
Non-Cell Autonomous and Epigenetic Mechanisms of Huntington’s Disease
by

Chaebin Kim
1,†, 
Ali Yousefian-Jazi
1,†,
Seung-Hye Choi
1,
Inyoung Chang
2,
Junghee Lee
3,4,5,* and
Hoon Ryu
1,* 1
Brain Science Institute, Korea Institute of Science and Technology, Seoul 02792, Korea
2
Department of Biology, Boston University, Boston, MA 02215, USA
3
Boston University Alzheimer’s Disease Research Center, Boston University, Boston, MA 02118, USA
4
Department of Neurology, Boston University School of Medicine, Boston, MA 02118, USA
5
VA Boston Healthcare System, Boston, MA 02130, USA
*
Authors to whom correspondence should be addressed.
†
These authors equally contributed to this review.
Int. J. Mol. Sci. 2021, 22(22), 12499; https://doi.org/10.3390/ijms222212499
Submission received: 5 October 2021
/
Revised: 10 November 2021
/
Accepted: 15 November 2021
/
Published: 19 November 2021
(This article belongs to the Special Issue Molecular Research on Huntington’s Disease)
Abstract
:Huntington’s disease (HD) is a rare neurodegenerative disorder caused by an expansion of CAG trinucleotide repeat located in the exon 1 of Huntingtin (HTT) gene in human chromosome 4. The HTT protein is ubiquitously expressed in the brain. Specifically, mutant HTT (mHTT) protein-mediated toxicity leads to a dramatic degeneration of the striatum among many regions of the brain. HD symptoms exhibit a major involuntary movement followed by cognitive and psychiatric dysfunctions. In this review, we address the conventional role of wild type HTT (wtHTT) and how mHTT protein disrupts the function of medium spiny neurons (MSNs). We also discuss how mHTT modulates epigenetic modifications and transcriptional pathways in MSNs. In addition, we define how non-cell autonomous pathways lead to damage and death of MSNs under HD pathological conditions. Lastly, we overview therapeutic approaches for HD. Together, understanding of precise neuropathological mechanisms of HD may improve therapeutic approaches to treat the onset and progression of HD.
1. Introduction
Huntington’s disease (HD) is a fatal progressive neurodegenerative disorder with a mid-life onset which ranges from infancy to the ninth decade. HD occurs in 5–10 cases per 100,000 persons worldwide, and characterized by chorea, emotional distress, and progressive cognitive decline [1]. Generally, in HD patients, there are more than 38 repeats of trinucleotide CAG within the Huntingtin (HTT) gene with an inverse relationship between the number of CAG repeats and the age of onset, indicating that the high number of CAG repeats cause the earlier phenotype of HD symptoms [2,3,4]. HTT has a crucial role in embryonic development by showing the death of embryos of huntingtin homozygous knockout mice by day 7.5 [5]. On the other hand, HTT plays an important role in cardiomyocytes cellular energy and nucleotides metabolism [6].
Currently, HD is considered as a multi-systemic neurodegenerative disease due to skeletal muscle and heart function disturbances with energy metabolism and mitochondrial alterations beyond the brain dysfunction [7]. Although most of studies show intracellular inclusions formed by mutant HTT (mHTT) protein in selected regions of the brain such as the striatum and cortex of HD brain [8,9], other studies find the expression and function of mHTT in skeletal muscle and heart as well [10]. The mHTT is involved in transcriptional alterations, disruption of intracellular transport, excitotoxicity, collapse of protein degradation mechanisms, mitochondrial dysfunction, and disorders of myelin, which make neurons more susceptible to generic stresses, eventually leading to neuronal death [11,12,13,14]. Yamanishi et al. have reported a novel cell death pathway known as transcriptional repression-induced atypical cell death of neurons (TRIAD) in the HD pathology, exhibiting that enlargement of endoplasmic reticulum (ER) contributes to neuronal cell damage without alteration of mitochondria and nuclei structure [15]. Another study has shown that inflammation may play a role in cardiac dysfunction in HD by overexpression of the inflammatory cytokine, tumor necrosis factor-α, in cardiomyocytes of R6/2 HD mice [16].
The epigenetic modifications are closely associated with the pathogenesis of HD [17,18]. mHTT sequesters specific transcription factors and impairs their function, resulting in disruption of their target genes transcription [19,20,21]. mHTT also regulates transcription by facilitating transcriptional factor interaction with protein complexes [22]. Otherwise, mHTT leads to histone hypoacetylation which can change genes expressions [23,24,25]. Moreover, several studies have shown that the biochemical defect and impairment of neuronal energy metabolism in HD patients are caused by mitochondrial dysfunction, where mitochondria is a major contributor of energy production and a regulator of intracellular signaling and survival [26,27]. The calcium-ion (Ca2+) buffering abnormalities and bioenergetic impairments in mitochondria can occur by interaction of mHTT with mitochondrial proteins [28,29,30]. Recently, researchers have become more interested in identifying the roles of non-neuronal cells in HD pathophysiology through elucidation of alterations in oligodendrocyte functions such as myelin formation [31,32,33] and astrocyte morphological changes and functions including impairment of glutamate metabolism and potassium homeostasis [34,35,36]. In this review, we discuss how astrocytes and oligodendrocytes contribute to HD pathogenesis via a non-cell autonomous pathway. Furthermore, we review the different epigenetic modifications which have key roles in neurotoxicity and neurogenesis impairment, and finally discuss potential therapeutic strategies for HD.
2. Non-Cell Autonomous Cell Death Pathway in HD
While the primary causes of neuronal damage and cell death are abnormalities within the damaged neuron itself, other neighboring cells such as glial cells also contribute to neuronal death [37,38]. In this regard, non-cell autonomous pathway is defined in this review as the mechanism of the neuronal damage caused by non-neuronal cells. Recent studies have proven the non-cell autonomous pathway in that reactive astrocytes lead to neuronal damage and neurodegeneration in Alzheimer’s disease (AD), amyotrophic lateral sclerosis (ALS), HD, and Parkinson’s disease (PD) [39,40,41,42]. Indeed, neuron-specific expression of mHTT in the striatum and cortex is not sufficient to fully induce pathological phenotypes of HD [43]. Otherwise, astrocyte-specific expression of mHTT does not induce neurodegeneration, but it shows neurological symptoms [44]. In the HD fly model (UAS HttQ100-mRFP) where mHTT is expressed in neurons, suppression of mHTT aggregation specifically in astrocytes expands the lifespan of HD fly [45]. On the other hand, white matter is degenerated and the oligodendrocyte differentiation is defective in HD [46]. mHTT-expressing microglia are hyperreactive to inflammatory stimuli to cause synaptic dysfunction in dendritic spines [47,48,49], where neurons try to control microglia activation but fail in HD [50]. Both mHTT and wtHTT are secreted from brain cells [51], where mHTT oligomers disrupt energy metabolism of neighboring cells [52]. mHTT not only affects brain metabolism, it also causes the dysfunction of other organs such as liver [53]. In the below sections, we discuss how mHTT affects astrocytes and oligodendrocytes in the neuropathogenesis of HD.
2.1. Alteration of Astrocyte Function
Astrocytes support neural functions by managing local environments including nutrients, ions, and neurotransmitters. In addition to the role as a generic supporter of neurons, astrocytes are involved in information processing [54], especially in the form of tripartite synapse [55]. Within the tripartite synapse, astrocytes tightly envelop synapses to clear residual neurotransmitters and ions after synaptic activities [56]. Failure in clearance of the residuals leads to accumulation of the neurotransmitters and ions. Excessive buildup of excitatory neurotransmitter such as glutamate is toxic to neurons and eventually causes neurodegenerative diseases [57]. High concentration of extracellular potassium ion depolarizes the membrane potential and contributes to the hyperexcitability of local neurons [58].
In the brain of HD patients, degeneration of medium spiny neurons (MSNs) in the striatum starts in the early stage of the disease before the widespread cell death in the striatum. The mechanism of the degeneration of MSNs is regulated by glutamate-mediated excitotoxicity via N-methyl-D-aspartate receptor (NMDAR) [40]. NMDAR is composed of three types of subunits (GluN1, GluN2A-D, and GluN3A-B), where NMDAR that contains GluN2A or GluN2B control synaptic dynamics [59]. Particularly, GluN2B-type NMDARs are phosphorylated by death-associated protein kinase to serve as toxic receptors [60]. Here, GluN2B-type NMDAR is highly expressed in mature MSNs of striatum, which is the reason behind the selective vulnerability of MSNs in HD [61,62]. Striatal neurons in HD show upregulated surface expression of GluN2B-type NMDAR due to the dysfunction of the huntingtin-interacting protein 14L [63]. Otherwise, gene silencing of a glutamate receptor subunit reverses HD phenotype [64]. Excitotoxicity is also studied systemically. Considering that most glutamatergic input of striatum is from the cortex, destruction of cortico-striatal pathway is a key process in presymptomatic phase of the disease [65]. Dysfunction of MSNs increases the excitation from the cortex in the form of positive feedback loop and exacerbates the excitotoxicity [66]. Interestingly, NMDAR response in the corticostriatal synapse is rescued to normal state by astrocyte-specific reduction of mHTT in BACHD mice [67]. In this context, astrocytes are recently reviewed as a player in excitotoxicity in HD and other neurodegenerative disorders [11,35,39,40,41,42,68,69,70,71,72,73,74,75,76,77].
In HD astrocytes, mRNA level of excitatory amino acid transporter 2 (EAAT2) is lower compared to that of the normal group [78,79]. In addition, protein level of EAAT2 is reduced throughout whole brain [34,80,81]. Low EAAT2 level is rendered as decreased glutamate uptake [82,83,84], which results in neuronal degeneration from chronic glutamate stimulation [85]. In addition, downregulation of an inwardly rectifying potassium channel, Kir4.1, in HD astrocytes results in increased extracellular K+ concentration, which subsequently increases the resting membrane potential of nearby neurons to make the neurons more excitable [86]. Interestingly, downregulation of EAAT2 and Kir4.1 in astrocytes not only induces hyperexcitability of neurons, but also induces evoked Ca2+ signaling in the astrocytes [87]. Increase of evoked Ca2+ level in astrocytes increases sodium pump activity which further increases extracellular K+ concentration [88] (Figure 1, Table 1).
2.2. Alteration of Oligodendrocyte Function
Oligodendrocytes are a type of glial cells in the brain and the spinal cord which produce myelin sheaths and play an important role in maintaining axonal integrity and function. Although oligodendrocytes are less explored in HD in previous studies, defective oligodendrocyte functions and deficient myelination are commonly observed in other neurodegenerative diseases [91]. Myers et al. are the first time to show an increase of oligodendrocytes in the striatum but no changes in astrocytes in postmortem HD brains [92]. Later, the other researchers identify myelin damage and breakdown in pre-symptomatic HD patients [93,94]. The full-length myelin regulatory factor (fMYRF) is self-cleaved to N-terminal myelin regulatory factor (nMYRF) which was transferred from the ER to the nucleus. In HD, binding of mHTT to nMYRF deficits normal bindings of nMYRF transcription factor in nucleus which leads to inhibition of myelination-related genes expression and oligodendrocyte dysfunction [95,96,97] (Figure 2). On the other hand, Cui et al. show that the expression of proliferator-activated receptor-gamma coactivator (PGC)-1α is significantly downregulated in HD striatal cells and tissues. In HD, mHTT interferes with promoter binding of cAMP response element binding protein (CREB) and TATA-binding protein-associated factor (TAF), which regulate the expression of PGC-1α. This mis-binding leads to the inhibition of PGC-1α expression which may cause reduction of myelin basic protein (MBP) expression and myelination deficit [98,99] (Figure 2). In parallel with these results, Xiang et al. also show the downregulation of MBP and deficient myelination in the oligodendrocytes of R6/2 transgenic mouse model of HD, and in the striatum of PGC-1α knockout mice as well [100]. Further study is necessary to verify whether PGC-1α rescues myelination in HD models in a cell-type-specific manner.
3. The Role of Epigenetic Modifications and Noncoding RNAs in the Pathogenesis of HD
Better understanding of epigenetic mechanisms may provide important insights, resulting in improved therapeutic approaches for treating HD [101]. In this section, we discuss the epigenetic changes and mechanisms that are associated with the pathogenesis of HD. We focus on two main epigenetic alterations that influence chromatin structure: DNA and histone modifications [102]. DNA methylation and hydroxymethylation have been involved in different neurodevelopmental and psychiatric disorders [103,104,105]. In DNA methylation, methyl groups are transferred to the cysteine 5 position of cytosine via the action of DNA methyltransferases [106]. Ng et al. propose that mHTT has a significant effect on changing the methylation of promoter regions of octamer-binding transcription factor 1 (OCT4), sex determining region Y-box 2 (SOX2), and Nanog homeobox (NANOG) as these genes are involved in neurogenesis. Therefore, inhibition of the expression level of these genes may lead to neurogenesis impairment and cognitive decline in HD [107] (Figure 3). In addition, histone modification is another major epigenetic mechanism which plays a special role in unraveling the pathogenesis of HD. CREB binding protein (CBP) interacts with several transcription factors such as specificity protein 1 (SP1), TAF, and RNA polymerase II, and acts as a co-activator or a repressor of transcription [108,109]. The CBP can also be considered as a histone acetyltransferase which acetylates histones to alter chromatin structure [110]. The mHTT interaction with CBP blocks its transcriptional co-activator function and inherent CBP histone acetyltransferase activity [111]. Therefore, CBP sequestration and depletion are accompanied by histone hypoacetylation, resulting in neuronal transcriptional dysfunction and neurotoxicity [112,113,114] (Figure 3).
Our group has found that SET domain bifurcated histone lysine methyltransferase 1 (SETDB1/ESET), a histone H3 at lysine 9 (H3K9)-specific methyltransferase, is elevated in the striatal neurons of HD patients and HD transgenic (R6/2) mice [115]. In parallel, the level of histone H3K9me3 is increased in the striatal neurons of HD patients and in HD transgenic (R6/2) mice. This study has proven that the SETDB1-H3K9me3 pathway is involved in silencing of genes in HD. Interestingly, not only SETDB1 modulates the nuclear gene transcription though heterochromatin remodeling, but it also down regulates the nucleolar gene transcription (ribosomal DNA components) by increasing methylation of upstream binding protein 1 (UBF1). SETDB1 interacts with UBF1 and trimethylates at lysine 232/235 in the nucleolus of striatal cells. As a result, trimethylated UBF1 leads to nucleolar chromatin condensation and down regulates the transcription of ribosomal DNA (rDNA) [12]. This study presents a novel epigenetic mechanism that SETDB1-UBF1 trimethylation pathway is associated the nucleolar chromatin remodeling and dysfunction of rDNA transcription in the pathogenesis of HD.
Moreover, several studies have focused on microRNAs (miRNAs) which are involved in the early differentiation, development, and function of neurons [116,117]. miR-146a is one of the major regulators of the NF-κB pathway which can also target human and mouse HTT gene [118,119]. Das et al. demonstrate that heat shock factor 1 is regulated by this miRNA, resulting suppression of mHTT aggregates in HD cells [120]. Another study confirmed that miR-214 directly targets the HTT gene which can suppress mHTT aggregation in an HD mouse striatal cell and HEK293T cell [120,121]. On the other hand, Bucha et al. showed the upregulation of miRNA-214 in HD cells could regulate mitofusin2, resulting in alteration of mitochondrial morphology [122]. Therefore, this miRNA can be considered as a critical node for therapeutic targets in HD pathogenesis.
4. Roles of Wild Type HTT (wtHTT) Versus mHTT in Vesicle Trafficking
Understanding the exact molecular and cellular functions of wtHTT and mHTT is crucial in further clarifying the pathogenesis of HD. wtHTT is involved in axonal transport, which is essential for neuronal synaptic activity [123]. Transport of cargo is important for neuron to work properly because of its unique morphology containing axons and dendrites. Vesicular transport is accelerated by overexpression of wtHTT [124]. There are emerging models to explain how wtHTT coordinates vesicular transport and ongoing studies to discover a more detailed mechanism of coordination and the HD pathology related to vesicle transport [125] (Figure 4).
wtHTT recruits glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to transport vesicles, where vesicular GAPDH produce adenosine 5′-triphosphate (ATP) to provide energy for the transport [126]. In HD pathogenesis, GAPDH is sequestered by mHTT [127,128], where the sequestration of GAPDH is rescued by high-affinity RNA aptamers that specifically recognize mHTT [129]. In addition, HTT forms complexes of motor proteins with huntingtin-associated protein-1 and p150Glued subunit of dynactin [130]. HTT-associated protein-1 binds to both kinesin-1 and vesicles to serve as an adaptor [131]. Huntingtin’s recruitment of kinesin-1 is governed by the phosphorylation of wtHTT at serine421 (Ser421), which stimulates anterograde transport [132]. Interestingly, phosphorylation of HTT (Ser421) protects against the mHTT toxicity, where the endogenous level of phosphorylated HTT (Ser421) is least in the striatum [133].
