
XRCC4 and MRE11 Roles and Transcriptional Response to Repair of TALEN-Induced Double-Strand DNA Breaks

 , , and
, , and 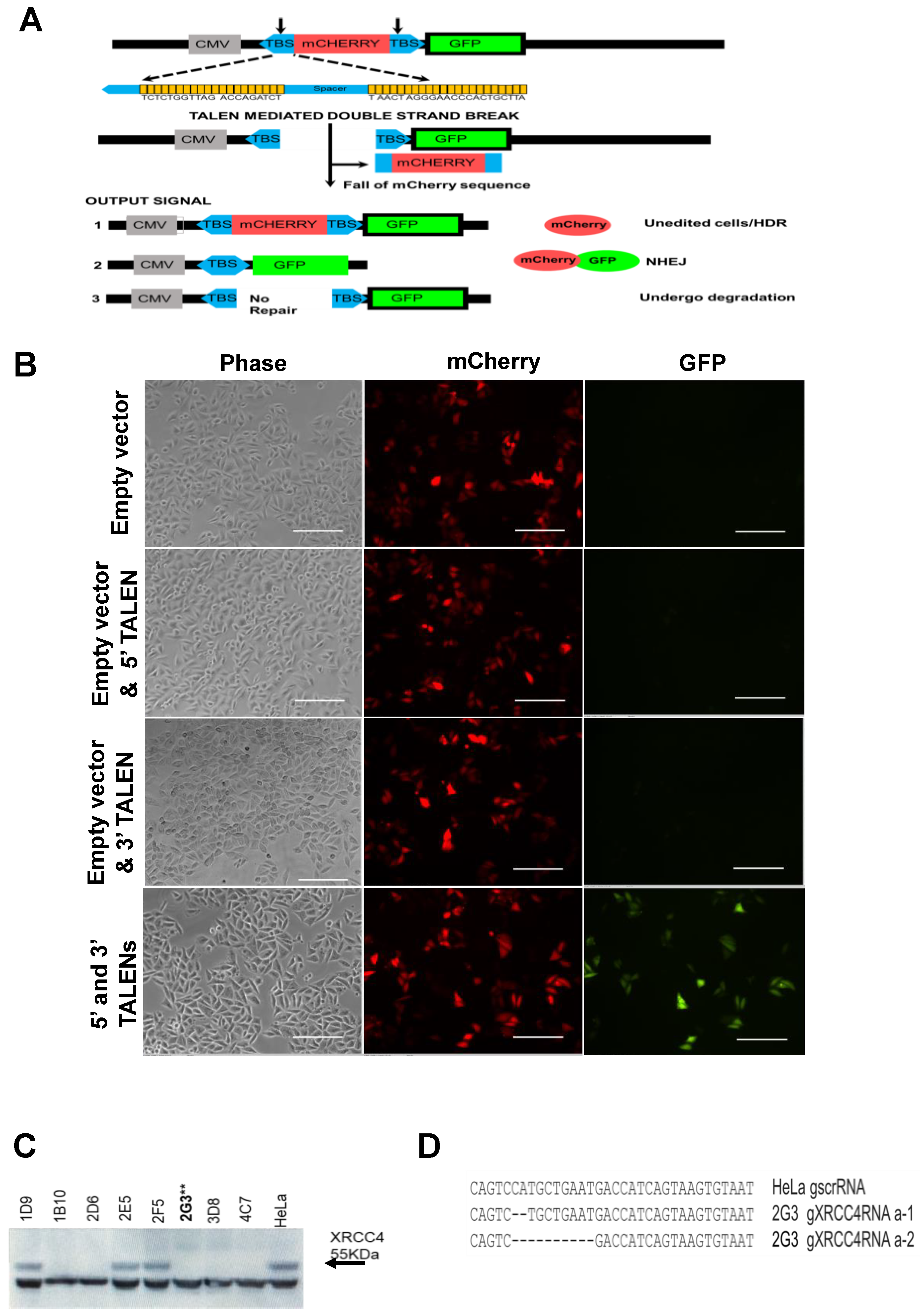{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Construction and Validation of a DNA Repair Reporter Assay
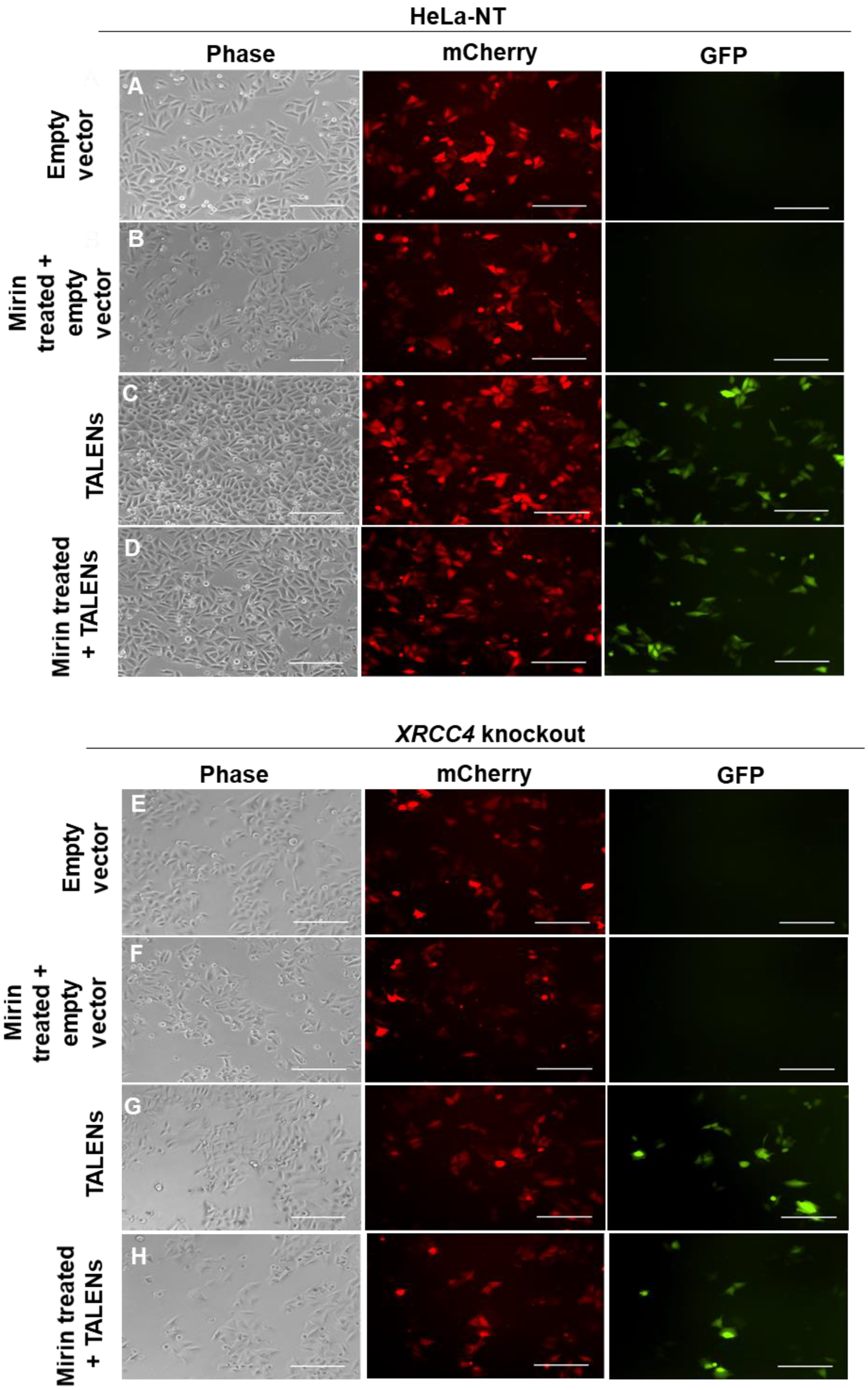
2.2. Inhibition of MRE11 Exonuclease Activity in an XRCC4-Deficient Background
2.3. XRCC4-Deficient Cells Treated with Mirin Still Repair TALEN-Induced Breaks
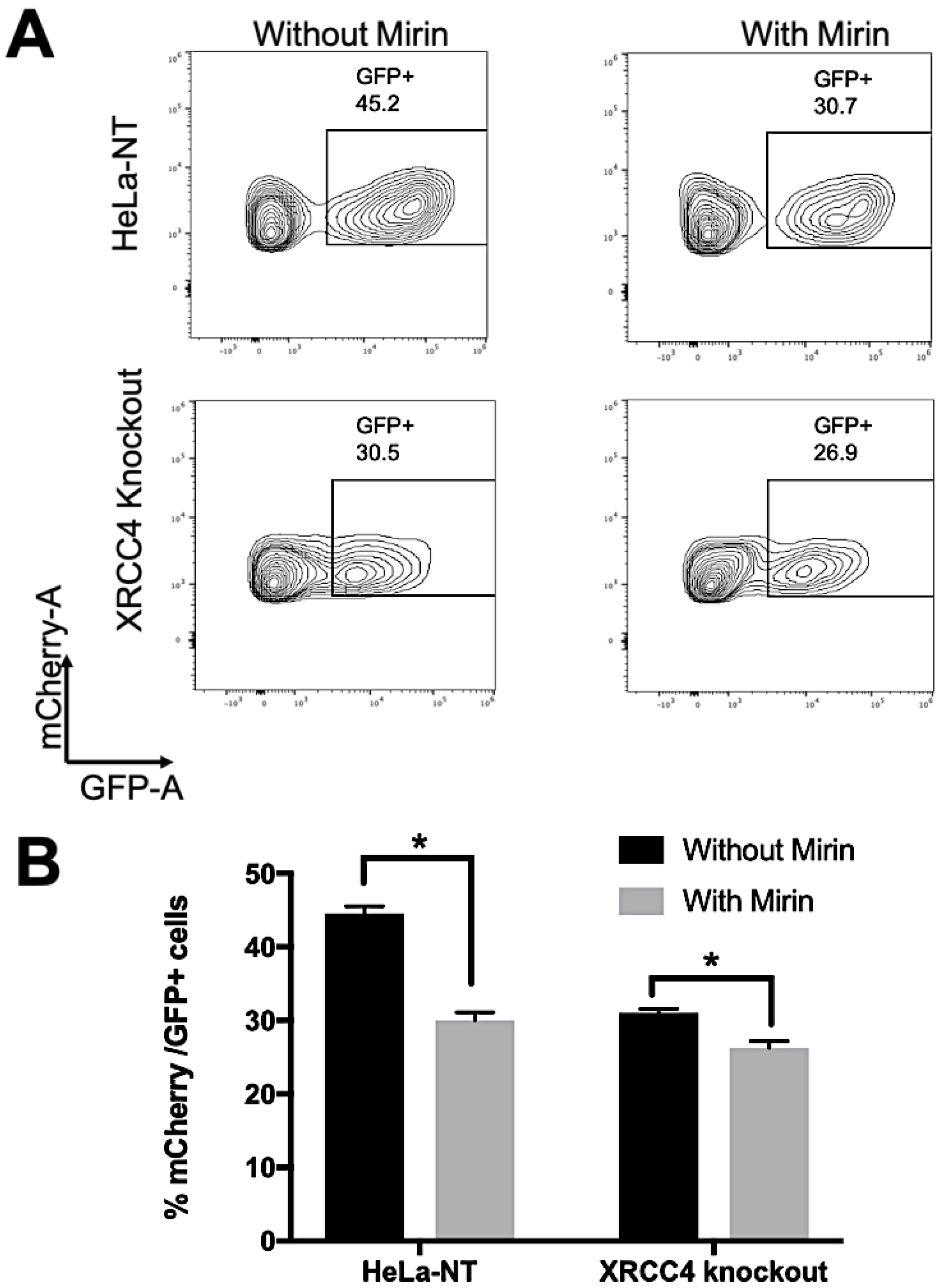
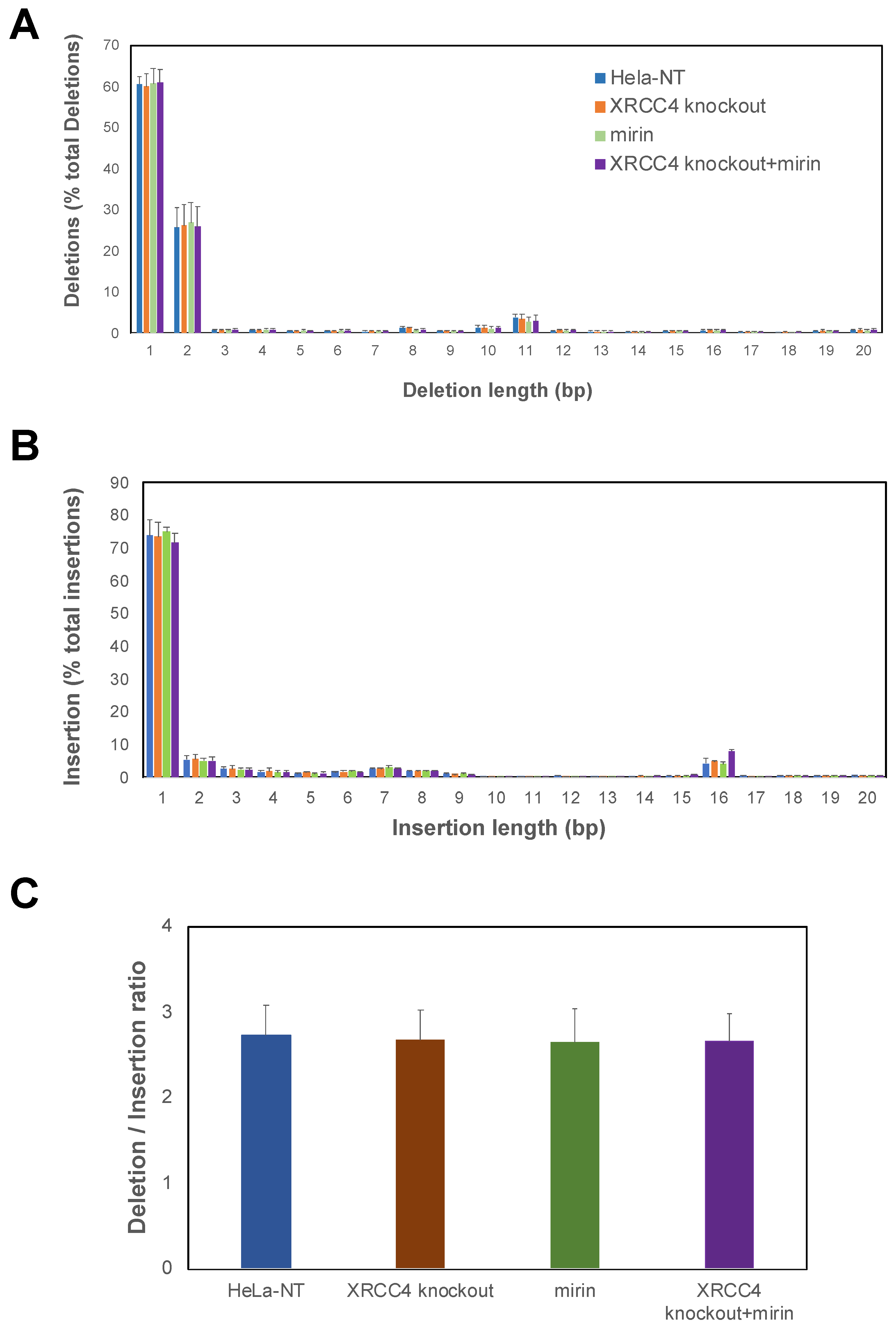
2.4. Repair Efficiency Is Lowered in XRCC4-Deficient Cells Treated with Mirin
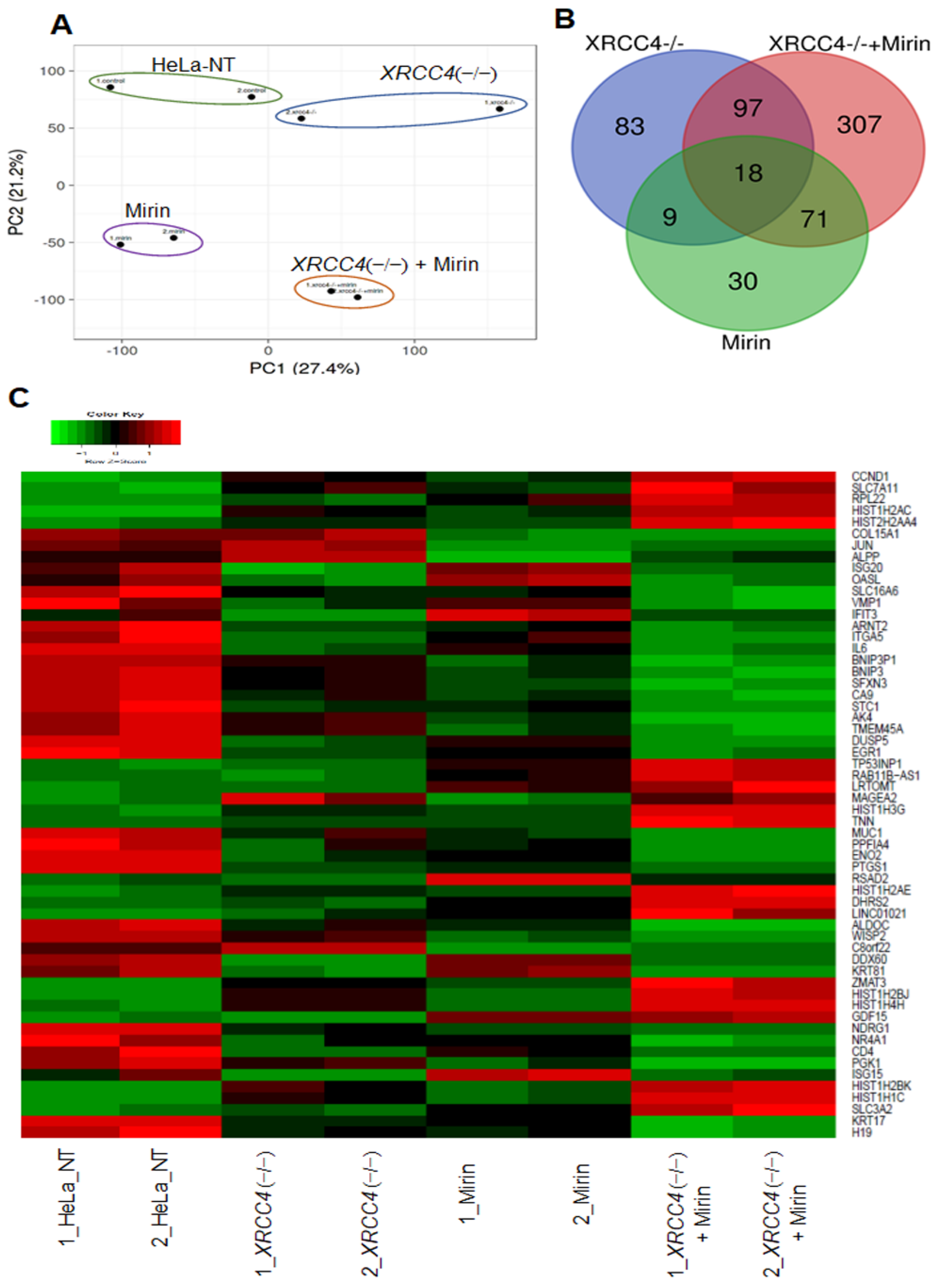
2.5. Altered Expression of Genes When Proteins Critical for Major Repair Pathways Are Blocked
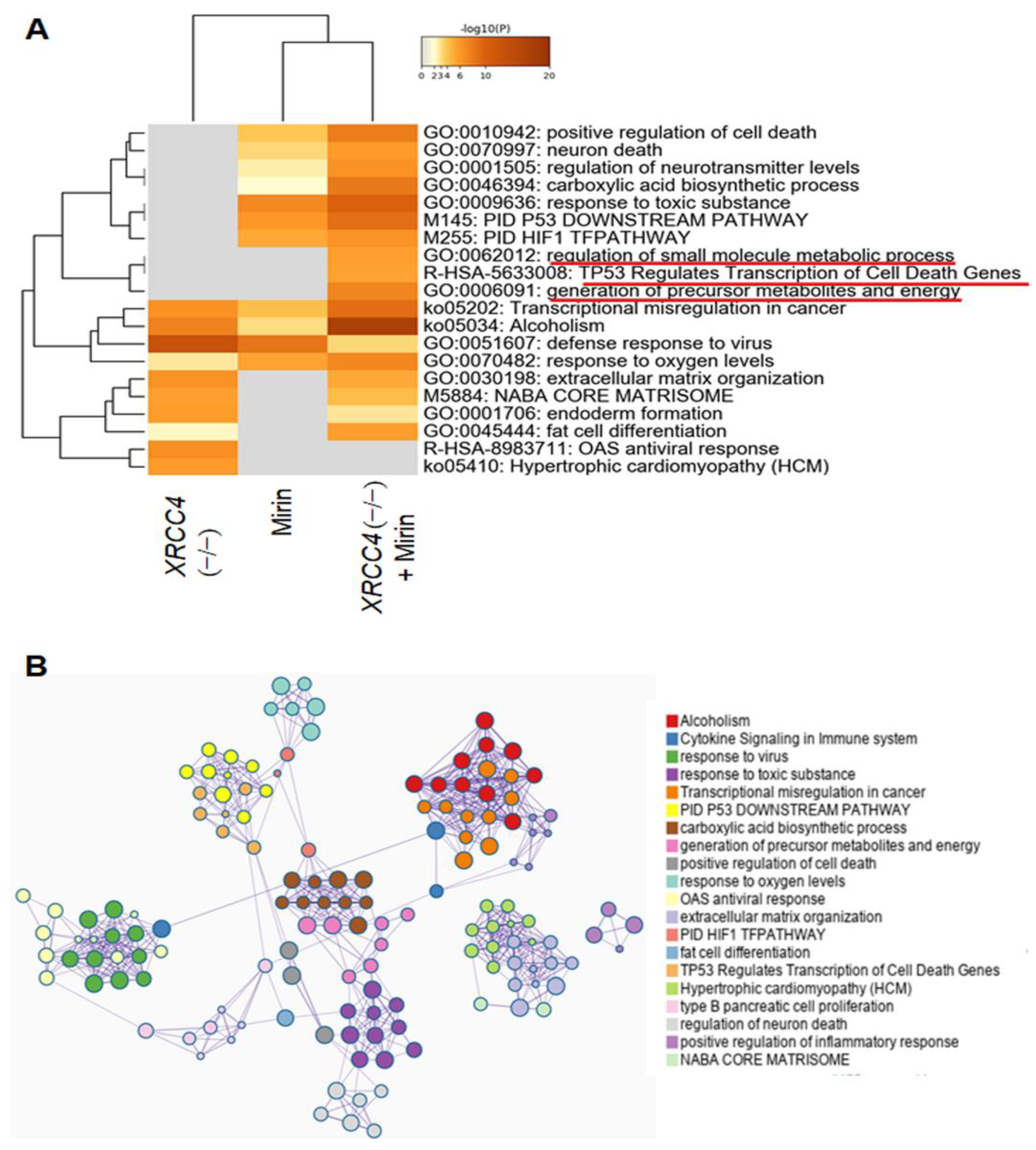
2.6. Pathway Analysis of Differentially Expressed Genes (DEGs)
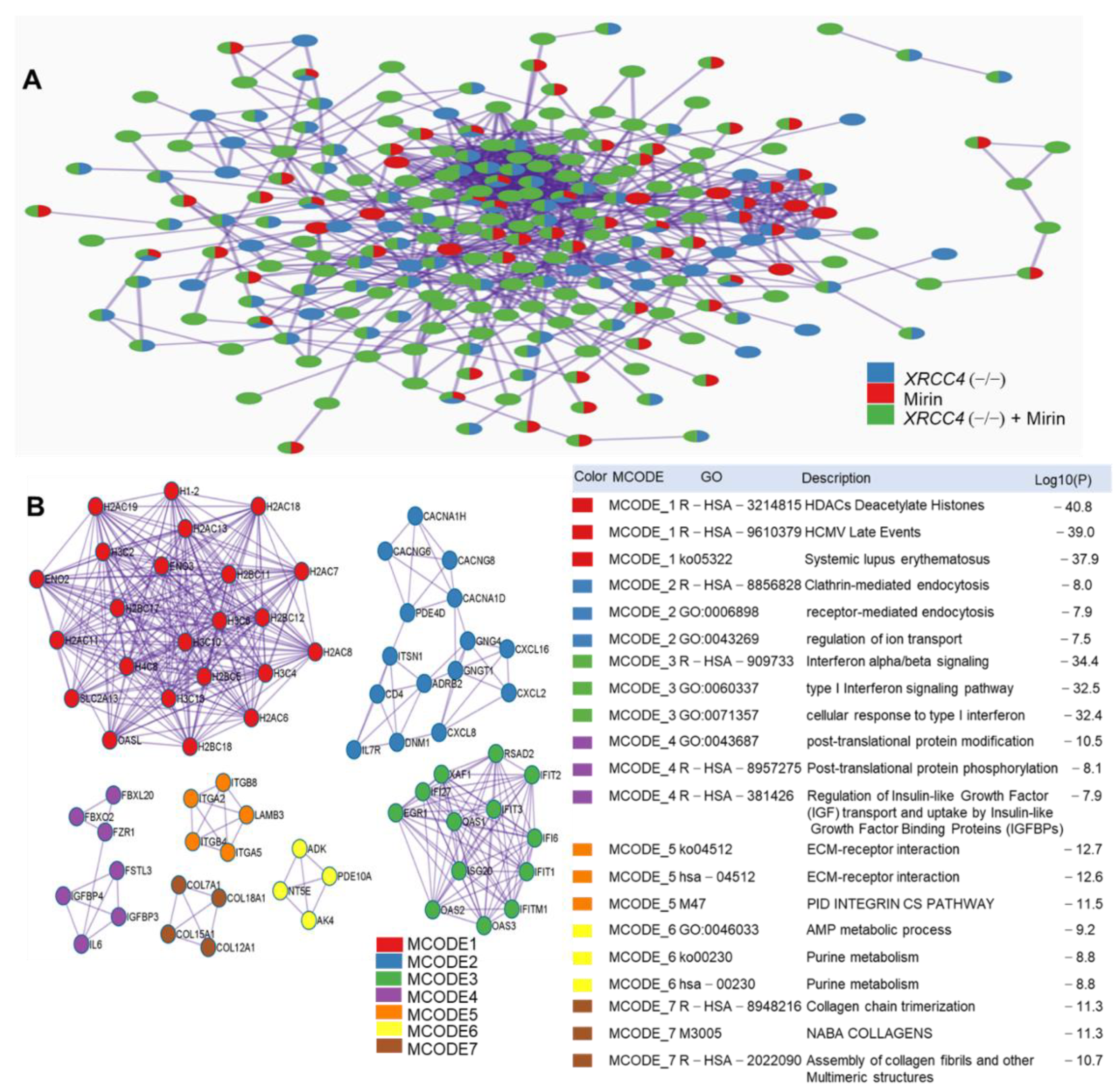
2.7. Protein-Protein Interaction (PPI) Network Construction and Pathways Interaction Analysis
3. Discussion
4. Materials and Methods
4.1. Repair Reporter Plasmid Construction
4.2. Cell Culture and Lentivirus Production
4.3. Assessment of DNA Editing
4.4. Generation of a XRCC4 Knockout Cell Line
4.5. Fluorescence Microscopy
4.6. Fluorescent Activated Cell Sorting (FACS)
4.7. RNA-seq and Data Analysis
4.8. DNA Editing Indel Analysis
4.9. Bioinformatic Analysis
4.10. Protein–Protein Interaction (PPI) Network Analysis and Pathway Interrelation Analysis
4.11. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
4.12. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA Double-Strand Breaks: Production, Fidelity of Repair, and Induction of Cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of Nonhomologous End Joining and Homologous Recombination in Human Cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [Green Version]
- Takata, M.; Sasaki, M.S.; Sonoda, E.; Morrison, C.; Hashimoto, M.; Utsumi, H.; Yamaguchi-Iwai, Y.; Shinohara, A.; Takeda, S. Homologous Recombination and Non-Homologous End-Joining Pathways of DNA Double-Strand Break Repair Have Overlapping Roles in the Maintenance of Chromosomal Integrity in Vertebrate Cells. EMBO J. 1998, 17, 5497–5508. [Google Scholar] [CrossRef]
