
Inhibition of Neuronal Necroptosis Mediated by RIPK1 Provides Neuroprotective Effects on Hypoxia and Ischemia In Vitro and In Vivo

 , ,
, ,
Abstract
:1. Introduction
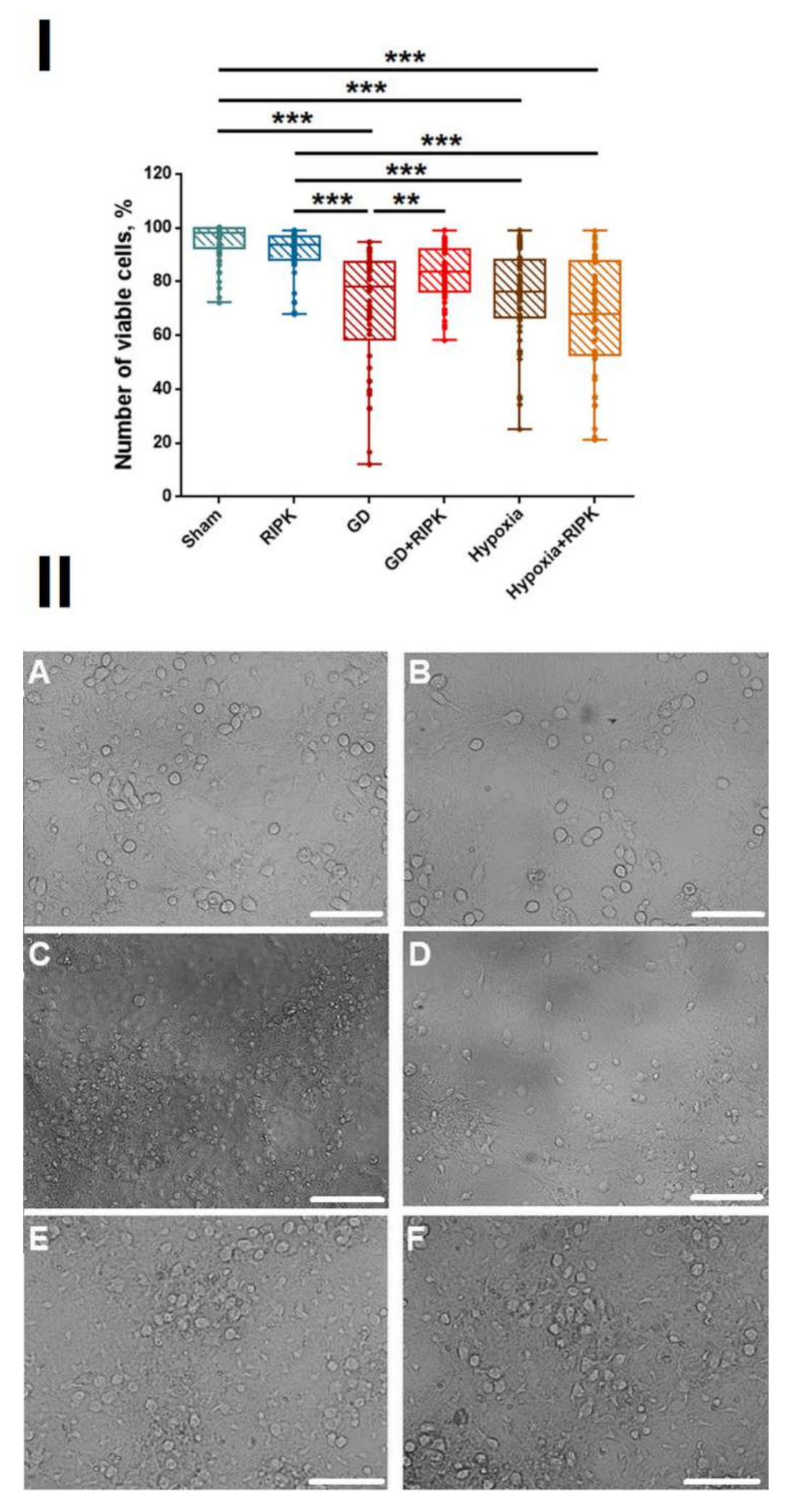
