
Expression and Functional Contribution of Different Organic Cation Transporters to the Cellular Uptake of Doxorubicin into Human Breast Cancer and Cardiac Tissue


Abstract
:1. Introduction
2. Results
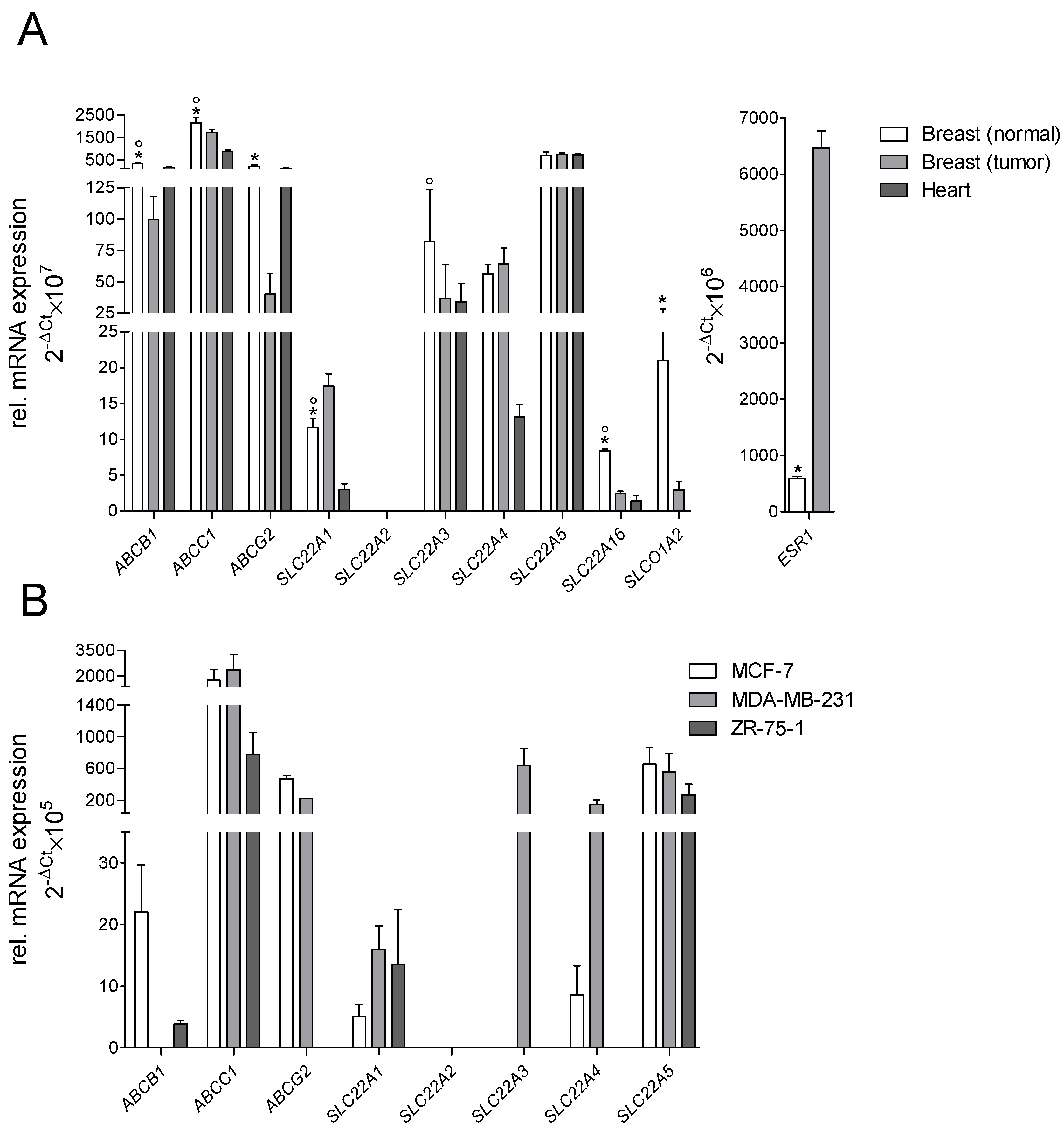
2.1. Gene Expression of Transporters
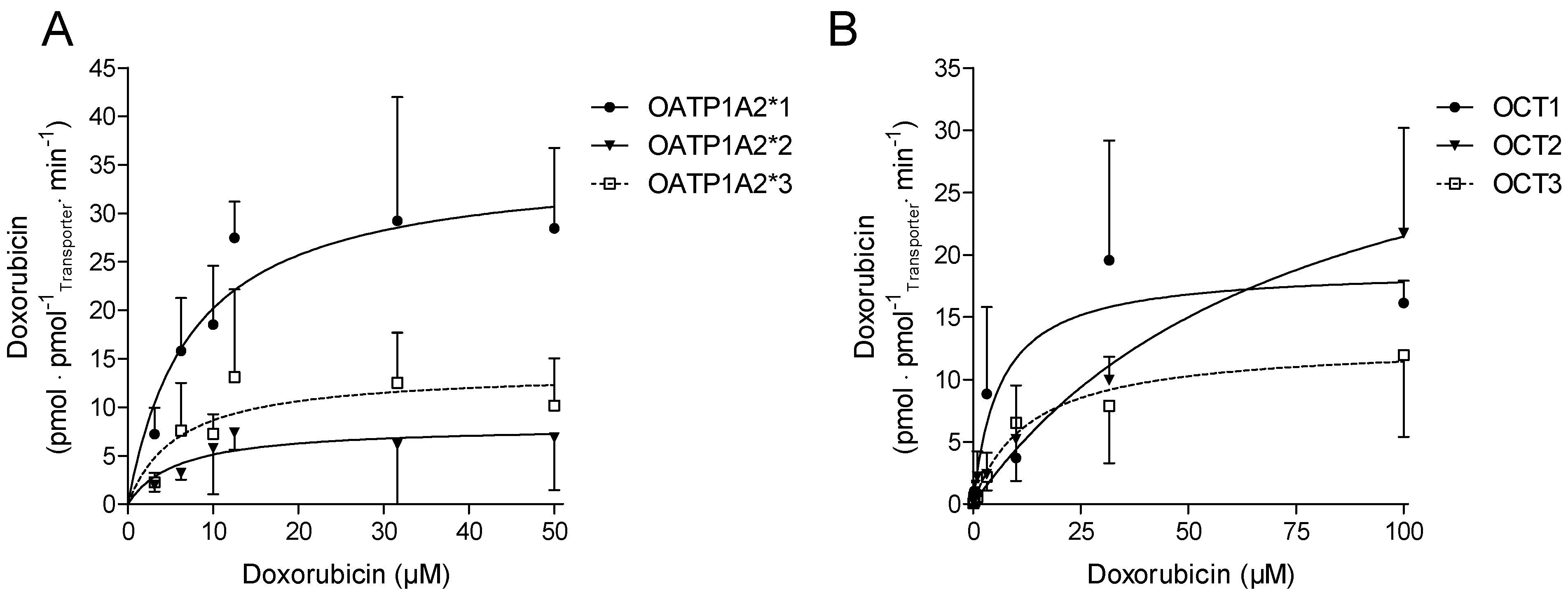
2.2. Cellular Transport of Doxorubicin
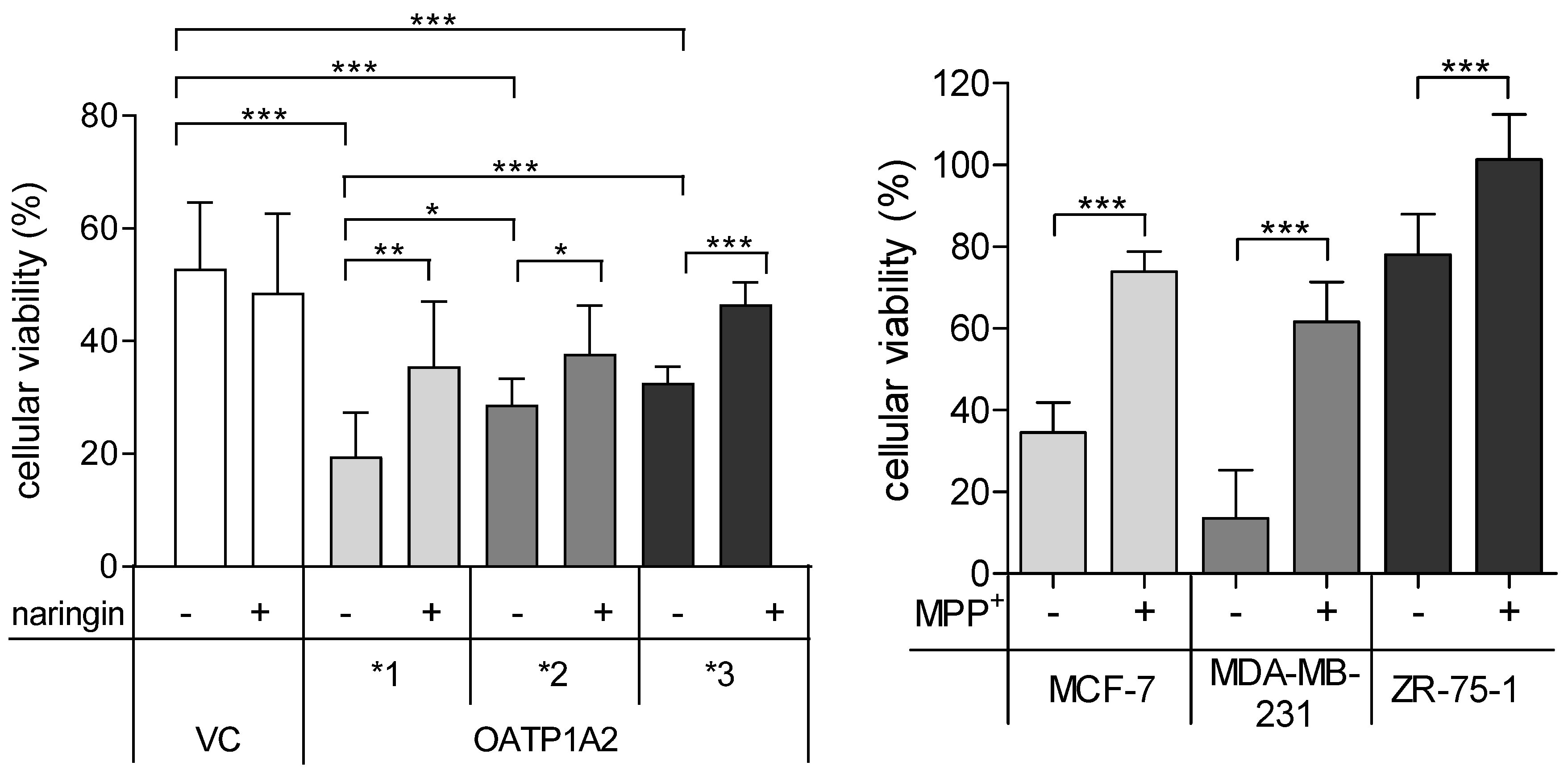
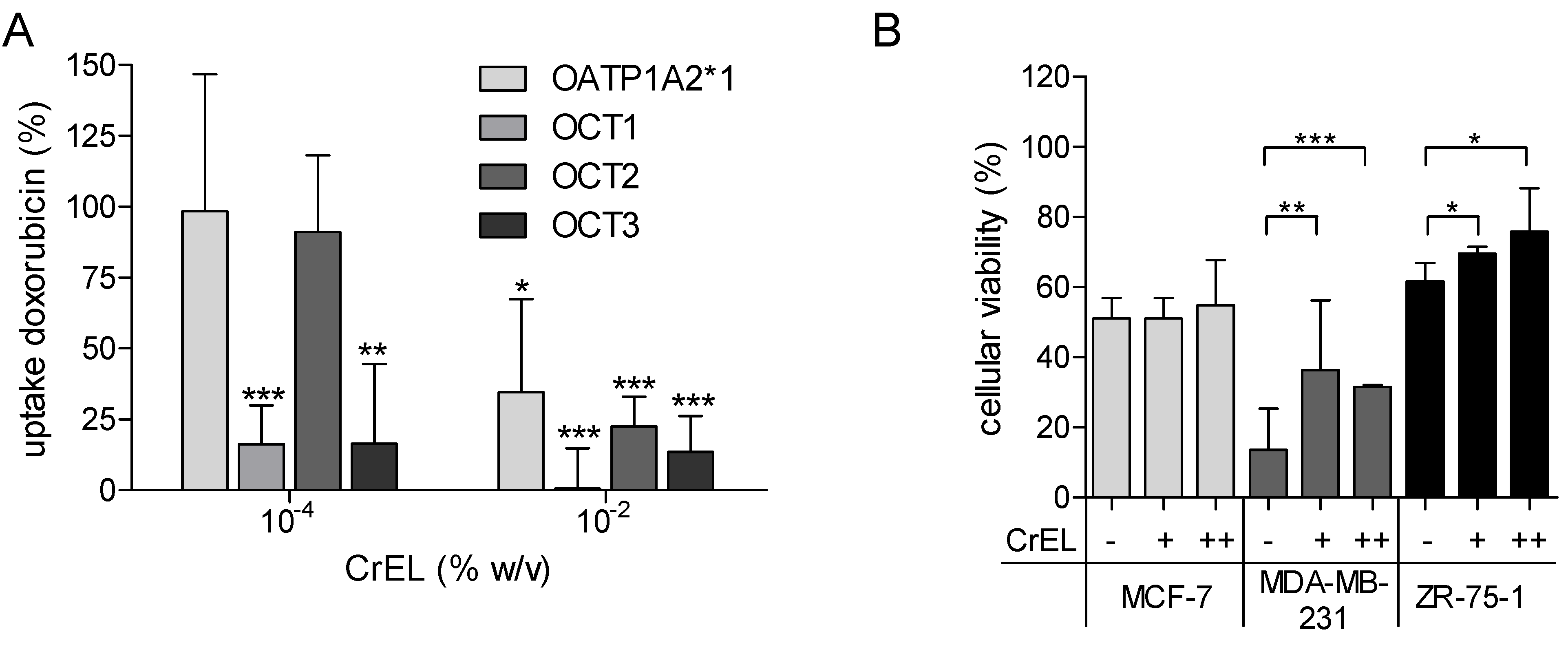
2.3. Cell Viability
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Weiss, R.B. The anthracyclines: Will we ever find a better doxorubicin? Semin. Oncol. 1992, 19, 670–686. [Google Scholar] [PubMed]
- Cortés-Funes, H.; Coronado, C. Role of anthracyclines in the era of targeted therapy. Cardiovasc. Toxicol. 2007, 7, 56–60. [Google Scholar] [CrossRef]
- Carvalho, C.; Santos, R.X.; Cardoso, S.; Correia, S.; Oliveira, P.J.; Santos, M.S.; Moreira, P.I. Doxorubicin: The good, the bad and the ugly effect. CMC 2009, 16, 3267–3285. [Google Scholar] [CrossRef]
- Thorn, C.F.; Oshiro, C.; Marsh, S.; Hernandez-Boussard, T.; McLeod, H.; Klein, T.E.; Altman, R.B. Doxorubicin pathways: Pharmacodynamics and adverse effects. Pharmacogenet. Genom. 2011, 21, 440–446. [Google Scholar] [CrossRef]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [Green Version]
- Renu, K.; Abilash, V.G.; Tirupathi Pichiah, P.B.; Arunachalam, S. Molecular mechanism of doxorubicin-induced cardiomyopathy—An update. Eur. J. Pharmacol. 2018, 818, 241–253. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, X.; Bawa-Khalfe, T.; Lu, L.-S.; Lyu, Y.L.; Liu, L.F.; Yeh, E.T.H. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012, 18, 1639–1642. [Google Scholar] [CrossRef]
- Chatterjee, K.; Zhang, J.; Honbo, N.; Karliner, J.S. Doxorubicin cardiomyopathy. Cardiology 2010, 115, 155–162. [Google Scholar] [CrossRef]
- Swain, S.M.; Whaley, F.S.; Ewer, M.S. Congestive heart failure in patients treated with doxorubicin: A retrospective analysis of three trials. Cancer 2003, 97, 2869–2879. [Google Scholar] [CrossRef] [PubMed]
- Dalmark, M.; Storm, H.H. A Fickian diffusion transport process with features of transport catalysis. Doxorubicin transport in human red blood cells. J. Gen. Physiol. 1981, 78, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.L.; Rastatter, J.C.; Colombo, T.; Long, M.E. Doxorubicin pharmacokinetics: Macromolecule binding, metabolism, and excretion in the context of a physiologic model. J. Pharm. Sci. 2002, 91, 1488–1501. [Google Scholar] [CrossRef]
