
Can Forest Trees Cope with Climate Change?—Effects of DNA Methylation on Gene Expression and Adaptation to Environmental Change


Abstract
:1. Introduction
2. Effects of DNA Methylation of Forest Trees on Gene Expression and Climate Adaptation
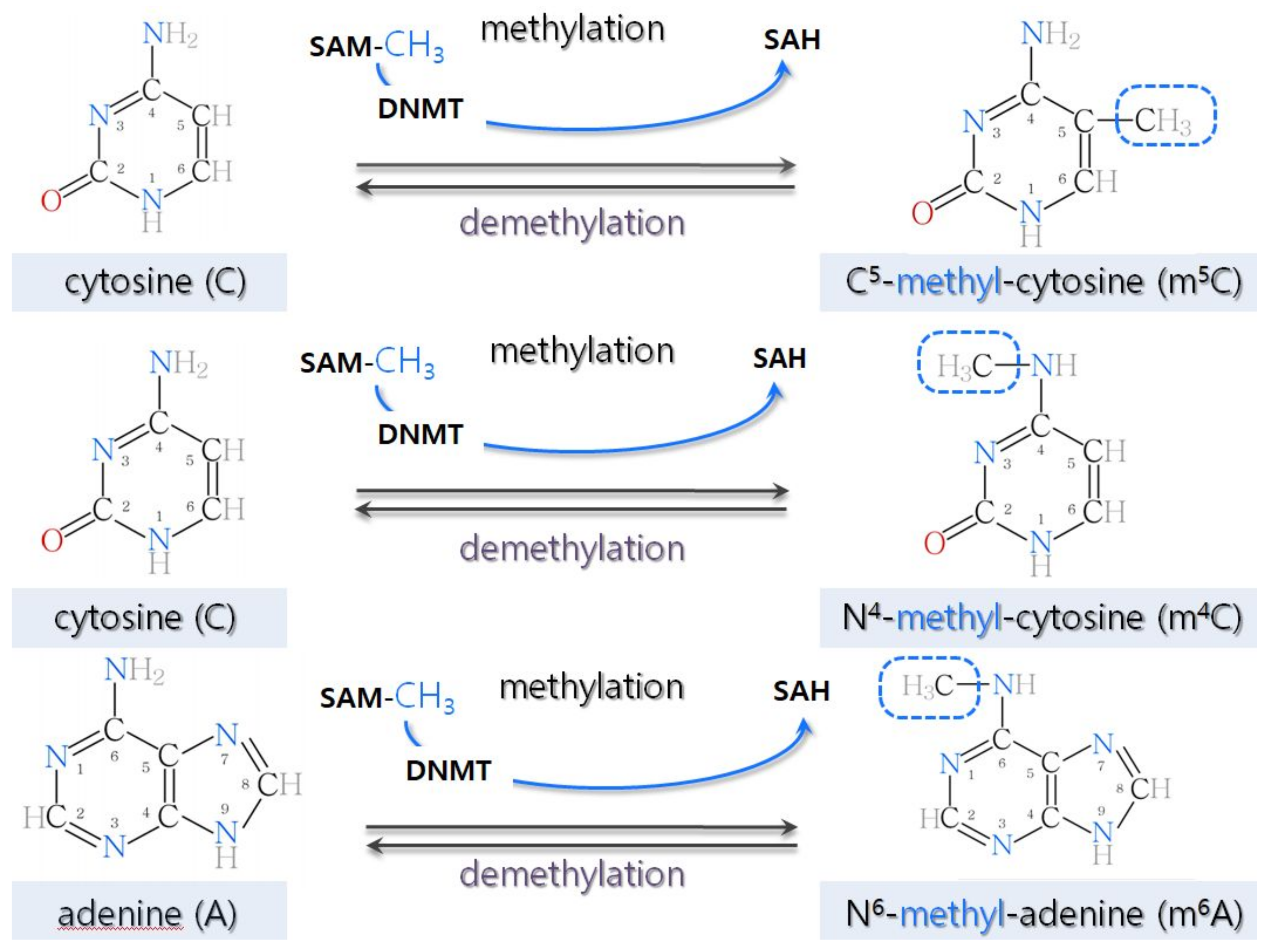
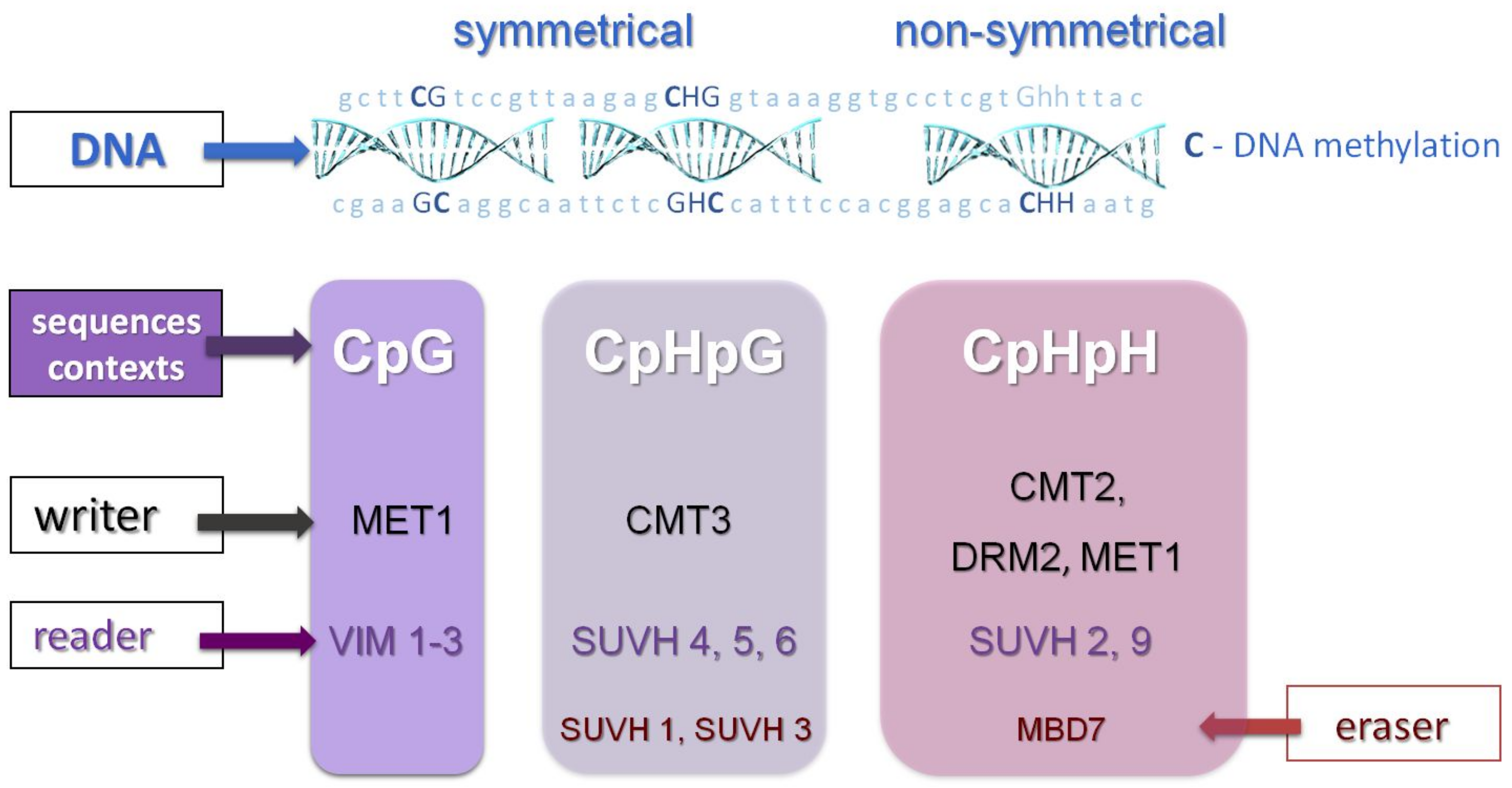
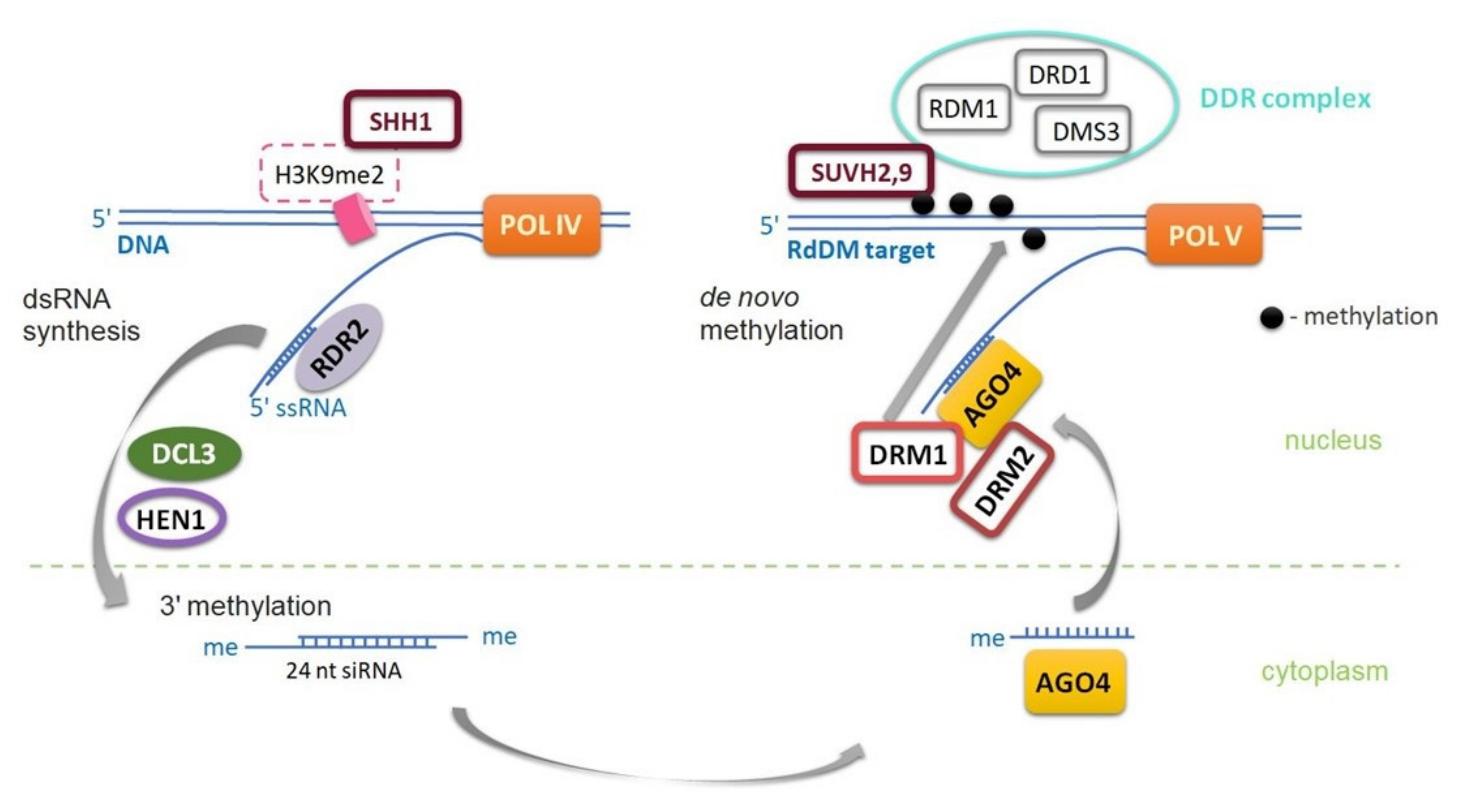
2.1. DNA Methylation in Plants
2.2. Forest Trees—Ecosystems Important to Humans
2.3. Effects of DNA Methylation on Adaptations of Forest Trees
2.4. Epigenetic Modifications of Trees and Environmental Conditions—A Review of Existing Research and the Current State of Knowledge
3. Reprogramming Genes
4. Editing the Epigenome of Trees, CRISPR/Cas9, and Other Molecular Tools
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amaral, J.; Ribeyre, Z.; Vigneaud, J.; Sow, M.D.; Fichot, R.; Messier, C.; Pinto, G.; Nolet, P.; Maury, S. Advances and Promises of Epigenetics for Forest Trees. Forests 2020, 11, 976. [Google Scholar] [CrossRef]
- Johnson, T.B.; Coghill, R.D. Researches on Pyrimidines. C111. The Discovery of 5-Methyl-Cytosine in Tuberculinic Acid, The Nucleic Acid of the Tubercle bacillus. J. Am. Chem. Soc. 1925, 47, 2838–2844. [Google Scholar] [CrossRef]
- Waddington, C.H. An Introduction to Modern Genetics; The Macmillan Company: New York, NY, USA, 1939. [Google Scholar]
- Yakovlev, I.; Fossdal, C.G.; Skrøppa, T.; Olsen, J.E.; Jahren, A.H.; Johnsen, Ø. An Adaptive Epigenetic Memory in Conifers with Important Implications for Seed Production. Seed Sci. Res. 2012, 22, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Alakärppä, E.; Salo, H.M.; Valledor, L.; Cañal, M.J.; Häggman, H.; Vuosku, J. Natural Variation of DNA Methylation and Gene Expression May Determine Local Adaptations of Scots Pine Populations. J. Exp. Bot. 2018, 69, 5293–5305. [Google Scholar] [CrossRef]
- Martínez-Pérez, M.; Aparicio, F.; López-Gresa, M.P.; Bellés, J.M.; Sánchez-Navarro, J.A.; Pallás, V. Arabidopsis M6A Demethylase Activity Modulates Viral Infection of a Plant Virus and the M6A Abundance in Its Genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.A.; Takai, D. The Role of DNA Methylation in Mammalian Epigenetics. Science 2001, 293, 1068–1070. [Google Scholar] [CrossRef]
- Matzke, M.; Kanno, T.; Daxinger, L.; Huettel, B.; Matzke, A.J. RNA-Mediated Chromatin-Based Silencing in Plants. Curr. Opin. Cell Biol. 2009, 21, 367–376. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Illingworth, R.S.; Bird, A.P. CpG Islands—‘A Rough Guide’. FEBS Lett. 2009, 583, 1713–1720. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Tollefsbol, T.O. DNA Methylation Methods: Global DNA Methylation and Methylomic Analyses. Methods 2021, 187, 28–43. [Google Scholar] [CrossRef]
- Nuñez, J.K.; Chen, J.; Pommier, G.C.; Cogan, J.Z.; Replogle, J.M.; Adriaens, C.; Ramadoss, G.N.; Shi, Q.; Hung, K.L.; Samelson, A.J.; et al. Genome-Wide Programmable Transcriptional Memory by CRISPR-Based Epigenome Editing. Cell 2021, 184, 2503–2519.e17. [Google Scholar] [CrossRef]
- Meyer, P. Epigenetic Variation and Environmental Change. J. Exp. Bot. 2015, 66, 3541–3548. [Google Scholar] [CrossRef] [Green Version]
- Niederhuth, C.E.; Schmitz, R.J. Putting DNA Methylation in Context: From Genomes to Gene Expression in Plants. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.; Ma, L.; Pang, H.; Wang, P.; Liu, L.; Cheng, Y.; Cheng, J.; Guo, Y.; Li, Q. Methionine Synthase1 Is Involved in Chromatin Silencing by Maintaining DNA and Histone Methylation. Plant Physiol. 2019, 181, 249–261. [Google Scholar] [CrossRef]