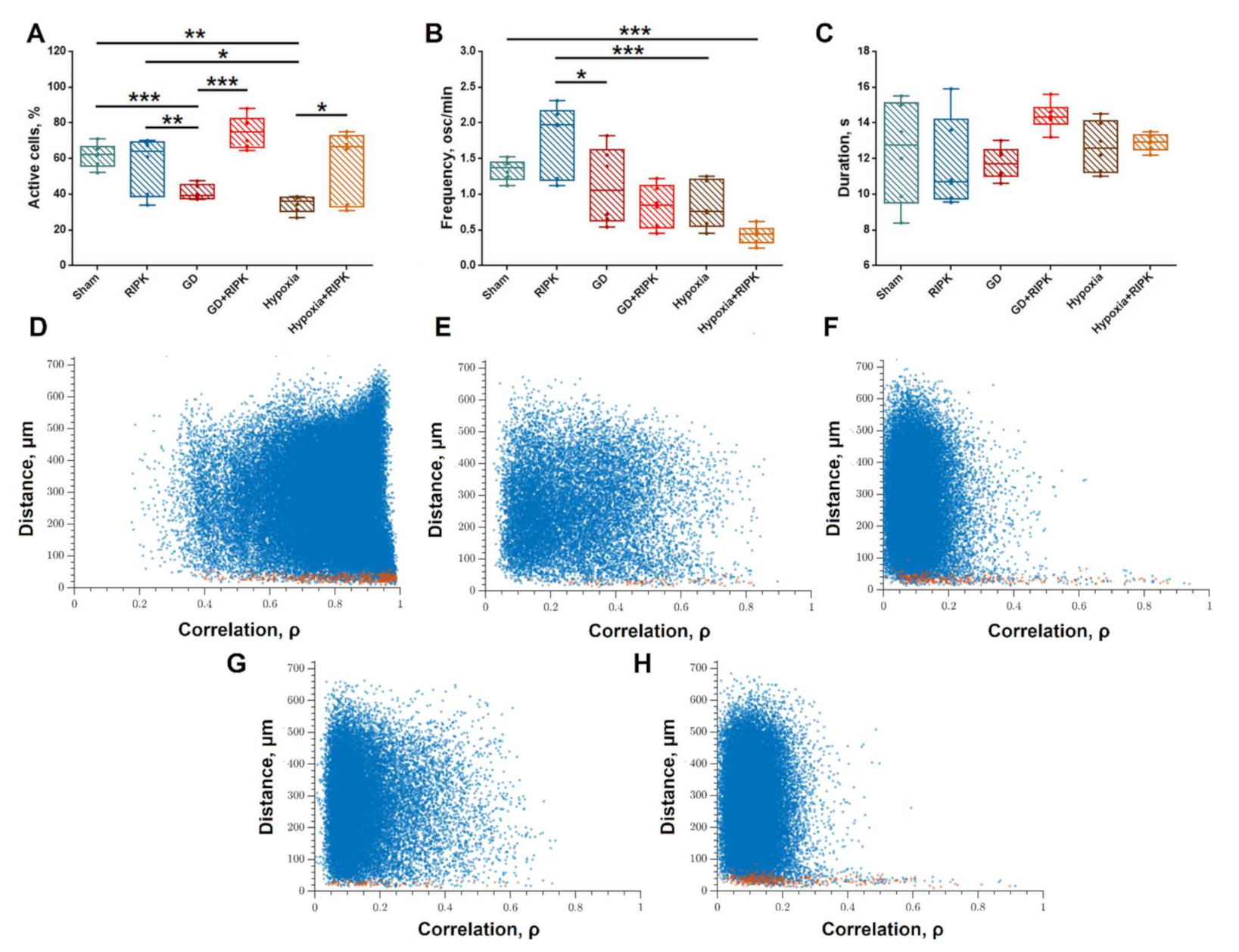
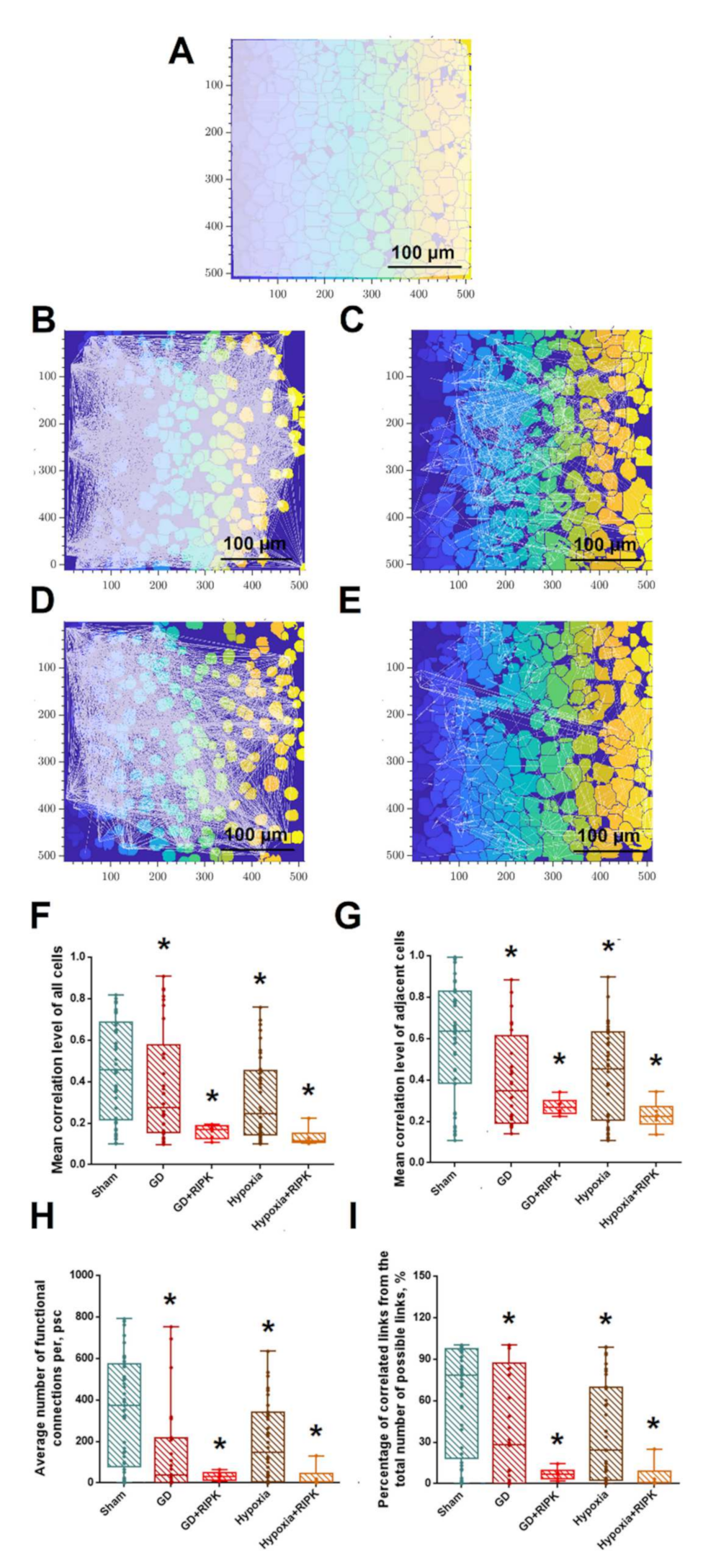
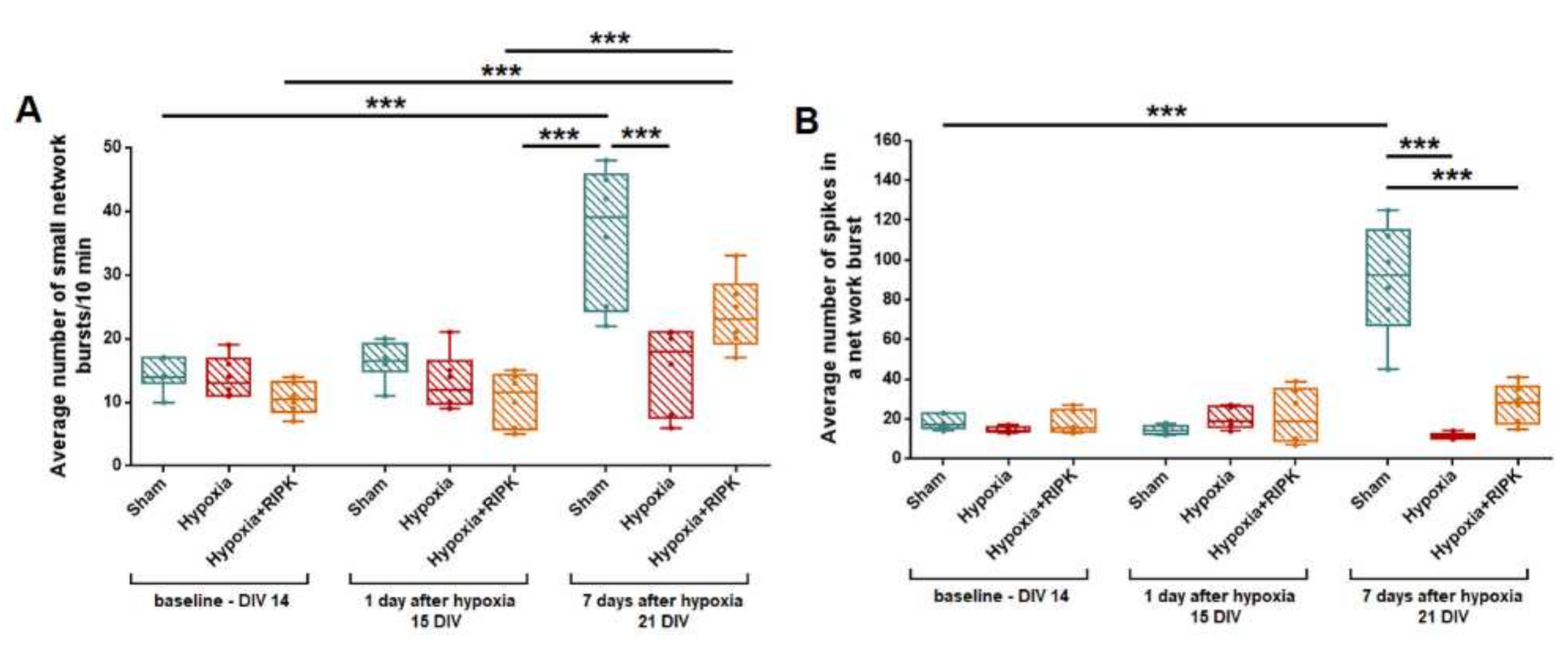
2. Results
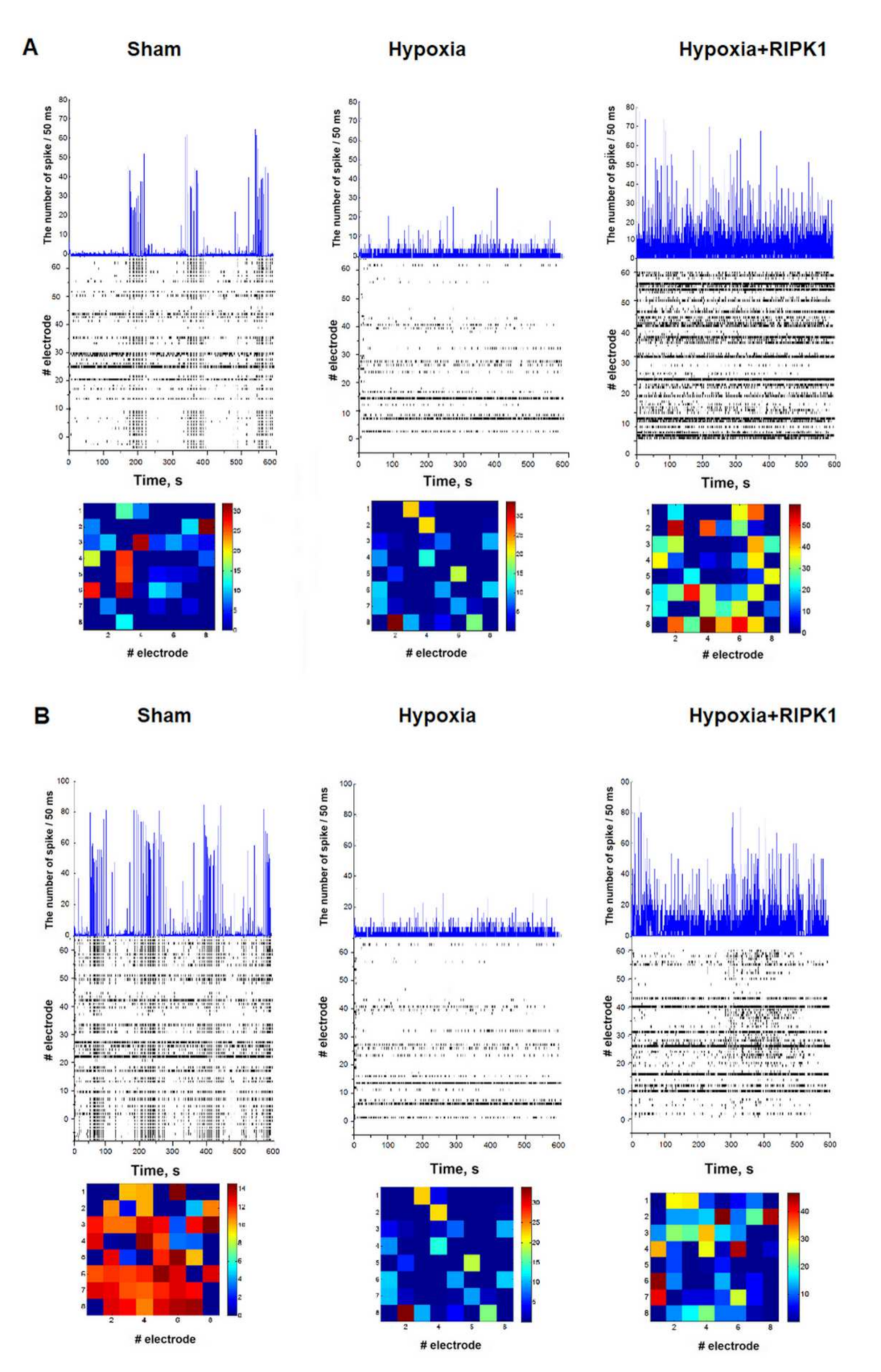