- Sasaya, M.; Wada, I.; Shida, M.; Sato, M.; Hatakeyama, Y.; Saitoh, H.; Takada, M. Uptake of doxorubicin by cultured kidney epithelial cells LLC-PK1. Biol. Pharm. Bull. 1998, 21, 527–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.D.; Brinkhuis, R.F.; Wijnholds, J.; Schinkel, A.H. The mouse Bcrp1/Mxr/Abcp gene: Amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999, 59, 4237–4241. [Google Scholar] [PubMed]
- van Asperen, J.; van Tellingen, O.; Beijnen, J.H. The role of mdr1a P-glycoprotein in the biliary and intestinal secretion of doxorubicin and vinblastine in mice. Drug Metab. Dispos. 2000, 28, 264–267. [Google Scholar]
- van Asperen, J.; van Tellingen, O.; Tijssen, F.; Schinkel, A.H.; Beijnen, J.H. Increased accumulation of doxorubicin and doxorubicinol in cardiac tissue of mice lacking mdr1a P-glycoprotein. Br. J. Cancer 1999, 79, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.M.; Hu, S.; Sparreboom, A. Drug transporters and anthracycline-induced cardiotoxicity. Pharmacogenomics 2018, 19, 883–888. [Google Scholar] [CrossRef] [PubMed]
- Vlaming, M.L.H.; Mohrmann, K.; Wagenaar, E.; de Waart, D.R.; Elferink, R.P.J.O.; Lagas, J.S.; van Tellingen, O.; Vainchtein, L.D.; Rosing, H.; Beijnen, J.H.; et al. Carcinogen and anticancer drug transport by Mrp2 in vivo: Studies using Mrp2 (Abcc2) knockout mice. J. Pharmacol. Exp. Ther. 2006, 318, 319–327. [Google Scholar] [CrossRef] [Green Version]
- Tada, Y.; Wada, M.; Migita, T.; Nagayama, J.; Hinoshita, E.; Mochida, Y.; Maehara, Y.; Tsuneyoshi, M.; Kuwano, M.; Naito, S. Increased expression of multidrug resistance-associated proteins in bladder cancer during clinical course and drug resistance to doxorubicin. Int. J. Cancer 2002, 98, 630–635. [Google Scholar] [CrossRef]
- Mechetner, E.; Kyshtoobayeva, A.; Zonis, S.; Kim, H.; Stroup, R.; Garcia, R.; Parker, R.J.; Fruehauf, J.P. Levels of multidrug resistance (MDR1) P-glycoprotein expression by human breast cancer correlate with in vitro resistance to taxol and doxorubicin. Clin. Cancer Res. 1998, 4, 389–398. [Google Scholar]
- Gillet, J.-P.; Gottesman, M.M. Mechanisms of multidrug resistance in cancer. Methods Mol. Biol. 2010, 596, 47–76. [Google Scholar] [CrossRef]