- Sishc, B.J.; Davis, A.J. The Role of the Core Non-Homologous End Joining Factors in Carcinogenesis and Cancer. Cancers 2017, 9, 81. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.R.; Corpina, R.A.; Goldberg, J. Structure of the Ku Heterodimer Bound to DNA and Its Implications for Double-Strand Break Repair. Nature 2001, 412, 607–614. [Google Scholar] [CrossRef]
- Xu, Y.; Xu, D. Repair Pathway Choice for Double-Strand Breaks. Essays Biochem. 2020, 64, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Bee, L.; Fabris, S.; Cherubini, R.; Mognato, M.; Celotti, L. The Efficiency of Homologous Recombination and Non-Homologous End Joining Systems in Repairing Double-Strand Breaks during Cell Cycle Progression. PLoS ONE 2013, 8, e69061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, K.; McVey, M. Error-Prone Repair of DNA Double-Strand Breaks. J. Cell. Physiol. 2016, 231, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef] [Green Version]
- Grawunder, U.; Wilm, M.; Wu, X.; Kulesza, P.; Wilson, T.E.; Mann, M.; Lieber, M.R. Activity of DNA Ligase IV Stimulated by Complex Formation with XRCC4 Protein in Mammalian Cells. Nature 1997, 388, 492–495. [Google Scholar] [CrossRef]
- Schulte-Uentrop, L.; El-Awady, R.A.; Schliecker, L.; Willers, H.; Dahm-Daphi, J. Distinct Roles of XRCC4 and Ku80 in Non-Homologous End-Joining of Endonuclease- and Ionizing Radiation-Induced DNA Double-Strand Breaks. Nucleic Acids Res. 2008, 36, 2561–2569. [Google Scholar] [CrossRef]
- Giaccia, A.J.; Denko, N.; MacLaren, R.; Mirman, D.; Waldren, C.; Hart, I.; Stamato, T.D. Human Chromosome 5 Complements the DNA Double-Strand Break-Repair Deficiency and Gamma-Ray Sensitivity of the XR-1 Hamster Variant. Am. J. Hum. Genet. 1990, 47, 459–469. [Google Scholar] [PubMed]