2.1. Studies of the Effect of RIP1 Kinase Blockade on the Functional Activity of Neural Networks in Modeling Ischemic Factors In Vitro
2.2. Effect of RIPK1 Blockade on Resistance of Laboratory Animals to Hypoxic and Ischemic Brain Damage
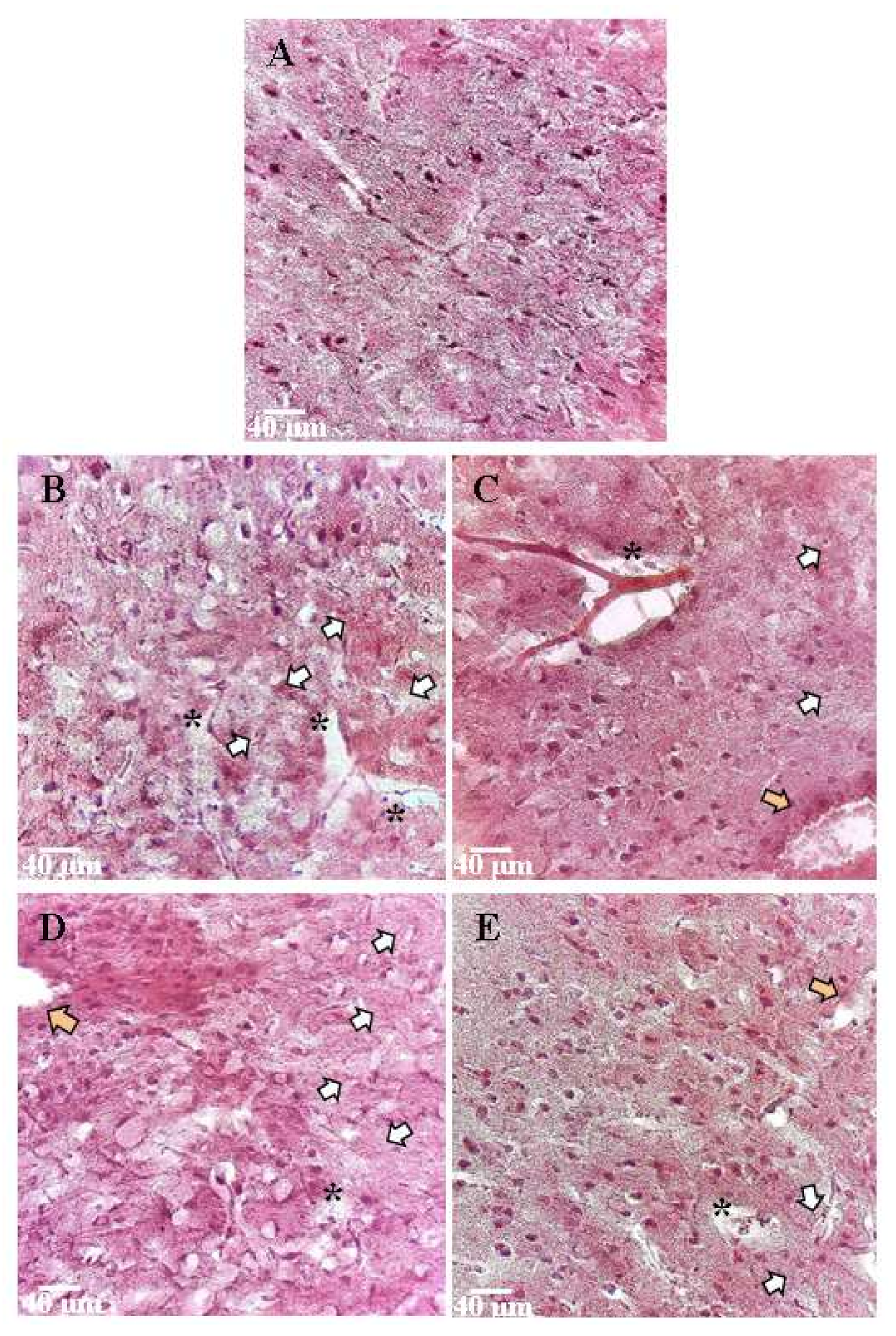
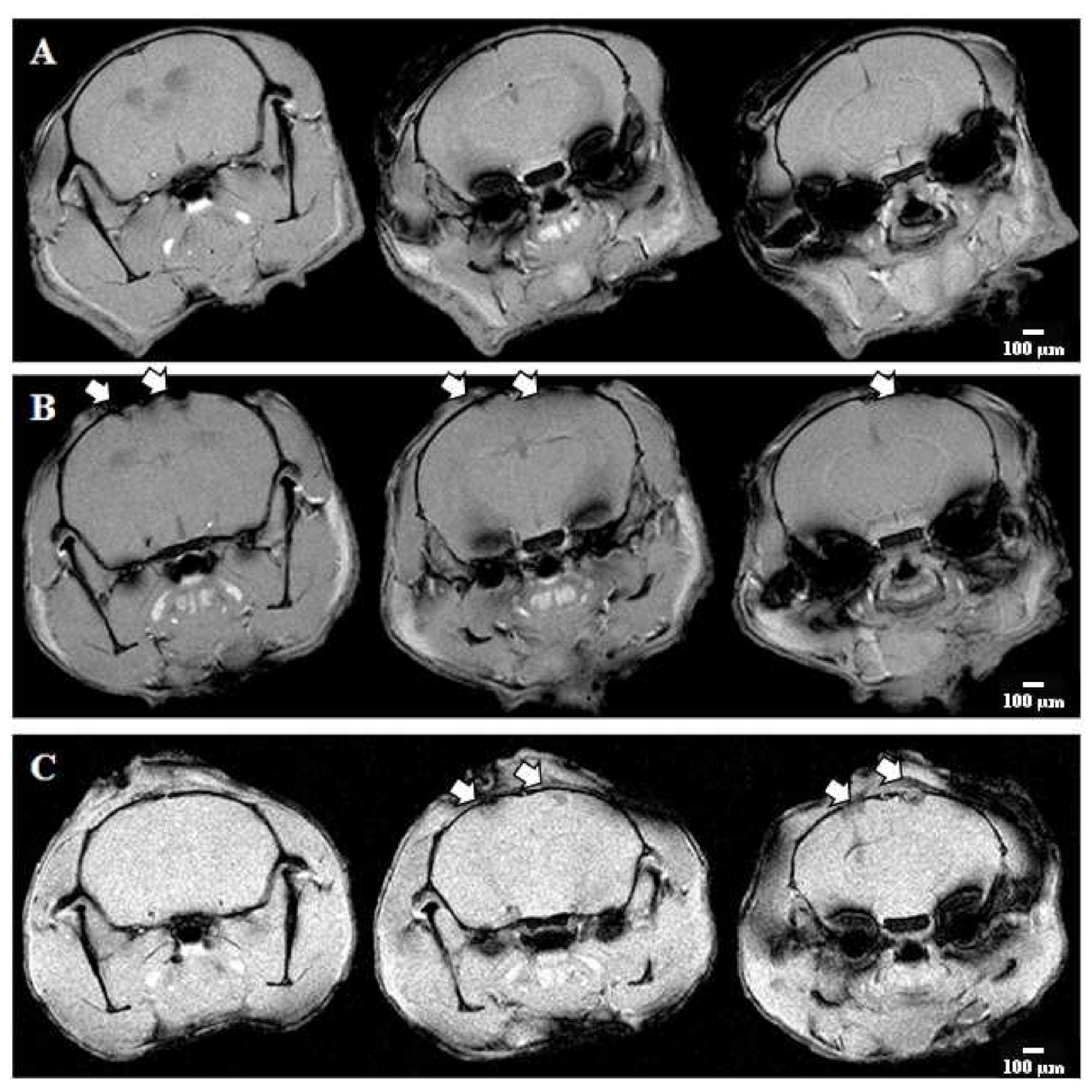
2.3. Histopathological Evaluation
3. Discussion
4. Materials and Methods
4.1. Research Object and Ethics Statement
4.2. Isolation of Murine Primary Hippocampal Cultures
4.3. Modeling of Ischemic Factors In Vitro
4.4. Assessment of Cell-Culture Viability
4.5. Calcium Imaging
- 1.