- Durmus, S.; Naik, J.; Buil, L.; Wagenaar, E.; van Tellingen, O.; Schinkel, A.H. In vivo disposition of doxorubicin is affected by mouse Oatp1a/1b and human OATP1A/1B transporters. Int. J. Cancer 2014, 135, 1700–1710. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Leake, B.F.; Kim, R.B.; Ho, R.H. Contribution of Organic Anion-Transporting Polypeptides 1A/1B to Doxorubicin Uptake and Clearance. Mol. Pharmacol. 2017, 91, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.M.; Zavorka Thomas, M.; Magdy, T.; Eisenmann, E.D.; Uddin, M.E.; DiGiacomo, D.F.; Pan, A.; Keiser, M.; Otter, M.; Xia, S.H.; et al. Targeting OCT3 attenuates doxorubicin-induced cardiac injury. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Okabe, M.; Unno, M.; Harigae, H.; Kaku, M.; Okitsu, Y.; Sasaki, T.; Mizoi, T.; Shiiba, K.; Takanaga, H.; Terasaki, T.; et al. Characterization of the organic cation transporter SLC22A16: A doxorubicin importer. Biochem. Biophys. Res. Commun. 2005, 333, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.J.; Asmann, Y.W.; Maurer, M.J.; Wang, C.; Slager, S.L.; Hodge, L.S.; Manske, M.; Price-Troska, T.; Yang, Z.-Z.; Zimmermann, M.T.; et al. Whole-exome analysis reveals novel somatic genomic alterations associated with outcome in immunochemotherapy-treated diffuse large B-cell lymphoma. Blood Cancer J. 2015, 5, e346. [Google Scholar] [CrossRef]
- Okabe, M.; Szakács, G.; Reimers, M.A.; Suzuki, T.; Hall, M.D.; Abe, T.; Weinstein, J.N.; Gottesman, M.M. Profiling SLCO and SLC22 genes in the NCI-60 cancer cell lines to identify drug uptake transporters. Mol. Cancer Ther. 2008, 7, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Lal, S.; Wong, Z.W.; Jada, S.R.; Xiang, X.; Chen Shu, X.; Ang, P.C.S.; Figg, W.D.; Lee, E.J.; Chowbay, B. Novel SLC22A16 polymorphisms and influence on doxorubicin pharmacokinetics in Asian breast cancer patients. Pharmacogenomics 2007, 8, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Rudek, M.A.; Sparreboom, A.; Garrett-Mayer, E.S.; Armstrong, D.K.; Wolff, A.C.; Verweij, J.; Baker, S.D. Factors affecting pharmacokinetic variability following doxorubicin and docetaxel-based therapy. Eur. J. Cancer 2004, 40, 1170–1178. [Google Scholar] [CrossRef]
- Kolitz, J.E.; George, S.L.; Marcucci, G.; Vij, R.; Powell, B.L.; Allen, S.L.; DeAngelo, D.J.; Shea, T.C.; Stock, W.; Baer, M.R.; et al. P-glycoprotein inhibition using valspodar (PSC-833) does not improve outcomes for patients younger than age 60 years with newly diagnosed acute myeloid leukemia: Cancer and Leukemia Group B study 19808. Blood 2010, 116, 1413–1421. [Google Scholar] [CrossRef]
