Scholar] [CrossRef]
- Lo, M.K.; Miller, D.; Aljofan, M.; Mungall, B.A.; Rollin, P.E.; Bellini, W.J.; Rota, P.A. Characterization of the antiviral and inflammatory responses against Nipah virus in endothelial cells and neurons. Virology 2010, 404, 78–88. [Google Scholar] [CrossRef] [Green Version]
- Satterfield, B.A.; Cross, R.W.; Fenton, K.A.; Agans, K.N.; Basler, C.F.; Geisbert, T.W.; Mire, C.E. The immunomodulating V and W proteins of Nipah virus determine disease course. Nat. Commun. 2015, 6, 7483. [Google Scholar] [CrossRef] [PubMed]
- Enchery, F. Etude de la Modulation de la Voie Canonique D’activation de NF-κB par les Protéines non Structurales du virus Nipah. Thèse de Doctorat, Université de Lyon, Lyon, France, 2017. [Google Scholar]
- Edwards, M.R.; Hoad, M.; Tsimbalyuk, S.; Menicucci, A.R.; Messaoudi, I.; Forwood, J.K.; Basler, C.F. Henipavirus W Proteins Interact with 14-3-3 To Modulate Host Gene Expression. J. Virol. 2020, 94, e00373-20. [Google Scholar] [CrossRef]
- Enchery, F.; Dumont, C.; Iampietro, M.; Pelissier, R.; Aurine, N.; Bloyet, L.M.; Carbonelle, C.; Mathieu, C.; Journo, C.; Gerlier, D.; et al. Nipah virus W protein harnesses nuclear 14-3-3 to inhibit NF-κB-induced proinflammatory response. Commun. Biol. 2021, in press. [Google Scholar] [CrossRef]
- Atkinson, S.C.; Audsley, M.D.; Lieu, K.G.; Marsh, G.A.; Thomas, D.R.; Heaton, S.M.; Paxman, J.J.; Wagstaff, K.M.; Buckle, A.M.; Moseley, G.W.; et al. Recognition by host nuclear transport proteins drives disorder-to-order transition in Hendra virus V. Sci. Rep. 2018, 8, 358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Campen, A.; Williams, R.M.; Brown, C.J.; Meng, J.; Uversky, V.N.; Dunker, A.K. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein. Pept. Lett. 2008, 15, 956–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. What does it mean to be natively unfolded? Eur. J. Biochem. 2002, 269, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Meszaros, B.; Erdos, G.; Dosztanyi, Z. IUPred2A: Context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 2018, 46, W329–W337. [Google Scholar] [CrossRef] [PubMed]
- Mohan, A.; Oldfield, C.J.; Radivojac, P.; Vacic, V.; Cortese, M.S.; Dunker, A.K.; Uversky, V.N. Analysis of Molecular Recognition Features (MoRFs). J. Mol. Biol. 2006, 362, 1043–1059. [Google Scholar] [CrossRef]
- Vacic, V.; Oldfield, C.J.; Mohan, A.; Radivojac, P.; Cortese, M.S.; Uversky, V.N.; Dunker, A.K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichmann, D.; Jakob, U. The roles of conditional disorder in redox proteins. Curr. Opin. Struct. Biol. 2013, 23, 436–442. [Google Scholar] [CrossRef] [Green Version]
- Mao, A.H.; Crick, S.L.; Vitalis, A.; Chicoine, C.L.; Pappu, R.V. Net charge per residue modulates conformational ensembles of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 8183–8188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Spath, S.; Soranno, A.; Hirschfeld, V.; Hofmann, H.; Ruegger, S.; Reymond, L.; Nettels, D.; Schuler, B. Charge interactions can dominate the dimensions of intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 14609–14614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [Green Version]