- The number of functional connections between pairs of cells
- 2.
- Average number of connections per cell
- 3.
- Average speed of signal propagation between cells
- 4.
- Average network correlation
- 5.
- Average level of neighboring cells’ correlation
4.6. Analysis of the Spontaneous Bioelectrical Activity of Primary Neuronal Cultures
4.7. Administration of the Investigated Inhibitors In Vivo
4.8. Modeling of Acute Hypobaric Hypoxia In Vivo
4.9. Modeling of Ischemic Brain Injury In Vivo
4.10. The Open Field Test
4.11. Morris Water-Maze Test
4.12. Histological Examinations
4.13. Magnetic Resonance Imaging
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Radak, D.; Katsiki, N.; Resanovic, I.; Jovanović, A.; Sudar-Milovanovic, E.; Zafirovic, S.; Mousa, S.; Isenovic, E.R. Apoptosis and Acute Brain Ischemia in Ischemic Stroke. Curr. Vasc. Pharmacol. 2017, 15, 115–122. [Google Scholar] [CrossRef]
- Lanzino, G.; Burrows, A.M.; Tymianski, M. Advances in Stroke. Stroke 2013, 44, 316–317. [Google Scholar] [CrossRef] [PubMed]
- Nagy, Z.; Nardai, S. Cerebral ischemia/repefusion injury: From bench space to bedside. Brain Res. Bull. 2017, 134, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Jayachandran, N.; Mejia, E.M.; Sheikholeslami, K.; Sher, A.A.; Hou, S.; Hatch, G.M.; Marshall, A.J. TAPP Adaptors Control B Cell Metabolism by Modulating the Phosphatidylinositol 3-Kinase Signaling Pathway: A Novel Regulatory Circuit Preventing Autoimmunity. J. Immunol. 2018, 201, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Turovsky, E.A.; Turovskaya, M.V.; Gaidin, S.G.; Zinchenko, V.P. Cytokine IL-10, activators of PI3-kinase, agonists of α-2 adrenoreceptor and antioxidants prevent ischemia-induced cell death in rat hippocampal cultures. Arch. Biochem. Biophys. 2017, 615, 35–43. [Google Scholar] [CrossRef]
- Mitroshina, E.V.; Mishchenko, T.A.; Loginova, M.M.; Tarabykin, V.S.; Vedunova, M.V. Identification of Kinome Representatives with Neuroprotective Activity. Neurochem. J. 2020, 14, 394–407. [Google Scholar] [CrossRef]
- Yu, Z.; Jiang, N.; Su, W.; Zhuo, Y. Necroptosis: A Novel Pathway in Neuroinflammation. Front. Pharmacol. 2021, 12, 701564. [Google Scholar] [CrossRef]
- Rinaldi, C.; Donato, L.; Alibrandi, S.; Scimone, C.; D’Angelo, R.; Sidoti, A. Oxidative Stress and the Neurovascular Unit. Life 2021, 11, 767. [Google Scholar] [CrossRef]
- Scimone, C.; Donato, L.; Alibrandi, S.; Vadalà, M.; Giglia, G.; Sidoti, A.; D’Angelo, R. N-retinylidene-N-retinylethanolamine adduct induces expression of chronic inflammation cytokines in retinal pigment epithelium cells. Exp. Eye Res. 2021, 209, 108641. [Google Scholar] [CrossRef]
- Donato, L.; Abdalla, E.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.; D’Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef]
- Deng, X.-X.; Li, S.-S.; Sun, F.-Y. Necrostatin-1 Prevents Necroptosis in Brains after Ischemic Stroke via Inhibition of RIPK1-Mediated RIPK3/MLKL Signaling. Aging Dis. 2019, 10, 807–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehn, A.C.; Khalin, I.; Duering, M.; Hellal, F.; Culmsee, C.; Vandenabeele, P.; Plesnila, N.; Terpolilli, N.A. RIPK1 or RIPK3 deletion prevents progressive neuronal cell death and improves memory function after traumatic brain injury. Acta Neuropathol. Commun. 2021, 9, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Mishchenko, T.A.; Mitroshina, E.V.; Usenko, A.V.; Voronova, N.V.; Astrakhanova, T.A.; Shirokova, O.M.; Kastalskiy, I.A.; Vedunova, M.V. Features of Neural Network Formation and Their Functions in Primary Hippocampal Cultures in the Context of Chronic TrkB Receptor System Influence. Front. Physiol. 2019, 9, 1925. [Google Scholar] [CrossRef] [Green Version]
- Yuste, R. From the neuron doctrine to neural networks. Nat. Rev. Neurosci. 2015, 16, 487–497. [Google Scholar] [CrossRef] [PubMed]