Defects in the axonal transport are associated with neurodegenerative diseases. For example, mutations in amyloid beta precursor protein obstruct motor protein activity of the hippocampal and cortical neurons in AD, and mutation in superoxide dismutase type-1 impede binding of motor proteins to neurofilaments of motor neurons in ALS [134]. In fact, fast axonal transport is commonly disrupted in polyglutamine-expansion diseases [135]. In HD, fast axonal transport is slowed down specifically in striatal neurons [136]. The impairment of the vesicular transport induces axonal degeneration, which is the early neuropathology of HD [137].
Pathogenic HTT disrupts the motility of vesicle complex, accessory proteins, and molecular motors [138], hence, the efficiency of vesicle trafficking [131,139] (Figure 4). Comparably, both HTT-depleted neurons and mHTT-expressing neurons suffer from defective axonal transport [140], where motor proteins are sequestered by mHTT [141]. Tubulin acetylation is also reduced in HD resulting in reduced binding of motor proteins to microtubules [142]. Vesicle trafficking related proteins such as HTT interacting protein 1, dynamin, and endophilin-A3 are depleted by mHTT bodies [143].
On the other hand, mHTT activates axonal c-Jun amino-terminal kinase3 via stress-signaling kinase [144], where inhibition of the c-Jun amino-terminal kinase/c-Jun partially restores striatal neurodegeneration in HD [85]. Consequently, kinesin-1 is phosphorylated at serine 176, which results in detachment of kinesin-1 and cargo from the microtubules [145].
Despite the growing evidence of mHTT and its effects on vesicle trafficking in neurons, further study is necessary to define why MSNs are much more susceptible to mHTT than other neuronal cell types. Additionally, precise cellular and molecular mechanism of mHTT oligomers versus mHTT aggregates-dependent vesicle trafficking should be determined.
In addition, wtHTT is also involved in autophagy as reviewed in [146]. wtHTT form complex with sequestosome 1 to enhance cargo recognition, where depletion of wtHTT results in empty autophagosome [147]. C-terminal domain of wtHTT has structural homology with yeast autophagy scaffold protein 11 and both proteins show similar protein–protein interaction patterns [148]. Interestingly, deletion of N-terminal domain of wtHTT in mouse suffers from DNA damage in striatum and cortex without any difference in autophagy function [149]. wtHTT is also associated with ER, where ER stress release the wtHTT to promote autophagy (reviewed in [150]). In addition, wtHTT has important role in homeostasis of presynaptic and postsynaptic terminal [151]. Loss of wtHTT lead to dysfunction of synaptic vesicle endocytosis in striatal neurons [152].
We need to provide attention to an important HD pathophysiology that the dysfunctions of central nervous system and other organs in HD are caused by mHTT accumulation as well as by the loss of functionality of wtHTT protein. Molecular simulation reveals that mHTT oligomer also sequester wtHTT [153]. Indeed, wtHTT protein expression level is inversely correlated to the age of onset [154]. In macrophage, reduced wtHTT level is associated with decreased cytokine and increased phagocytosis [155]. Research is ongoing to reveal the wtHTT function and structure further. RNA-seq of wtHTT knockout neural cell shows that wtHTT has a role in development of neurons and neurotransmission [156]. Cryo-electron microscopy structure of wtHTT confirms its role in protein–protein interaction [157]. The importance of the loss of functionality of wtHTT is associated with clinical safety of HTT gene therapy as reviewed previously [158].
5. Mitochondria Dysfunction in HD
A growing body of evidence show that mitochondrial dysfunctions, including membrane potential and respiratory function deficits, Ca2+ buffering capacity reduction, and mitochondrial number and morphology alteration, play a critical role in HD pathogenesis [159,160,161,162,163,164,165]. The mHTT aggregation reduces the mitochondrial membrane potential and increases the level of mitochondrial matrix Ca2+ loading that leads to decreased ATP level and enhanced reactive oxygen species (ROS) [160,166,167]. Moreover, the release of cytochrome c from dysfunctional mitochondria leads to activation of caspases 9 and 3 which are involved in apoptosis, resulting in neuronal cell death [168,169] (Figure 5). On the other hand, Yablonska et al. show mHTT binding with high affinity to translocase of mitochondrial inner membrane 23 (TIM23) complex in mitochondrial intermembrane space leads to inhibition of import of nuclear-encoded proteins through TIM23 [14]. Therefore, the mHTT–TIM23 complex interaction alters mitochondrial proteome, resulting in mitochondrial dysfunction in HD [170] (Figure 5). In addition, Guo et al. showed that valosin-containing protein (VCP) is bound to mHTT as a binding protein on the mitochondria. Mitochondria-accumulated VCP works as a mitophagy adaptor to bind to the autophagosome component, microtubule-associated proteins 1A/1B light chain 3B (LC3), leading to enhanced mitophagy, reduced mitochondrial mass, and ultimately, neuronal cell death [28,171] (Figure 5). Moreover, the previous studies demonstrated that down regulation of wtHTT is related to mitochondria dysfunction by inability of the mitochondria to generate ATP [172] and diminished purines and inosine monophosphate [6].
6. Therapeutic Approaches for Huntington’s Disease
Despite remarkable efforts to overcome the symptoms of HD, effective therapeutic targets are still very limited in HD. Furthermore, no standard treatment has been established for HD. HD transgenic mouse models have been used for translational study with many candidate drugs before conducting clinical trials with patients, but the efficacy of most drugs is lower than expected [173]. Accordingly, the benefits of translating the therapeutic efficacy from the HD transgenic mouse models to human patients are not clear.
Gene editing method and strategy have been attempted for treating various genetic disorders including HD. Clustered regularly interspaced short palindromic repeats and (CRIPSR) and CRISPR-associated genes (CRISPR/Cas9) has been applied haplotype-specifically to common promoter-local single-nucleotide polymorphisms (SNPs) for the selective deletion of mHTT [174,175,176,177]. Otherwise, Zinc finger proteins containing the Kruppel associated box (KRAB-ZFPs), short hairpin RNA (shRNA), small interfering RNA (siRNA), and miRNA have been examined to impair transcription of mHTT [178,179,180,181,182,183,184,185]. To use siRNA or antisense oligonucleotide to knock-down mRNA of mutant Hunting requires repetitive administration. shRNA treatment lasts relatively longer than siRNA treatment; however, the dosage control of shRNA treatment is limited [186]. miRNA therapy suffers from off-target effect in general, which is recently overcome using in silico analysis [187]. Glutamine repeat-binding [188] and deletion [189] on mHTT gene are also effective in HTT lowering and alleviate HD phenotypes. For further information, gene targeting approaches are reviewed in [190,191,192,193,194,195,196,197,198,199,200,201].
Among many small compounds, epigenetic modulators have been used for rescuing transcriptional dysfunction in HD. For example, phenylbutyrate, sodium butyrate, histone deacetylases inhibitor (HDACi) 4b and LBH589, Tubastatin A, and CKD-504 hinder histone deacetylase increase the acetylation of H3K9, and improve neuropathology, behaviors, and survival of HD transgenic mice [202,203,204,205,206,207,208]. Importantly, epigenetic compounds exhibit transgenerational effect in HD animal models [205]. Mithramycin, a DNA binding drug, inhibits expression of histone methyltransferase, reduces H3K9me3 level and heterochromatin condensation, and ameliorates symptoms of HD [209]. In order to improve the efficacy of epigenetic compounds, further efforts to reduce the side effect of these drugs need to be made.
In addition to HD genetic and epigenetic targets, pathologic phenotypes including HTT fragmentation and aggregates, transcriptional dysfunction, oxidative stress, apoptosis, autophagy dysfunction, and excitotoxicity appear to be reasonable drug targets [101] (Table 2). The most effective therapeutic strategy in HD is to target the inhibition of aggregation or fragmentation of mHTT, because mHTT is directly responsible for the pathogenesis of HD. Cystamine, Congo red, Chrysamine G, direct fast yellow, and trehalose are drugs that bind to polyglutamine or block oligomerization, and consequently inhibit the aggregation of mHTT. Congo red, an organic compound and diazo dye, binds to β–sheets of protein structure of mHTT and prevents polyglutamate oligomerization. Since Congo red cannot cross the blood–brain barrier, compounds with similar structure of Congo red are discovered as potential drugs. Chrysamine G and direct fast yellow are found to effectively inhibit mHTT aggregation [210]. Saccharides including trehalose also bind directly to the polyglutamate region of mHTT to suppress the mHTT aggregation effectively [211]. Both antibodies which binds polyproline domain of mHTT and DnaJ heat shock protein family member B6 also reduce mHTT aggregation [212]. Otherwise, insulin, exendin-4, GM1, RCAN1-IL, and SGK block mHTT aggregation by increasing mHTT phosphorylation and modifying mHTT toxicity through post-translational modification. Increasing the phosphorylation of mHTT enhances solubility and decreases aggregation. Surprisingly, phosphorylation of mHTT on Ser 421 is known to be neuroprotective [213,214]. Formation of mHTT aggregate is exacerbated by transglutaminase, which cross-links mHTT. Inhibition of transglutaminase with cystamine reduces abnormal behavior, extends lifespan, and prevents weight loss of HD transgenic (R6/2) mice. In addition, cystamine injected HD mice have higher expression level of Dnajb1, which catalyze ATP hydrolysis. In addition to the mHTT aggregation, fragmentation of mHTT has been a plausible therapeutic target for HD because the pathology of HD is exacerbated by mHTT fragments including polyglutamate region [215]. Minocycline and Z-VAD-FMK inhibit caspases to prevent the proteolysis of mHTT and improve neuropathology of HD transgenic mice [216].
Interestingly, Rieux et al. (2020) tested whether a parabiosis therapy, an in vivo blood transfusion via surgical linking of two bodies, can reduce mHTT propagation and pathology in HD transgenic mice (zQ175 mice) [217]. It is concluded that blood transfusion improves mitochondrial activity in peripheral organs and ameliorates neuropathology in MSNs of striatum. This study indicates that healthy blood can diminish the pathogenicity of circulating mHTT. If the concentration of mHTT exceeds a certain concentration in the body, it is likely to cause a disease onset of HD systemically. In this paradigm, reducing the concentration of mHTT by removing circulating mHTT with blood transfusion can be another treatment. However, application of the parabiosis therapy for HD may need further verification in regard to unexpected adaptive immune reactions in vivo.
Mitochondrial dysfunction is also one of the therapeutic targets in HD. Creatine is applied to restore mitochondrial dysfunction as it deactivates mitochondrial permeability transition [218]. Coenzyme Q10 promotes electron transport chain activity, which in turn improves mitochondrial respiration [218,219,220]. Both creatine and coenzyme Q10 have been used as beneficial compounds in HD and progressed up to Phase II clinical trials. Mitochondrial dysfunction induces oxidative stress, which can be managed by antioxidants (reviewed in [221]). PGC-1α is associated with transcriptional regulation of mitochondria-related genes and is also the target of HD therapy (reviewed in [222]). rhIGF-1 increases glucose uptake and regulates energy metabolism in striatal neurons, and its therapeutic effect has been tested in HD transgenic mouse models (R6/2 and YAC128) [223]. Autophagy is also involved in the clearing and recycling of mHTT in MSNs and its function is impaired in HD [224]. Niclosamide reduces mHTT by increasing autophagy activity [225]. It seems likely that niclosamide is therapeutically more effective in increasing lysosomal degradation of ubiquitinated molecules including ubiquitinated mHTT rather than activating proteasomal activity [226]. Apoptotic cell death of MSNs has been a therapeutic target in HD [227]. MAP4343, 17EE2, and isoquercitrin are known to control stress responses and reduce apoptosis of MSNs in HD [228]. Z-VAD-FMK, Z-DEVD-FMK, Z-LEHD-FMK, PG3d, and lithium chloride are well-known apoptosis inhibitors and used to treat HD animal models (C. elegans and Rat) [216,226,227,229,230]. Laquinimod increase the brain-derived neurotrophic factor level in striatum of R6/2 mouse model and has the neuroprotective effect [231].
Continuous stimulation by excitatory or inhibitory neurotransmitter can damage MSNs in HD. Notably, controlling glutamate-induced neurotoxicity is one of many therapeutic strategies for treating HD and other neurodegenerative disorders (reviewed in [232]). Silencing of a glutamate receptor subunit could reverse HD phenotype [64]. Activation of NMDA receptors and cation channels elevates intracellular Ca2+ flux, impairs mitochondria function, and triggers neuronal cell death pathways. Memantine acts as an inhibitor of NMDA receptor, draws attention in HD therapy, and its clinical trials are on-going at phase 2 and phase 4, respectively. Otherwise, necrostatin-1, an inhibitor of receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and necrosis, shows positive effects for delaying the onset and improving motor behaviors while the survival extension is not improved in HD transgenic (R6/2) mice [77,233,234].
7. Conclusions
Since the mutation of HTT gene at exon 1 with glutamine repeats was identified as the cause of HD in 1993 [2], many studies have shown that mHTT proteins directly cause the neuropathogenesis of HD. wtHTT plays an important role in vesicular transport, which is an essential cellular event in MSNs, whereas mHTT disrupts vesicle transport by sequestrating motor proteins. Understanding of exact mechanisms on the mHTT-induced selective neuronal damage in the neostriatum is pivotal to develop beneficial therapeutic targets or strategies to ameliorate the neurodegeneration in HD. In this context, further investigations about effective clearance or detoxification of mHTT remain to be performed.
The brain is a multicellular organ. Accordingly, it is possible that mHTT-induced cellular dysfunctions are varied and differentially modulated in specific brain regions and cell-type specific manner. In terms of autonomous versus non-cell autonomous neuronal damage, it is also critical to determine which brain cell-types (e.g., excitatory neurons, inhibitory neurons, astrocytes, and oligodendrocytes) are vulnerable to mHTT and contribute to the pathogenesis of HD [11,70]. Importantly, gliosis, production of new astrocytes, microglia, and oligodendrocytes, is a prominent pathology in HD as well as in other neurodegenerative disorders (reviewed in [74,77]). Therefore, it is necessary to define how mHTT affects the fate of neuron and glia, and whether therapeutic targets can selectively modulate and rescue cell-type specific functions in HD.
The scope of this review is to briefly introduce the previous and recent studies about mechanisms of HD pathologies and therapeutic strategies and that our review has limitation in the scope. Just in case, for the readers need further information, we recommend the previous reviews dealt with the specific aspects of striatal vulnerability [248], white matter phenotype [249], cerebellar dysfunction [250], progression of cell type-specific phenotype [251], microglial activation [252], synapse [253], intracellular transmission of mHTT [254], protein–protein interactions [255], biochemical alterations and HTT dynamics [256,257], posttranslational modifications [258], proteostasis [259], autophagy [260,261], redox homeostasis [262], metabolism [263,264], HTT mRNA [265,266], Ca2+ and dopamine signaling [61], inflammation [267], in vitro modelling of HD [268,269], striatal neurogenesis [270], stem cell treatment [271,272,273,274,275,276,277,278,279], electric stimulation therapy [280], network connectivity in presymptomatic HD brain [281], non-motor symptoms [282], gut microbiome [283], human immunodeficiency virus [284], diagnosis [285,286], clinical progression [287], treatment for the symptoms [288], physical therapy [289], psychological interventions [290,291], and management of agitation [292]. Collectively, the previous studies have potential to reveal spatiotemporal and cell-type specific mechanism of HD pathology. The future challenges in HD research are brought by the complexity of the pathology from biochemical level [293,294,295,296,297,298,299,300,301,302,303] to system level [304,305,306,307,308]. Accordingly, the ultimate mechanisms of HD pathology can be further scrutinized by state-of-the art research methods such as multi-omics approach combining transcriptome, proteome, and interactome [309], big data analysis with machine learning [310], and meta-analysis combining the publicly available data [311]. On the other hand, the potential HD therapeutics should specifically modulate the function of the striatal neurons while they prevent the adverse behavior of glial cells. High-throughput in silico and in vitro screening of chemical libraries [312,313,314,315,316] are expected to expedite the designing of beneficial compounds for HD.
Previous studies indicate that epigenetic cellular events have been emerged as potential therapeutic targets in HD [12,109]. The reversible characters of epigenetic modifications during the pathogenesis of HD are reasonable therapeutic targets. It is highly expected that we can prevent neuronal damage more efficiently by balancing the epigenetic disequilibrium in HD before the pathogenesis becomes irreversible and degenerative under HD stress condition. In this regard, future therapeutic strategies and agents to treat HD should consider appropriate epigenetic targets and cell-type specificity. On the other hand, identification of blood cell-derived epigenetic markers that can mimic the brain molecular pathology, will facilitate the advanced diagnosis and treatment of HD. Taken together, development of cell-type specific epigenetic therapeutic targets will pave a way to slow down the onset and progress of HD.