- Kabotyanski, E.B.; Gomelsky, L.; Han, J.-O.; Roth, D.B.; Stamato, T.D. Double-Strand Break Repair in Ku86- and XRCC4-Deficient Cells. Nucleic Acids Res. 1998, 26, 5333–5342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deriano, L.; Roth, D.B. Modernizing the Nonhomologous End-Joining Repertoire: Alternative and Classical NHEJ Share the Stage. Annu. Rev. Genet. 2013, 47, 433–455. [Google Scholar] [CrossRef]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative End-Joining Pathway(s): Bricolage at DNA Breaks. DNA Repair 2014, 17, 81–97. [Google Scholar] [CrossRef] [Green Version]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA Double-Strand Breaks by Mammalian Alternative End-Joining Pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [Green Version]
- McVey, M.; Lee, S.E. MMEJ Repair of Double-Strand Breaks (Director’s Cut): Deleted Sequences and Alternative Endings. Trends Genet. TIG 2008, 24, 529–538. [Google Scholar] [CrossRef] [Green Version]
- Ruis, B.; Molan, A.; Takasugi, T.; Hendrickson, E.A. Absence of XRCC4 and Its Paralogs in Human Cells Reveal Differences in Outcomes for DNA Repair and V(D)J Recombination. DNA Repair 2020, 85, 102738. [Google Scholar] [CrossRef]
- Xie, A.; Kwok, A.; Scully, R. Role of Mammalian Mre11 in Classical and Alternative Nonhomologous End Joining. Nat. Struct. Mol. Biol. 2009, 16, 814–818. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.S.; Moncalian, G.; Williams, J.S.; Yamada, Y.; Limbo, O.; Shin, D.S.; Groocock, L.M.; Cahill, D.; Hitomi, C.; Guenther, G.; et al. Mre11 Dimers Coordinate DNA End Bridging and Nuclease Processing in Double-Strand-Break Repair. Cell 2008, 135, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Paull, T.T. The Mre11/Rad50/Xrs2 Complex and Non-Homologous End-Joining of Incompatible Ends in S. Cerevisiae. DNA Repair 2005, 4, 1281–1294. [Google Scholar] [CrossRef] [PubMed]
- Dupré, A.; Boyer-Chatenet, L.; Sattler, R.M.; Modi, A.P.; Lee, J.-H.; Nicolette, M.L.; Kopelovich, L.; Jasin, M.; Baer, R.; Paull, T.T.; et al. A Forward Chemical Genetic Screen Reveals an Inhibitor of the Mre11–Rad50–Nbs1 Complex. Nat. Chem. Biol. 2008, 4, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Vriend, L.E.M.; Jasin, M.; Krawczyk, P.M. Assaying Break and Nick-Induced Homologous Recombination in Mammalian Cells Using the DR-GFP Reporter and Cas9 Nucleases. Methods Enzymol. 