
PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc

 , ,
, ,  ,
,  , , , , , , , and
, , , , , , , and 
Abstract
:1. Introduction
2. Results
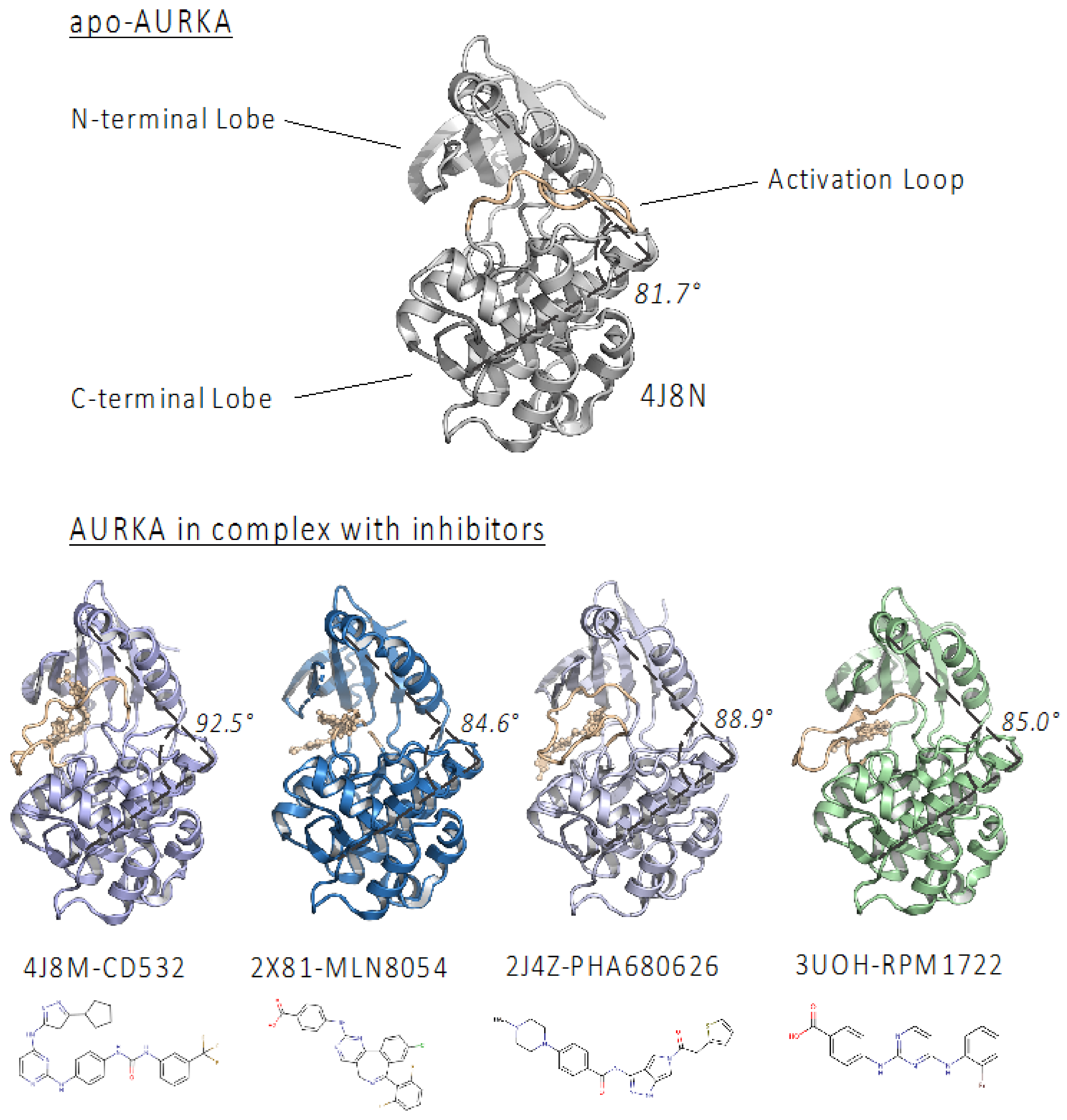
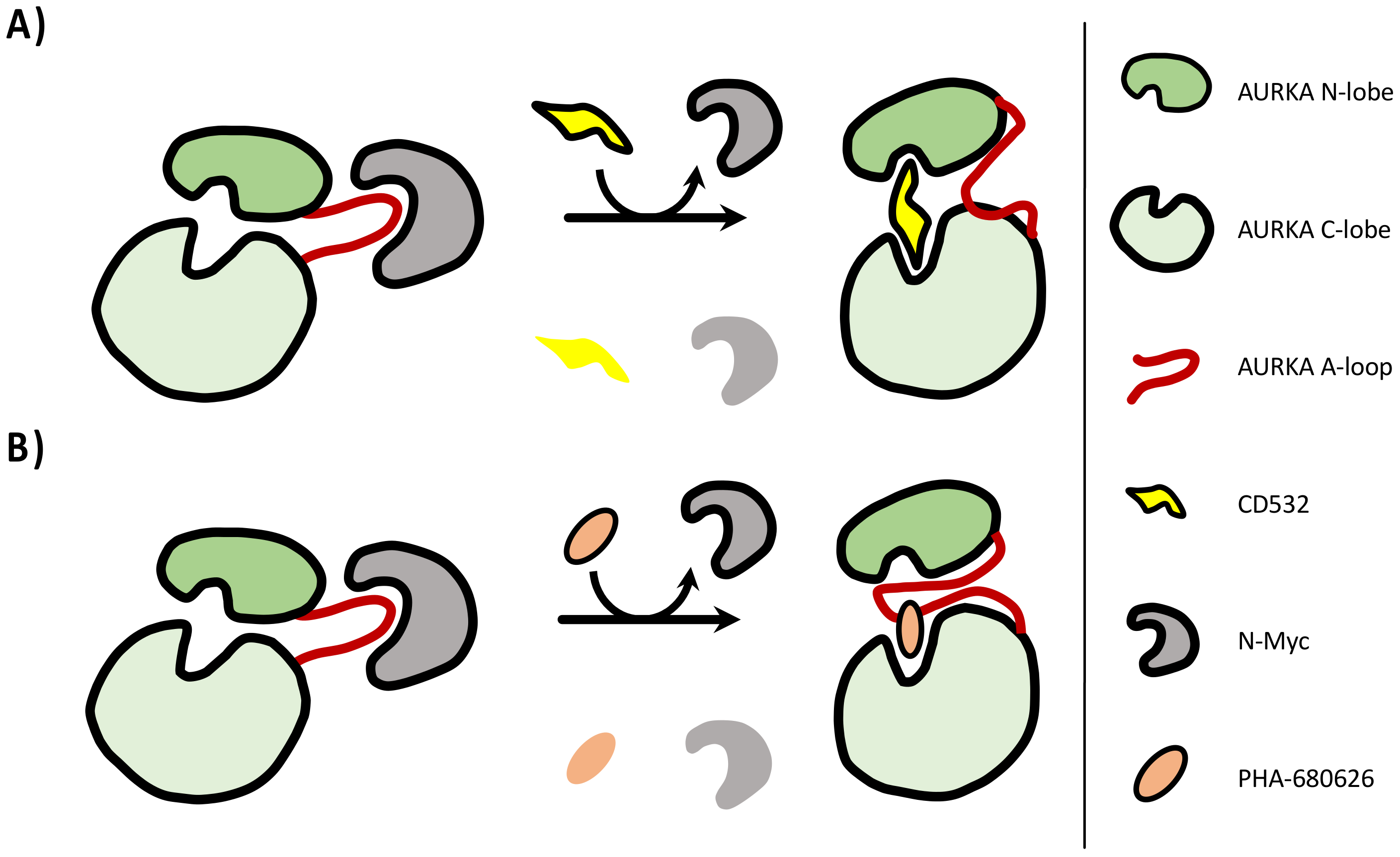
2.1. PHA-680626 and RPM1722 Are Predicted to Prevent N-Myc Binding to AURKA
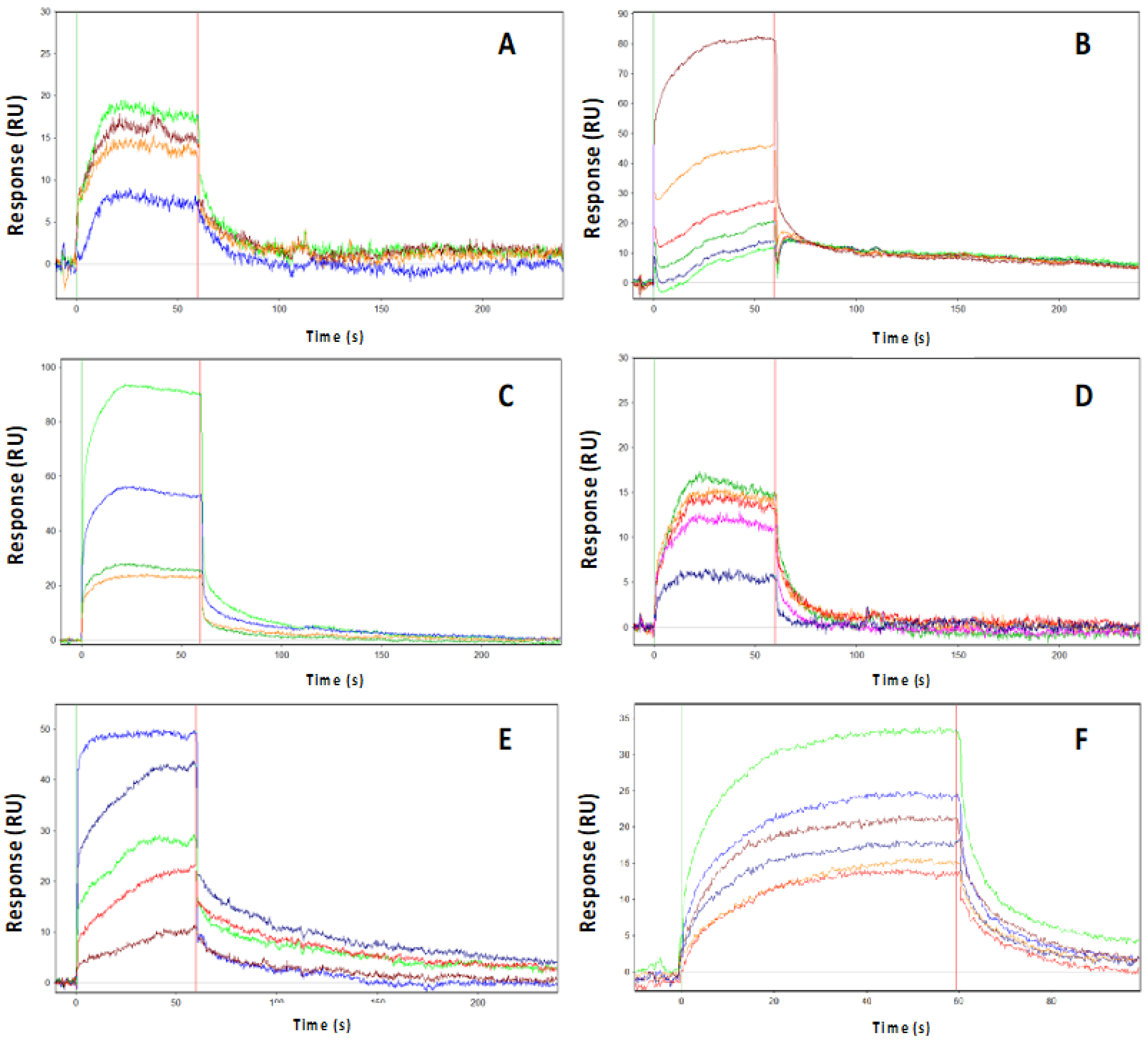
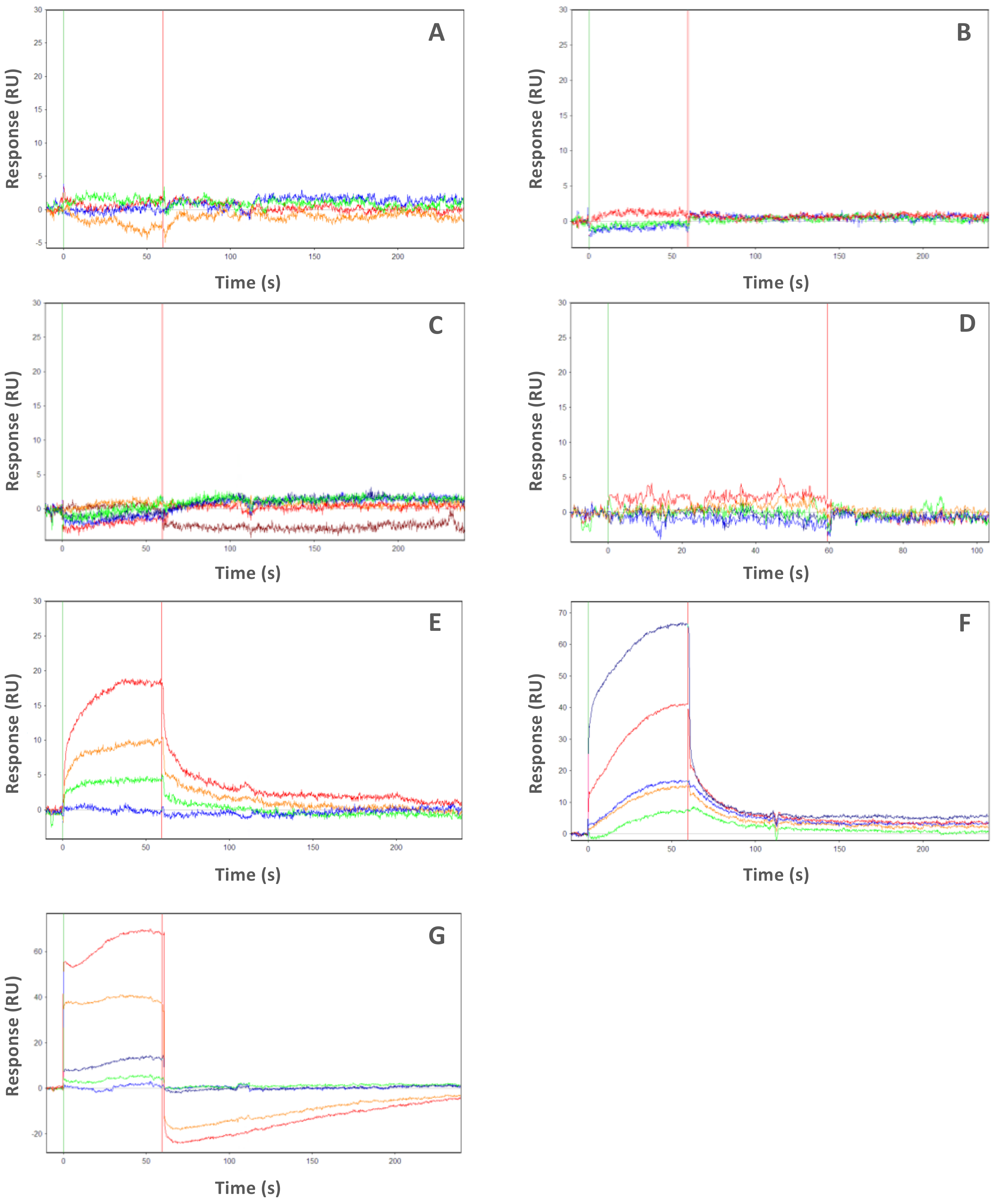
2.2. PHA-680626 and RPM1722 Behave as CD Inhibitors In Vitro
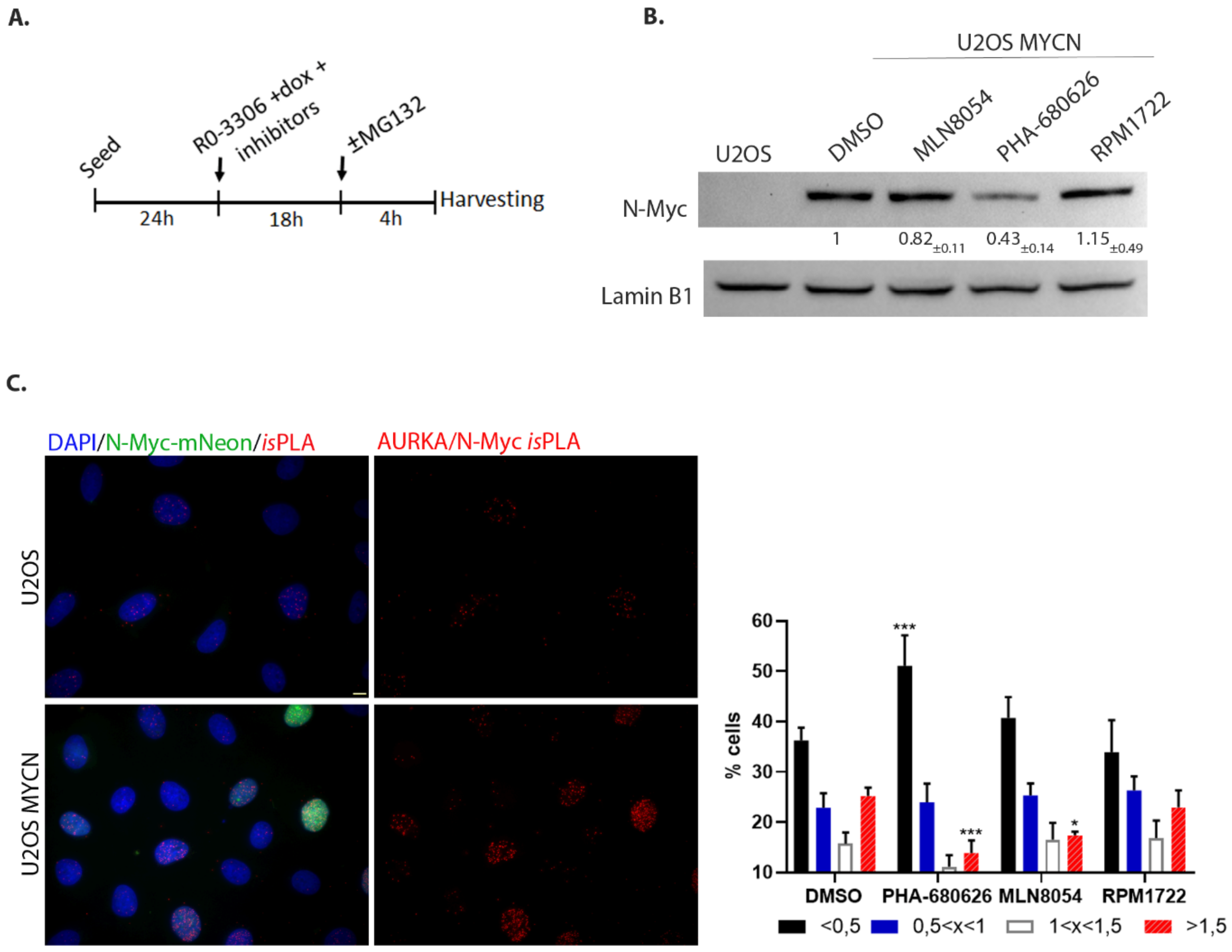
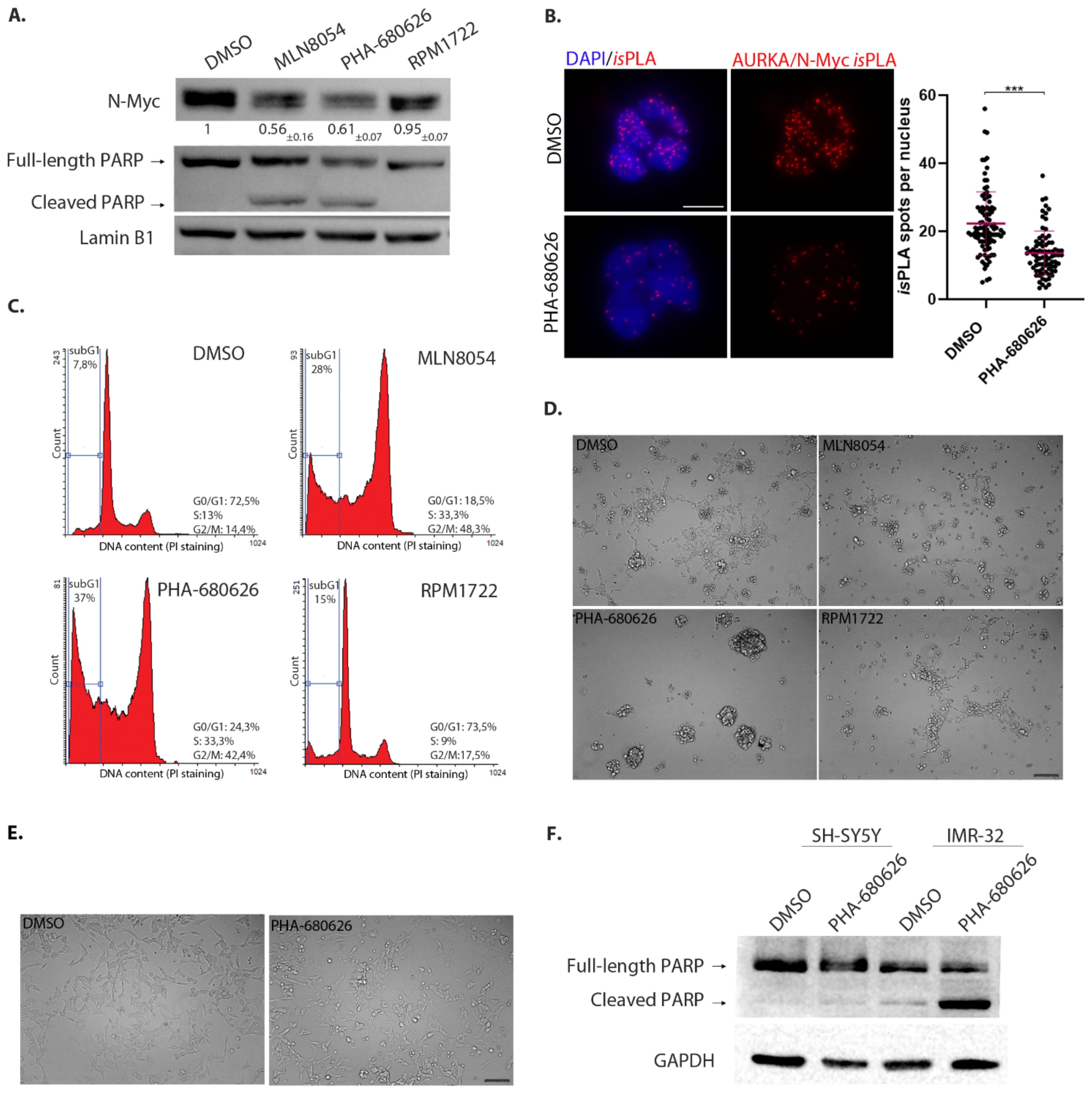
2.3. PHA-680626 Behaves as a CD Inhibitor in MYCN Overexpressing Cell Lines
3. Discussion
4. Materials and Methods
4.1. Screening on Protein Data Bank and Homology Modeling
4.2. Purification of AURKA Catalytic Domain
4.3. Chemistry
4.4. Kinase Activity Assays
4.5. Surface Plasmon Resonance
4.6. Cell Lines, Synchronization Protocols and Treatments
4.7. In Situ Proximity Ligation Assays (isPLA)
4.8. Immunofluorescence
4.9. Western Blotting
4.10. FACS Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Joukov, V.; De Nicolo, A. Aurora-PLK1 cascades as key signaling modules in the regulation of mitosis. Sci. Signal. 2018, 11, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnaghi-Jaulin, L.; Eot-Houllier, G.; Gallaud, E.; Giet, R. Aurora a protein kinase: To the centrosome and beyond. Biomolecules 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bischoff, J.R.; Anderson, L.; Zhu, Y.; Mossie, K.; Ng, L.; Souza, B.; Schryver, B.; Flanagan, P.; Clairvoyant, F.; Ginther, C.; et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998, 17, 3052–3065. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Zhou, H.; Zhang, R.; Yoon, D.S.; Vakar-lopez, F.; Ito, S.; Jiang, F.; Johnston, D.; Grossman, H.B.; Ruifrok, A.C.; et al. Amplification/Overexpression of a Mitotic Kinase Gene in Human Bladder Cancer. J. Natl. Cancer Inst. 2002, 94, 1320–1329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, R.; Huang, C.; Liu, K.; Li, X.; Dong, Z. Targeting AURKA in Cancer: Molecular mechanisms and opportunities for Cancer therapy. Mol. Cancer 2021, 20, 1–27. [Google Scholar] [CrossRef]
- Johnsen, J.I.; Dyberg, C.; Wickström, M. Neuroblastoma—A neural crest derived embryonal malignancy. Front. Mol. Neurosci. 2019, 12, 1–11. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S.; Brockmann, M.; Eilers, U.; Schüttrumpf, L.; Popov, N.; Kenney, A.M.; Schulte, J.H.; Beijersbergen, R.; Christiansen, H.; et al. Stabilization of N-Myc Is a Critical Function of Aurora A in Human Neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Ann. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef]
- Albihn, A.; Johnsen, J.I.; Henriksson, M.A. MYC in oncogenesis and as a target for cancer therapies. Adv. Cancer Res. 2010, 107, 163–224. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—the grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2021, 1–14. [Google Scholar] [CrossRef]
- Fladvad, M.; Zhou, K.; Moshref, A.; Pursglove, S.; Säfsten, P.; Sunnerhagen, M. N and C-terminal sub-regions in the c-Myc transactivation region and their joint role in creating versatility in folding and binding. J. Mol. Biol. 2005, 346, 175–189. [Google Scholar] [CrossRef]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [Green Version]
- Sjostrom, S.K.; Finn, G.; Hahn, W.C.; Rowitch, D.H.; Kenney, A.M. The Cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev. Cell 2005, 9, 327–338. [Google Scholar] [CrossRef] [Green Version]