- Das, R.K.; Ruff, K.M.; Pappu, R.V. Relating sequence encoded information to form and function of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2015, 32, 102–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callebaut, I.; Labesse, G.; Durand, P.; Poupon, A.; Canard, L.; Chomilier, J.; Henrissat, B.; Mornon, J.P. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): Current status and perspectives. Cell. Mol. Life Sci. 1997, 53, 621–645. [Google Scholar] [CrossRef] [PubMed]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, W488–W493. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The PSIPRED protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Cruz, C.D.; Horvath, C.M. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J. Virol. 2004, 78, 5358–5367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Tompa, P. Intrinsically unstructured proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Schramm, A.; Bignon, C.; Brocca, S.; Grandori, R.; Santambrogio, C.; Longhi, S. An arsenal of methods for the experimental characterization of intrinsically disordered proteins—How to choose and combine them? Arch. Biochem. Biophys. 2019, 676, 108055. [Google Scholar] [CrossRef]
- Hames, B. Gel Electrophoresis of Proteins: A Practical Approach, 3rd ed.; Oxford University Press: Oxford, NY, USA, 1998. [Google Scholar]
- Frottin, F.; Martinez, A.; Peynot, P.; Mitra, S.; Holz, R.C.; Giglione, C.; Meinnel, T. The proteomics of N-terminal methionine cleavage. Mol. Cell. Proteom. 2006, 5, 2336–2349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Receveur-Bréchot, V.; Bourhis, J.M.; Uversky, V.N.; Canard, B.; Longhi, S. Assessing protein disorder and induced folding. Proteins Struct. Funct. Bioinform. 2006, 62, 24–45. [Google Scholar] [CrossRef]
- Uversky, V.N. Natively unfolded proteins: A point where biology waits for physics. Protein Sci. 2002, 11, 739–756. [Google Scholar] [CrossRef] [Green Version]
- Hua, Q.X.; Jia, W.H.; Bullock, B.P.; Habener, J.F.; Weiss, M.A. Transcriptional activator-coactivator recognition: Nascent folding of a kinase-inducible transactivation domain predicts its structure on coactivator binding. Biochemistry 1998, 37, 5858–5866. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Mercurio, F.A.; Leone, M. About TFE: Old and New Findings. Curr. Protein Pept. Sci. 2019, 20, 425–451. [Google Scholar] [CrossRef] [PubMed]
- Bernado, P. Effect of interdomain dynamics on the structure determination of modular proteins by small-angle scattering. Eur. Biophys. J. 2010, 39, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Gast, K.; Damaschun, H.; Misselwitz, R.; Muller-Frohne, M.; Zirwer, D.; Damaschun, G. Compactness of protein molten globules: Temperature-induced structural changes of the apomyoglobin folding intermediate. Eur. Biophys. J. 1994, 23, 297–305. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L. The PyMOL molecular graphics system. Proteins Struct. Funct. Bioinform. 2002, 30, 442–454. [Google Scholar]
- Franke, D.; Jeffries, C.M.; Svergun, D. Correlation Map, a goodness-of-fit test for one-dimensional X-ray scattering spectra. Nat. Methods 2015, 12, 419–422. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Pappu, R.V. Functional Implications of Intracellular Phase Transitions. Biochemistry 2018, 57, 2415–2423. [Google Scholar] [CrossRef]
- Alberti, S.; Hyman, A.A. Biomolecular condensates at the nexus of cellular stress, protein aggregation disease and ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 196–213. [Google Scholar] [CrossRef] [PubMed]
- Fare, C.M.; Villani, A.; Drake, L.E.; Shorter, J. Higher-order organization of biomolecular condensates. Open Biol. 2021, 11, 210137. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Brangwynne, C.P. Liquid phase condensation in cell physiology and disease. Science 2017, 357, eaaf4382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, B.; Pritišanac, I.; Scherer, S.W.; Moses, A.M.; Forman-Kay, J.D. Phase Separation as a Missing Mechanism for Interpretation of Disease Mutations. Cell 2020, 183, 1742–1756. [Google Scholar] [CrossRef] [PubMed]