2014, 546, 175–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosbach, V.; Poggi, L.; Viterbo, D.; Charpentier, M.; Richard, G.-F. TALEN-Induced Double-Strand Break Repair of CTG Trinucleotide Repeats. Cell Rep. 2018, 22, 2146–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, C.L.; Guerra, H.P.; Mathew, K.R.; Roy, N.; Simpson, L.R.; Schiller, M.R. Damaging the Integrated HIV Proviral DNA with TALENs. PLoS ONE 2015, 10, e0125652. [Google Scholar] [CrossRef] [PubMed]
- Rahal, E.A.; Henricksen, L.A.; Li, Y.; Williams, R.S.; Tainer, J.A. ATM Regulates Mre11-Dependent DNA End-Degradation and Microhomology-Mediated End Joining. Cell Cycle 2010, 9, 2866–2877. [Google Scholar] [CrossRef] [Green Version]
- Edwards, T.G.; Vidmar, T.J.; Koeller, K.; Bashkin, J.K.; Fisher, C. DNA Damage Repair Genes Controlling Human Papillomavirus (HPV) Episome Levels under Conditions of Stability and Extreme Instability. PLoS ONE 2013, 8, e75406. [Google Scholar] [CrossRef] [Green Version]
- Rass, E.; Grabarz, A.; Plo, I.; Gautier, J.; Bertrand, P.; Lopez, B.S. Role of Mre11 in Chromosomal Nonhomologous End Joining in Mammalian Cells. Nat. Struct. Mol. Biol. 2009, 16, 819–824. [Google Scholar] [CrossRef]
- Roques, C.; Coulombe, Y.; Delannoy, M.; Vignard, J.; Grossi, S.; Brodeur, I.; Rodrigue, A.; Gautier, J.; Stasiak, A.Z.; Stasiak, A.; et al. MRE11–RAD50–NBS1 Is a Critical Regulator of FANCD2 Stability and Function during DNA Double-Strand Break Repair. EMBO J. 2009, 28, 2400–2413. [Google Scholar] [CrossRef] [Green Version]
- Garcia, P.A.A.; Lovejoy, C.M.; Nagarajan, P.; Park, D.; Popova, L.V.; Freitas, M.A.; Parthun, M.R. Histone Acetyltransferase 1 Is Required for DNA Replication Fork Function and Stability. J. Biol. Chem. 2020, 295, 8363–8373. [Google Scholar] [CrossRef]
- Christian, M.L.; Demorest, Z.L.; Starker, C.G.; Osborn, M.J.; Nyquist, M.D.; Zhang, Y.; Carlson, D.F.; Bradley, P.; Bogdanove, A.J.; Voytas, D.F. Targeting G with TAL Effectors: A Comparison of Activities of TALENs Constructed with NN and NK Repeat Variable Di-Residues. PLoS ONE 2012, 7, e45383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]