- Chesler, L.; Schlieve, C.; Goldenberg, D.D.; Kenney, A.; Kim, G.; Mcmillan, A.; Matthay, K.K.; Rowitch, D.; Weiss, W.A. Inhibition of Phosphatidylinositol 3-Kinase Destabilizes Mycn Protein and Blocks Malignant Progression in Neuroblastoma. Cancer Res. 2006, 66, 8139–8146. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Orian, A.; Grim, J.A.; Eisenman, R.N.; Clurman, B.E. A nucleolar isoform of the Fbw7 ubiquitin ligase regulates c-Myc and cell size. Curr. Biol. 2004, 14, 1852–1857. [Google Scholar] [CrossRef] [Green Version]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Richards, M.W.; Burgess, S.G.; Poon, E.; Carstensen, A.; Eilers, M.; Chesler, L.; Bayliss, R. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Ignacio, P. Aurora kinase A inhibitors: Promising agents in antitumoral therapy. Expert Opin. Ther. Targets 2014, 18, 1377–1393. [Google Scholar]
- Levinson, N.M. The multifaceted allosteric regulation of Aurora kinase A. Biochem. J. 2018, 475, 2025–2042. [Google Scholar] [CrossRef] [Green Version]
- Brockmann, M.; Poon, E.; Berry, T.; Carstensen, A.; Deubzer, H.E.; Rycak, L.; Jamin, Y.; Thway, K.; Robinson, S.P.; Roels, F.; et al. Small Molecule Inhibitors of Aurora-A Induce Proteasomal Degradation of N-Myc in Childhood Neuroblastoma. Cancer Cell 2013, 24, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Gustafson, W.C.; Meyerowitz, J.G.; Nekritz, E.A.; Chen, J.; Benes, C.; Charron, E.; Simonds, E.F.; Seeger, R.; Matthay, K.K.; Hertz, N.T.; et al. Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [Green Version]
- Meyerowitz, J.G.; Weiss, W.A.; Gustafson, W.C. A new “angle” on kinase inhibitor design: Prioritizing amphosteric activity above kinase inhibition. Mol. Cell. Oncol. 2015, 2, 10–12. [Google Scholar] [CrossRef] [Green Version]