- Zhang, Y.; Harris, C.J.; Liu, Q.; Liu, W.; Ausin, I.; Long, Y.; Xiao, L.; Feng, L.; Chen, X.; Xie, Y.; et al. Large-Scale Comparative Epigenomics Reveals Hierarchical Regulation of Non-CG Methylation in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E1069–E1074. [Google Scholar] [CrossRef] [Green Version]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for Maintenance of CpXpG Methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef] [Green Version]
- Zemach, A.; Kim, M.Y.; Hsieh, P.-H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis Nucleosome Remodeler DDM1 Allows DNA Methyltransferases to Access H1-Containing Heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nat. Commun. 2019, 10, 729. [Google Scholar] [CrossRef]
- Grimanelli, D.; Ingouff, M. DNA Methylation Readers in Plants. J. Mol. Biol. 2020, 432, 1706–1717. [Google Scholar] [CrossRef]
- Yu, Z.; Zhang, G.; Teixeira da Silva, J.A.; Li, M.; Zhao, C.; He, C.; Si, C.; Zhang, M.; Duan, J. Genome-Wide Identification and Analysis of DNA Methyltransferase and Demethylase Gene Families in Dendrobium officinale Reveal their Potential Functions in Polysaccharide Accumulation. BMC Plant Biol. 2021, 21, 21. [Google Scholar] [CrossRef]
- Gallego-Bartolomé, J. DNA Methylation in Plants: Mechanisms and Tools for Targeted Manipulation. New Phytol. 2020, 227, 38–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Liu, H.-L.; Daxinger, L.; Pontes, O.; He, X.; Qian, W.; Lin, H.; Xie, M.; Lorkovic, Z.J.; Zhang, S.; et al. An RNA Polymerase II- and AGO4-Associated Protein Acts in RNA-Directed DNA Methylation. Nature 2010, 465, 106–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzke, M.A.; Kanno, T.; Matzke, A.J.M. RNA-Directed DNA Methylation: The Evolution of a Complex Epigenetic Pathway in Flowering Plants. Annu. Rev. Plant Biol. 2015, 66, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Williams, B.P.; Gehring, M. Principles of Epigenetic Homeostasis Shared Between Flowering Plants and Mammals. Trends Genet. 2020, 36, 751–763. [Google Scholar] [CrossRef]
- Beech, E.; Rivers, M.; Oldfield, S.; Smith, P.P. GlobalTreeSearch: The First Complete Global Database of Tree Species and Country Distributions. J. Sustain. For. 2017, 36, 454–489. [Google Scholar] [CrossRef]
- Li, D.-Z.; Pritchard, H.W. The Science and Economics of Ex Situ Plant Conservation. Trends Plant Sci. 2009, 14, 614–621. [Google Scholar] [CrossRef]
- Guerrant, E.O.; Havens, K.; Vitt, P. Sampling for Effective Ex Situ Plant Conservation. Int. J. Plant Sci. 2014, 175, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Cochrane, A. Are We Underestimating the Impact of Rising Summer Temperatures on Dormancy Loss in Hard-Seeded Species? Aust. J. Bot. 2017, 65, 248–256. [Google Scholar] [CrossRef]
- O’Donnell, K.; Sharrock, S. The Contribution of Botanic Gardens to Ex Situ Conservation through Seed Banking. Plant Divers. 2017, 39, 373–378. [Google Scholar] [CrossRef]