- Alberti, S.; Gladfelter, A.; Mittag, T. Considerations and Challenges in Studying Liquid-Liquid Phase Separation and Biomolecular Condensates. Cell 2019, 176, 419–434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeynaems, S.; Alberti, S.; Fawzi, N.L.; Mittag, T.; Polymenidou, M.; Rousseau, F.; Schymkowitz, J.; Shorter, J.; Wolozin, B.; Van Den Bosch, L.; et al. Protein Phase Separation: A New Phase in Cell Biology. Trends Cell Biol. 2018, 28, 420–435. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Fuxreiter, M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell 2016, 165, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Fuxreiter, M.; Vendruscolo, M. Generic nature of the condensed states of proteins. Nat. Cell Biol. 2021, 23, 587–594. [Google Scholar] [CrossRef]
- Salladini, E.; Debarnot, C.; Delauzun, V.; Murrali, M.G.; Sutto-Ortiz, P.; Spinelli, S.; Pierattelli, R.; Bignon, C.; Longhi, S. Phase transition and amyloid formation by a viral protein as an additional molecular mechanism of virus-induced cell toxicity. BioRxiv 2018, 497024. [Google Scholar] [CrossRef]
- Salladini, E.; Gondelaud, F.; Nilsson, J.; Pesce, G.; Bignon, C.; Murrali, M.G.; Horvat, B.; Fabre, R.; Pierattelli, R.; Kajava, A.V.; et al. Identification of a region in the common ammino-terminal domain of Hendra virus P, V and W proteins responsible for 3 phase transition and amyloid formation. Biomolecules 2021, in press. 1324. [Google Scholar] [CrossRef]
- Halfmann, R.; Lindquist, S. Screening for amyloid aggregation by Semi-Denaturing Detergent-Agarose Gel Electrophoresis. J. Vis. Exp. 2008, 838. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.V.; Ishtikhar, M.; Rabbani, G.; Zaman, M.; Abdelhameed, A.S.; Khan, R.H. Polyols (Glycerol and Ethylene glycol) mediated amorphous aggregate inhibition and secondary structure restoration of metalloproteinase-conalbumin (ovotransferrin). Int. J. Biol. Macromol. 2017, 94, 290–300. [Google Scholar] [CrossRef]
- LeVine, H., 3rd. Thioflavine T interaction with synthetic Alzheimer’s disease beta-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993, 2, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Boyer, D.R.; Mynhier, N.A.; Saway, M.R. Why amyloid fibrils have a limited width. BioRxiv 2021. [Google Scholar] [CrossRef]
- Livernois, A.M.; Hnatchuk, D.J.; Findlater, E.E.; Graether, S.P. Obtaining highly purified intrinsically disordered protein by boiling lysis and single step ion exchange. Anal. Biochem. 2009, 392, 70–76. [Google Scholar] [CrossRef] [PubMed]
- KrishnaKumar, V.G.; Gupta, S. Simplified method to obtain enhanced expression of tau protein from E. coli and one-step purification by direct boiling. Prep. Biochem. Biotechnol. 2017, 47, 530–538. [Google Scholar] [CrossRef]
- Marsh, J.A.; Forman-Kay, J.D. Sequence determinants of compaction in intrinsically disordered proteins. Biophys. J. 2010, 98, 2383–2390. [Google Scholar] [CrossRef] [Green Version]
- Van Hoy, M.; Leuther, K.K.; Kodadek, T.; Johnston, S.A. The acidic activation domains of the GCN4 and GAL4 proteins are not alpha helical but form beta sheets. Cell 1993, 72, 587–594. [Google Scholar] [CrossRef]