- Lake, E.W.; Muretta, J.M.; Thompson, A.R.; Rasmussen, D.M.; Majumdar, A.; Faber, E.B.; Ruff, E.F.; Thomas, D.D.; Levinson, N.M. Quantitative conformational profiling of kinase inhibitors reveals origins of selectivity for Aurora kinase activation states. Proc. Natl. Acad. Sci. USA 2018, 115, E11894–E11903. [Google Scholar] [CrossRef] [Green Version]
- Schindler, T.; Bornmann, W.; Pellicena, P.; Miller, W.T.; Clarkson, B.; Kuriyan, J. Structural mechanism for STI-571 inhibition of Abelson tyrosine kinase. Science 2000, 289, 1938–1942. [Google Scholar] [CrossRef] [Green Version]
- Pargellis, C.; Tong, L.; Churchill, L.; Cirillo, P.F.; Gilmore, T.; Graham, A.G.; Grob, P.M.; Hickey, E.R.; Moss, N.; Pav, S.; et al. Inhibition of p38 MAP kinase by utilizing a novel allosteric binding site. Nat. Struct. Biol. 2002, 9, 268–272. [Google Scholar] [CrossRef]
- Wan, P.T.C.; Garnett, M.J.; Roe, S.M.; Lee, S.; Niculescu-Duvaz, D.; Good, V.M.; Project, C.G.; Jones, C.M.; Marshall, C.J.; Springer, C.J.; et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004, 116, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Heron, N.M.; Anderson, M.; Blowers, D.P.; Breed, J.; Eden, J.M.; Green, S.; Hill, G.B.; Johnson, T.; Jung, F.H.; McMiken, H.H.J.; et al. SAR and inhibitor complex structure determination of a novel class of potent and specific Aurora kinase inhibitors. Biorgan. Med. Chem. Lett. 2006, 16, 1320–1323. [Google Scholar] [CrossRef]
- De Groot, C.O.; Hsia, J.E.; Anzola, J.V.; Motamedi, A.; Yoon, M.; Wong, Y.L.; Jenkins, D.; Lee, H.J.; Martinez, M.B.; Davis, R.L.; et al. A cell biologist’s field guide to aurora kinase inhibitors. Front. Oncol. 