- Miller, K.M.; Tjeertes, J.V.; Coates, J.; Legube, G.; Polo, S.E.; Britton, S.; Jackson, S.P. Human HDAC1 and HDAC2 Function in the DNA-Damage Response to Promote DNA Nonhomologous End-Joining. Nat. Struct. Mol. Biol. 2010, 17, 1144–1151. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa-Yokoi, A.; Cermak, T.; Hoshino, T.; Sugimoto, K.; Saika, H.; Mori, A.; Osakabe, K.; Hamada, M.; Katayose, Y.; Starker, C.; et al. A Defect in DNA Ligase4 Enhances the Frequency of TALEN-Mediated Targeted Mutagenesis in Rice. Plant Physiol. 2016, 170, 653–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular Basis of Microhomology-Mediated End-Joining by Purified Full-Length Polθ. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef] [Green Version]
- Xing, M.; Yang, M.; Huo, W.; Feng, F.; Wei, L.; Jiang, W.; Ning, S.; Yan, Z.; Li, W.; Wang, Q.; et al. Interactome Analysis Identifies a New Paralogue of XRCC4 in Non-Homologous End Joining DNA Repair Pathway. Nat. Commun. 2015, 6, 6233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmann, M.; Kaina, B. Transcriptional Regulation of Human DNA Repair Genes Following Genotoxic Stress: Trigger Mechanisms, Inducible Responses and Genotoxic Adaptation. Nucleic Acids Res. 2013, 41, 8403–8420. [Google Scholar] [CrossRef] [Green Version]
- Daley, J.M.; Sung, P. 53BP1, BRCA1, and the Choice between Recombination and End Joining at DNA Double-Strand Breaks. Mol. Cell. Biol. 2014, 34, 1380–1388. [Google Scholar] [CrossRef] [Green Version]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA Double-Strand Break Repair Pathway Choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Xia, W.; Ci, S.; Li, M.; Wang, M.; Dianov, G.L.; Ma, Z.; Li, L.; Hua, K.; Alagamuthu, K.K.; Qing, L.; et al. Two-Way Crosstalk between BER and c-NHEJ Repair Pathway Is Mediated by Pol-β and Ku70. FASEB J. 2019, 33, 11668–11681. [Google Scholar] [CrossRef] [Green Version]