- Shirokova, O.M.; Frumkina, L.E.; Vedunova, M.V.; Mitroshina, E.V.; Zakharov, Y.N.; Khaspekov, L.G.; Mukhina, I.V. Morphofunctional Patterns of Neuronal Network Developing in Dissociated Hippocampal Cell Cultures. Sovrem. Tehnol. Med. 2013, 5, 6–12. [Google Scholar]
- Carrillo-Reid, L.; Yang, W.; Miller, J.-E.K.; Peterka, D.S.; Yuste, R. Imaging and Optically Manipulating Neuronal Ensembles. Annu. Rev. Biophys. 2017, 46, 271–293. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.; Loginova, M.; Savyuk, M.; Krivonosov, M.; Mishchenko, T.; Tarabykin, V.; Ivanchenko, M.; Vedunova, M. Neuroprotective Effect of Kinase Inhibition in Ischemic Factor Modeling In Vitro. Int. J. Mol. Sci. 2021, 22, 1885. [Google Scholar] [CrossRef] [PubMed]
- Mitroshina, E.V.; Krivonosov, M.I.; Burmistrov, D.E.; Savyuk, M.O.; Mishchenko, T.A.; Ivanchenko, M.V.; Vedunova, M.V. Signatures of the Consolidated Response of Astrocytes to Ischemic Factors In Vitro. Int. J. Mol. Sci. 2020, 21, 7952. [Google Scholar] [CrossRef]
- Savyuk, M.; Krivonosov, M.; Mishchenko, T.; Gazaryan, I.; Ivanchenko, M.; Khristichenko, A.; Poloznikov, A.; Hushpulian, D.; Nikulin, S.; Tonevitsky, E.; et al. Neuroprotective Effect of HIF Prolyl Hydroxylase Inhibition in an In Vitro Hypoxia Model. Antioxidants 2020, 9, 662. [Google Scholar] [CrossRef]
- Zhu, J.; Yang, L.-K.; Wang, Q.-H.; Lin, W.; Feng, Y.; Xu, Y.-P.; Chen, W.-L.; Xiong, K.; Wang, Y.-H. NDRG2 attenuates ischemia-induced astrocyte necroptosis via the repression of RIPK1. Mol. Med. Rep. 2020, 22, 3103–3110. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, A.; Htike, T.T.; James, R.; Picon, C.; Reynolds, R. TNF-mediated neuroinflammation is linked to neuronal necroptosis in Alzheimer’s disease hippocampus. Acta Neuropathol. Commun. 2021, 9, 1–21. [Google Scholar] [CrossRef]
- Ofengeim, D.; Yuan, J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 2013, 14, 727–736. [Google Scholar] [CrossRef]
- Naito, M.G.; Xu, D.; Amin, P.; Lee, J.; Wang, H.; Li, W.; Kelliher, M.; Pasparakis, M.; Yuan, J. Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. USA 2020, 117, 4959–4970. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, J.; Chen, J.-R.; Zhu, Y.-M.; Gao, X.; Ni, Y.; Lin, B.; Li, H.; Qiao, S.-G.; Wang, C.; et al. Neuroprotective Effects of DTIO, A Novel Analog of Nec-1, in Acute and Chronic Stages After Ischemic Stroke. Neuroscience 2018, 390, 12–29. [Google Scholar] [CrossRef]
- Arrázola, M.S.; Saquel, C.; Catalán, R.J.; Barrientos, S.A.; Hernandez, D.E.; Martínez, N.W.; Catenaccio, A.; Court, F.A. Axonal Degeneration Is Mediated by Necroptosis Activation. J. Neurosci. 2019, 39, 3832–3844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soeandy, C.D.; Elia, A.J.; Cao, Y.; Rodgers, C.; Huang, S.; Elia, A.C.; Henderson, J.T. Necroptotic–Apoptotic Regulation in an Endothelin-1 Model of Cerebral Ischemia. Cell. Mol. Neurobiol. 2020, 41, 1727–1742. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hu, H.; Wu, B. RIPK1 inhibitor ameliorates the MPP+/MPTP-induced Parkinson’s disease through the ASK1/JNK signalling pathway. Brain Res. 2021, 1757, 147310. [Google Scholar] [CrossRef]
- Vedunova, M.V.; Mishchenko, T.; Mitroshina, E.V.; Mukhina, I.V. TrkB-Mediated Neuroprotective and Antihypoxic Properties of Brain-Derived Neurotrophic Factor. Oxidative Med. Cell. Longev. 2015, 2015, 1–9. [Google Scholar] [CrossRef]
- Jercog, P.; Rogerson, T.; Schnitzer, M.J. Large-Scale Fluorescence Calcium-Imaging Methods for Studies of Long-Term Memory in Behaving Mammals. Cold Spring Harb. Perspect. Biol. 2016, 8, a021824. [Google Scholar] [CrossRef] [Green Version]
- Pimashkin, A.; Kastalskiy, I.; Simonov, A.; Koryagina, E.; Mukhina, I.; Kazantsev, V. Spiking Signatures of Spontaneous Activity Bursts in Hippocampal Cultures. Front. Comput. Neurosci. 2011, 5, 46. [Google Scholar] [CrossRef] [Green Version]