2015, 5, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Fancelli, D.; Moll, J.; Varasi, M.; Bravo, R.; Artico, R.; Berta, D.; Bindi, S.; Cameron, A.; Candiani, I.; Cappella, P.; et al. 1,4,5,6-Tetrahydropyrrolo[3,4-c]pyrazoles: Identification of a potent aurora kinase inhibitor with a favorable antitumor kinase inhibition profile. J. Med. Chem. 2006, 49, 7247–7251. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.P.; Zhu, J.Y.; Lawrence, H.R.; Pireddu, R.; Luo, Y.; Alam, R.; Ozcan, S.; Sebti, S.M.; Lawrence, N.J.; Schönbrunn, E. A novel mechanism by which small molecule inhibitors induce the DFG flip in Aurora A. ACS Chem. Biol. 2012, 7, 698–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soncini, C.; Carpinelli, P.; Gianellini, L.; Fancelli, D.; Vianello, P.; Rusconi, L.; Storici, P.; Zugnoni, P.; Pesenti, E.; Croci, V.; et al. PHA-680632, a Novel Aurora Kinase Inhibitor with Potent Antitumoral Activity. Clin. Cancer Res. 2006, 12, 4080–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodson, C.A.; Bayliss, R. Activation of Aurora-A kinase by protein partner binding and phosphorylation are independent and synergistic. J. Biol. Chem. 2012, 287, 1150–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgieva, I.; Koychev, D.; Wang, Y.; Holstein, J.; Hopfenmüller, W.; Zeitz, M.; Grabowski, P. ZM447439, a novel promising aurora kinase inhibitor, provokes antiproliferative and proapoptotic effects alone and in combination with bio- and chemotherapeutic agents in gastroenteropancreatic neuroendocrine tumor cell lines. Neuroendocrinology 2010, 91, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, A.; Chowdhury, S.; Tsai, M.Y. A novel Aurora kinase A inhibitor MK-8745 predicts TPX2 as a therapeutic biomarker in non-Hodgkin lymphoma cell lines. Leuk. Lymphoma 2012, 53, 462–471. [Google Scholar] [CrossRef]