Author Contributions
C.K., A.Y.-J., S.-H.C. and H.R. wrote and revised the manuscript; C.K, A.Y.-J. and H.R. drew the figures; I.C. and J.L. edited the manuscript; and J.L. and H.R. read, reviewed, and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the National Research Foundation (NRF) grants (NRF-2018M3C7A1056894, and 2020M3E5D9079742), the National Research Council of Science & Technology (NST) grant (No. CRC-15-04-KIST) from Korea Ministry of Science, ICT and Future Planning (MSIP), and grants (2E30951, 2E30954 and 2E30962) from Korea Institute of Science and Technology of South Korea. This study was also supported by NIH grants (R01AG054156 to H.R. and R01NS109537 to J.L.).
Acknowledgments
The authors would like to thank Go Woon Choi in the BSI of KIST for her kind administrative support.
Conflicts of Interest
The authors have no conflict of interest to declare.
References
- Exuzides, A.; Crowell, V.; Reddy, S.R.; Chang, E.; Yohrling, G. Epidemiology of Huntington’s disease (HD) in the US medicare population. Neurology 2020, 94, 670. [Google Scholar]
- The Huntington’s disease collaborative research group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Di Prospero, N.A.; Fischbeck, K.H. Therapeutics development for triplet repeat expansion diseases. Nat. Rev. Genet. 2005, 6, 756–766. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Polyglutamine pathogenesis: Emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron 2002, 35, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Cattaneo, E.; Rigamonti, D.; Goffredo, D.; Zuccato, C.; Squitieri, F.; Sipione, S. Loss of normal huntingtin function: New developments in Huntington’s disease research. Trends Neurosci. 2001, 24, 182–188. [Google Scholar] [CrossRef]
- Tomczyk, M.; Glaser, T.; Ulrich, H.; Slominska, E.M.; Smolenski, R.T. Huntingtin protein maintains balanced energetics in mouse cardiomyocytes. Nucleosides Nucleotides Nucleic Acids 2020. accepted. [Google Scholar] [CrossRef]
- Rüb, U.; Seidel, K.; Heinsen, H.; Vonsattel, J.P.; den Dunnen, W.F.; Korf, H.W. Huntington’s disease (HD): The neuropathology of a multisystem neurodegenerative disorder of the human brain. Brain Pathol. 2016, 26, 726–740. [Google Scholar] [CrossRef] [PubMed]
- DiFiglia, M.; Sapp, E.; Chase, K.O.; Davies, S.W.; Bates, G.P.; Vonsattel, J.P.; Aronin, N. Aggregation of Huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 1997, 277, 1990–1993. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A. Intranuclear neuronal inclusions: A common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 1997, 19, 1147–1150. [Google Scholar] [CrossRef] [Green Version]
- Kojer, K.; Hering, T.; Bazenet, C.; Weiss, A.; Herrmann, F.; Taanman, J.-W.; Orth, M. Huntingtin aggregates and mitochondrial pathology in skeletal muscle but not heart of late-stage R6/2 mice. J. Huntingt. Dis. 2019, 8, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Creus-Muncunill, J.; Ehrlich, M.E. Cell-autonomous and non-cell-autonomous pathogenic mechanisms in Huntington’s disease: Insights from in vitro and in vivo models. Neurotherapeutics 2019, 16, 957–978. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Kim, K.Y.; Kowall, N.W.; Ryu, H. Epigenetic mechanisms of neurodegeneration in Huntington’s disease. Neurotherapeutics 2013, 10, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Trushina, E.; Dyer, R.B.; Badger, J.D.; Ure, D.; Eide, L.; Tran, D.D.; Vrieze, B.T.; Legendre-Guillemin, V.; McPherson, P.S.; Mandavilli, B.S. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol. Cell. Biol. 2004, 24, 8195–8209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yablonska, S.; Ganesan, V.; Ferrando, L.M.; Kim, J.; Pyzel, A.; Baranova, O.V.; Khattar, N.K.; Larkin, T.M.; Baranov, S.V.; Chen, N.; et al. Mutant huntingtin disrupts mitochondrial proteostasis by interacting with TIM23. Proc. Natl. Acad. Sci. USA 2019, 116, 16593–16602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanishi, E.; Hasegawa, K.; Fujita, K.; Ichinose, S.; Yagishita, S.; Murata, M.; Tagawa, K.; Akashi, T.; Eishi, Y.; Okazawa, H. A novel form of necrosis, TRIAD, occurs in human Huntington’s disease. Acta Neuropathol. Commun. 2017, 5, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.-L.; Wang, S.-E.; Hsu, C.-H.; Sheu, S.-J.; Wu, C.-H. Oral treatment with herbal formula B307 alleviates cardiac failure in aging R6/2 mice with Huntington’s disease via suppressing oxidative stress, inflammation, and apoptosis. Clin. Interv. Aging 2015, 10, 1173–1187. [Google Scholar] [PubMed] [Green Version]
- Bassi, S.; Tripathi, T.; Monziani, A.; Di Leva, F.; Biagioli, M. Epigenetics of Huntington’s disease. Adv. Exp. Med. Biol. 2017, 978, 277–299. [Google Scholar] [PubMed]
- Valor, L.M. Understanding histone deacetylation in Huntington’s disease. Oncotarget 2017, 8, 5660–5661. [Google Scholar] [CrossRef] [PubMed]
- Li, S.H.; Li, X.J. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004, 20, 146–154. [Google Scholar] [CrossRef]
- Riley, B.E.; Orr, H.T. Polyglutamine neurodegenerative diseases and regulation of transcription: Assembling the puzzle. Genes Dev. 2006, 20, 2183–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamanaka, T.; Miyazaki, H.; Oyama, F.; Kurosawa, M.; Washizu, C.; Doi, H.; Nukina, N. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. Embo. J. 2008, 27, 827–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmer-Bensch, G. Epigenomic remodeling in Huntington’s disease—master or servant? Epigenomes 2020, 4, 15. [Google Scholar] [CrossRef]
- Gray, S.G. Targeting Huntington’s disease through histone deacetylases. Clin. Epigenetics 2011, 2, 257–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadri-Vakili, G.; Cha, J.H. Mechanisms of disease: Histone modifications in Huntington’s disease. Nat. Clin. Pr. Neurol. 2006, 2, 330–338. [Google Scholar] [CrossRef]
- Vashishtha, M.; Ng, C.W.; Yildirim, F.; Gipson, T.A.; Kratter, I.H.; Bodai, L.; Song, W.; Lau, A.; Labadorf, A.; Vogel-Ciernia, A.; et al. Targeting H3K4 trimethylation in Huntington disease. Proc. Natl. Acad. Sci. USA 2013, 110, E3027–E3036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, B.G.; Koroshetz, W.J.; Beal, M.F.; Rosen, B.R. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology 1993, 43, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- McBride, H.M.; Neuspiel, M.; Wasiak, S. Mitochondria: More than just a powerhouse. Curr. Biol. 2006, 16, R551–R560. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Sun, X.; Hu, D.; Wang, Y.J.; Fujioka, H.; Vyas, R.; Chakrapani, S.; Joshi, A.U.; Luo, Y.; Mochly-Rosen, D.; et al. VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington’s disease. Nat. Commun. 2016, 7, 12646. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.M. Mitochondrial bioenergetics and dynamics in Huntington’s disease: Tripartite synapses and selective striatal degeneration. J. Bioenerg. Biomembr. 2010, 42, 227–234. [Google Scholar] [CrossRef]
- Yano, H.; Baranov, S.V.; Baranova, O.V.; Kim, J.; Pan, Y.; Yablonska, S.; Carlisle, D.L.; Ferrante, R.J.; Kim, A.H.; Friedlander, R.M. Inhibition of mitochondrial protein import by mutant huntingtin. Nat. Neurosci. 2014, 17, 822–831. [Google Scholar] [CrossRef] [PubMed]
- Faria, A.V.; Ratnanather, J.T.; Tward, D.J.; Lee, D.S.; van den Noort, F.; Wu, D.; Brown, T.; Johnson, H.; Paulsen, J.S.; Ross, C.A.; et al. Linking white matter and deep gray matter alterations in premanifest Huntington disease. Neuroimage Clin. 2016, 11, 450–460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.; Peng, Q.; Hou, Z.; Jiang, M.; Wang, X.; Langseth, A.J.; Tao, M.; Barker, P.B.; Mori, S.; Bergles, D.E.; et al. Early white matter abnormalities, progressive brain pathology and motor deficits in a novel knock-in mouse model of Huntington’s disease. Hum. Mol. Genet. 2015, 24, 2508–2527. [Google Scholar] [CrossRef] [PubMed]
- Teo, R.T.; Hong, X.; Yu-Taeger, L.; Huang, Y.; Tan, L.J.; Xie, Y.; To, X.V.; Guo, L.; Rajendran, R.; Novati, A.; et al. Structural and molecular myelination deficits occur prior to neuronal loss in the YAC128 and BACHD models of Huntington disease. Hum. Mol. Genet. 2016, 25, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Faideau, M.; Kim, J.; Cormier, K.; Gilmore, R.; Welch, M.; Auregan, G.; Dufour, N.; Guillermier, M.; Brouillet, E.; Hantraye, P. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: A correlation with Huntington’s disease subjects. Hum. Mol. Genet. 2010, 19, 3053–3067. [Google Scholar] [CrossRef]
- Khakh, B.S.; Beaumont, V.; Cachope, R.; Munoz-Sanjuan, I.; Goldman, S.A.; Grantyn, R. Unravelling and exploiting astrocyte dysfunction in Huntington’s disease. Trends Neurosci. 2017, 40, 422–437. [Google Scholar] [CrossRef]
- Meunier, C.; Merienne, N.; Jollé, C.; Déglon, N.; Pellerin, L. Astrocytes are key but indirect contributors to the development of the symptomatology and pathophysiology of Huntington’s disease. Glia 2016, 64, 1841–1856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [Green Version]
- Möller, T. Neuroinflammation in Huntington’s disease. J. Neural. Transm. 2010, 117, 1001–1008. [Google Scholar] [CrossRef] [PubMed]
- Chun, H.; Im, H.; Kang, Y.J.; Kim, Y.; Shin, J.H.; Won, W.; Lim, J.; Ju, Y.; Park, Y.M.; Kim, S. Severe reactive astrocytes precipitate pathological hallmarks of Alzheimer’s disease via H2O2− production. Nat. Neurosci. 2020, 23, 1555–1566. [Google Scholar] [CrossRef]
- Heng, M.Y.; Detloff, P.J.; Wang, P.L.; Tsien, J.Z.; Albin, R.L. In vivo evidence for NMDA receptor-mediated excitotoxicity in a murine genetic model of Huntington disease. J. Neurosci. 2009, 29, 3200–3205. [Google Scholar] [CrossRef] [Green Version]
- Heo, J.Y.; Nam, M.-H.; Yoon, H.H.; Kim, J.; Hwang, Y.J.; Won, W.; Woo, D.H.; Lee, J.A.; Park, H.-J.; Jo, S. Aberrant tonic inhibition of dopaminergic neuronal activity causes motor symptoms in animal models of Parkinson’s disease. Curr. Biol. 2020, 30, 276–291.e9. [Google Scholar] [CrossRef]
- Lee, J.; Hyeon, S.J.; Im, H.; Ryu, H.; Kim, Y.; Ryu, H. Astrocytes and microglia as non-cell autonomous players in the pathogenesis of ALS. Exp. Neurobiol. 2016, 25, 233–240. [Google Scholar] [CrossRef]
- Gu, X.; André, V.M.; Cepeda, C.; Li, S.-H.; Li, X.-J.; Levine, M.S.; Yang, X.W. Pathological cell-cell interactions are necessary for striatal pathogenesis in a conditional mouse model of Huntington’s disease. Mol. Neurodegener. 2007, 2, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, L.; Cheng, S.; Pan, Y.; Liu, Q.; Yang, W.; Li, S.; Li, X.-J. Accumulation of Endogenous Mutant Huntingtin in Astrocytes Exacerbates Neuropathology of Huntington Disease in Mice. Mol. Neurobiol. 2021, 58, 5112–5126. [Google Scholar] [CrossRef]
- Bason, M.; Meister-Broekema, M.; Alberts, N.; Dijkers, P.; Bergink, S.; Sibon, O.C.M.; Kampinga, H.H. Astrocytic expression of the chaperone DNAJB6 results in non-cell autonomous protection in Huntington’s disease. Neurobiol. Dis. 2019, 124, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Benraiss, A.; Mariani, J.N.; Tate, A.; Solly, R.; Capellano, L.; de Mesy Bentley, K.L.; Chandler-Militello, D.; Goldman, S. Huntington Disease Mice Exhibit a TCF7L2-Responsive Suppression of Both Homeostatic and Compensatory Remyelination. SSRN Electron. J. 2021. under review. [Google Scholar] [CrossRef]
- Savage, J.C.; St-Pierre, M.-K.; Carrier, M.; El Hajj, H.; Novak, S.W.; Sanchez, M.G.; Cicchetti, F.; Tremblay, M.-È. Microglial physiological properties and interactions with synapses are altered at presymptomatic stages in a mouse model of Huntington’s disease pathology. J. Neuroinflammation 2020, 17, 98. [Google Scholar] [CrossRef]
- Kim, A.; García-García, E.; Straccia, M.; Comella-Bolla, A.; Miguez, A.; Masana, M.; Alberch, J.; Canals, J.M.; Rodríguez, M.J. Reduced Fractalkine Levels Lead to Striatal Synaptic Plasticity Deficits in Huntington’s Disease. Front. Cell. Neurosci. 2020, 14, 163. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.C.; Farag, S.H.; Casey, C.S.; Wood-Kaczmar, A.; Pocock, J.M.; Tabrizi, S.J.; Andre, R. Human Huntington’s disease pluripotent stem cell-derived microglia develop normally but are abnormally hyper-reactive and release elevated levels of reactive oxygen species. J. Neuroinflammation 2021, 18, 94. [Google Scholar] [CrossRef]
- Bolla, A.C.; Valente, T.; Miguez, A.; Brito, V.; Gines, S.; Solà, C.; Straccia, M.; Canals, J.M. CD200 is up-regulated in R6/1 transgenic mouse model of Huntington’s disease. PLoS ONE 2019, 14, e0224901. [Google Scholar]
- Caron, N.S.; Banos, R.; Yanick, C.; Aly, A.E.; Byrne, L.M.; Smith, E.D.; Xie, Y.; Smith, S.E.; Potluri, N.; Black, H.F.; et al. Mutant huntingtin is cleared from the brain via active mechanisms in Huntington disease. J. Neurosci. 2021, 41, 780–796. [Google Scholar] [CrossRef] [PubMed]
- Sameni, S.; Zhang, R.; Digman, M.A. The phasor FLIM method reveals a link between a change in energy metabolism and mHtt protein spread in healthy Mammalian cells when co-cultured with Huntington diseased cells. Methods Appl. Fluoresc. 2021, 9, 015005. [Google Scholar] [CrossRef]
- Singh, A.; Agrawal, N. Deciphering the key mechanisms leading to alteration of lipid metabolism in Drosophila model of Huntington’s disease. Biochim. Et. Biophys. Acta (BBA)-Mol. Basis Dis. 2021, 1867, 166127. [Google Scholar] [CrossRef] [PubMed]
- Sancho, L.; Contreras, M.; Allen, N.J. Glia as sculptors of synaptic plasticity. Neurosci. Res. 2020, 167, 17–29. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Brymer, K.J.; Barnes, J.R.; Parsons, M.P. Entering a new era of quantifying glutamate clearance in health and disease. J. Neurosci. Res. 2021, 99, 1598–1617. [Google Scholar] [CrossRef]
- Florence, G.; Pereira, T.; Kurths, J. Extracellular potassium dynamics in the hyperexcitable state of the neuronal ictal activity. Commun. Nonlinear Sci. Numer. Simul. 2012, 17, 4700–4706. [Google Scholar] [CrossRef]
- Radiske, A.; Gonzalez, M.C.; Nôga, D.A.; Rossato, J.I.; Bevilaqua, L.R.M.; Cammarota, M. GluN2B and GluN2A-containing NMDAR are differentially involved in extinction memory destabilization and restabilization during reconsolidation. Sci. Rep. 2021, 11, 186. [Google Scholar] [CrossRef]
- Schmidt, M.E.; Caron, N.S.; Aly, A.E.; Lemarié, F.L.; Dal Cengio, L.; Ko, Y.; Lazic, N.; Anderson, L.; Nguyen, B.; Raymond, L.A. DAPK1 promotes extrasynaptic GluN2B phosphorylation and striatal spine instability in the YAC128 mouse model of Huntington disease. Front. Cell. Neurosci. 2020, 14, 348. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.R.; Bezprozvanny, I. Corticostriatal circuit dysfunction in Huntington’s disease: Intersection of glutamate, dopamine and calcium. Future Neurol. 2010, 5, 735–756. [Google Scholar] [CrossRef] [Green Version]