- Gladkov, A.; Grinchuk, O.; Pigareva, Y.; Mukhina, I.; Kazantsev, V.; Pimashkin, A. Theta rhythm-like bidirectional cycling dynamics of living neuronal networks in vitro. PLoS ONE 2018, 13, e0192468. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group/Test Score | Survival Time at Altitude, min | Survival Rate, % | Loss of Posture Time, s | Posture Recovery Time, s | Resistance, % |
|---|---|---|---|---|---|
| AHH (n = 19) | 5.43 ± 0.35 | 21.1% | 86.11 ± 1.89 | 679.67 ± 13.46 | HR—21 MR—74 LR—5 |
| PBS intraventricularly (n = 12) | 3.8 ± 0.71 | 25% | 76.83 ± 2.56 | 674.8 ± 53.13 | HR—8.3 MR—58.3 LR—33.3 |
| RIPK1 inhibitor intraventricularly (n = 9) | 7.65 ± 0.41 *# | 55.6% | 79.4 ± 3.38 | 796.2 ± 64.36 | HR—33.3 MR—66.0 LR—0 |
| Group | Total Distance (mm) | Distance in the Central Zone (mm) | Distance in Peripheral Zone (mm) | Time Spent in the Central Zone (s) | Time Spent in the Peripheral Zone (s) | Number of Upright Postures |
|---|---|---|---|---|---|---|
| Intact (n = 6) | 1606.1 [1456.7; 1611.2] | 73.1 [63.7; 163.8] | 1442.2 [1414.1; 1547.7] | 16.4 [9.2; 16.4] | 283.6 [283.6; 290.8] | 40 [30; 40] |
| AHH (n = 5) | 1636.7 [1399.1; 2167.4] | 247.5 [108.2; 285.7] | 1389.2 [1290.9; 1881.7] | 24.0 [17.6; 39.6] | 276 [260.4; 282.4] | 38 [16; 43] |
| AHH + PBS intraventricularly (n = 5) | 1515.6 [973.25; 2002.5] | 359.1 [160.15; 480.65] | 1355.45 [591.4; 1743.35] | 49.5 [31.6; 152.1] | 251 [147.9; 268.4] | 22 [9; 27] |
| AHH + RIPK1 inhibitor intraventricularly (n = 6) | 1339.4 [1314.3; 1633.4] | 134.8 [128.7; 292.9] | 1269.8 [1179.4; 1504.7] | 24.92 ± 8.782 16.8 [11.6; 35] | 283.2 [265; 288.4] | 25 [11; 26] |
| Ischemia (n = 11) | 1468.3 [296.6; 1558.1] | 250.2[166.8; 250.2] | 1302.4 [161.2; 1375.2] | 24.4 [18; 38] | 275.6 [262; 282] | 20 [5; 22] * F = 3.37; p = 0.02 |
| Ischemia + PBS intraventricularly (n = 6) | 1468.3 [296.6; 1558.1] | 262.1[147.9; 281.2] | 1348.1 [482.7; 1444.2] | 31.41 [24; 33] | 243.3 [202; 271] | 21 [4.5; 25.1] * F = 3.37; p = 0.044 |
| Ischemia+ RIPK1 inhibitor intraventricularly (n = 6) | 930.4 [756.2; 1555.5] | 127. ± 32.73 109.3 [74.4; 166] | 875.5 [674.6; 1445.7] | 43.2 ± 28.09 16.8 [14.8; 21.6] | 283.2 [278.4; 285.2] | 17.6 ± 7.440 10 [9;25] |
| Group/Test Score | Distance Covered before Platform Was Detected (cm) | Total Distance Covered (cm) | Delayed Coefficient of Retention (%) |
|---|---|---|---|
| Intact (n = 6) | 13.21 [9.85; 15.2] | 147.5 [135.94; 149.8] | 42.68 [38.36; 43.14] |
| AHH (n = 5) | 24.2 [13.45; 32.1] | 117.48 [115.6; 124.44] * F = 7.256; p = 0.005 | 35.25 [32;38.36] |
| AHH + RIPK1 inhibitor (n = 6) | 23.4 [12.9; 30.4] | 141.47 [141.36; 150.09] | 42 [41.87; 48] |
| Ischemia (n = 12) | 28.5 [20.1; 34,1] | 129.8 [125.64; 136.7] | 29.14 [24.59; 33.09] * F = 4.66; p = 0.039 |
| Ischemia + RIPK1 inhibitor (n = 6) | 26.8 [16.7; 33.5] | 147.28 [145.46; 151.15] # F = 7.256; p = 0.047 | 32.81 [24.53; 39.82] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitroshina, E.V.; Loginova, M.M.; Yarkov, R.S.; Urazov, M.D.; Novozhilova, M.O.; Krivonosov, M.I.; Ivanchenko, M.V.; Vedunova, M.V. Inhibition of Neuronal Necroptosis Mediated by RIPK1 Provides Neuroprotective Effects on Hypoxia and Ischemia In Vitro and In Vivo. Int. J. Mol. Sci. 2022, 23, 735. https://doi.org/10.3390/ijms23020735
Mitroshina EV, Loginova MM, Yarkov RS, Urazov MD, Novozhilova MO, Krivonosov MI, Ivanchenko MV, Vedunova MV. Inhibition of Neuronal Necroptosis Mediated by RIPK1 Provides Neuroprotective Effects on Hypoxia and Ischemia In Vitro and In Vivo. International Journal of Molecular Sciences. 2022; 23(2):735. https://doi.org/10.3390/ijms23020735
Chicago/Turabian StyleMitroshina, Elena V., Maria M. Loginova, Roman S. Yarkov, Mark D. Urazov, Maria O. Novozhilova, Mikhail I. Krivonosov, Mikhail V. Ivanchenko, and Maria V. Vedunova. 2022. "Inhibition of Neuronal Necroptosis Mediated by RIPK1 Provides Neuroprotective Effects on Hypoxia and Ischemia In Vitro and In Vivo" International Journal of Molecular Sciences 23, no. 2: 735. https://doi.org/10.3390/ijms23020735