- Schwab, M.; Westermann, F.; Hero, B.; Berthold, F. Neuroblastoma: Biology and molecular and chromosomal pathology. Lancet Oncol. 2003, 4, 472–480. [Google Scholar] [CrossRef]
- Ferguson, F.M.; Doctor, Z.M.; Chaikuad, A.; Sim, T.; Kim, N.D.; Knapp, S.; Gray, N.S. Characterization of a highly selective inhibitor of the Aurora kinases. Biorgan. Med. Chem. Lett. 2017, 27, 4405–4408. [Google Scholar] [CrossRef]
- Gilburt, J.A.H.; Sarkar, H.; Sheldrake, P.; Blagg, J.; Ying, L.; Dodson, C.A. Dynamic Equilibrium of the Aurora A Kinase Activation Loop Revealed by Single-Molecule Spectroscopy. Angew. Chem.-Int. Ed. 2017, 56, 11409–11414. [Google Scholar] [CrossRef] [Green Version]
- Tsuchiya, Y.; Byrne, D.P.; Burgess, S.G.; Bormann, J.; Baković, J.; Huang, Y.; Zhyvoloup, A.; Yu, B.Y.K.; Peak-Chew, S.; Tran, T.; et al. Covalent Aurora A regulation by the metabolic integrator coenzyme A. Redox Biol. 2020, 28, 101318. [Google Scholar] [CrossRef]
- Sloane, D.A.; Trikic, M.Z.; Chu, M.L.H.; Lamers, M.B.A.C.; Mason, C.S.; Mueller, I.; Savory, W.J.; Williams, D.H.; Eyers, P.A. Drug-Resistant Aurora A Mutants for Cellular Target Validation of the Small Molecule Kinase Inhibitors MLN8054 and MLN8237. ACS Chem. Biol. 2010, 5, 563–576. [Google Scholar] [CrossRef]
- Janson, G.; Paiardini, A. PyMod 3: A complete suite for structural bioinformatics in PyMOL. Bioinformatics 2020, 37, 1471–1472. [Google Scholar] [CrossRef]
- Lawrence, H.R.; Martin, M.P.; Luo, Y.; Pireddu, R.; Yang, H.; Gevariya, H.; Ozcan, S.; Zhu, J.Y.; Kendig, R.; Rodriguez, M.; et al. Development of o-chlorophenyl substituted pyrimidines as exceptionally potent aurora kinase inhibitors. J. Med. Chem. 2012, 55, 7392–7416. [Google Scholar] [CrossRef] [Green Version]
- Lundström, I. Real-time biospecific interaction analysis. Biosens. Bioelectron. 1994, 9, 725–736. [Google Scholar] [CrossRef]