- Raymond, L.A.; André, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, R.; Wang, L.; Sanders, S.S.; Zuo, K.; Hayden, M.R.; Raymond, L.A. Altered regulation of striatal neuronal N-methyl-D-aspartate receptor trafficking by palmitoylation in Huntington disease mouse model. Front. Synaptic Neurosci. 2019, 11, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marco, S.; Murillo, A.; Pérez-Otaño, I. RNAi-based GluN3A silencing prevents and reverses disease phenotypes induced by mutant huntingtin. Mol. Ther. 2018, 26, 1965–1972. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, C.; Wu, N.; André, V.M.; Cummings, D.M.; Levine, M.S. The corticostriatal pathway in Huntington’s disease. Prog. Neurobiol. 2007, 81, 253–271. [Google Scholar] [CrossRef] [Green Version]
- Bunner, K.D.; Rebec, G.V. Corticostriatal dysfunction in Huntington’s disease: The basics. Front. Hum. Neurosci. 2016, 10, 317. [Google Scholar] [CrossRef] [Green Version]
- Wood, T.E.; Barry, J.; Yang, Z.; Cepeda, C.; Levine, M.S.; Gray, M. Mutant huntingtin reduction in astrocytes slows disease progression in the BACHD conditional Huntington’s disease mouse model. Hum. Mol. Genet. 2019, 28, 487–500. [Google Scholar] [CrossRef]
- Blumenstock, S.; Dudanova, I. Cortical and striatal circuits in Huntington’s disease. Front. Neurosci. 2020, 14, 82. [Google Scholar] [CrossRef]
- Chan, C.S.; Surmeier, D.J. Astrocytes go awry in Huntington’s disease. Nat. Neurosci. 2014, 17, 641–642. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, M.E. Huntington’s disease and the striatal medium spiny neuron: Cell-autonomous and non-cell-autonomous mechanisms of disease. Neurotherapeutics 2012, 9, 270–284. [Google Scholar] [CrossRef] [Green Version]
- Estrada-Sánchez, A.M.; Rebec, G.V. Corticostriatal dysfunction and glutamate transporter 1 (GLT1) in Huntington’s disease: Interactions between neurons and astrocytes. Basal. Ganglia 2012, 2, 57–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gray, M. The role of astrocytes in Huntington’s disease. In Pathological Potential of Neuroglia; Springer: Berlin/Heidelberg, Germany, 2014; pp. 213–229. [Google Scholar]
- Gray, M. Astrocytes in Huntington’s disease. Neurogl. Neurodegener. Dis. 2019, 1175, 355–381. [Google Scholar]
- Palpagama, T.H.; Waldvogel, H.J.; Faull, R.L.; Kwakowsky, A. The role of microglia and astrocytes in Huntington’s disease. Front. Mol. Neurosci. 2019, 12, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proft, J.; Weiss, N. Rectifying rectifier channels in Huntington disease. Commun. Integr. Biol. 2014, 7, 694–703. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, A.M.E.; Mejía-Toiber, J.; Massieu, L. Excitotoxic neuronal death and the pathogenesis of Huntington’s disease. Arch. Med. Res. 2008, 39, 265–276. [Google Scholar] [CrossRef]
- Wilton, D.K.; Stevens, B. The contribution of glial cells to Huntington’s disease pathogenesis. Neurobiol. Dis. 2020, 143, 104963. [Google Scholar] [CrossRef]
- Al-Dalahmah, O.; Sosunov, A.A.; Shaik, A.; Ofori, K.; Liu, Y.; Vonsattel, J.P.; Adorjan, I.; Menon, V.; Goldman, J.E. Single-nucleus RNA-seq identifies Huntington disease astrocyte states. Acta Neuropathol. Commun. 2020, 8, 19. [Google Scholar] [CrossRef] [Green Version]
- Arzberger, T.; Krampfl, K.; Leimgruber, S.; Weindl, A. Changes of NMDA receptor subunit (NR1, NR2B) and glutamate transporter (GLT1) mRNA expression in Huntington’s disease—an in situ hybridization study. J. Neuropathol. Exp. Neurol. 1997, 56, 440–454. [Google Scholar] [CrossRef] [Green Version]
- Petr, G.T.; Schultheis, L.A.; Hussey, K.C.; Sun, Y.; Dubinsky, J.M.; Aoki, C.; Rosenberg, P.A. Decreased expression of GLT-1 in the R6/2 model of Huntington’s disease does not worsen disease progression. Eur. J. Neurosci. 2013, 38, 2477–2490. [Google Scholar] [CrossRef] [Green Version]
- Skotte, N.H.; Andersen, J.V.; Santos, A.; Aldana, B.I.; Willert, C.W.; Nørremølle, A.; Waagepetersen, H.S.; Nielsen, M.L. Integrative characterization of the R6/2 mouse model of Huntington’s disease reveals dysfunctional astrocyte metabolism. Cell Rep. 2018, 23, 2211–2224. [Google Scholar] [CrossRef]
- Cho, I.K.; Yang, B.; Forest, C.; Qian, L.; Chan, A.W. Amelioration of Huntington’s disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington’s disease monkeys. PLoS ONE 2019, 14, e0214156. [Google Scholar]
- Dvorzhak, A.; Helassa, N.; Török, K.; Schmitz, D.; Grantyn, R. Single synapse indicators of impaired glutamate clearance derived from fast iGluu imaging of cortical afferents in the striatum of normal and Huntington (Q175) mice. J. Neurosci. 2019, 39, 3970–3982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassel, B.; Tessler, S.; Faull, R.L.; Emson, P.C. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem. Res. 2008, 33, 232–237. [Google Scholar] [CrossRef]
- Garcia, M.; Charvin, D.; Caboche, J. Expanded huntingtin activates the C-Jun N terminal kinase/c-Jun pathway prior to aggregate formation in striatal neurons in culture. Neuroscience 2004, 127, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Ao, Y.; Faas, G.C.; Nwaobi, S.E.; Xu, J.; Haustein, M.D.; Anderson, M.A.; Mody, I.; Olsen, M.L.; Sofroniew, M.V. Astrocyte Kir4. 1 ion channel deficits contribute to neuronal dysfunction in Huntington’s disease model mice. Nat. Neurosci. 2014, 17, 694–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional calcium and glutamate signaling in striatal astrocytes from Huntington’s disease model mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [Green Version]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef]
- Garcia, V.J.; Rushton, D.J.; Tom, C.M.; Allen, N.D.; Kemp, P.J.; Svendsen, C.N.; Mattis, V.B. Huntington’s disease patient-derived astrocytes display electrophysiological impairments and reduced neuronal support. Front. Neurosci. 2019, 13, 669. [Google Scholar] [CrossRef]
- Shin, J.-Y.; Fang, Z.-H.; Yu, Z.-X.; Wang, C.-E.; Li, S.-H.; Li, X.-J. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J. Cell Biol. 2005, 171, 1001–1012. [Google Scholar] [CrossRef]
- Bankston, A.N.; Mandler, M.D.; Feng, Y. Oligodendroglia and neurotrophic factors in neurodegeneration. Neurosci. Bull. 2013, 29, 216–228. [Google Scholar] [CrossRef]
- Myers, R.H.; Vonsattel, J.P.; Paskevich, P.A.; Kiely, D.K.; Stevens, T.J.; Cupples, L.A.; Richardson, E.P., Jr.; Bird, E.D. Decreased neuronal and increased oligodendroglial densities in Huntington’s disease caudate nucleus. J. Neuropathol. Exp. Neurol. 1991, 50, 729–742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartzokis, G.; Lu, P.H.; Tishler, T.A.; Fong, S.M.; Oluwadara, B.; Finn, J.P.; Huang, D.; Bordelon, Y.; Mintz, J.; Perlman, S. Myelin breakdown and iron changes in Huntington’s disease: Pathogenesis and treatment implications. Neurochem. Res. 2007, 32, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Phillips, O.; Squitieri, F.; Sanchez-Castaneda, C.; Elifani, F.; Caltagirone, C.; Sabatini, U.; Di Paola, M. Deep white matter in Huntington’s disease. PLoS ONE 2014, 9, e109676. [Google Scholar]
- Bardile, C.F.; Garcia-Miralles, M.; Caron, N.S.; Rayan, N.A.; Langley, S.R.; Harmston, N.; Rondelli, A.M.; Teo, R.T.Y.; Waltl, S.; Anderson, L.M.; et al. Intrinsic mutant HTT-mediated defects in oligodendroglia cause myelination deficits and behavioral abnormalities in Huntington disease. Proc. Natl. Acad. Sci. USA 2019, 116, 9622–9627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Wei, W.; Wang, G.; Gaertig, M.A.; Feng, Y.; Wang, W.; Li, X.J.; Li, S. Mutant huntingtin downregulates myelin regulatory factor-mediated myelin gene expression and affects mature oligodendrocytes. Neuron 2015, 85, 1212–1226. [Google Scholar] [CrossRef] [Green Version]
- Wang, N.; Yang, X.W. Huntington disease’s glial progenitor cells hit the pause button in the mouse brain. Cell Stem. Cell 2019, 24, 3–4. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [Green Version]
- Johri, A.; Chandra, A.; Beal, M.F. PGC-1α, mitochondrial dysfunction, and Huntington’s disease. Free Radic. Biol. Med. 2013, 62, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Xiang, Z.; Valenza, M.; Cui, L.; Leoni, V.; Jeong, H.K.; Brilli, E.; Zhang, J.; Peng, Q.; Duan, W.; Reeves, S.A.; et al. Peroxisome-proliferator-activated receptor gamma coactivator 1 α contributes to dysmyelination in experimental models of Huntington’s disease. J. Neurosci. 2011, 31, 9544–9553. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Ferrante, R.J. Emerging chemotherapeutic strategies for Huntington’s disease. Expert Opin. Emerg. Drugs 2005, 10, 345–363. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef]
- Kuehner, J.N.; Bruggeman, E.C.; Wen, Z.; Yao, B. Epigenetic regulations in neuropsychiatric disorders. Front. Genet. 2019, 10, 268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Jiao, C.; Wang, K.; Yuan, N. DNA methylation and psychiatric disorders. Prog Mol. Biol. Transl. Sci. 2018, 157, 175–232. [Google Scholar] [PubMed]
- Grayson, D.R.; Guidotti, A. The dynamics of DNA methylation in Schizophrenia and related psychiatric disorders. Neuropsychopharmacology 2013, 38, 138–166. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.W.; Yildirim, F.; Yap, Y.S.; Dalin, S.; Matthews, B.J.; Velez, P.J.; Labadorf, A.; Housman, D.E.; Fraenkel, E. Extensive changes in DNA methylation are associated with expression of mutant huntingtin. Proc. Natl. Acad. Sci. USA 2013, 110, 2354–2359. [Google Scholar] [CrossRef] [Green Version]
- Beal, M.F.; Ferrante, R.J. Experimental therapeutics in transgenic mouse models of Huntington’s disease. Nat. Rev. Neurosci. 2004, 5, 373–384. [Google Scholar] [CrossRef]
- Lee, J.; Hwang, Y.J.; Ryu, H.; Kowall, N.W.; Ryu, H. Nucleolar dysfunction in Huntington’s disease. Biochim. Biophys Acta 2014, 1842, 785–790. [Google Scholar] [CrossRef] [Green Version]
- Korzus, E.; Rosenfeld, M.G.; Mayford, M. CBP histone acetyltransferase activity is a critical component of memory consolidation. Neuron 2004, 42, 961–972. [Google Scholar] [CrossRef] [Green Version]
- Glajch, K.E.; Sadri-Vakili, G. Epigenetic mechanisms involved in Huntington’s disease pathogenesis. J. Huntingt. Dis. 2015, 4, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Poirier, M.A.; Liang, Y.; Pei, Z.; Weiskittel, C.E.; Smith, W.W.; DeFranco, D.B.; Ross, C.A. Depletion of CBP is directly linked with cellular toxicity caused by mutant huntingtin. Neurobiol. Dis. 2006, 23, 543–551. [Google Scholar] [CrossRef] [PubMed]
- McFarland, K.N.; Das, S.; Sun, T.T.; Leyfer, D.; Xia, E.; Sangrey, G.R.; Kuhn, A.; Luthi-Carter, R.; Clark, T.W.; Sadri-Vakili, G.; et al. Genome-wide histone acetylation is altered in a transgenic mouse model of Huntington’s disease. PLoS ONE 2012, 7, e41423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadri-Vakili, G.; Bouzou, B.; Benn, C.L.; Kim, M.O.; Chawla, P.; Overland, R.P.; Glajch, K.E.; Xia, E.; Qiu, Z.; Hersch, S.M.; et al. Histones associated with downregulated genes are hypo-acetylated in Huntington’s disease models. Hum. Mol. Genet. 2007, 16, 1293–1306. [Google Scholar] [CrossRef] [Green Version]
- Ryu, H.; Lee, J.; Hagerty, S.W.; Soh, B.Y.; McAlpin, S.E.; Cormier, K.A.; Smith, K.M.; Ferrante, R.J. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 19176–19181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Pietri Tonelli, D.; Pulvers, J.N.; Haffner, C.; Murchison, E.P.; Hannon, G.J.; Huttner, W.B. miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development 2008, 135, 3911–3921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, X.; Cong, S. MicroRNAs in Huntington’s disease: Diagnostic biomarkers or therapeutic agents? Front. Cell. Neurosci. 2021, 15, 313. [Google Scholar] [CrossRef]
- Juźwik, C.A.; Drake, S.S.; Zhang, Y.; Paradis-Isler, N.; Sylvester, A.; Amar-Zifkin, A.; Douglas, C.; Morquette, B.; Moore, C.S.; Fournier, A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019, 182, 101664. [Google Scholar] [CrossRef] [PubMed]
- Sinha, M.; Ghose, J.; Bhattarcharyya, N.P. Micro RNA -214,-150,-146a and-125b target Huntingtin gene. RNA Biol. 2011, 8, 1005–1021. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Bhattacharyya, N.P. Heat shock factor 1-regulated miRNAs can target Huntingtin and suppress aggregates of mutant huntingtin. Microrna 2015, 4, 185–193. [Google Scholar] [CrossRef]
- Kozlowska, E.; Krzyzosiak, W.J.; Koscianska, E. Regulation of huntingtin gene expression by miRNA-137, -214, -148a, and their respective isomiRs. Int. J. Mol. Sci. 2013, 14, 16999–17016. [Google Scholar] [CrossRef]
- Bucha, S.; Mukhopadhyay, D.; Bhattacharyya, N.P. Regulation of mitochondrial morphology and cell cycle by microRNA-214 targeting Mitofusin2. Biochem. Biophys Res. Commun. 2015, 465, 797–802. [Google Scholar] [CrossRef]
- Ma, B.; Savas, J.N.; Yu, M.-S.; Culver, B.P.; Chao, M.V.; Tanese, N. Huntingtin mediates dendritic transport of β-actin mRNA in rat neurons. Sci. Rep. 2011, 1, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caviston, J.P.; Ross, J.L.; Antony, S.M.; Tokito, M.; Holzbaur, E.L. Huntingtin facilitates dynein/dynactin-mediated vesicle transport. Proc. Natl. Acad. Sci. USA 2007, 104, 10045–10050. [Google Scholar] [CrossRef] [Green Version]
- Caviston, J.P.; Holzbaur, E.L. Huntingtin as an essential integrator of intracellular vesicular trafficking. Trends Cell Biol. 2009, 19, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zala, D.; Hinckelmann, M.-V.; Yu, H.; Da Cunha, M.M.L.; Liot, G.; Cordelières, F.P.; Marco, S.; Saudou, F. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell 2013, 152, 479–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.R.; Enghild, J.J.; Martin, M.E.; Jou, Y.-S.; Myers, R.M.; Roses, A.D.; Vance, J.M.; Strittmatter, W.J. Huntingtin and DRPLA proteins selectively interact with the enzyme GAPDH. Nat. Med. 1996, 2, 347–350. [Google Scholar] [CrossRef]
- Wu, J.; Lin, F.; Qin, Z. Sequestration of glyceraldehyde-3-phosphate dehydrogenase to aggregates formed by mutant huntingtin. Acta Biochim. Et Biophys. Sin. 2007, 39, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Chaudhary, R.K.; Patel, K.A.; Patel, M.K.; Joshi, R.H.; Roy, I. Inhibition of aggregation of mutant huntingtin by nucleic acid aptamers in vitro and in a yeast model of Huntington’s disease. Mol. Ther. 2015, 23, 1912–1926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauthier, L.R.; Charrin, B.C.; Borrell-Pagès, M.; Dompierre, J.P.; Rangone, H.; Cordelières, F.P.; De Mey, J.; MacDonald, M.E.; Leßmann, V.; Humbert, S. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004, 118, 127–138. [Google Scholar] [CrossRef] [Green Version]
- Twelvetrees, A.E.; Lesept, F.; Holzbaur, E.L.; Kittler, J.T. The adaptor proteins HAP1a and GRIP1 collaborate to activate the kinesin-1 isoform KIF5C. J. Cell Sci. 2019, 132, jcs215822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colin, E.; Zala, D.; Liot, G.; Rangone, H.; Borrell-Pagès, M.; Li, X.J.; Saudou, F.; Humbert, S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008, 27, 2124–2134. [Google Scholar] [CrossRef] [Green Version]