- Friedenberg, W.R.; Rue, M.; Blood, E.A.; Dalton, W.S.; Shustik, C.; Larson, R.A.; Sonneveld, P.; Greipp, P.R. Phase III study of PSC-833 (valspodar) in combination with vincristine, doxorubicin, and dexamethasone (valspodar/VAD) versus VAD alone in patients with recurring or refractory multiple myeloma (E1A95): A trial of the Eastern Cooperative Oncology Group. Cancer 2006, 106, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Brosseau, N.; Andreev, E.; Ramotar, D. Complementation of the Yeast Model System Reveals that Caenorhabditis elegans OCT-1 Is a Functional Transporter of Anthracyclines. PLoS ONE 2015, 10, e0133182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budde, T.; Haney, J.; Bien, S.; Schwebe, M.; Riad, A.; Tschöpe, C.; Staudt, A.; Jedlitschky, G.; Felix, S.B.; Kroemer, H.K.; et al. Acute exposure to doxorubicin results in increased cardiac P-glycoprotein expression. J. Pharm. Sci. 2011, 100, 3951–3958. [Google Scholar] [CrossRef]
- Solbach, T.F.; Paulus, B.; Weyand, M.; Eschenhagen, T.; Zolk, O.; Fromm, M.F. ATP-binding cassette transporters in human heart failure. Naunyn Schmiedebergs Arch. Pharmacol. 2008, 377, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Grube, M.; Ameling, S.; Noutsias, M.; Köck, K.; Triebel, I.; Bonitz, K.; Meissner, K.; Jedlitschky, G.; Herda, L.R.; Reinthaler, M.; et al. Selective regulation of cardiac organic cation transporter novel type 2 (OCTN2) in dilated cardiomyopathy. Am. J. Pathol. 2011, 178, 2547–2559. [Google Scholar] [CrossRef] [Green Version]
- McBride, B.F.; Yang, T.; Liu, K.; Urban, T.J.; Giacomini, K.M.; Kim, R.B.; Roden, D.M. The organic cation transporter, OCTN1, expressed in the human heart, potentiates antagonism of the HERG potassium channel. J. Cardiovasc. Pharmacol. 2009, 54, 63–71. [Google Scholar] [CrossRef] [Green Version]
- Doyle, L.A.; Ross, D.D. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2). Oncogene 2003, 22, 7340–7358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, H.; Zhang, Y.; Han, T.K.; Everett, R.S.; Thakker, D.R. Cation-selective transporters are critical to the AMPK-mediated antiproliferative effects of metformin in human breast cancer cells. Int. J. Cancer 2016, 138, 2281–2292. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Uray, I.P.; Mazumdar, A.; Mayer, J.A.; Brown, P.H. SLC22A5/OCTN2 expression in breast cancer is induced by estrogen via a novel intronic estrogen-response element (ERE). Breast Cancer Res. Treat. 2012, 134, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Meyer zu Schwabedissen, H.E.; Tirona, R.G.; Yip, C.S.; Ho, R.H.; Kim, R.B. Interplay between the nuclear receptor pregnane X receptor and the uptake transporter organic anion transporter polypeptide 1A2 selectively enhances estrogen effects in breast cancer. Cancer Res. 2008, 68, 9338–9347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Obaidat, A.; Roth, M.; Hagenbuch, B. The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 135–151. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Tatsumi, S.; Takeda, R.; Naka, A.; Ogane, N.; Kameda, Y.; Kawachi, K.; Shimizu, S.; Sakai, M.; Kamoshida, S. Expression of organic anion-transporting polypeptide 1A2 and organic cation transporter 6 as a predictor of pathologic response to neoadjuvant chemotherapy in triple negative breast cancer. Breast Cancer Res. Treat. 2014, 145, 101–111. [Google Scholar] [CrossRef]
- Bao, L.; Hazari, S.; Mehra, S.; Kaushal, D.; Moroz, K.; Dash, S. Increased expression of P-glycoprotein and doxorubicin chemoresistance of metastatic breast cancer is regulated by miR-298. Am. J. Pathol. 2012, 180, 2490–2503. [Google Scholar] [CrossRef] [Green Version]
- Larkin, A.; O’Driscoll, L.; Kennedy, S.; Purcell, R.; Moran, E.; Crown, J.; Parkinson, M.; Clynes, M. Investigation of MRP-1 protein and MDR-1 P-glycoprotein expression in invasive breast cancer: A prognostic study. Int. J. Cancer 2004, 112, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Faneyte, I.F.; Kristel, P.M.; van de Vijver, M.J. Determining MDR1/P-glycoprotein expression in breast cancer. Int. J. Cancer 2001, 93, 114–122. [Google Scholar] [CrossRef]
- Wlcek, K.; Svoboda, M.; Thalhammer, T.; Sellner, F.; Krupitza, G.; Jaeger, W. Altered expression of organic anion transporter polypeptide (OATP) genes in human breast carcinoma. Cancer Biol. Ther. 2008, 7, 1450–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirota, T.; Tanaka, T.; Takesue, H.; Ieiri, I. Epigenetic regulation of drug transporter expression in human tissues. Expert Opin. Drug Metab. Toxicol. 2017, 13, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Yuan, J.; Li, Z.; Wang, Z.; Cheng, D.; Du, Y.; Li, W.; Kan, Q.; Zhang, W. Genetic polymorphisms and function of the organic anion-transporting polypeptide 1A2 and its clinical relevance in drug disposition. Pharmacology 2015, 95, 201–208. [Google Scholar] [CrossRef]