- Mouillon, J.M.; Gustafsson, P.; Harryson, P. Structural investigation of disordered stress proteins. Comparison of full-length dehydrins with isolated peptides of their conserved segments. Plant Physiol. 2006, 141, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Belle, V.; Rouger, S.; Costanzo, S.; Liquiere, E.; Strancar, J.; Guigliarelli, B.; Fournel, A.; Longhi, S. Mapping alpha-helical induced folding within the intrinsically disordered C-terminal domain of the measles virus nucleoprotein by site-directed spin-labeling EPR spectroscopy. Proteins Struct. Funct. Bioinform. 2008, 73, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Martinho, M.; Habchi, J.; El Habre, Z.; Nesme, L.; Guigliarelli, B.; Belle, V.; Longhi, S. Assessing induced folding within the intrinsically disordered C-terminal domain of the Henipavirus nucleoproteins by site directed spin labeling EPR spectroscopy. J. Biomol. Struct. Dyn. 2013, 31, 453–471. [Google Scholar] [CrossRef] [PubMed]
- Tayeb-Fligelman, E.; Tabachnikov, O.; Moshe, A.; Goldshmidt-Tran, O.; Sawaya, M.R.; Coquelle, N.; Colletier, J.P.; Landau, M. The cytotoxic Staphylococcus aureus PSMα3 reveals a cross-α amyloid-like fibril. Science 2017, 355, 831–833. [Google Scholar] [CrossRef] [PubMed]
- Engelberg, Y.; Landau, M. The Human LL-37(17-29) antimicrobial peptide reveals a functional supramolecular structure. Nat. Commun. 2020, 11, 3894. [Google Scholar] [CrossRef] [PubMed]
- Fuxreiter, M.; Simon, I.; Friedrich, P.; Tompa, P. Preformed structural elements feature in partner recognition by intrinsically unstructured proteins. J. Mol. Biol. 2004, 338, 1015–1026. [Google Scholar] [CrossRef]
- Lacy, E.R.; Filippov, I.; Lewis, W.S.; Otieno, S.; Xiao, L.; Weiss, S.; Hengst, L.; Kriwacki, R.W. p27 binds cyclin-CDK complexes through a sequential mechanism involving binding-induced protein folding. Nat. Struct. Mol. Biol. 2004, 11, 358–364. [Google Scholar] [CrossRef]
- Sivakolundu, S.G.; Bashford, D.; Kriwacki, R.W. Disordered p27Kip1 exhibits intrinsic structure resembling the Cdk2/cyclin A-bound conformation. J. Mol. Biol. 2005, 353, 1118–1128. [Google Scholar] [CrossRef] [PubMed]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; del-Toro, N.; et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Nevers, Q.; Albertini, A.A.; Lagaudrière-Gesbert, C.; Gaudin, Y. Negri bodies and other virus membrane-less replication compartments. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118831. [Google Scholar] [CrossRef] [PubMed]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Pesce, G.; Brocca, S.; Grandori, R.; Longhi, S.; Uversky, A.V. Droplets of life: Role of phase separation in virus replication and compartmentalization. In Droplets of Life; Uversky, A.V., Ed.; Elsevier: Amsterdam, The Netherlands, in press.
- Nikolic, J.; Le Bars, R.; Lama, Z.; Scrima, N.; Lagaudriere-Gesbert, C.; Gaudin, Y.; Blondel, D. Negri bodies are viral factories with properties of liquid organelles. Nat. Commun. 2017, 8, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinrich, B.S.; Maliga, Z.; Stein, D.A.; Hyman, A.A.; Whelan, S.P.J. Phase Transitions Drive the Formation of Vesicular Stomatitis Virus Replication Compartments. MBio 2018, 9, e02290-17. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Su, J.M.; Samuel, C.E.; Ma, D. Measles Virus Forms Inclusion Bodies with Properties of Liquid Organelles. J. Virol. 2019, 93, e00948-19. [Google Scholar] [CrossRef] [PubMed]