- Frostell-Karlsson, Å.; Remaeus, A.; Roos, H.; Andersson, K.; Borg, P.; Hämäläinen, M.; Karlsson, R. Biosensor analysis of the interaction between immobilized human serum albumin and drug compounds for prediction of human serum albumin binding levels. J. Med. Chem. 2000, 43, 1986–1992. [Google Scholar] [CrossRef]
- Myszka, D.G.; Morton, T.A. Clamp: A biosensor kinetic data analysis program. Trends Biochem. Sci. 1998, 23, 149–150. [Google Scholar] [CrossRef]
- Myszka, D.G. Improving biosensor analysis. J. Mol. Recognit. 1999, 12, 279–284. [Google Scholar] [CrossRef]
- Khmelinskii, A.; Keller, P.J.; Bartosik, A.; Meurer, M.; Barry, J.D.; Mardin, B.R.; Kaufmann, A.; Trautmann, S.; Wachsmuth, M.; Pereira, G.; et al. Tandem fluorescent protein timers for in vivo analysis of protein dynamics. Nat. Biotechnol. 2012, 30, 708–714. [Google Scholar] [CrossRef]
- Ferrara, M.; Sessa, G.; Fiore, M.; Bernard, F.; Asteriti, I.A.; Cundari, E.; Colotti, G.; Ferla, S.; Desideri, M.; Buglioni, S.; et al. Small molecules targeted to the microtubule-Hec1 interaction inhibit cancer cell growth through microtubule stabilization. Oncogene 2018, 37, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Roeschert, I.; Poon, E.; Henssen, A.G.; Dorado Garcia, H.; Gatti, M.; Giansanti, C.; Jamin, Y.; Ade, C.P.; Gallant, P.; Schülein-Völk, C.; et al. Combined inhibition of Aurora-A and ATR kinases results in regression of MYCN-amplified neuroblastoma. Nat. Cancer 2021, 2, 312–326. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB Code | Inhibitor | PDB ID | Selectivity | IC50 (nM) | Angle (°) |
|---|---|---|---|---|---|
| 4J8M | CD532 | CJ5 | AURKA | 35 | 92.9 |
| 2X81 | MLN8054 | ZZL | AURKA | 6 | 84.6 |