- Lee, W.; Glaeser, H.; Smith, L.H.; Roberts, R.L.; Moeckel, G.W.; Gervasini, G.; Leake, B.F.; Kim, R.B. Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): Implications for altered drug disposition and central nervous system drug entry. J. Biol. Chem. 2005, 280, 9610–9617. [Google Scholar] [CrossRef] [Green Version]
- Schulte, R.R.; Ho, R.H. Organic Anion Transporting Polypeptides: Emerging Roles in Cancer Pharmacology. Mol. Pharmacol. 2019, 95, 490–506. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.R.; Prasad, B.; Bhatt, D.K.; Mathialagan, S.; Varma, M.V.S.; Unadkat, J.D. In Vivo-to-In Vitro Extrapolation of Transporter-Mediated Renal Clearance: Relative Expression Factor Versus Relative Activity Factor Approach. Drug Metab. Dispos. 2020, 49, 470–478. [Google Scholar] [CrossRef]
- Andreev, E.; Brosseau, N.; Carmona, E.; Mes-Masson, A.-M.; Ramotar, D. The human organic cation transporter OCT1 mediates high affinity uptake of the anticancer drug daunorubicin. Sci. Rep. 2016, 6, 20508. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Piquette-Miller, M.; Polli, J.W.; Russel, F.G.M.; Sprowl, J.A.; Tohyama, K.; Ware, J.A.; de Wildt, S.N.; Xie, W.; Brouwer, K.L.R. Disease-Associated Changes in Drug Transporters May Impact the Pharmacokinetics and/or Toxicity of Drugs: A White Paper From the International Transporter Consortium. Clin. Pharmacol. Ther. 2018, 104, 900–915. [Google Scholar] [CrossRef] [PubMed]
- Koepsell, H. Organic Cation Transporters in Health and Disease. Pharmacol. Rev. 2020, 72, 253–319. [Google Scholar] [CrossRef]
- Kalliokoski, A.; Niemi, M. Impact of OATP transporters on pharmacokinetics. Br. J. Pharmacol. 2009, 158, 693–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gelderblom, H.; Verweij, J.; Nooter, K.; Sparreboom, A. Cremophor EL: The drawbacks and advantages of vehicle selection for drug formulation. Eur. J. Cancer 2001, 37, 1590–1598. [Google Scholar] [CrossRef]
- Perez, E.A. Doxorubicin and paclitaxel in the treatment of advanced breast cancer: Efficacy and cardiac considerations. Cancer Investig. 2001, 19, 155–164. [Google Scholar] [CrossRef]