- Guseva, S.; Milles, S.; Ringkjøbing Jensen, M.; Salvi, N.; Kleman, J.; Maurin, D.; Ruigrok, R.W.; Blackledge, M. Measles virus nucleo- and phosphoproteins form liquid-like phase-separated compartments that promote nucleocapsid assembly. Sci. Adv. 2020, 6, eaaz7095. [Google Scholar] [CrossRef] [Green Version]
- Guseva, S.; Milles, S.; Jensen, M.R.; Schoehn, G.; Ruigrok, R.W.; Blackledge, M. Structure, dynamics and phase separation of measles virus RNA replication machinery. Curr. Opin. Virol. 2020, 41, 59–67. [Google Scholar] [CrossRef]
- Etibor, T.A.; Yamauchi, Y.; Amorim, M.J. Liquid Biomolecular Condensates and Viral Lifecycles: Review and Perspectives. Viruses 2021, 13, 366. [Google Scholar] [CrossRef]
- Dolnik, O.; Gerresheim, G.; Biedenkopf, N. New Perspectives on the Biogenesis of Viral Inclusion Bodies in Negative-Sense RNA Virus Infections. Cells 2021, 10, 1460. [Google Scholar] [CrossRef]
- Galloux, M.; Risso-Ballester, J.; Richard, C.A.; Fix, J.; Rameix-Welti, M.A.; Eléouët, J.F. Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. mBio 2020, 11, e01202-20. [Google Scholar] [CrossRef]
- Peng, Q.; Wang, L.; Qin, Z.; Wang, J.; Zheng, X.; Wei, L.; Zhang, X.; Zhang, X.; Liu, C.; Li, Z.; et al. Phase Separation of Epstein-Barr Virus EBNA2 and Its Coactivator EBNALP Controls Gene Expression. J. Virol. 2020, 94, e01771-19. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, N.C.; Wang, Y.D.; Scarborough, E.A.; Moore, J.; Diaz, Z.; MacLea, K.S.; Freibaum, B.; Li, S.; Molliex, A.; et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature 2013, 495, 467–473. [Google Scholar] [CrossRef] [PubMed]
- White, J.P.; Lloyd, R.E. Regulation of stress granules in virus systems. Trends Microbiol. 2012, 20, 175–183. [Google Scholar] [CrossRef]
- Tayeb-Fligelman, E.; Cheng, X.; Tai, C.; Bowler, J.T.; Griner, S.; Sawaya, M.R.; Seidler, P.M.; Jiang, Y.X.; Lu, J.; Rosenberg, G.M.; et al. Inhibition of amyloid formation of the Nucleoprotein of SARS-CoV-2. bioRxiv 2021. [Google Scholar] [CrossRef]
- Vidic, J.; Richard, C.A.; Péchoux, C.; Da Costa, B.; Bertho, N.; Mazerat, S.; Delmas, B.; Chevalier, C. Amyloid Assemblies of Influenza A Virus PB1-F2 Protein Damage Membrane and Induce Cytotoxicity. J. Biol. Chem. 2016, 291, 739–751. [Google Scholar] [CrossRef] [Green Version]
- Pham, C.L.; Shanmugam, N.; Strange, M.; O’Carroll, A.; Brown, J.W.; Sierecki, E.; Gambin, Y.; Steain, M.; Sunde, M. Viral M45 and necroptosis-associated proteins form heteromeric amyloid assemblies. EMBO Rep. 2019, 20, e46518. [Google Scholar] [CrossRef]
- Steain, M.; Baker, M.; Pham, C.L.L.; Shanmugam, N.; Gambin, Y.; Sierecki, E.; McSharry, B.P.; Avdic, S.; Slobedman, B.; Sunde, M.; et al. Varicella zoster virus encodes a viral decoy RHIM to inhibit cell death. PLoS Pathog. 2020, 16, e1008473. [Google Scholar] [CrossRef]
- Shanmugam, N.; Baker, M.; Sanz-Hernandez, M.; Sierecki, E.; Gambin, Y.; Steain, M.; Pham, C.L.L.; Sunde, M. Herpes simplex virus encoded ICP6 protein forms functional amyloid assemblies with necroptosis-associated host proteins. Biophys. Chem. 2021, 269, 106524. [Google Scholar] [CrossRef]
- Le May, N.; Dubaele, S.; Proietti De Santis, L.; Billecocq, A.; Bouloy, M.; Egly, J.M. TFIIH transcription factor, a target for the Rift Valley hemorrhagic fever virus. Cell 2004, 116, 541–550. [Google Scholar] [CrossRef] [Green Version]
- Léger, P.; Nachman, E.; Richter, K.; Tamietti, C.; Koch, J.; Burk, R.; Kummer, S.; Xin, Q.; Stanifer, M.; Bouloy, M.; et al. NSs amyloid formation is associated with the virulence of Rift Valley fever virus in mice. Nat. Commun. 2020, 11, 3281. [Google Scholar] [CrossRef]