- Daws, M.I.; Cleland, H.; Chmielarz, P.; Gorian, F.; Leprince, O.; Mullins, C.E.; Thanos, C.A.; Vandvik, V.; Pritchard, H.W. Variable Desiccation Tolerance in Acer Pseudoplatanus Seeds in Relation to Developmental Conditions: A Case of Phenotypic Recalcitrance? Funct. Plant Biol. 2006, 33, 59–66. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P. Cryopreservation of Dormant Orthodox Seeds of Forest Trees: Mazzard Cherry (Prunus avium L.). Ann. For. Sci. 2009, 66, 405. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P.; Michalak, M.; Pałucka, M.; Wasileńczyk, U. Successful Cryopreservation of Quercus robur Plumules. Plant Cell Rep. 2011, 30, 1405–1414. [Google Scholar] [CrossRef]
- Suszka, J.; Plitta, B.P.; Michalak, M.; Bujarska-Borkowska, B.; Tylkowski, T.; Chmielarz, P. Optimal Seed Water Content and Storage Temperature for Preservation of Populus nigra L. Germplasm. Ann. For. Sci. 2014, 71, 543–549. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, H.W.; Moat, J.F.; Ferraz, J.B.S.; Marks, T.R.; Camargo, J.L.C.; Nadarajan, J.; Ferraz, I.D.K. Innovative Approaches to the Preservation of Forest Trees. For. Ecol. Manag. 2014, 333, 88–98. [Google Scholar] [CrossRef] [Green Version]
- Mattana, E.; Picciau, R.; Puddu, S.; Lombraña, A.C.; Bacchetta, G. Effect of Temperature and Cold Stratification on Seed Germination of the Mediterranean Wild Aromatic Clinopodium sandalioticum (Lamiaceae). Plant. Biosyst. Int. J. Deal. All Asp. Plant. Biol. 2016, 150, 846–850. [Google Scholar] [CrossRef]
- Potter, K.M.; Jetton, R.M.; Bower, A.; Jacobs, D.F.; Man, G.; Hipkins, V.D.; Westwood, M. Banking on the Future: Progress, Challenges and Opportunities for the Genetic Conservation of Forest Trees. New For. 2017, 48, 153–180. [Google Scholar] [CrossRef]
- Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.Z.; Kotlarski, S.; Tomaszewski, D.; Tylkowski, T.; Barciszewski, J.; Chmielarz, P.; Michalak, M. Changes in Genomic 5-Methylcytosine Level Mirror the Response of Orthodox (Acer platanoides L.) and Recalcitrant (Acer pseudoplatanus L.) Seeds to Severe Desiccation. Tree Physiol. 2018, 38, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Pascual, E.; Mattana, E.; Pritchard, H.W. Seeds of Future Past: Climate Change and the Thermal Memory of Plant Reproductive Traits. Biol. Rev. 2019, 94, 439–456. [Google Scholar] [CrossRef]
- Ballesteros, D.; Pritchard, H.W. The Cryobiotechnology of Oaks: An Integration of Approaches for the Long-Term Ex Situ Conservation of Quercus Species. Forests 2020, 11, 1281. [Google Scholar] [CrossRef]
- Theilade, I.; Petri, L. Conservation of Tropical Trees Ex Situ through Storage and Use; Danida Forest Seed Centre: Humlebaek, Denmark, 2003. [Google Scholar]
- Phartyal, S.S.; Thapliyal, R.C.; Koedam, N.; Godefroid, S. Ex Situ Conservation of Rare and Valuable Forest Tree Species through Seed-Gene Bank. Curr. Sci. 2002, 83, 1351–1357. [Google Scholar]
- Hamilton, K.N.; Offord, C.A.; Cuneo, P.; Deseo, M.A. A Comparative Study of Seed Morphology in Relation to Desiccation Tolerance and Other Physiological Responses in 71 Eastern Australian Rainforest Species. Plant Species Biol. 2013, 28, 51–62. [Google Scholar] [CrossRef]
- Dürr, C.; Dickie, J.B.; Yang, X.-Y.; Pritchard, H.W. Ranges of Critical Temperature and Water Potential Values for the Germination of Species Worldwide: Contribution to a Seed Trait Database. Agric. For. Meteorol. 2015, 200, 222–232. [Google Scholar] [CrossRef]
- Espinas, N.A.; Saze, H.; Saijo, Y. Epigenetic Control of Defense Signaling and Priming in Plants. Front. Plant Sci. 2016, 7, 1201. [Google Scholar] [CrossRef] [PubMed]
- Vozzo, J.A.; United States Forest Service. Tropical Tree Seed Manual; U.S. Department of Agriculture, Forest Service: Washington, DC, USA, 2002.