- Lavin, M.F. ATM and the Mre11 Complex Combine to Recognize and Signal DNA Double-Strand Breaks. Oncogene 2007, 26, 7749–7758. [Google Scholar] [CrossRef] [Green Version]
- Shamanna, R.A.; Lu, H.; de Freitas, J.K.; Tian, J.; Croteau, D.L.; Bohr, V.A. WRN Regulates Pathway Choice between Classical and Alternative Non-Homologous End Joining. Nat. Commun. 2016, 7, 13785. [Google Scholar] [CrossRef] [Green Version]
- Tang, K.-F.; Ren, H. The Role of Dicer in DNA Damage Repair. Int. J. Mol. Sci. 2012, 13, 16769–16778. [Google Scholar] [CrossRef]
- Igoucheva, O.; Alexeev, V.; Yoon, K. Differential Cellular Responses to Exogenous DNA in Mammalian Cells and Its Effect on Oligonucleotide-Directed Gene Modification. Gene Ther. 2006, 13, 266–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hristova, D.B.; Lauer, K.B.; Ferguson, B.J. Viral Interactions with Non-Homologous End-Joining: A Game of Hide-and-Seek. J. Gen. Virol. 2020, 101, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Collins, L.B.; Chen, T.-H.; Herr, N.; Takeda, S.; Sun, W.; Swenberg, J.A.; Nakamura, J. Oxidative Stress at Low Levels Can Induce Clustered DNA Lesions Leading to NHEJ Mediated Mutations. Oncotarget 2016, 7, 25377–25390. [Google Scholar] [CrossRef]
- Huangyang, P.; Simon, M.C. Hidden Features: Exploring the Non-Canonical Functions of Metabolic Enzymes. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatt, A.N.; Chauhan, A.; Khanna, S.; Rai, Y.; Singh, S.; Soni, R.; Kalra, N.; Dwarakanath, B.S. Transient Elevation of Glycolysis Confers Radio-Resistance by Facilitating DNA Repair in Cells. BMC Cancer 2015, 15, 335. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.I.; Najaf-Panah, M.J.; Sena, J.; Schilkey, F.D.; Ashley, A.K. Comparative Gene Expression in Cells Competent in or Lacking DNA-PKcs Kinase Activity Following Etoposide Exposure Reveal Differences in Gene Expression Associated with Histone Modifications, Inflammation, Cell Cycle Regulation, Wnt Signaling, and Differentiation. bioRxiv 2020. [Google Scholar] [CrossRef]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of P53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Lakin, N.D.; Jackson, S.P. Regulation of P53 in Response to DNA Damage. Oncogene 1999, 18, 7644–7655. [Google Scholar] [CrossRef] [Green Version]