- Shiell, B.J.; Gardner, D.R.; Crameri, G.; Eaton, B.T.; Michalski, W.P. Sites of phosphorylation of P and V proteins from Hendra and Nipah viruses: Newly emerged members of Paramyxoviridae. Virus Res. 2003, 92, 55–65. [Google Scholar] [CrossRef]
- Darling, A.L.; Uversky, V.N. Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front. Genet. 2018, 9, 158. [Google Scholar] [CrossRef]
- Lieutaud, P.; Canard, B.; Longhi, S. MeDor: A metaserver for predicting protein disorder. BMC Genom. 2008, 9, S25. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Noguere, C.; Larsson, A.M.; Guyot, J.C.; Bignon, C. Fractional factorial approach combining 4 Escherichia coli strains, 3 culture media, 3 expression temperatures and 5 N-terminal fusion tags for screening the soluble expression of recombinant proteins. Protein Expr. Purif. 2012, 84, 204–213. [Google Scholar] [CrossRef]
- Vincentelli, R.; Canaan, S.; Campanacci, V.; Valencia, C.; Maurin, D.; Frassinetti, F.; Scappucini-Calvo, L.; Bourne, Y.; Cambillau, C.; Bignon, C. High-throughput automated refolding screening of inclusion bodies. Protein Sci. 2004, 13, 2782–2792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brocca, S.; Testa, L.; Sobott, F.; Samalikova, M.; Natalello, A.; Papaleo, E.; Lotti, M.; De Gioia, L.; Doglia, S.M.; Alberghina, L.; et al. Compaction properties of an intrinsically disordered protein: Sic1 and its kinase-inhibitor domain. Biophys. J. 2011, 100, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Hamdi, K.; Salladini, E.; O’Brien, D.P.; Brier, S.; Chenal, A.; Yacoubi, I.; Longhi, S. Structural disorder and induced folding within two cereal, ABA stress and ripening (ASR) proteins. Sci. Rep. 2017, 7, 15544. [Google Scholar] [CrossRef] [PubMed]
- Yacoubi, I.; Hamdi, K.; Fourquet, P.; Bignon, C.; Longhi, S. Structural and Functional Characterization of the ABA-Water Deficit Stress Domain from Wheat and Barley: An Intrinsically Disordered Domain behind the Versatile Functions of the Plant Abscissic Acid, Stress and Ripening Protein Family. Int. J. Mol. Sci. 2021, 22, 2314. [Google Scholar] [CrossRef] [PubMed]
- Whitmore, L.; Wallace, B.A. DICHROWEB, an online server for protein secondary structure analyses from circular dichroism spectroscopic data. Nucleic Acids Res. 2004, 32, W668–W673. [Google Scholar] [CrossRef] [Green Version]
- Brookes, E.; Rocco, M. Recent advances in the UltraScan SOlution MOdeller (US-SOMO) hydrodynamic and small-angle scattering data analysis and simulation suite. Eur. Biophys. J. 2018, 47, 855–864. [Google Scholar] [CrossRef]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Guinier, A. La diffraction des rayons X aux tres petits angles; application a l’etude de phenomenes ultramicroscopiques. Ann. Phys. 1939, 12, 161–237. [Google Scholar] [CrossRef]
- Guinier, A.; Fournet, F. Small angle Scattering of X-rays; Wiley Interscience: New York, NY, USA, 1955. [Google Scholar] [CrossRef]
- Svergun, D. Determination of the regularization parameters in indirect-trasform methods using perceptual criteria. J. Appl. Cryst. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Bernado, P.; Blackledge, M. A self-consistent description of the conformational behavior of chemically denatured proteins from NMR and small angle scattering. Biophys. J. 2009, 97, 2839–2845. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, D.K.; Grimshaw, S.B.; Receveur, V.; Dobson, C.M.; Jones, J.A.; Smith, L.J. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry 1999, 38, 16424–16431. [Google Scholar] [CrossRef]