- Sautu, A.; Baskin, J.M.; Baskin, C.C.; Deago, J.; Condit, R. Classification and Ecological Relationships of Seed Dormancy in a Seasonal Moist Tropical Forest, Panama, Central America. Seed Sci. Res. 2007, 17, 127–140. [Google Scholar] [CrossRef] [Green Version]
- Małecka, A.; Ciszewska, L.; Staszak, A.; Ratajczak, E. Relationship between Mitochondrial Changes and Seed Aging as a Limitation of Viability for the Storage of Beech Seed (Fagus sylvatica L.). PeerJ 2021, 9, e10569. [Google Scholar] [CrossRef]
- Kijowska-Oberc, J.; Staszak, A.M.; Ratajczak, E. Climate Change Affects Seed Aging? Initiation Mechanism and Consequences of Loss of Forest Tree Seed Viability. Trees 2021, 35, 1099–1108. [Google Scholar] [CrossRef]
- Michalak, M.; Plitta-Michalak, B.P.; Naskręt-Barciszewska, M.; Barciszewski, J.; Bujarska-Borkowska, B.; Chmielarz, P. Global 5-Methylcytosine Alterations in DNA during Ageing of Quercus Robur Seeds. Ann. Bot. 2015, 116, 369–376. [Google Scholar] [CrossRef] [Green Version]
- Technical Guidelines for Genetic Conservation of Norway Spruce (Picea abies (L.) Karst.); Koski, V.; International Plant Genetic Resources Institute (Eds.) IPGRI: Rome, Italy, 1997. [Google Scholar]
- Selås, V.; Piovesan, G.; Adams, J.M.; Bernabei, M. Climatic Factors Controlling Reproduction and Growth of Norway Spruce in Southern Norway. Can. J. For. Res. 2002, 32, 217–225. [Google Scholar] [CrossRef]
- Jansen, S.; Konrad, H.; Geburek, T. The Extent of Historic Translocation of Norway Spruce Forest Reproductive Material in Europe. Ann. For. Sci. 2017, 74, 56. [Google Scholar] [CrossRef]
- Klopčič, M.; Mina, M.; Bugmann, H.; Bončina, A. The Prospects of Silver Fir (Abies alba Mill.) and Norway Spruce (Picea abies (L.) Karst.) in Mixed Mountain Forests under Various Management Strategies, Climate Change and High Browsing Pressure. Eur. J. For. Res. 2017, 136, 1071–1090. [Google Scholar] [CrossRef]
- Theilade, I.; Graudal, L.; Kjær, E.D.; Hald, S. Conservation of Genetic Resources of Pinus merkusii in Thailand; Technical Note; Danida Forest Seed Centre: Humlebaek, Denmark, 2000. [Google Scholar]
- Mansor, M. Diversity and Conservation of Tropical Forestry Species in Southeast Asia. In Conservation of Tropical Plant Species; Normah, M.N., Chin, H.F., Reed, B.M., Eds.; Springer: New York, NY, USA, 2013; pp. 317–345. [Google Scholar] [CrossRef]
- Das, A.; Singha, L.B.; Khan, M.L. Community Structure and Species Diversity of Pinus merkusii Jungh & de Vriese Forest along an Altitudinal Gradient in Eastern Himalaya, Arunachal Pradesh, India. Available online: https://www.semanticscholar.org/paper/Community-structure-and-species-diversity-of-Pinus-Das-Singha/c7621bf87087a10f03d4505d4020bc1d8ce582f5 (accessed on 2 July 2021).