- Warby, S.C.; Chan, E.Y.; Metzler, M.; Gan, L.; Singaraja, R.R.; Crocker, S.F.; Robertson, H.A.; Hayden, M.R. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum. Mol. Genet. 2005, 14, 1569–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Dittlau, K.S.; Van Den Bosch, L. Axonal transport defects and neurodegeneration: Molecular mechanisms and therapeutic implications. Semin. Cell Dev. Biol. 2020, 99, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Morfini, G.; Pigino, G.; Brady, S.T. Polyglutamine expansion diseases: Failing to deliver. Trends Mol. Med. 2005, 11, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Her, L.-S.; Goldstein, L.S. Enhanced sensitivity of striatal neurons to axonal transport defects induced by mutant huntingtin. J. Neurosci. 2008, 28, 13662–13672. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Li, S.-H.; Yu, Z.-X.; Shelbourne, P.; Li, X.-J. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 2001, 21, 8473–8481. [Google Scholar] [CrossRef] [Green Version]
- White, J.A.; Krzystek, T.J.; Hoffmar-Glennon, H.; Thant, C.; Zimmerman, K.; Iacobucci, G.; Vail, J.; Thurston, L.; Rahman, S.; Gunawardena, S. Excess Rab4 rescues synaptic and behavioral dysfunction caused by defective HTT-Rab4 axonal transport in Huntington’s disease. Acta Neuropathol. Commun. 2020, 8, 97. [Google Scholar] [CrossRef]
- Liot, G.; Zala, D.; Pla, P.; Mottet, G.; Piel, M.; Saudou, F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J. Neurosci. 2013, 33, 6298–6309. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L. The regulation of autophagosome dynamics by huntingtin and HAP1 is disrupted by expression of mutant huntingtin, leading to defective cargo degradation. J. Neurosci. 2014, 34, 1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gunawardena, S.; Her, L.-S.; Brusch, R.G.; Laymon, R.A.; Niesman, I.R.; Gordesky-Gold, B.; Sintasath, L.; Bonini, N.M.; Goldstein, L.S. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 2003, 40, 25–40. [Google Scholar] [CrossRef] [Green Version]
- Dompierre, J.P.; Godin, J.D.; Charrin, B.C.; Cordelieres, F.P.; King, S.J.; Humbert, S.; Saudou, F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington’s disease by increasing tubulin acetylation. J. Neurosci. 2007, 27, 3571–3583. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.-H.; Wang, Y.; Sapp, E.; Cuiffo, B.; Wanker, E.; Hayden, M.R.; Kegel, K.B.; Aronin, N.; DiFiglia, M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004, 24, 269–281. [Google Scholar] [CrossRef]
- Liu, Y.F. Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J. Biol. Chem. 1998, 273, 28873–28877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morfini, G.A.; You, Y.-M.; Pollema, S.L.; Kaminska, A.; Liu, K.; Yoshioka, K.; Björkblom, B.; Coffey, E.T.; Bagnato, C.; Han, D. Pathogenic huntingtin inhibits fast axonal transport by activating JNK3 and phosphorylating kinesin. Nat. Neurosci. 2009, 12, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Gelman, A.; Rawet-Slobodkin, M.; Elazar, Z. Huntingtin facilitates selective autophagy. Nat. Cell Biol. 2015, 17, 214–215. [Google Scholar] [CrossRef]
- Rui, Y.N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J.; et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochaba, J.; Lukacsovich, T.; Csikos, G.; Zheng, S.; Margulis, J.; Salazar, L.; Mao, K.; Lau, A.L.; Yeung, S.Y.; Humbert, S.; et al. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 16889–16894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braatz, E.M.; André, E.A.; Liu, J.P.; Zeitlin, S.O. Characterization of a knock-in mouse model with a huntingtin exon 1 deletion. J. Huntingt. Dis. 2021, 10, 435–454. [Google Scholar] [CrossRef] [PubMed]
- Atwal, R.S.; Truant, R. A stress sensitive ER membrane-association domain in Huntingtin protein defines a potential role for Huntingtin in the regulation of autophagy. Autophagy 2008, 4, 91–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barron, J.C.; Hurley, E.P.; Parsons, M.P. Huntingtin and the synapse. Front. Cell Neurosci. 2021, 15, 225. [Google Scholar] [CrossRef]
- McAdam, R.L.; Morton, A.; Gordon, S.L.; Alterman, J.F.; Khvorova, A.; Cousin, M.A.; Smillie, K.J. Loss of huntingtin function slows synaptic vesicle endocytosis in striatal neurons from the htt(Q140/Q140) mouse model of Huntington’s disease. Neurobiol. Dis. 2020, 134, 104637. [Google Scholar] [CrossRef]
- Bonfanti, S.; Lionetti, M.C.; Fumagalli, M.R.; Chirasani, V.R.; Tiana, G.; Dokholyan, N.V.; Zapperi, S.; La Porta, C.A.M. Molecular mechanisms of heterogeneous oligomerization of huntingtin proteins. Sci. Rep. 2019, 9, 7615. [Google Scholar] [CrossRef]
- Bečanović, K.; Nørremølle, A.; Neal, S.J.; Kay, C.; Collins, J.A.; Arenillas, D.; Lilja, T.; Gaudenzi, G.; Manoharan, S.; Doty, C.N.; et al. A SNP in the HTT promoter alters NF-κB binding and is a bidirectional genetic modifier of Huntington disease. Nat. Neurosci. 2015, 18, 807–816. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, G.C.; Farag, S.H.; Ostroff, G.R.; Tabrizi, S.J.; Andre, R. Wild-type huntingtin regulates human macrophage function. Sci Rep. 2020, 10, 17269. [Google Scholar] [CrossRef]
- Bensalel, J.; Xu, H.; Lu, M.L.; Capobianco, E.; Wei, J. RNA-seq analysis reveals significant transcriptome changes in huntingtin-null human neuroblastoma cells. BMC Med. Genom. 2021, 14, 176. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Huang, B.; Cheng, J.; Seefelder, M.; Engler, T.; Pfeifer, G.; Oeckl, P.; Otto, M.; Moser, F.; Maurer, M. The cryo-electron microscopy structure of huntingtin. Nature 2018, 555, 117–120. [Google Scholar] [CrossRef]
- Kaemmerer, W.F.; Grondin, R.C. The effects of huntingtin-lowering: What do we know so far? Degener Neurol. Neuromuscul. Dis. 2019, 9, 3–17. [Google Scholar] [CrossRef] [Green Version]
- Burtscher, J.; Di Pardo, A.; Maglione, V.; Schwarzer, C.; Squitieri, F. Mitochondrial respiration changes in R6/2 Huntington’s disease model mice during aging in a brain region specific manner. Int. J. Mol. Sci. 2020, 21, 5412. [Google Scholar] [CrossRef]
- Cherubini, M.; Lopez-Molina, L.; Gines, S. Mitochondrial fission in Huntington’s disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca2+ efflux and reactive oxygen species (ROS) homeostasis. Neurobiol. Dis. 2020, 136, 104741. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Giacomello, M.; Hudec, R.; Lopreiato, R.; Ermak, G.; Lim, D.; Malorni, W.; Davies, K.J.; Carafoli, E.; Scorrano, L. Mitochondrial fission and cristae disruption increase the response of cell models of Huntington’s disease to apoptotic stimuli. EMBO Mol. Med. 2010, 2, 490–503. [Google Scholar] [CrossRef]
- Damiano, M.; Galvan, L.; Déglon, N.; Brouillet, E. Mitochondria in Huntington’s disease. Biochim. Biophys Acta 2010, 1802, 52–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jędrak, P.; Krygier, M.; Tońska, K.; Drozd, M.; Kaliszewska, M.; Bartnik, E.; Sołtan, W.; Sitek, E.J.; Stanisławska-Sachadyn, A.; Limon, J.; et al. Mitochondrial DNA levels in Huntington disease leukocytes and dermal fibroblasts. Metab. Brain Dis. 2017, 32, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Moody, J.P.; Edgerly, C.K.; Bordiuk, O.L.; Cormier, K.; Smith, K.; Beal, M.F.; Ferrante, R.J. Mitochondrial loss, dysfunction and altered dynamics in Huntington’s disease. Hum. Mol. Genet. 2010, 19, 3919–3935. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Chen, J.; Petrilli, A.; Liot, G.; Klinglmayr, E.; Zhou, Y.; Poquiz, P.; Tjong, J.; Pouladi, M.A.; Hayden, M.R.; et al. Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity. Nat. Med. 2011, 17, 377–382. [Google Scholar] [CrossRef] [Green Version]
- Jodeiri Farshbaf, M.; Ghaedi, K. Huntington’s disease and mitochondria. Neurotox. Res. 2017, 32, 518–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.-Q.; Chen, Q.; Wang, X.; Wang, Q.-C.; Wang, Y.; Cheng, H.-P.; Guo, C.; Sun, Q.; Chen, Q.; Tang, T.-S. Dysregulation of mitochondrial calcium signaling and superoxide flashes cause mitochondrial genomic DNA damage in Huntington disease. J. Biol. Chem. 2013, 288, 3070–3084. [Google Scholar] [CrossRef] [Green Version]
- Elena-Real, C.A.; Díaz-Quintana, A.; González-Arzola, K.; Velázquez-Campoy, A.; Orzáez, M.; López-Rivas, A.; Gil-Caballero, S.; De la Rosa, M.Á.; Díaz-Moreno, I. Cytochrome c speeds up caspase cascade activation by blocking 14-3-3ε-dependent Apaf-1 inhibition. Cell Death Dis. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Rosas, H.D.; Hersch, S.M.; Ferrante, R.J. The therapeutic role of creatine in Huntington’s disease. Pharm 2005, 108, 193–207. [Google Scholar] [CrossRef]
- Palmer, C.S.; Anderson, A.J.; Stojanovski, D. Mitochondrial protein import dysfunction: Mitochondrial disease, neurodegenerative disease and cancer. FEBS Lett. 2021, 595, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Plaza-Zabala, A.; Montpeyo, M.; Sebastian, D.; Vila, M.; Martinez-Vicente, M. Mutant HTT (huntingtin) impairs mitophagy in a cellular model of Huntington disease. Autophagy 2021, 17, 672–689. [Google Scholar] [CrossRef] [PubMed]
- Ismailoglu, I.; Chen, Q.; Popowski, M.; Yang, L.; Gross, S.S.; Brivanlou, A.H. Huntingtin protein is essential for mitochondrial metabolism, bioenergetics and structure in murine embryonic stem cells. Dev. Biol. 2014, 391, 230–240. [Google Scholar] [CrossRef] [Green Version]
- Kim, A.; Lalonde, K.; Truesdell, A.; Gomes Welter, P.; Brocardo, P.S.; Rosenstock, T.R.; Gil-Mohapel, J. New avenues for the treatment of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 8363. [Google Scholar] [CrossRef]
- Shin, J.W.; Kim, K.-H.; Chao, M.J.; Atwal, R.S.; Gillis, T.; MacDonald, M.E.; Gusella, J.F.; Lee, J.-M. Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum. Mol. Genet. 2016, 25, 4566–4576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteys, A.M.; Ebanks, S.A.; Keiser, M.S.; Davidson, B.L. CRISPR/Cas9 editing of the mutant huntingtin allele in vitro and in vivo. Mol. Ther. 2017, 25, 12–23. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [Green Version]
- Ekman, F.K.; Ojala, D.S.; Adil, M.M.; Lopez, P.A.; Schaffer, D.V.; Gaj, T. CRISPR-Cas9-mediated genome editing increases lifespan and improves motor deficits in a Huntington’s disease mouse model. Mol. Ther.-Nucleic Acids 2019, 17, 829–839. [Google Scholar] [CrossRef] [Green Version]
- Agustín-Pavón, C.; Mielcarek, M.; Garriga-Canut, M.; Isalan, M. Deimmunization for gene therapy: Host matching of synthetic zinc finger constructs enables long-term mutant Huntingtin repression in mice. Mol. Neurodegener 2016, 11, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garriga-Canut, M.; Agustín-Pavón, C.; Herrmann, F.; Sánchez, A.; Dierssen, M.; Fillat, C.; Isalan, M. Synthetic zinc finger repressors reduce mutant huntingtin expression in the brain of R6/2 mice. Proc. Natl. Acad. Sci. USA 2012, 109, E3136–E3145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, B.; Schiefer, J.; Sass, C.; Landwehrmeyer, G.B.; Kosinski, C.M.; Kochanek, S. High-capacity adenoviral vector-mediated reduction of huntingtin aggregate load in vitro and in vivo. Hum. Gene Ther. 2007, 18, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Staber, P.D.; He, X.; Eliason, S.L.; Martins, I.H.; Mao, Q.; Yang, L.; Kotin, R.M.; Paulson, H.L.; Davidson, B.L. RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA 2005, 102, 5820–5825. [Google Scholar] [CrossRef] [Green Version]
- Drouet, V.; Perrin, V.; Hassig, R.; Dufour, N.; Auregan, G.; Alves, S.; Bonvento, G.; Brouillet, E.; Luthi-Carter, R.; Hantraye, P. Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 2009, 65, 276–285. [Google Scholar]
- DiFiglia, M.; Sena-Esteves, M.; Chase, K.; Sapp, E.; Pfister, E.; Sass, M.; Yoder, J.; Reeves, P.; Pandey, R.K.; Rajeev, K.G. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. USA 2007, 104, 17204–17209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, D.; Pendergraff, H.; Liu, J.; Kordasiewicz, H.B.; Cleveland, D.W.; Swayze, E.E.; Lima, W.F.; Crooke, S.T.; Prakash, T.P.; Corey, D.R. Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 2012, 150, 895–908. [Google Scholar] [CrossRef] [Green Version]
- McBride, J.L.; Boudreau, R.L.; Harper, S.Q.; Staber, P.D.; Monteys, A.M.; Martins, I.; Gilmore, B.L.; Burstein, H.; Peluso, R.W.; Polisky, B. Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. USA 2008, 105, 5868–5873. [Google Scholar] [CrossRef] [Green Version]
- Aguiar, S.; van der Gaag, B.; Cortese, F.A.B. RNAi mechanisms in Huntington’s disease therapy: siRNA versus shRNA. Transl. Neurodegener. 2017, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keskin, S.; Brouwers, C.C.; Sogorb-Gonzalez, M.; Martier, R.; Depla, J.A.; Vallès, A.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT lowers huntingtin mRNA and protein without off-target effects in patient-derived neuronal cultures and astrocytes. Mol. Ther.-Methods Clin. Dev. 2019, 15, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Matthes, F.; Massari, S.; Bochicchio, A.; Schorpp, K.; Schilling, J.; Weber, S.; Offermann, N.; Desantis, J.; Wanker, E.; Carloni, P.; et al. Reducing mutant huntingtin protein expression in living cells by a newly identified RNA CAG binder. ACS Chem. Neurosci. 2018, 9, 1399–1408. [Google Scholar] [CrossRef] [Green Version]
- Lopes, C.; Tang, Y.; Anjo, S.I.; Manadas, B.; Onofre, I.; De Almeida, L.P.; Daley, G.Q.; Schlaeger, T.M.; Rego, A.C.C. Mitochondrial and redox modifications in huntington disease induced pluripotent stem cells rescued by CRISPR/Cas9 CAGs targeting. Front. Cell Dev. Biol. 2020, 8, 967. [Google Scholar] [CrossRef]
- Dos Santos, N.T.H.; do Bomfim, F.R.C. Gene editing by CRISPR/CAS9 for treatment of Huntington disease. Int. J. Dev. Res. 2020, 10, 38631–38635. [Google Scholar]
- Jamwal, S.; Elsworth, J.D.; Rahi, V.; Kumar, P. Gene therapy and immunotherapy as promising strategies to combat Huntington’s disease-associated neurodegeneration: Emphasis on recent updates and future perspectives. Expert Rev. Neurother. 2020, 20, 1123–1141. [Google Scholar] [CrossRef] [PubMed]
- Marxreiter, F.; Stemick, J.; Kohl, Z. Huntingtin lowering strategies. Int. J. Mol. Sci. 2020, 21, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leavitt, B.R.; Kordasiewicz, H.B.; Schobel, S.A. Huntingtin-lowering therapies for Huntington disease: A review of the evidence of potential benefits and risks. JAMA Neurol. 2020, 77, 764–772. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Flower, M.D.; Ross, C.A.; Wild, E.J. Huntington disease: New insights into molecular pathogenesis and therapeutic opportunities. Nat. Rev. Neurol. 2020, 16, 529–546. [Google Scholar] [CrossRef]