- Pluth, J.M.; Fried, L.M.; Kirchgessner, C.U. Severe Combined Immunodeficient Cells Expressing Mutant HRAD54 Exhibit a Marked DNA Double-Strand Break Repair and Error-Prone Chromosome Repair Defect. Cancer Res. 2001, 61, 2649–2655. [Google Scholar] [PubMed]
- Robert, F.; Barbeau, M.; Éthier, S.; Dostie, J.; Pelletier, J. Pharmacological Inhibition of DNA-PK Stimulates Cas9-Mediated Genome Editing. Genome Med. 2015, 7, 93. [Google Scholar] [CrossRef] [Green Version]
- Piekna-Przybylska, D.; Bambara, R.A.; Balakrishnan, L. Acetylation Regulates DNA Repair Mechanisms in Human Cells. Cell Cycle Georget. Tex 2016, 15, 1506–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamburini, B.A.; Tyler, J.K. Localized Histone Acetylation and Deacetylation Triggered by the Homologous Recombination Pathway of Double-Strand DNA Repair. Mol. Cell. Biol. 2005, 25, 4903–4913. [Google Scholar] [CrossRef] [Green Version]
- Jazayeri, A.; McAinsh, A.D.; Jackson, S.P. Saccharomyces Cerevisiae Sin3p Facilitates DNA Double-Strand Break Repair. Proc. Natl. Acad. Sci. USA 2004, 101, 1644–1649. [Google Scholar] [CrossRef] [Green Version]
- Martin, S.G.; Laroche, T.; Suka, N.; Grunstein, M.; Gasser, S.M. Relocalization of Telomeric Ku and SIR Proteins in Response to DNA Strand Breaks in Yeast. Cell 1999, 97, 621–633. [Google Scholar] [CrossRef] [Green Version]
- Mills, K.D.; Sinclair, D.A.; Guarente, L. MEC1-Dependent Redistribution of the Sir3 Silencing Protein from Telomeres to DNA Double-Strand Breaks. Cell 1999, 97, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Munshi, A.; Kurland, J.F.; Nishikawa, T.; Tanaka, T.; Hobbs, M.L.; Tucker, S.L.; Ismail, S.; Stevens, C.; Meyn, R.E. Histone Deacetylase Inhibitors Radiosensitize Human Melanoma Cells by Suppressing DNA Repair Activity. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 4912–4922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.C.; Tan, S.; Qiao, G.; Barlow, K.A.; Wang, J.; Xia, D.F.; Meng, X.; Paschon, D.E.; Leung, E.; Hinkley, S.J.; et al. A TALE Nuclease Architecture for Efficient Genome Editing. Nat. Biotechnol. 2011, 29, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Ewing, B.; Hillier, L.; Wendl, M.C.; Green, P. Base-Calling of Automated Sequencer Traces Using Phred. I. Accuracy Assessment. Genome Res. 1998, 8, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Huber, W.; Liaw, A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; et al. Gplots: Various R Programming Tools for Plotting Data; ScienceOpen: Burlington, MA, USA, 2020. [Google Scholar]
- Blighe, K.; Rana, S.; Turkes, E.; Ostendorf, B.; Grioni, A.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling; Bioconductor version: Release (3.14). 2022. Available online: https://www.bioconductor.org/packages/release/bioc/vignettes/EnhancedVolcano/inst/doc/EnhancedVolcano.html (accessed on 13 January 2021).
- Martin, M. Cutadapt Removes Adapter Sequences from High-Throughput Sequencing Reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:13033997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benjamin, R.; Banerjee, A.; Wu, X.; Geurink, C.; Buczek, L.; Eames, D.; Trimidal, S.G.; Pluth, J.M.; Schiller, M.R. XRCC4 and MRE11 Roles and Transcriptional Response to Repair of TALEN-Induced Double-Strand DNA Breaks. Int. J. Mol. Sci. 2022, 23, 593. https://doi.org/10.3390/ijms23020593
Benjamin R, Banerjee A, Wu X, Geurink C, Buczek L, Eames D, Trimidal SG, Pluth JM, Schiller MR. XRCC4 and MRE11 Roles and Transcriptional Response to Repair of TALEN-Induced Double-Strand DNA Breaks. International Journal of Molecular Sciences. 2022; 23(2):593. https://doi.org/10.3390/ijms23020593
Chicago/Turabian StyleBenjamin, Ronald, Atoshi Banerjee, Xiaogang Wu, Corey Geurink, Lindsay Buczek, Danielle Eames, Sara G. Trimidal, Janice M. Pluth, and Martin R. Schiller. 2022. "XRCC4 and MRE11 Roles and Transcriptional Response to Repair of TALEN-Induced Double-Strand DNA Breaks" International Journal of Molecular Sciences 23, no. 2: 593. https://doi.org/10.3390/ijms23020593