- Pouli, T.; Alatimu, T.; Thomson, L. Conserving the Pacific Island’s Unique Trees: Terminalia richii and Manilkara samoensis in Samoa. Int. For. Rev. 2002, 4, 286–291. [Google Scholar] [CrossRef]
- Feng, S.; Jacobsen, S.E. Epigenetic Modifications in Plants: An Evolutionary Perspective. Curr. Opin. Plant Biol. 2011, 14, 179–186. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.J.; Ecker, J.R. Epigenetic and Epigenomic Variation in Arabidopsis thaliana. Trends Plant Sci. 2012, 17, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Colomé-Tatché, M.; Cortijo, S.; Wardenaar, R.; Morgado, L.; Lahouze, B.; Sarazin, A.; Etcheverry, M.; Martin, A.; Feng, S.; Duvernois-Berthet, E.; et al. Features of the Arabidopsis Recombination Landscape Resulting from the Combined Loss of Sequence Variation and DNA Methylation. Proc. Natl. Acad. Sci. USA 2012, 109, 16240–16245. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Virlouvet, L.; Liu, N.; Riethoven, J.-J.; Fromm, M.; Avramova, Z. Dehydration Stress Memory Genes of Zea mays; Comparison with Arabidopsis thaliana. BMC Plant. Biol. 2014, 14, 141. [Google Scholar] [CrossRef] [Green Version]
- Yong-Villalobos, L.; González-Morales, S.I.; Wrobel, K.; Gutiérrez-Alanis, D.; Cervantes-Peréz, S.A.; Hayano-Kanashiro, C.; Oropeza-Aburto, A.; Cruz-Ramírez, A.; Martínez, O.; Herrera-Estrella, L. Methylome Analysis Reveals an Important Role for Epigenetic Changes in the Regulation of the Arabidopsis Response to Phosphate Starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E7293–E7302. [Google Scholar] [CrossRef] [Green Version]
- Kawakatsu, T.; Huang, S.C.; Jupe, F.; Sasaki, E.; Schmitz, R.J.; Urich, M.A.; Castanon, R.; Nery, J.R.; Barragan, C.; He, Y.; et al. Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions. Cell 2016, 166, 492–505. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Zhai, K.; Xie, Z.; Yang, D.; Zhu, X.; Liu, J.; Wang, X.; Qin, P.; Yang, Y.; Zhang, G.; et al. Epigenetic Regulation of Antagonistic Receptors Confers Rice Blast Resistance with Yield Balance. Science 2017, 355, 962–965. [Google Scholar] [CrossRef]
- Bhat, S.S.; Bielewicz, D.; Jarmolowski, A.; Szweykowska-Kulinska, Z. N6-Methyladenosine (M6A): Revisiting the Old with Focus on New, an Arabidopsis thaliana Centered Review. Genes 2018, 9, 596. [Google Scholar] [CrossRef] [Green Version]
- Boquete, M.T.; Muyle, A.; Alonso, C. Plant Epigenetics: Phenotypic and Functional Diversity beyond the DNA Sequence. Am. J. Bot. 2021, 108, 553–558. [Google Scholar] [CrossRef]
- Liu, Q.A. The Impact of Climate Change on Plant Epigenomes. Trends Genet. 2013, 29, 503–505. [Google Scholar] [CrossRef]
- Keller, T.E.; Lasky, J.R.; Yi, S.V. The Multivariate Association between Genomewide DNA Methylation and Climate across the Range of Arabidopsis thaliana. Mol. Ecol. 2016, 25, 1823–1837. [Google Scholar] [CrossRef]
- Sork, V.L. Genomic Studies of Local Adaptation in Natural Plant Populations. J. Hered. 2018, 109, 3–15. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Li, Z. Epigenetic Environmental Memories in Plants: Establishment, Maintenance, and Reprogramming. Trends Genet. 2018, 34, 856–866. [Google Scholar] [CrossRef]
- Perrone, A.; Martinelli, F. Plant Stress Biology in Epigenomic Era. Plant Sci. 2020, 294, 110376. [Google Scholar] [CrossRef]
- Srikant, T.; Drost, H.-G. How Stress Facilitates Phenotypic Innovation Through Epigenetic Diversity. Front. Plant Sci. 2021, 11, 606800. [Google Scholar] [CrossRef]
- Boyko, A.; Kovalchuk, I. Epigenetic control of plant stress response. Environ. Mol. Mutagenes. 2008, 49, 61–72. [Google Scholar] [CrossRef]
- Fleta-Soriano, E.; Munné-Bosch, S. Stress Memory and the Inevitable Effects of Drought: A Physiological Perspective. Front. Plant Sci. 2016, 7, 143. [Google Scholar] [CrossRef] [Green Version]
- Thiebaut, F.; Hemerly, A.S.; Ferreira, P.C.G. A Role for Epigenetic Regulation in the Adaptation and Stress Responses of Non-Model Plants. Front. Plant Sci. 2019, 10, 246. [Google Scholar] [CrossRef] [Green Version]
- Pascual, J.; Cañal, M.; Correia, B.; Escandón, M.; Hasbun, R.; Meijón, M.; Pinto, G.; Valledor, L. Can Epigenetics Help Forest Plants to Adapt to Climate Change? In Epigenetics in Plants of Agronomic Importance: Fundamentals and Applications; Springer: Berlin/Heidelberg, Germany, 2014; pp. 125–146. [Google Scholar] [CrossRef]
- Gugger, P.F.; Fitz-Gibbon, S.; PellEgrini, M.; Sork, V.L. Species-Wide Patterns of DNA Methylation Variation in Quercus lobata and their Association with Climate Gradients. Mol. Ecol. 2016, 25, 1665–1680. [Google Scholar] [CrossRef]
- Le Gac, A.-L.; Lafon-Placette, C.; Delaunay, A.; Maury, S. Developmental, Genetic and Environmental Variations of Global DNA Methylation in the First Leaves Emerging from the Shoot Apical Meristem in Poplar Trees. Plant Signal. Behav. 2019, 14, 1596717. [Google Scholar] [CrossRef]
- Jacinto Pereira, W.; de Castro Rodrigues Pappas, M.; Camargo Campoe, O.; Stape, J.L.; Grattapaglia, D.; Joannis Pappas, G., Jr. Patterns of DNA Methylation Changes in Elite Eucalyptus Clones across Contrasting Environments. For. Ecol. Manag. 2020, 474, 118319. [Google Scholar] [CrossRef]
- Tuskan, G.A.; DiFazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The Genome of Black Cottonwood, Populus trichocarpa (Torr. & Gray). Science 2006, 313, 1596–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nystedt, B.; Street, N.R.; Wetterbom, A.; Zuccolo, A.; Lin, Y.-C.; Scofield, D.G.; Vezzi, F.; Delhomme, N.; Giacomello, S.; Alexeyenko, A.; et al. The Norway Spruce Genome Sequence and Conifer Genome Evolution. Nature 2013, 497, 579–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birol, I.; Raymond, A.; Jackman, S.D.; Pleasance, S.; Coope, R.; Taylor, G.A.; Yuen, M.M.S.; Keeling, C.I.; Brand, D.; Vandervalk, B.P.; et al. Assembling the 20 Gb White Spruce (Picea glauca) Genome from Whole-Genome Shotgun Sequencing Data. Bioinformatics 2013, 29, 1492–1497. [Google Scholar] [CrossRef]