- Tria, G.; Mertens, H.D.T.; Kachala, M.; Svergun, D. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015, 2, 202–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentini, E.; Kikhney, A.G.; Previtali, G.; Jeffries, C.M.; Svergun, D.I. SASBDB, a repository for biological small-angle scattering data. Nucleic Acids Res. 2015, 43, D357–D363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazar, T.; Martínez-Pérez, E.; Quaglia, F.; Hatos, A.; Chemes, L.B.; Iserte, J.A.; Méndez, N.A.; Garrone, N.A.; Saldaño, T.E.; Marchetti, J.; et al. PED in 2021: A major update of the protein ensemble database for intrinsically disordered proteins. Nucleic Acids Res. 2021, 49, D404–D411. [Google Scholar] [CrossRef]
- Longhi, S.; Receveur-Brechot, V.; Karlin, D.; Johansson, K.; Darbon, H.; Bhella, D.; Yeo, R.; Finet, S.; Canard, B. The C-terminal domain of the measles virus nucleoprotein is intrinsically disordered and folds upon binding to the C-terminal moiety of the phosphoprotein. J. Biol. Chem. 2003, 278, 18638–18648. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins | N | pI | f+ | f- | FCR | NCPR | k | Hydropathy | Disorder Promoting | PDR |
|---|---|---|---|---|---|---|---|---|---|---|
| HeV W | 448 | 4.93 | 0.123 | 0.170 | 0.292 | −0.047 | 0.223 | 3.545 | 0.721 | 2 |
| HeV WNTD | 404 | 4.64 | 0.111 | 0.186 | 0.297 | −0.074 | 0.200 | 3.500 | 0.730 | 2 |
| HeV WCTD | 44 | 11.89 | 0.227 | 0.023 | 0.250 | 0.205 | 0.209 | 3.952 | 0.636 | 2 |
| NiV W | 450 | 4.84 | 0.117 | 0.169 | 0.287 | −0.051 | 0.194 | 3.674 | 0.696 | 2 |
| NiV WNTD | 406 | 4.53 | 0.106 | 0.183 | 0.288 | −0.076 | 0.167 | 3.677 | 0.697 | 2 |
| NiV WCTD | 44 | 11.89 | 0.227 | 0.046 | 0.273 | 0.182 | 0.292 | 3.645 | 0.682 | 2 |
| Proteins | Mass | RSobs | RSNF | RSPMG | RSU | RSIDP | RSobs/RSNF | RSobs/RSPMG | RSobs/RSU | RSobs/RSIDP | CI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HeV W | 52,706 | 51.0 ± 0.9 | 30.3 | 46.1 | 64.7 | 57.4 | 1.68 | 1.11 | 0.79 | 0.89 | 0.40 ± 0.03 |
| NiV W | 52,875 | 50.1 ± 0.8 | 30.3 | 46.2 | 64.8 | 57.5 | 1.65 | 1.08 | 0.77 | 0.87 | 0.43 ± 0.03 |
| Proteins | I(0) cm−1 | Rg (Å) Guinier | Rg P(r) (Å) | Rg (ave) EOM (Å) | Dmax (Å) | Rflex (%) (pool) | Rflex (%) (ens) | χ2 | p-Value CorMap | RgIDP (Å) | CI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| HeV W | 0.030 ± 2.4 × 10−4 | 72.01 ± 0.89 | 73.62 ± 0.40 | 71.7 | 240 | 84.7 | 82.5 | 0.52 | 0.530 | 63.5 | 0.089 ± 0.016 |
| NiV W | 0.024 ± 3.1 × 10−4 | 70.96 ± 1.27 | 73.62 ± 0.62 | 70.7 | 245 | 84.5 | 82.0 | 0.44 | 0.014 | 63.6 | 0.111 ± 0.023 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pesce, G.; Gondelaud, F.; Ptchelkine, D.; Nilsson, J.F.; Bignon, C.; Cartalas, J.; Fourquet, P.; Longhi, S. Experimental Evidence of Intrinsic Disorder and Amyloid Formation by the Henipavirus W Proteins. Int. J. Mol. Sci. 2022, 23, 923. https://doi.org/10.3390/ijms23020923
Pesce G, Gondelaud F, Ptchelkine D, Nilsson JF, Bignon C, Cartalas J, Fourquet P, Longhi S. Experimental Evidence of Intrinsic Disorder and Amyloid Formation by the Henipavirus W Proteins. International Journal of Molecular Sciences. 2022; 23(2):923. https://doi.org/10.3390/ijms23020923
Chicago/Turabian StylePesce, Giulia, Frank Gondelaud, Denis Ptchelkine, Juliet F. Nilsson, Christophe Bignon, Jérémy Cartalas, Patrick Fourquet, and Sonia Longhi. 2022. "Experimental Evidence of Intrinsic Disorder and Amyloid Formation by the Henipavirus W Proteins" International Journal of Molecular Sciences 23, no. 2: 923. https://doi.org/10.3390/ijms23020923