- Tabrizi, S.J.; Ghosh, R.; Leavitt, B.R. Huntingtin lowering strategies for disease modification in Huntington’s disease. Neuron 2019, 101, 801–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estevez-Fraga, C.; Flower, M.D.; Tabrizi, S.J. Therapeutic strategies for Huntington’s disease. Curr. Opin. Neurol. 2020, 33, 508–518. [Google Scholar] [CrossRef]
- Evers, M.M.; Konstantinova, P. AAV5-miHTT gene therapy for Huntington disease: Lowering both huntingtins. Expert Opin. Biol. Ther. 2020, 20, 1121–1124. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.; Fujimaki, M.; Rogers, P.; Rubinsztein, D. Huntingtin-lowering strategies for Huntington’s disease. Expert Opin. Investig. Drugs 2020, 29, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.V.; Tabrizi, S.J. Therapeutic antisense targeting of huntingtin. DNA Cell Biol. 2020, 39, 154–158. [Google Scholar] [CrossRef]
- Fields, E.; Vaughan, E.; Tripu, D.; Lim, I.; Shrout, K.; Conway, J.; Salib, N.; Lee, Y.; Dhamsania, A.; Jacobsen, M. Gene targeting techniques for Huntington’s disease. Ageing Res. Rev. 2021, 70, 101385. [Google Scholar] [CrossRef]
- Wild, E.J.; Tabrizi, S.J. Therapies targeting DNA and RNA in Huntington’s disease. Lancet Neurol. 2017, 16, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Gardian, G.; Browne, S.E.; Choi, D.-K.; Klivenyi, P.; Gregorio, J.; Kubilus, J.K.; Ryu, H.; Langley, B.; Ratan, R.R.; Ferrante, R.J. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J. Biol. Chem. 2005, 280, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, R.J.; Kubilus, J.K.; Lee, J.; Ryu, H.; Beesen, A.; Zucker, B.; Smith, K.; Kowall, N.W.; Ratan, R.R.; Luthi-Carter, R. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J. Neurosci. 2003, 23, 9418–9427. [Google Scholar] [CrossRef]
- Thomas, E.A.; Coppola, G.; Desplats, P.A.; Tang, B.; Soragni, E.; Burnett, R.; Gao, F.; Fitzgerald, K.M.; Borok, J.F.; Herman, D. The HDAC inhibitor 4b ameliorates the disease phenotype and transcriptional abnormalities in Huntington’s disease transgenic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 15564–15569. [Google Scholar] [CrossRef] [Green Version]
- Jia, H.; Morris, C.D.; Williams, R.M.; Loring, J.F.; Thomas, E.A. HDAC inhibition imparts beneficial transgenerational effects in Huntington’s disease mice via altered DNA and histone methylation. Proc. Natl. Acad. Sci. USA 2015, 112, E56–E64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Pallos, J.; Jacques, V.; Lau, A.; Tang, B.; Cooper, A.; Syed, A.; Purcell, J.; Chen, Y.; Sharma, S. Histone deacetylase (HDAC) inhibitors targeting HDAC3 and HDAC1 ameliorate polyglutamine-elicited phenotypes in model systems of Huntington’s disease. Neurobiol. Dis. 2012, 46, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siebzehnrübl, F.A.; Raber, K.A.; Urbach, Y.K.; Schulze-Krebs, A.; Canneva, F.; Moceri, S.; Habermeyer, J.; Achoui, D.; Gupta, B.; Steindler, D.A. Early postnatal behavioral, cellular, and molecular changes in models of Huntington disease are reversible by HDAC inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E8765–E8774. [Google Scholar] [CrossRef] [Green Version]
- Guedes-Dias, P.; de Proença, J.; Soares, T.R.; Leitão-Rocha, A.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M. HDAC6 inhibition induces mitochondrial fusion, autophagic flux and reduces diffuse mutant huntingtin in striatal neurons. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2015, 1852, 2484–2493. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Ryu, H.; Kubilus, J.K.; D’Mello, S.; Sugars, K.L.; Lee, J.; Lu, P.; Smith, K.; Browne, S.; Beal, M.F. Chemotherapy for the brain: The antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington’s disease. J. Neurosci. 2004, 24, 10335–10342. [Google Scholar] [CrossRef]
- Heiser, V.; Scherzinger, E.; Boeddrich, A.; Nordhoff, E.; Lurz, R.; Schugardt, N.; Lehrach, H.; Wanker, E.E. Inhibition of huntingtin fibrillogenesis by specific antibodies and small molecules: Implications for Huntington’s disease therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 6739–6744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, M.; Machida, Y.; Niu, S.; Ikeda, T.; Jana, N.R.; Doi, H.; Kurosawa, M.; Nekooki, M.; Nukina, N. Trehalose alleviates polyglutamine-mediated pathology in a mouse model of Huntington disease. Nat. Med. 2004, 10, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Y.D.; Wang, N.; Shen, K.; Stricos, M.; Langfelder, P.; Cheon, K.H.; Cortés, E.P.; Vinters, H.V.; Vonsattel, J.P.; Wexler, N.S.; et al. Disease-related Huntingtin seeding activities in cerebrospinal fluids of Huntington’s disease patients. Sci. Rep. 2020, 10, 20295. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Ng, B.; Sim, B.; Radulescu, C.I.; Yusof, N.A.B.M.; Goh, W.I.; Lin, S.; Lim, J.S.Y.; Cha, Y.; Kusko, R.; et al. pS421 huntingtin modulates mitochondrial phenotypes and confers neuroprotection in an HD hiPSC model. Cell Death Dis. 2020, 11, 809. [Google Scholar] [CrossRef] [PubMed]
- Kratter, I.H.; Zahed, H.; Lau, A.; Tsvetkov, A.S.; Daub, A.C.; Weiberth, K.F.; Gu, X.; Saudou, F.; Humbert, S.; Yang, X.W.; et al. Serine 421 regulates mutant huntingtin toxicity and clearance in mice. J. Clin. Investig. 2016, 126, 3585–3597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.J.; Kloock, S.J.; Nagel, M.; Ortiz-Rios, M.M.; Hofmann, J.; Riess, O.; Nguyen, H.P. Calpastatin ablation aggravates the molecular phenotype in cell and animal models of Huntington disease. Neuropharmacology 2018, 133, 94–106. [Google Scholar] [CrossRef]
- Kim, M.; Lee, H.; LaForet, G.; McIntyre, C.; Martin, E.J.; Chang, P.; Kim, T.W.; Williams, M.; Reddy, P.; Tagle, D. Mutant huntingtin expression in clonal striatal cells: Dissociation of inclusion formation and neuronal survival by caspase inhibition. J. Neurosci. 1999, 19, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieux, M.; Alpaugh, M.; Sciacca, G.; Saint-Pierre, M.; Masnata, M.; Denis, H.L.; Lévesque, S.A.; Herrmann, F.; Bazenet, C.; Garneau, A.P. Shedding a new light on Huntington’s disease: How blood can both propagate and ameliorate disease pathology. Mol. Psychiatry 2020, 26, 5441–5463. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, R.J.; Andreassen, O.A.; Jenkins, B.G.; Dedeoglu, A.; Kuemmerle, S.; Kubilus, J.K.; Kaddurah-Daouk, R.; Hersch, S.M.; Beal, M.F. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J. Neurosci. 2000, 20, 4389–4397. [Google Scholar] [CrossRef] [Green Version]
- Koroshetz, W.J.; Jenkins, B.G.; Rosen, B.R.; Beal, M.F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child. Neurol. Soc. 1997, 41, 160–165. [Google Scholar] [CrossRef]
- Investigators, H.S.G.P.C. Safety and tolerability of high-dosage coenzyme Q10 in Huntington’s disease and healthy subjects. Mov. Disord. 2010, 25, 1924–1928. [Google Scholar]
- Essa, M.M.; Moghadas, M.; Ba-Omar, T.; Qoronfleh, M.W.; Guillemin, G.J.; Manivasagam, T.; Justin-Thenmozhi, A.; Ray, B.; Bhat, A.; Chidambaram, S.B. Protective effects of antioxidants in Huntington’s disease: An extensive review. Neurotox. Res. 2019, 35, 739–774. [Google Scholar] [CrossRef]
- McMeekin, L.J.; Fox, S.N.; Boas, S.M.; Cowell, R.M. Dysregulation of PGC-1α-dependent transcriptional programs in neurological and developmental disorders: Therapeutic challenges and opportunities. Cells 2021, 10, 352. [Google Scholar] [CrossRef]
- Lopes, C.; Ribeiro, M.; Duarte, A.I.; Humbert, S.; Saudou, F.; De Almeida, L.P.; Hayden, M.; Rego, A.C. IGF-1 intranasal administration rescues Huntington’s disease phenotypes in YAC128 mice. Mol. Neurobiol. 2014, 49, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Pandey, N.K.; Lim, C.K.-W.; Ding, Z.; Tao, M.; Thomas, D.D.; Langen, R.; Sachs, J.N. Discovery of small molecule inhibitors of huntingtin exon 1 aggregation by FRET-Based high-throughput screening in living cells. ACS Chem. Neurosci. 2020, 11, 2286–2295. [Google Scholar] [CrossRef] [PubMed]
- Gies, E.; Wilde, I.; Winget, J.M.; Brack, M.; Rotblat, B.; Novoa, C.A.; Balgi, A.D.; Sorensen, P.H.; Roberge, M.; Mayor, T. Niclosamide prevents the formation of large ubiquitin-containing aggregates caused by proteasome inhibition. PLoS ONE 2010, 5, e14410. [Google Scholar] [CrossRef] [Green Version]
- Tang, T.-S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinás, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef] [Green Version]
- Farina, F.; Lambert, E.; Commeau, L.; Lejeune, F.-X.; Roudier, N.; Fonte, C.; Parker, J.A.; Boddaert, J.; Verny, M.; Baulieu, E.-E. The stress response factor daf-16/FOXO is required for multiple compound families to prolong the function of neurons with Huntington’s disease. Sci. Rep. 2017, 7, 4014. [Google Scholar] [CrossRef] [Green Version]
- Ehrnhoefer, D.E.; Skotte, N.H.; Reinshagen, J.; Qiu, X.; Windshügel, B.; Jaishankar, P.; Ladha, S.; Petina, O.; Khankischpur, M.; Nguyen, Y.T.N.; et al. Activation of caspase-6 is promoted by a mutant huntingtin fragment and blocked by an allosteric inhibitor compound. Cell Chem. Biol. 2019, 26, 1295–1305.e1296. [Google Scholar] [CrossRef]
- Wei, H.; Qin, Z.-H.; Senatorov, V.; Wei, W.; Wang, Y.; Qian, Y.; Chuang, D.-M. Lithium suppresses excitotoxicity-induced striatal lesions in a rat model of Huntington’s disease. Neuroscience 2001, 106, 603–612. [Google Scholar] [CrossRef]
- Ellrichmann, G.; Blusch, A.; Fatoba, O.; Brunner, J.; Reick, C.; Hayardeny, L.; Hayden, M.; Sehr, D.; Winklhofer, K.F.; Saft, C.; et al. Laquinimod treatment in the R6/2 mouse model. Sci. Rep. 2017, 7, 4947. [Google Scholar] [CrossRef]
- Megahed, M.; El-Azab, M.F.; El Sayed, M.I.; Moustafa, Y. Huntington’s disease and NMDA receptors; a new arena for therapeutic development. Rec. Pharm. Biomed. Sci. 2018, 2, 1–13. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Y.; Bai, G.; Li, H. Necrostatin-1 ameliorates symptoms in R6/2 transgenic mouse model of Huntington’s disease. Cell Death Dis. 2011, 2, e115. [Google Scholar] [CrossRef] [PubMed]
- Beconi, M.G.; Howland, D.; Park, L.; Lyons, K.; Giuliano, J.; Dominguez, C.; Munoz-Sanjuan, I.; Pacifici, R. Pharmacokinetics of memantine in rats and mice. PLoS Curr. 2011, 3, RRN1291. [Google Scholar] [CrossRef] [PubMed]
- Reilmann, R.; Ross, C.; Testa, C.; Frank, S.; Evers, M.; de Haan, M.; Valles-Sanchez, A.; Konstantinova, P.; van Deventer, S.; Higgins, J. Translation of AMT-130 preclinical data to inform the design of the first FDA-approved human AAV gene therapy clinical trial in adults with early manifest Huntington’s disease (4531). Neurology 2020, 94, 4531. [Google Scholar]
- Tabrizi, S.J.; Leavitt, B.R.; Landwehrmeyer, G.B.; Wild, E.J.; Saft, C.; Barker, R.A.; Blair, N.F.; Craufurd, D.; Priller, J.; Rickards, H. Targeting huntingtin expression in patients with Huntington’s disease. New Engl. J. Med. 2019, 380, 2307–2316. [Google Scholar] [CrossRef] [PubMed]
- Kordasiewicz, H.B.; Stanek, L.M.; Wancewicz, E.V.; Mazur, C.; McAlonis, M.M.; Pytel, K.A.; Artates, J.W.; Weiss, A.; Cheng, S.H.; Shihabuddin, L.S. Sustained therapeutic reversal of Huntington’s disease by transient repression of huntingtin synthesis. Neuron 2012, 74, 1031–1044. [Google Scholar] [CrossRef] [Green Version]
- Karpuj, M.V.; Becher, M.W.; Springer, J.E.; Chabas, D.; Youssef, S.; Pedotti, R.; Mitchell, D.; Steinman, L. Prolonged survival and decreased abnormal movements in transgenic model of Huntington disease, with administration of the transglutaminase inhibitor cystamine. Nat. Med. 2002, 8, 143–149. [Google Scholar] [CrossRef]
- Dedeoglu, A.; Kubilus, J.K.; Jeitner, T.M.; Matson, S.A.; Bogdanov, M.; Kowall, N.W.; Matson, W.R.; Cooper, A.J.; Ratan, R.R.; Beal, M.F. Therapeutic effects of cystamine in a murine model of Huntington’s disease. J. Neurosci. 2002, 22, 8942–8950. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, I.; Mahlke, C.; Yuan, J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature 2003, 421, 373–379. [Google Scholar] [CrossRef]
- Chen, M.; Ona, V.O.; Li, M.; Ferrante, R.J.; Fink, K.B.; Zhu, S.; Bian, J.; Guo, L.; Farrell, L.A.; Hersch, S.M. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000, 6, 797–801. [Google Scholar] [CrossRef]
- Rea, S.; Della-Morte, D.; Pacifici, F.; Capuani, B.; Pastore, D.; Coppola, A.; Arriga, R.; Andreadi, A.; Donadel, G.; Di Daniele, N. Insulin and exendin-4 reduced mutated Huntingtin accumulation in neuronal cells. Front. Pharmacol. 2020, 11, 779. [Google Scholar] [CrossRef]
- Di Pardo, A.; Maglione, V.; Alpaugh, M.; Horkey, M.; Atwal, R.S.; Sassone, J.; Ciammola, A.; Steffan, J.S.; Fouad, K.; Truant, R. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3528–3533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ermak, G.; Hench, K.J.; Chang, K.T.; Sachdev, S.; Davies, K.J. Regulator of calcineurin (RCAN1-1L) is deficient in Huntington disease and protective against mutant huntingtin toxicity in vitro. J. Biol. Chem. 2009, 284, 11845–11853. [Google Scholar] [CrossRef] [Green Version]
- Rangone, H.; Poizat, G.; Troncoso, J.; Ross, C.A.; MacDonald, M.E.; Saudou, F.; Humbert, S. The serum-and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur. J. Neurosci. 2004, 19, 273–279. [Google Scholar] [CrossRef]
- Dickey, A.S.; Pineda, V.V.; Tsunemi, T.; Liu, P.P.; Miranda, H.C.; Gilmore-Hall, S.K.; Lomas, N.; Sampat, K.R.; Buttgereit, A.; Torres, M.-J.M. PPAR-δ is repressed in Huntington’s disease, is required for normal neuronal function and can be targeted therapeutically. Nat. Med. 2016, 22, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Li, J. Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 2013, 4, e716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morigaki, R.; Goto, S. Striatal vulnerability in Huntington’s disease: Neuroprotection versus neurotoxicity. Brain Sci. 2017, 7, 63. [Google Scholar] [CrossRef] [PubMed]
- Casella, C.; Lipp, I.; Rosser, A.; Jones, D.K.; Metzler-Baddeley, C. A critical review of white matter changes in huntington’s disease. Mov. Disord. 2020, 35, 1302–1311. [Google Scholar] [CrossRef] [PubMed]
- Franklin, G.L.; Camargo, C.H.F.; Meira, A.T.; Lima, N.S.; Teive, H.A. The role of the cerebellum in Huntington’s disease: A systematic review. Cerebellum 2021, 20, 254–265. [Google Scholar] [CrossRef]
- Reiner, A.; Deng, Y.P. Disrupted striatal neuron inputs and outputs in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 250–280. [Google Scholar] [CrossRef]
- Yang, H.-M.; Yang, S.; Huang, S.-S.; Tang, B.-S.; Guo, J.-F. Microglial Activation in the Pathogenesis of Huntington’s Disease. Front. Aging Neurosci. 2017, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Levine, M.S. Synaptic dysfunction in Huntington’s disease: Lessons from genetic animal models. Neuroscience 2020, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Unconventional secretion and intercellular transfer of mutant huntingtin. Cells 2018, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Wanker, E.E.; Ast, A.; Schindler, F.; Trepte, P.; Schnoegl, S. The pathobiology of perturbed mutant huntingtin protein–protein interactions in Huntington’s disease. J. Neurochem. 2019, 151, 507–519. [Google Scholar] [CrossRef]