- Myburg, A.A.; Grattapaglia, D.; Tuskan, G.A.; Hellsten, U.; Hayes, R.D.; Grimwood, J.; Jenkins, J.; Lindquist, E.; Tice, H.; Bauer, D.; et al. The Genome of Eucalyptus grandis. Nature 2014, 510, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.; Stevens, K.A.; Crepeau, M.W.; Holtz-Morris, A.; Koriabine, M.; Marçais, G.; Puiu, D.; Roberts, M.; Wegrzyn, J.L.; de Jong, P.J.; et al. Sequencing and Assembly of the 22-Gb Loblolly Pine Genome. Genetics 2014, 196, 875–890. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Ibeas, D.; Martinez-Garcia, P.J.; Famula, R.A.; Delfino-Mix, A.; Stevens, K.A.; Loopstra, C.A.; Langley, C.H.; Neale, D.B.; Wegrzyn, J.L. Assessing the Gene Content of the Megagenome: Sugar Pine (Pinus lambertiana). G3 Genes Genomes Genet. 2016, 6, 3787–3802. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Li, S.-G.; Fan, X.-F.; Su, Z.-H. Application of Somatic Embryogenesis in Woody Plants. Front. Plant Sci. 2016, 7, 938. [Google Scholar] [CrossRef] [Green Version]
- Sollars, E.S.A.; Harper, A.L.; Kelly, L.J.; Sambles, C.M.; Ramirez-Gonzalez, R.H.; Swarbreck, D.; Kaithakottil, G.; Cooper, E.D.; Uauy, C.; Havlickova, L.; et al. Genome Sequence and Genetic Diversity of European Ash Trees. Nature 2017, 541, 212–216. [Google Scholar] [CrossRef]
- Neale, D.B.; McGuire, P.E.; Wheeler, N.C.; Stevens, K.A.; Crepeau, M.W.; Cardeno, C.; Zimin, A.V.; Puiu, D.; Pertea, G.M.; Sezen, U.U.; et al. The Douglas-Fir Genome Sequence Reveals Specialization of the Photosynthetic Apparatus in Pinaceae. G3 Genes Genomes Genet. 2017, 7, 3157–3167. [Google Scholar] [CrossRef] [Green Version]
- Salojärvi, J.; Smolander, O.-P.; Nieminen, K.; Rajaraman, S.; Safronov, O.; Safdari, P.; Lamminmäki, A.; Immanen, J.; Lan, T.; Tanskanen, J.; et al. Genome Sequencing and Population Genomic Analyses Provide Insights into the Adaptive Landscape of Silver Birch. Nat. Genet. 2017, 49, 904–912. [Google Scholar] [CrossRef]
- Bondar, E.I.; Putintseva, Y.A.; Oreshkova, N.V.; Krutovsky, K.V. Siberian Larch (Larix sibirica Ledeb.) Chloroplast Genome and Development of Polymorphic Chloroplast Markers. BMC Bioinform. 2019, 20, 38. [Google Scholar] [CrossRef]
- Mishra, B.; Gupta, D.K.; Pfenninger, M.; Hickler, T.; Langer, E.; Nam, B.; Paule, J.; Sharma, R.; Ulaszewski, B.; Warmbier, J.; et al. A Reference Genome of the European Beech (Fagus sylvatica L.). GigaScience 2018, 7, giy063. [Google Scholar] [CrossRef]
- Mosca, E.; Cruz, F.; Gómez-Garrido, J.; Bianco, L.; Rellstab, C.; Brodbeck, S.; Csilléry, K.; Fady, B.; Fladung, M.; Fussi, B.; et al. A Reference Genome Sequence for the European Silver Fir (Abies alba Mill.): A Community-Generated Genomic Resource. G3 Genes Genomes Genet. 2019, 9, 2039–2049. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Das, A.; Kainer, D.; Schalamun, M.; Morales-Suarez, A.; Schwessinger, B.; Lanfear, R. The Draft Nuclear Genome Assembly of Eucalyptus pauciflora: A Pipeline for Comparing de Novo Assemblies. GigaScience 2020, 9, giz160. [Google Scholar] [CrossRef]
- Raj, S.; Bräutigam, K.; Hamanishi, E.T.; Wilkins, O.; Thomas, B.R.; Schroeder, W.; Mansfield, S.D.; Plant, A.L.; Campbell, M.M. Clone History Shapes Populus Drought Responses. Proc. Natl. Acad. Sci. USA 2011, 108, 12521–12526. [Google Scholar] [CrossRef] [Green Version]
- Lafon-Placette, C.; Le Gac, A.-L.; Chauveau, D.; Segura, V.; Delaunay, A.; Lesage-Descauses, M.-C.; Hummel, I.; Cohen, D.; Jesson, B.; Le Thiec, D.; et al. Changes in the Epigenome and Transcriptome of the Poplar Shoot Apical Meristem in Response to Water Availability Affect Preferentially Hormone Pathways. J. Exp. Bot. 2018, 69, 537–551. [Google Scholar] [CrossRef]
- Fox, H.; Doron-Faigenboim, A.; Kelly, G.; Bourstein, R.; Attia, Z.; Zhou, J.; Moshe, Y.; Moshelion, M.; David-Schwartz, R. Transcriptome Analysis of Pinus Halepensis under Drought Stress and during Recovery. Tree Physiol. 2018, 38, 423–441. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Rana, K.; Xiong, Z.; Ge, X.; Li, Z.; Song, H.; Qian, W. Genetic and Epigenetic Alterations in Hybrid and Derived Hexaploids between Brassica napus and B. oleracea Revealed by SSR and MSAP Analysis. Acta Physiol. Plant. 2019, 41, 61. [Google Scholar] [CrossRef]
- Cicatelli, A.; Todeschini, V.; Lingua, G.; Biondi, S.; Torrigiani, P.; Castiglione, S. Epigenetic Control of Heavy Metal Stress Response in Mycorrhizal versus Non-Mycorrhizal Poplar Plants. Environ. Sci. Pollut. Res. 2014, 21, 1723–1737. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Asante, D.K.A.; Fossdal, C.G.; Junttila, O.; Johnsen, Ø. Differential Gene Expression Related to an Epigenetic Memory Affecting Climatic Adaptation in Norway Spruce. Plant Sci. 2011, 180, 132–139. [Google Scholar] [CrossRef]
- Conde, D.; Gac, A.-L.L.; Perales, M.; Dervinis, C.; Kirst, M.; Maury, S.; González-Melendi, P.; Allona, I. Chilling-Responsive DEMETER-LIKE DNA Demethylase Mediates in Poplar Bud Break. Plant Cell Environ. 2017, 40, 2236–2249. [Google Scholar] [CrossRef]
- Deng, X.; Wang, J.; Li, Y.; Wu, S.; Yang, S.; Chao, J.; Chen, Y.; Zhang, S.; Shi, M.; Tian, W. Comparative Transcriptome Analysis Reveals Phytohormone Signalings, Heat Shock Module and ROS Scavenger Mediate the Cold-Tolerance of Rubber Tree. Sci. Rep. 2018, 8, 4931. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, M.; Fu, R.; Qian, X.; Rong, P.; Zhang, Y.; Jiang, P.; Wang, J.; Lu, X.; Wang, D.; et al. Epigenetic Mechanisms of Salt Tolerance and Heterosis in Upland Cotton (Gossypium hirsutum L.) Revealed by Methylation-Sensitive Amplified Polymorphism Analysis. Euphytica 2016, 208, 477–491. [Google Scholar] [CrossRef]
- Liu, C.; Li, H.; Lin, J.; Wang, Y.; Xu, X.; Cheng, Z.-M.; Chang, Y. Genome-Wide Characterization of DNA Demethylase Genes and Their Association with Salt Response in Pyrus. Genes 2018, 9, 398. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.-G.; Han, X.; Yang, T.; Cui, W.-H.; Wu, A.-M.; Fu, C.-X.; Wang, B.-C.; Liu, L.-J. Genome-Wide Transcriptional Adaptation to Salt Stress in Populus. BMC Plant. Biol. 2019, 19, 367. [Google Scholar] [CrossRef]