- Sparreboom, A.; Verweij, J.; van der Burg, M.E.; Loos, W.J.; Brouwer, E.; Viganò, L.; Locatelli, A.; de Vos, A.I.; Nooter, K.; Stoter, G.; et al. Disposition of Cremophor EL in humans limits the potential for modulation of the multidrug resistance phenotype in vivo. Clin. Cancer Res. 1998, 4, 1937–1942. [Google Scholar]
- Gianni, L.; Viganò, L.; Locatelli, A.; Capri, G.; Giani, A.; Tarenzi, E.; Bonadonna, G. Human pharmacokinetic characterization and in vitro study of the interaction between doxorubicin and paclitaxel in patients with breast cancer. J. Clin. Oncol. 1997, 15, 1906–1915. [Google Scholar] [CrossRef]
- Gehl, J.; Boesgaard, M.; Paaske, T.; Jensen, B.V.; Dombernowsky, P. Combined doxorubicin and paclitaxel in advanced breast cancer: Effective and cardiotoxic. Ann. Oncol. 1996, 7, 687–693. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Tichopad, A.; Prgomet, C.; Neuvians, T.P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--Excel-based tool using pair-wise correlations. Biotechnol. Lett. 2004, 26, 509–515. [Google Scholar] [CrossRef]
- Otter, M.; Oswald, S.; Siegmund, W.; Keiser, M. Effects of frequently used pharmaceutical excipients on the organic cation transporters 1-3 and peptide transporters 1/2 stably expressed in MDCKII cells. Eur. J. Pharm. Biopharm. 2017, 112, 187–195. [Google Scholar] [CrossRef]
- Mandery, K.; Bujok, K.; Schmidt, I.; Keiser, M.; Siegmund, W.; Balk, B.; König, J.; Fromm, M.F.; Glaeser, H. Influence of the flavonoids apigenin, kaempferol, and quercetin on the function of organic anion transporting polypeptides 1A2 and 2B1. Biochem. Pharmacol. 2010, 80, 1746–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bexten, M.; Oswald, S.; Grube, M.; Jia, J.; Graf, T.; Zimmermann, U.; Rodewald, K.; Zolk, O.; Schwantes, U.; Siegmund, W.; et al. Expression of drug transporters and drug metabolizing enzymes in the bladder urothelium in man and affinity of the bladder spasmolytic trospium chloride to transporters likely involved in its pharmacokinetics. Mol. Pharm. 2015, 12, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Seithel, A.; Eberl, S.; Singer, K.; Auge, D.; Heinkele, G.; Wolf, N.B.; Dörje, F.; Fromm, M.F.; König, J. The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab. Dispos. 2007, 35, 779–786. [Google Scholar] [CrossRef] [Green Version]