- Pandey, M.; Rajamma, U. Huntington’s disease: The coming of age. J. Genet. 2018, 97, 649–664. [Google Scholar] [CrossRef]
- Tellone, E.; Galtieri, A.; Ficarra, S. Reviewing biochemical implications of normal and mutated Huntingtin in Huntington’s disease. Curr. Med. Chem. 2020, 27, 5137–5158. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Sutton, L.; Hayden, M.R. Small changes, big impact: Posttranslational modifications and function of huntingtin in Huntington disease. Neurosci. 2011, 17, 475–492. [Google Scholar] [CrossRef]
- Soares, T.R.; Reis, S.D.; Pinho, B.R.; Duchen, M.R.; Oliveira, J.M.A. Targeting the proteostasis network in Huntington’s disease. Ageing Res. Rev. 2019, 49, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Cuervo, A.M.; Zhang, S. HTT/Huntingtin in selective autophagy and Huntington disease: A foe or a friend within? Autophagy 2015, 11, 858–860. [Google Scholar] [CrossRef] [Green Version]
- Valionyte, E.; Yang, Y.; Roberts, S.L.; Kelly, J.; Lu, B.; Luo, S. Lowering mutant huntingtin levels and toxicity: Autophagy-endolysosome pathways in Huntington’s disease. J. Mol. Biol. 2020, 432, 2673–2691. [Google Scholar] [CrossRef]
- Paul, B.D.; Snyder, S.H. Impaired redox signaling in Huntington’s disease: Therapeutic implications. Front. Mol. Neurosci. 2019, 12, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubinsky, J.M. Towards an understanding of energy impairment in Huntington’s disease brain. J. Huntingt. Dis. 2017, 6, 267–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, A.; Agrawal, N. Metabolism in Huntington’s disease: A major contributor to pathology. Metab. Brain Dis. 2021, 1–15. [Google Scholar] [CrossRef]
- Romo, L.; Mohn, E.S.; Aronin, N. A fresh look at Huntingtin mRNA processing in Huntington’s disease. J. Huntingt. Dis. 2018, 7, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Heinz, A.; Nabariya, D.K.; Krauss, S. Huntingtin and its role in mechanisms of RNA-mediated toxicity. Toxins 2021, 13, 487. [Google Scholar] [CrossRef]
- Valadão, P.A.C.; Santos, K.B.S.; E Vieira, T.H.F.; E Cordeiro, T.M.; Teixeira, A.L.; Guatimosim, C.; de Miranda, A.S. Inflammation in Huntington’s disease: A few new twists on an old tale. J. Neuroimmunol. 2020, 348, 577380. [Google Scholar] [CrossRef]
- Mattis, V.B.; Svendsen, C.N. Modeling Huntington’s disease with patient-derived neurons. Brain Res. 2017, 1656, 76–87. [Google Scholar] [CrossRef]
- Monk, R.; Connor, B. Cell Reprogramming to Model Huntington’s Disease: A Comprehensive Review. Cells 2021, 10, 1565. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M.; Aigner, L. Reactive Neuroblastosis in Huntington’s Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain. Front. Cell. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Connor, B. Concise review: The use of stem cells for understanding and treating Huntington’s disease. Stem Cells 2018, 36, 146–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colpo, G.D.; Stimming, E.F.; Teixeira, A.L. Stem cells in animal models of Huntington disease: A systematic review. Mol. Cell. Neurosci. 2019, 95, 43–50. [Google Scholar] [CrossRef]
- Gorantla, V.R.; Bhat, A.; Ghosh, A.R.; Bolla, S.R.; Bhojaraj, S.; Mohan, S.K.; Veeraraghavan, V.P.; Chidambaram, S.B.; Essa, M.M.; Qoronfleh, M.W. Stem cells therapy: A ray of hope for Huntington disease. Int. J. Nutr. Pharmacol. Neurol. Dis. 2021, 11, 95. [Google Scholar]
- Albin, R.L.; Kordower, J.H. A. Failed Future. Mov. Disord. 2020, 35, 1299–1301. [Google Scholar] [CrossRef] [PubMed]
- Rosser, A.E.; Busse, M.; Aron Badin, R.; Canals, J.M.; Wheelock, V.; Perrier, A.L.; Gray, W.; Thompson, L.; Goldman, S. Cell therapy for Huntington’s disease: Learning from failure. Mov. Disord. 2021, 36, 787–788. [Google Scholar] [CrossRef]
- Csobonyeiova, M.; Polak, S.; Danisovic, L. Recent overview of the use of iPSCs Huntington’s disease modeling and therapy. Int. J. Mol. Sci. 2020, 21, 2239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinageshwar, B.; Petersen, R.B.; Dunbar, G.L.; Rossignol, J. Prion-like mechanisms in neurodegenerative disease: Implications for Huntington’s disease therapy. Stem Cells Transl. Med. 2020, 9, 559–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachoud-Lévi, A.C.; Massart, R.; Rosser, A. Cell therapy in Huntington’s disease: Taking stock of past studies to move the field forward. Stem Cells 2021, 39, 144–155. [Google Scholar]
- Choi, K.-A.; Choi, Y.; Hong, S. Stem cell transplantation for Huntington’s diseases. Methods 2018, 133, 104–112. [Google Scholar] [CrossRef]
- Wojtecki, L.; Groiss, S.J.; Hartmann, C.J.; Elben, S.; Omlor, S.; Schnitzler, A.; Vesper, J. Deep brain stimulation in Huntington’s disease—preliminary evidence on pathophysiology, efficacy and safety. Brain Sci. 2016, 6, 38. [Google Scholar] [CrossRef]
- Pini, L.; Jacquemot, C.; Cagnin, A.; Meneghello, F.; Semenza, C.; Mantini, D.; Vallesi, A. Aberrant brain network connectivity in presymptomatic and manifest Huntington’s disease: A systematic review. Hum. Brain Mapp. 2020, 41, 256–269. [Google Scholar] [CrossRef]
- Cardoso, F. Chapter fifty-Nonmotor symptoms in Huntington disease. In International Review of Neurobiology; Chaudhuri, K.R., Titova, N., Eds.; Academic Press: Cambridge, MA, 2017; Volume 134, pp. 1397–1408. [Google Scholar]
- Patil, R.S.; Vyas, S.G.; Quazi, W.T.; Tembhurnikar, H.J.; Milmile, P.S.; Umekar, M.J. The gut microbiome in Huntington disease: A review. GSC Biol. Pharm. Sci. 2021, 15, 317–326. [Google Scholar] [CrossRef]
- Schultz, J.L.; Nopoulos, P.C.; Gonzalez-Alegre, P. Human Immunodeficiency Virus Infection in Huntington’s Disease is Associated with an Earlier Age of Symptom Onset. J. Huntingt. Dis. 2018, 7, 163–166. [Google Scholar] [CrossRef] [PubMed]
- McCusker, E.A.; Loy, C.T. Huntington disease: The complexities of making and disclosing a clinical diagnosis after premanifest genetic testing. Tremor Other Hyperkinet Mov. 2017, 7, 467. [Google Scholar] [CrossRef]
- Przybyl, L.; Wozna-Wysocka, M.; Kozlowska, E.; Fiszer, A. What, when and how to measure—peripheral biomarkers in therapy of Huntington’s disease. Int. J. Mol. Sci. 2021, 22, 1561. [Google Scholar] [CrossRef] [PubMed]
- Schobel, S.A.; Palermo, G.; Auinger, P.; Long, J.D.; Ma, S.; Khwaja, O.S.; Trundell, D.; Cudkowicz, M.; Hersch, S.; Sampaio, C.; et al. Motor, cognitive, and functional declines contribute to a single progressive factor in early HD. Neurology 2017, 89, 2495–2502. [Google Scholar] [CrossRef]
- Claassen, D.O.; Iyer, R.G.; Shah-Manek, B.; DiBonaventura, M.; Abler, V.; Sung, V.W. Tetrabenazine treatment patterns and outcomes for chorea associated with Huntington disease: A retrospective chart review. J. Huntingt. Dis. 2018, 7, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.; Kegelmeyer, D.; Kloos, A.; Rao, A.K.; Busse, M.; Fritz, N.E. Clinical recommendations to guide physical therapy practice for Huntington disease. Neurology 2020, 94, 217–228. [Google Scholar] [CrossRef] [Green Version]
- Zarotti, N.; Dale, M.; Eccles, F.; Simpson, J. Psychological Interventions for People with Huntington’s Disease: A Call to Arms. J. Huntingt. Dis. 2020, 9, 231–243. [Google Scholar] [CrossRef]
- Eccles, F.J.R.; Craufurd, D.; Smith, A.; Davies, R.; Glenny, K.; Homberger, M.; Peeren, S.; Rogers, D.; Rose, L.; Skitt, Z.; et al. A feasibility investigation of mindfulness-based cognitive therapy for people with Huntington’s disease. Pilot Feasibility Stud. 2020, 6, 90. [Google Scholar] [CrossRef]
- Rossi, G.; Oh, J.C. Management of agitation in Huntington’s disease: A review of the literature. Cureus 2020, 12, e9748. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Cheng, S.; Yang, H.; Zhu, L.; Pan, Y.; Jing, L.; Tang, B.; Li, S.; Li, X.-J. Loss of Hap1 selectively promotes striatal degeneration in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2020, 117, 20265–20273. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, S.; Jing, L.; Huang, L.; Chen, L.; Zhao, X.; Yang, W.; Pan, Y.; Yin, P.; Qin, Z.S.; et al. Truncation of mutant huntingtin in knock-in mice demonstrates exon1 huntingtin is a key pathogenic form. Nat. Commun. 2020, 11, 2582. [Google Scholar] [CrossRef]
- Sharma, M.; Subramaniam, S. Rhes travels from cell to cell and transports Huntington disease protein via TNT-like protrusion. J. Cell Biol. 2019, 218, 1972–1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, M.; Rajendrarao, S.; Shahani, N.; Ramírez-Jarquín, U.N.; Subramaniam, S. Cyclic GMP-AMP synthase promotes the inflammatory and autophagy responses in Huntington disease. Proc. Natl. Acad. Sci. USA 2020, 117, 15989–15999. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Noh, J.-Y.; Oh, Y.; Kim, Y.; Chang, J.-W.; Chung, C.-W.; Lee, S.-T.; Kim, M.; Ryu, H.; Jung, Y.-K. IRE1 plays an essential role in ER stress-mediated aggregation of mutant huntingtin via the inhibition of autophagy flux. Hum. Mol. Genet. 2011, 21, 101–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Kosaras, B.; Del Signore, S.J.; Cormier, K.; McKee, A.; Ratan, R.R.; Kowall, N.W.; Ryu, H. Modulation of lipid peroxidation and mitochondrial function improves neuropathology in Huntington’s disease mice. Acta Neuropathol. 2011, 121, 487–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, G.S.; Kim, K.Y.; Hwang, Y.J.; Jung, M.-K.; An, S.; Ouchi, M.; Ouchi, T.; Kowall, N.; Lee, J.; Ryu, H. Deregulation of BRCA1 leads to impaired spatiotemporal dynamics of γ-H2AX and DNA damage responses in Huntington’s disease. Mol. Neurobiol. 2012, 45, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Hyeon, S.J.; Park, J.; Yoo, J.; Kim, S.-H.; Hwang, Y.J.; Kim, S.-C.; Liu, T.; Shim, H.S.; Kim, Y.; Cho, Y.; et al. Dysfunction of X-linked inhibitor of apoptosis protein (XIAP) triggers neuropathological processes via altered p53 activity in Huntington’s disease. Prog. Neurobiol. 2021, 204, 102110. [Google Scholar] [CrossRef] [PubMed]
- Mario Isas, J.; Pandey, N.K.; Xu, H.; Teranishi, K.; Okada, A.K.; Fultz, E.K.; Rawat, A.; Applebaum, A.; Meier, F.; Chen, J.; et al. Huntingtin fibrils with different toxicity, structure, and seeding potential can be interconverted. Nat. Commun. 2021, 12, 4272. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, O.; Chen, C.; Bingham, R.; Argyrou, A.; Buxton, R.; Jönsson, C.P.; Jones, E.; Bridges, A.; Gatfield, K.; Krauß, S. Pharmacological disruption of the MID1/α4 interaction reduces mutant Huntingtin levels in primary neuronal cultures. Neurosci. Lett. 2018, 673, 44–50. [Google Scholar] [CrossRef] [Green Version]
- Miyazaki, H.; Yamanaka, T.; Oyama, F.; Kino, Y.; Kurosawa, M.; Yamada-Kurosawa, M.; Yamano, R.; Shimogori, T.; Hattori, N.; Nukina, N. FACS-array–based cell purification yields a specific transcriptome of striatal medium spiny neurons in a murine Huntington disease model. J. Biol. Chem. 2020, 295, 9768–9785. [Google Scholar] [CrossRef]
- Critchley, B.J.; Isalan, M.; Mielcarek, M. Neuro-cardio mechanisms in Huntington’s disease and other neurodegenerative disorders. Front. Physiol. 2018, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.T.; Chang, Y.G.; Chern, Y. Insights into GABA(A)ergic system alteration in Huntington’s disease. Open Biol. 2018, 8, 180165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgold, J.; Schulz-Trieglaff, E.K.; Voelkl, K.; Gutiérrez-Ángel, S.; Bader, J.M.; Hosp, F.; Mann, M.; Arzberger, T.; Klein, R.; Liebscher, S.; et al. Cortical circuit alterations precede motor impairments in Huntington’s disease mice. Sci. Rep. 2019, 9, 6634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, A.; Powell, F. Network model of pathology spread recapitulates neurodegeneration and selective vulnerability in Huntington’s Disease. NeuroImage 2021, 235, 118008. [Google Scholar] [CrossRef] [PubMed]
- Lebouc, M.; Richard, Q.; Garret, M.; Baufreton, J. Striatal circuit development and its alterations in Huntington’s disease. Neurobiol. Dis. 2020, 145, 105076. [Google Scholar] [CrossRef] [PubMed]
- Federspiel, J.D.; Greco, T.M.; Lum, K.K.; Cristea, I.M. Hdac4 interactions in Huntington’s disease viewed through the prism of multiomics. Mol. Cell Proteom. 2019, 18, S92–S113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.; Liu, H.P.; Lin, W.Y.; Tsai, F.J. Identification of contributing genes of Huntington’s disease by machine learning. BMC Med. Genom. 2020, 13, 176. [Google Scholar] [CrossRef]
- Seefelder, M.; Kochanek, S. A meta-analysis of transcriptomic profiles of Huntington’s disease patients. PLoS ONE 2021, 16, e0253037. [Google Scholar]
- Imamura, T.; Fujita, K.; Tagawa, K.; Ikura, T.; Chen, X.; Homma, H.; Tamura, T.; Mao, Y.; Taniguchi, J.B.; Motoki, K.; et al. Identification of hepta-histidine as a candidate drug for Huntington’s disease by in silico-in vitro- in vivo-integrated screens of chemical libraries. Sci. Rep. 2016, 6, 33861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Panwar, S.; Sharma, M.K.; Sharma, M.K. Genes to drug: An in-silico approach to design a drug for Huntington disease (HD) in Homo sapiens. Int. J. Comput. Biol. Drug Des. 2021, 14, 190–201. [Google Scholar] [CrossRef]
- Kohli, H.; Kumar, P.; Ambasta, R.K. In silico designing of putative peptides for targeting pathological protein Htt in Huntington’s disease. Heliyon 2021, 7, e06088. [Google Scholar] [CrossRef] [PubMed]
- Sundaram, J.R.; Wu, Y.; Lee, I.C.; George, S.E.; Hota, M.; Ghosh, S.; Kesavapany, S.; Ahmed, M.; Tan, E.-K.; Shenolikar, S. PromISR-6, a guanabenz analogue, improves cellular survival in an experimental model of Huntington’s disease. ACS Chem. Neurosci. 2019, 10, 3575–3589. [Google Scholar] [CrossRef] [PubMed]
- Deepa, S.; Rymbai, E.; Praveen, T.; Saravanan, J. Neuroprotective effects of farnesol on motor and cognitive impairment against 3-nitropropionic acid-induced Huntington’s disease. Thai J. Pharm. Sci. 2021, 45, 16–23. [Google Scholar]
Figure 1.
Dysfunction of cortico-striatal tripartite synapse in HD. mHTT aggregation deregulates transcription of EAAT2 and Kir4.1 in astrocytes. Deregulation of EAAT2 leads to low expression level of astrocyte glutamate transporter and impairment of the glutamate uptake by astrocytes in the tripartite synapse. As a result, excess glutamate in synapse induces hyper-excitability of the post-synaptic neuron (medial spiny neuron in cortico-striatal synapse). Deregulation of Kir4.1 leads to impaired potassium buffering of astrocytes. As a result, elevated potassium ion concentration increases the membrane potential of neurons, where high membrane potential induces hyper-excitability. Prolonged hyper-excitability is rendered as cellular toxicity.