- Lira-Medeiros, C.F.; Parisod, C.; Fernandes, R.A.; Mata, C.S.; Cardoso, M.A.; Ferreira, P.C.G. Epigenetic Variation in Mangrove Plants Occurring in Contrasting Natural Environment. PLoS ONE 2010, 5, e10326. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and Challenges of Genome-Wide DNA Methylation Analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Li, Y. Modern Epigenetics Methods in Biological Research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef]
- Moradi, N.; Rezaei, A.; Bahramnejad, B.; Goodwin, P.H. Methylation-Sensitive Amplified Polymorphism Analysis of Resistant and Susceptible Interactions of Cucumber with Podosphaera xanthii. Physiol. Mol. Plant Pathol. 2019, 106, 64–73. [Google Scholar] [CrossRef]
- Ramirez-Prado, J.S.; Abulfaraj, A.A.; Rayapuram, N.; Benhamed, M.; Hirt, H. Plant Immunity: From Signaling to Epigenetic Control of Defense. Trends Plant Sci. 2018, 23, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Skrøppa, T.; Tollefsrud, M.M.; Sperisen, C.; Johnsen, Ø. Rapid Change in Adaptive Performance from One Generation to the next in Picea abies—Central European Trees in a Nordic Environment. Tree Genet. Genomes 2010, 6, 93–99. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Lee, Y.; Rotter, B.; Olsen, J.E.; Skrøppa, T.; Johnsen, Ø.; Fossdal, C.G. Temperature-Dependent Differential Transcriptomes during Formation of an Epigenetic Memory in Norway Spruce Embryogenesis. Tree Genet. Genomes 2014, 10, 355–366. [Google Scholar] [CrossRef]
- Yakovlev, I.A.; Carneros, E.; Lee, Y.; Olsen, J.E.; Fossdal, C.G. Transcriptional Profiling of Epigenetic Regulators in Somatic Embryos during Temperature Induced Formation of an Epigenetic Memory in Norway Spruce. Planta 2016, 243, 1237–1249. [Google Scholar] [CrossRef]
- Wang, P.; Xia, H.; Zhang, Y.; Zhao, S.; Zhao, C.; Hou, L.; Li, C.; Li, A.; Ma, C.; Wang, X. Genome-Wide High-Resolution Mapping of DNA Methylation Identifies Epigenetic Variation across Embryo and Endosperm in Maize (Zea may). BMC Genom. 2015, 16, 21. [Google Scholar] [CrossRef] [Green Version]
- Lamelas, L.; Valledor, L.; Escandón, M.; Pinto, G.; Cañal, M.J.; Meijón, M. Integrative Analysis of the Nuclear Proteome in Pinus Radiata Reveals Thermopriming Coupled to Epigenetic Regulation. J. Exp. Bot. 2020, 71, 2040–2057. [Google Scholar] [CrossRef] [Green Version]
- Le Gac, A.-L.; Lafon-Placette, C.; Chauveau, D.; Segura, V.; Delaunay, A.; Fichot, R.; Marron, N.; Le Jan, I.; Berthelot, A.; Bodineau, G.; et al. Winter-Dormant Shoot Apical Meristem in Poplar Trees Shows Environmental Epigenetic Memory. J. Exp. Bot. 2018, 69, 4821–4837. [Google Scholar] [CrossRef] [Green Version]
- Forestan, C.; Farinati, S.; Zambelli, F.; Pavesi, G.; Rossi, V.; Varotto, S. Epigenetic Signatures of Stress Adaptation and Flowering Regulation in Response to Extended Drought and Recovery in Zea mays. Plant. Cell Environ. 2020, 43, 55–75. [Google Scholar] [CrossRef]
- Sow, M.D.; Segura, V.; Chamaillard, S.; Jorge, V.; Delaunay, A.; Lafon-Placette, C.; Fichot, R.; Faivre-Rampant, P.; Villar, M.; Brignolas, F.; et al. Narrow-Sense Heritability and PST Estimates of DNA Methylation in Three Populus nigra L. Populations under Contrasting Water Availability. Tree Genet. Genomes 2018, 14, 78. [Google Scholar] [CrossRef]
- Guarino, F.; Cicatelli, A.; Brundu, G.; Heinze, B.; Castiglione, S. Epigenetic Diversity of Clonal White Poplar (Populus alba L.) Populations: Could Methylation Support the Success of Vegetative Reproduction Strategy? PLoS ONE 2015, 10, e0131480. [Google Scholar] [CrossRef] [Green Version]
- Thellier, M.; Lüttge, U. Plant Memory: A Tentative Model. Plant Biol. 2013, 15, 1–12. [Google Scholar] [CrossRef]
- Rico, L.; Ogaya, R.; Barbeta, A.; Peñuelas, J. Changes in DNA Methylation Fingerprint of Quercus Ilex Trees in Response to Experimental Field Drought Simulating Projected Climate Change. Plant Biol. 2014, 16, 419–427. [Google Scholar] [CrossRef]
- Correia, B.; Valledor, L.; Meijón, M.; Rodriguez, J.L.; Dias, M.C.; Santos, C.; Cañal, M.J.; Rodriguez, R.; Pinto, G. Is the Interplay between Epigenetic Markers Related to the Acclimation of Cork Oak Plants to High Temperatures? PLoS ONE 2013, 8, e53543. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, T.; Seki, M. Epigenetic Memory for Stress Response and Adaptation in Plants. Plant Cell Physiol. 2014, 55, 1859–1863. [Google Scholar] [CrossRef]
- Pastor, V.; Luna, E.; Mauch-Mani, B.; Ton, J.; Flors, V. Primed Plants Do Not Forget. Environ. Exp. Bot. 2013, 94, 46–56. [Google Scholar] [CrossRef]
- Walter, J.; Jentsch, A.; Beierkuhnlein, C.; Kreyling, J. Ecological Stress Memory and Cross Stress Tolerance in Plants in the Face of Climate Extremes. Environ. Exp. Bot. 2013, 94, 3–8. [Google Scholar] [CrossRef]
- Martinez-Medina, A.; Flors, V.; Heil, M.; Mauch-Mani, B.; Pieterse, C.M.J.; Pozo, M.J.; Ton, J.; van Dam, N.M.; Conrath, U. Recognizing Plant Defense Priming. Trends Plant Sci. 2016, 21, 818–822. [Google Scholar] [CrossRef] [Green Version]
- Lambers, H.; Chapin, F.S.; Pons, T.L. Life Cycles: Environmental Influences and Adaptations. In Plant Physiological Ecology; Springer: New York, NY, USA, 2008; pp. 375–402. [Google Scholar] [CrossRef]
- Bruce, T.J.A.; Matthes, M.C.; Napier, J.A.; Pickett, J.A. Stressful “Memories” of Plants: Evidence and Possible Mechanisms. Plant Sci. 2007, 173, 603–608. [Google Scholar] [CrossRef]
- Bossdorf, O.; Richards, C.L.; Pigliucci, M. Epigenetics for Ecologists. Ecol. Lett. 2008, 11, 106–115. [Google Scholar] [CrossRef]
- Xu, X.; Tao, Y.; Gao, X.; Zhang, L.; Li, X.; Zou, W.; Ruan, K.; Wang, F.; Xu, G.; Hu, R. A CRISPR-Based Approach for Targeted DNA Demethylation. Cell Discov. 2016, 2, 16009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, J.I.; Celik, H.; Rois, L.E.; Fishberger, G.; Fowler, T.; Rees, R.; Kramer, A.; Martens, A.; Edwards, J.R.; Challen, G.A. Reprogrammable CRISPR/Cas9-Based System for Inducing Site-Specific DNA Methylation. Biol. Open 2016, 5, 866–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, A.; Fisher, D.I. Regulation of Gene Expression and the Elucidative Role of CRISPR-Based Epigenetic Modifiers and CRISPR-Induced Chromosome Conformational Changes. CRISPR J. 2021, 4, 43–57. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.M.; Tollefsbol, T.O. Targeting Cancer Epigenetics with CRISPR-DCAS9: Principles and Prospects. Methods 2021, 187, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.; Zhao, J.; Wei, S.; Li, Y.; Zhi, J.; Wang, H.; Hou, X.; Hu, L.; Zheng, X.; Gao, M. A CRISPR-Cas9-Based Near-Infrared Upconversion-Activated DNA Methylation Editing System. ACS Appl. Mater. Interfaces 2021, 13, 6043–6052. [Google Scholar] [CrossRef] [PubMed]