- Gröer, C.; Brück, S.; Lai, Y.; Paulick, A.; Busemann, A.; Heidecke, C.D.; Siegmund, W.; Oswald, S. LC-MS/MS-based quantification of clinically relevant intestinal uptake and efflux transporter proteins. J. Pharm. Biomed. Anal. 2013, 85, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Kisser, B.; Mangelsen, E.; Wingolf, C.; Partecke, L.I.; Heidecke, C.-D.; Tannergren, C.; Oswald, S.; Keiser, M. The Ussing Chamber Assay to Study Drug Metabolism and Transport in the Human Intestine. Curr. Protoc. Pharmacol. 2017, 77, 7.17.1–7.17.19. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Transporter | Km (µmol/L) | Vmax (pmol/mg × min) | Clint (µL/mg × min) |
|---|---|---|---|
| OATP1A2*1 | 7.49 ± 3.41 | 35.3 ± 5.23 | 4.71 ± 1.53 |
| OATP1A2*2 | 5.84 ± 3.95 | 8.09 ± 2.55 | 1.39 ± 0.65 |
| OATP1A2*3 | 5.94 ± 3.07 | 13.8 ± 3.40 | 2.32 ± 0.99 |
| OCT1 | 4.66 ± 3.43 | 19.4 ± 3.78 | 4.16 ± 1.10 |
| OCT2 | 76.1 ± 42.8 | 37.8 ± 11.1 | 0.50 ± 0.26 |
| OCT3 | 13.0 ± 10.7 | 12.9 ± 3.60 | 0.99 ± 0.33 |
| Gen | Protein | Assay-ID |
|---|---|---|
| GAPDH | GAPDH | Hs99999905_m1 |
| 18S | 18S | Hs99999901_s1 |
| ESR1 | ERα | Hs01046816_m1 |
| ABCB1 | P-gp | Hs01067802_m1 |
| ABCC1 | MRP1 | HS01561502_m1 |
| ABCG2 | BCRP | Hs01053790_m1 |
| SLC22A1 | OCT1 | Hs00427552_m1 |
| SLC22A2 | OCT2 | Hs01010723_m1 |
| SLC22A3 | OCT3 | Hs01009568_m1 |
| SLC22A4 | OCTN1 | HS01548718_m1 |
| SLC22A5 | OCTN2 | HS00929869_m1 |
| SLC22A16 | OCT6 | HS00263925_m1 |
| SLCO1A2 | OAPT1A2 | Hs00366488_m1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Otter, M.; Csader, S.; Keiser, M.; Oswald, S. Expression and Functional Contribution of Different Organic Cation Transporters to the Cellular Uptake of Doxorubicin into Human Breast Cancer and Cardiac Tissue. Int. J. Mol. Sci. 2022, 23, 255. https://doi.org/10.3390/ijms23010255
Otter M, Csader S, Keiser M, Oswald S. Expression and Functional Contribution of Different Organic Cation Transporters to the Cellular Uptake of Doxorubicin into Human Breast Cancer and Cardiac Tissue. International Journal of Molecular Sciences. 2022; 23(1):255. https://doi.org/10.3390/ijms23010255
Chicago/Turabian StyleOtter, Marcus, Susanne Csader, Markus Keiser, and Stefan Oswald. 2022. "Expression and Functional Contribution of Different Organic Cation Transporters to the Cellular Uptake of Doxorubicin into Human Breast Cancer and Cardiac Tissue" International Journal of Molecular Sciences 23, no. 1: 255. https://doi.org/10.3390/ijms23010255