| 2J4Z | PHA-680626 | 626 | PAN | 99 | 88.9 |
| 3UOH | RPM1722 | 0C4 | AURKA | 19 | 85.5 |
| - | MLN8237 * | A5B | AURKA | 6 | 85.0 ** |
| Inhibitors | Kon(M−1s−1) × 106 | Koff(s−1) × 10−3 | Kd(nM) | Res SD | Kd Myc-AIR(μM) |
|---|---|---|---|---|---|
| CD532 | 3.58 ± 0.05 | 93.7 ± 0.70 | 26.2 ± 0.4 | 0.83 | No binding |
| MLN8054 | 20.2 ± 0.03 | 46.1 ± 0.40 | 2.28 ± 0.03 | 1.254 | No binding |
| PHA-680626 | 0.91 ± 0.04 | 7.83 ± 0.02 | 8.56 ± 0.04 | 3.7 | No binding |
| RPM1722 | 2.61 ± 0.06 | 13.0 ± 0.10 | 5.0 ± 0.2 | 3 | No binding |
| Alisertib | 0.082 ± 0.1 | 5.04 ± 0.04 | 60 ± 1.0 | 2.3 | 48 |
| MK8754 | 7.8 ± 0.1 | 12.0 ± 0.09 | 1.55 ± 0.03 | 1.65 | 0.7 |
| ZM447439 | 0.096 ± 0.06 | 9.07 ± 0.04 | 92 ± 0.7 | 1.09 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boi, D.; Souvalidou, F.; Capelli, D.; Polverino, F.; Marini, G.; Montanari, R.; Pochetti, G.; Tramonti, A.; Contestabile, R.; Trisciuoglio, D.; et al. PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc. Int. J. Mol. Sci. 2021, 22, 13122. https://doi.org/10.3390/ijms222313122
Boi D, Souvalidou F, Capelli D, Polverino F, Marini G, Montanari R, Pochetti G, Tramonti A, Contestabile R, Trisciuoglio D, et al. PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc. International Journal of Molecular Sciences. 2021; 22(23):13122. https://doi.org/10.3390/ijms222313122
Chicago/Turabian StyleBoi, Dalila, Fani Souvalidou, Davide Capelli, Federica Polverino, Grazia Marini, Roberta Montanari, Giorgio Pochetti, Angela Tramonti, Roberto Contestabile, Daniela Trisciuoglio, and et al. 2021. "PHA-680626 Is an Effective Inhibitor of the Interaction between Aurora-A and N-Myc" International Journal of Molecular Sciences 22, no. 23: 13122. https://doi.org/10.3390/ijms222313122