- Replogle, J.M.; Norman, T.M.; Xu, A.; Hussmann, J.A.; Chen, J.; Cogan, J.Z.; Meer, E.J.; Terry, J.M.; Riordan, D.P.; Srinivas, N.; et al. Combinatorial Single-Cell CRISPR Screens by Direct Guide RNA Capture and Targeted Sequencing. Nat. Biotechnol. 2020, 38, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA Demethylation of the Arabidopsis Genome Using the Human TET1 Catalytic Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- Mlambo, T.; Nitsch, S.; Hildenbeutel, M.; Romito, M.; Müller, M.; Bossen, C.; Diederichs, S.; Cornu, T.I.; Cathomen, T.; Mussolino, C. Designer Epigenome Modifiers Enable Robust and Sustained Gene Silencing in Clinically Relevant Human Cells. Nucleic Acids Res. 2018, 46, 4456–4468. [Google Scholar] [CrossRef]
- Mercé, C.; Bayer, P.E.; Tay Fernandez, C.; Batley, J.; Edwards, D. Induced Methylation in Plants as a Crop Improvement Tool: Progress and Perspectives. Agronomy 2020, 10, 1484. [Google Scholar] [CrossRef]
- Neale, D.B.; Kremer, A. Forest Tree Genomics: Growing Resources and Applications. Nat. Rev. Genet. 2011, 12, 111–122. [Google Scholar] [CrossRef]
- Plomion, C.; Bastien, C.; Bogeat-Triboulot, M.-B.; Bouffier, L.; Déjardin, A.; Duplessis, S.; Fady, B.; Heuertz, M.; Le Gac, A.-L.; Le Provost, G.; et al. Forest Tree Genomics: 10 Achievements from the Past 10 Years and Future Prospects. Ann. For. Sci. 2016, 73, 77–103. [Google Scholar] [CrossRef] [Green Version]
- Balao, F.; Paun, O.; Alonso, C. Uncovering the Contribution of Epigenetics to Plant Phenotypic Variation in Mediterranean Ecosystems. Plant Biol. 2018, 20, 38–49. [Google Scholar] [CrossRef]
- Sollars, E.S.A.; Buggs, R.J.A. Genome-Wide Epigenetic Variation among Ash Trees Differing in Susceptibility to a Fungal Disease. BMC Genom. 2018, 19, 502. [Google Scholar] [CrossRef]
- Morselli, M.; Farrell, C.; Rubbi, L.; Fehling, H.L.; Henkhaus, R.; Pellegrini, M. Targeted Bisulfite Sequencing for Biomarker Discovery. Methods 2021, 187, 13–27. [Google Scholar] [CrossRef]
- Sow, M.D.; Allona, I.; Ambroise, C.; Conde, D.; Fichot, R.; Gribkova, S.; Jorge, V.; Le-Provost, G.; Pâques, L.; Plomion, C.; et al. Chapter Twelve—Epigenetics in Forest Trees: State of the Art and Potential Implications for Breeding and Management in a Context of Climate Change. In Advances in Botanical Research; Mirouze, M., Bucher, E., Gallusci, P., Eds.; Plant Epigenetics Coming of Age for Breeding Applications; Academic Press: Cambridge, MA, USA, 2018; Volume 88, pp. 387–453. [Google Scholar] [CrossRef]
- Neale, D.B.; Langley, C.H.; Salzberg, S.L.; Wegrzyn, J.L. Open Access to Tree Genomes: The Path to a Better Forest. Genome Biol. 2013, 14, 120. [Google Scholar] [CrossRef] [Green Version]
- Parrilla-Doblas, J.T.; Roldán-Arjona, T.; Ariza, R.R.; Córdoba-Cañero, D. Active DNA Demethylation in Plants. Int. J. Mol. Sci. 2019, 20, 4683. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Species | Type of Modification | Stress Condition | Method | Literature |
|---|---|---|---|---|
| Pinus radiata | changes in tissue DNA methylation dynamics | heat stress and priming | quantification of nuclear proteins by nLC-MS/MS | [115] |
| Picea abies | epigenetic memory—increase in overall DNA methylation levels induced by external stimuli | climate adaptation | expression analysis of 32 genes by qRT-PCR | [4,100,113] |
| Pinus nigra | decrease in global DNA methylation | drought stress | genome-wide SNPs | [118] |
| Pinus sylvestris | effect of DNA methylation on expression of 11 genes | environmental adaptation | DNA global methylation, GC/MS | [5] |
| Eucalyptus grandis × Eucalyptus urophylla and Eucalyptus urophylla | a stronger correlation between DNA methylation and genetic background than between DNA methylation and location | environment and growth characteristics | MS-DArT-seq, methyl Sensitive DArT-seq sequencing | [80] |
| Populus alba L. | DNA methylation dynamics—changes in methylation in relation to geographical location | climate adaptation | MSAP, methylation-sensitive amplified polymorphism | [119] |
| Quercus lobata | 43 single-methylation variants were significantly associated with climatic factors, such as mean maximum temperature | climate adaptation | RRBS, reduced-representation bisulphite sequencing | [78] |
| Quercus ilex | DNA methylation dynamics—the percentage of fully methylated loci was significantly higher | heat stress | MSAP, methylation-sensitive amplified polymorphism | [121] |
| Quercus suber | increase in DNA methylation at higher tepmeratures | heat stress | MS-RAPD, methylation-sensitive random-amplified polymorphic DNA | [122] |
| Laguncularia racemosa | variability of DNA methylation relative to populations—for all MSAP markers, identified 67 loci with CpG-methylation, 116 non-methylated loci and 26 hemimethylated loci | climate adaptation | MSAP, methylation-sensitive amplified polymorphism | [106] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klupczyńska, E.A.; Ratajczak, E. Can Forest Trees Cope with Climate Change?—Effects of DNA Methylation on Gene Expression and Adaptation to Environmental Change. Int. J. Mol. Sci. 2021, 22, 13524. https://doi.org/10.3390/ijms222413524
Klupczyńska EA, Ratajczak E. Can Forest Trees Cope with Climate Change?—Effects of DNA Methylation on Gene Expression and Adaptation to Environmental Change. International Journal of Molecular Sciences. 2021; 22(24):13524. https://doi.org/10.3390/ijms222413524
Chicago/Turabian StyleKlupczyńska, Ewelina A., and Ewelina Ratajczak. 2021. "Can Forest Trees Cope with Climate Change?—Effects of DNA Methylation on Gene Expression and Adaptation to Environmental Change" International Journal of Molecular Sciences 22, no. 24: 13524. https://doi.org/10.3390/ijms222413524