Figure 1.
Dysfunction of cortico-striatal tripartite synapse in HD. mHTT aggregation deregulates transcription of EAAT2 and Kir4.1 in astrocytes. Deregulation of EAAT2 leads to low expression level of astrocyte glutamate transporter and impairment of the glutamate uptake by astrocytes in the tripartite synapse. As a result, excess glutamate in synapse induces hyper-excitability of the post-synaptic neuron (medial spiny neuron in cortico-striatal synapse). Deregulation of Kir4.1 leads to impaired potassium buffering of astrocytes. As a result, elevated potassium ion concentration increases the membrane potential of neurons, where high membrane potential induces hyper-excitability. Prolonged hyper-excitability is rendered as cellular toxicity.

Figure 2.
Oligodendrocyte dysfunction in HD. In the first pathway, full-length MYRF is self-cleaved to nMYRF which detaches from ER and is translocated to the nucleus to regulate the expression of myelin related genes. In HD, N-terminal mHTT binds nMYRF causing abnormal binding of nMYRF and deficit myelin genes expression. Second pathway shows inhibition of PGC-1α expression by interference of mHTT in co-binding of CREB and TAF4, leading to reduced activity in the cholesterol biosynthesis pathway and myelination deficit. Created with BioRender.com.
Figure 2.
Oligodendrocyte dysfunction in HD. In the first pathway, full-length MYRF is self-cleaved to nMYRF which detaches from ER and is translocated to the nucleus to regulate the expression of myelin related genes. In HD, N-terminal mHTT binds nMYRF causing abnormal binding of nMYRF and deficit myelin genes expression. Second pathway shows inhibition of PGC-1α expression by interference of mHTT in co-binding of CREB and TAF4, leading to reduced activity in the cholesterol biosynthesis pathway and myelination deficit. Created with BioRender.com.

Figure 3.
Epigenetic modifications associated with HD. The promoter regions of neurogenesis-related genes, OCT4, SOX2, and NANOG, are methylated in cells expressing mHTT which can lead to impaired neurogenesis. On the other hand, mHTT sequestrates CBP in nuclear inclusions which causes the hypermethylation and hypoacetylation of histone proteins and CBP depletion. Depletion of CBP from the nucleus of cells leads to histone hypoacetylation, nuclear and nucleolar transcriptional dysfunction and increase in neurotoxicity. Created with BioRender.com.
Figure 3.
Epigenetic modifications associated with HD. The promoter regions of neurogenesis-related genes, OCT4, SOX2, and NANOG, are methylated in cells expressing mHTT which can lead to impaired neurogenesis. On the other hand, mHTT sequestrates CBP in nuclear inclusions which causes the hypermethylation and hypoacetylation of histone proteins and CBP depletion. Depletion of CBP from the nucleus of cells leads to histone hypoacetylation, nuclear and nucleolar transcriptional dysfunction and increase in neurotoxicity. Created with BioRender.com.

Figure 4.
Role of HTT in normal vesicular transport and role of mHTT in disturbed vesicular transport in HD. In normal conditions, HTT participates in motor protein complex with dynactin, dynein, and kinesin. In addition, HTT recruits GAPDH to vesicles to supply energy, ATP to motor proteins. In HD, polyglutamine expansions of the mHTT sequester GAPDH and motor proteins. Microtubules are acetylated by mHTT to hinder binding of motor proteins on the microtubules.
Figure 4.
Role of HTT in normal vesicular transport and role of mHTT in disturbed vesicular transport in HD. In normal conditions, HTT participates in motor protein complex with dynactin, dynein, and kinesin. In addition, HTT recruits GAPDH to vesicles to supply energy, ATP to motor proteins. In HD, polyglutamine expansions of the mHTT sequester GAPDH and motor proteins. Microtubules are acetylated by mHTT to hinder binding of motor proteins on the microtubules.

Figure 5.
Mitochondria dysfunction associated with HD. mHTT aggregation disrupts the mitochondrial membrane potential and increases excitotoxin-induced Ca2+ influx which leads to decreased ATP generation and increased ROS. Mitochondria dysfunction results in release of cytochrome C from the mitochondria which triggers the activation of apoptotic cascade via caspases 9 and 3, and neuronal injury. On the other hand, in HD, mHTT binds with high-affinity to TIM23 in mitochondrial intermembrane space, causing diminished levels of nuclear-encoded proteins imported through TIM23 and subsequently, neuronal death. Finally, VCP is selectively translocated to the mitochondria, where it is bound to mHTT and LC3 to enhance mitophagosome production, and reduce mitochondrial mass and energy supply, causing neuronal cell death. Created with BioRender.com.
Figure 5.
Mitochondria dysfunction associated with HD. mHTT aggregation disrupts the mitochondrial membrane potential and increases excitotoxin-induced Ca2+ influx which leads to decreased ATP generation and increased ROS. Mitochondria dysfunction results in release of cytochrome C from the mitochondria which triggers the activation of apoptotic cascade via caspases 9 and 3, and neuronal injury. On the other hand, in HD, mHTT binds with high-affinity to TIM23 in mitochondrial intermembrane space, causing diminished levels of nuclear-encoded proteins imported through TIM23 and subsequently, neuronal death. Finally, VCP is selectively translocated to the mitochondria, where it is bound to mHTT and LC3 to enhance mitophagosome production, and reduce mitochondrial mass and energy supply, causing neuronal cell death. Created with BioRender.com.


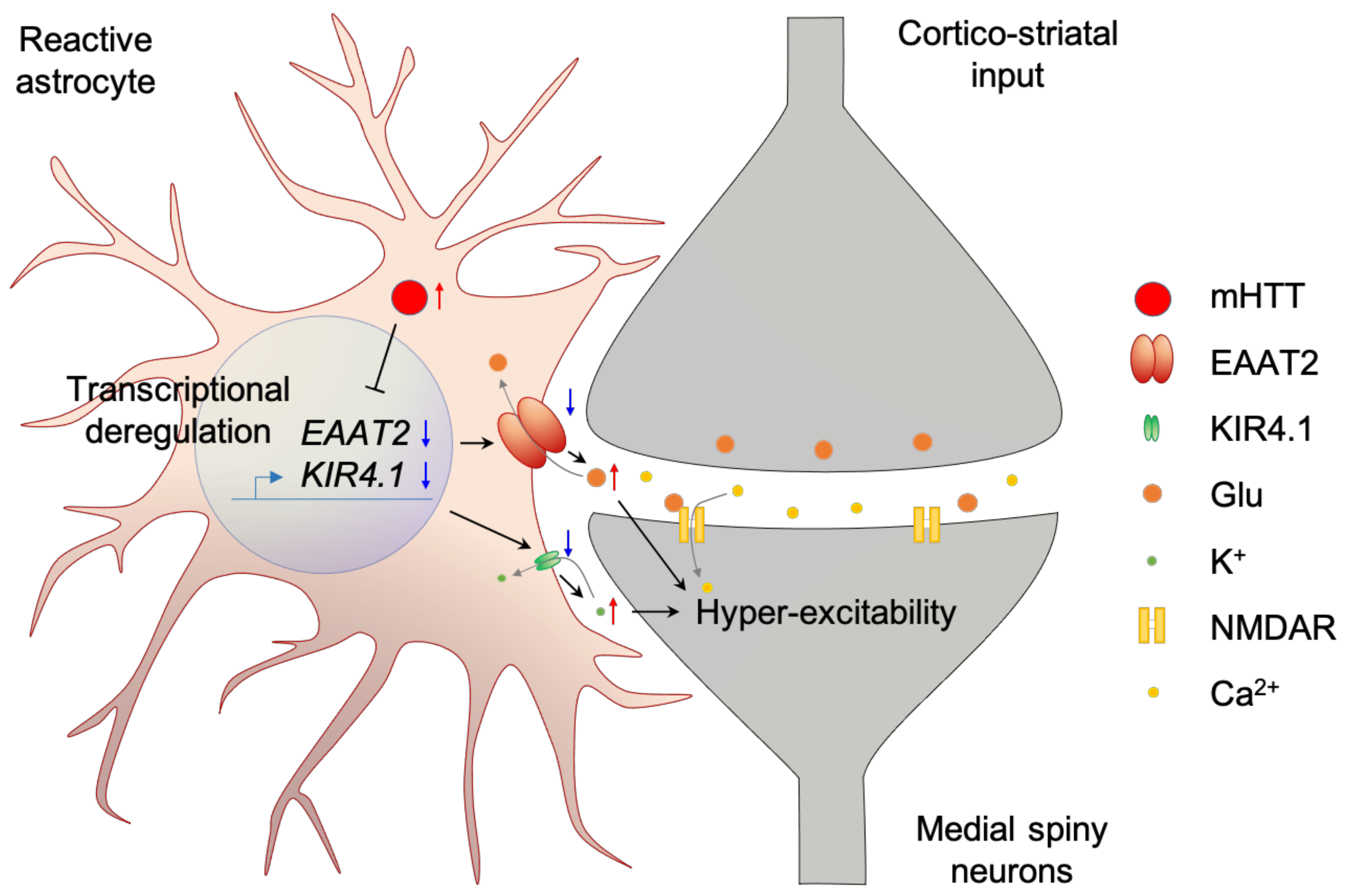{kind=link}
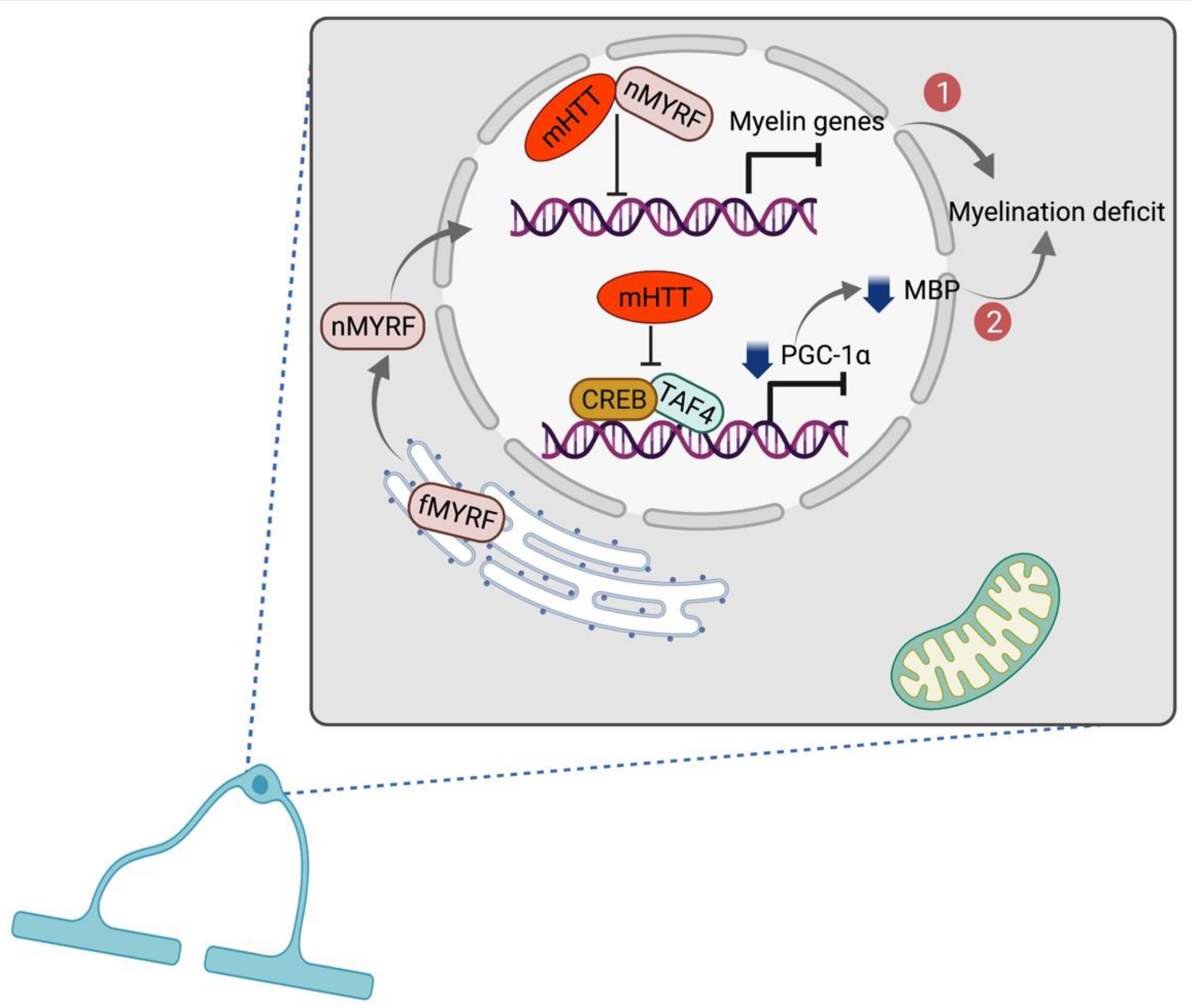{kind=link}
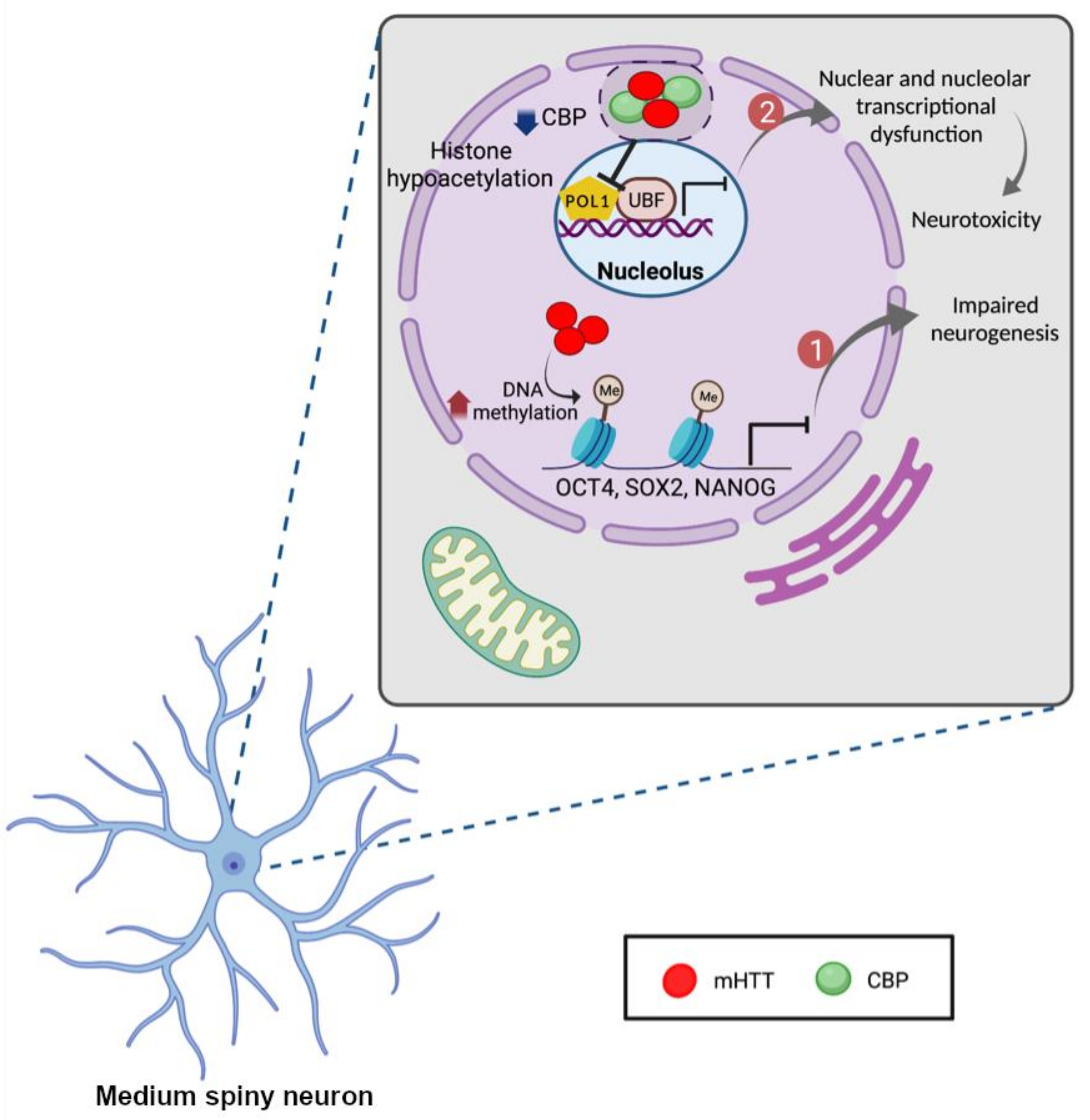{kind=link}
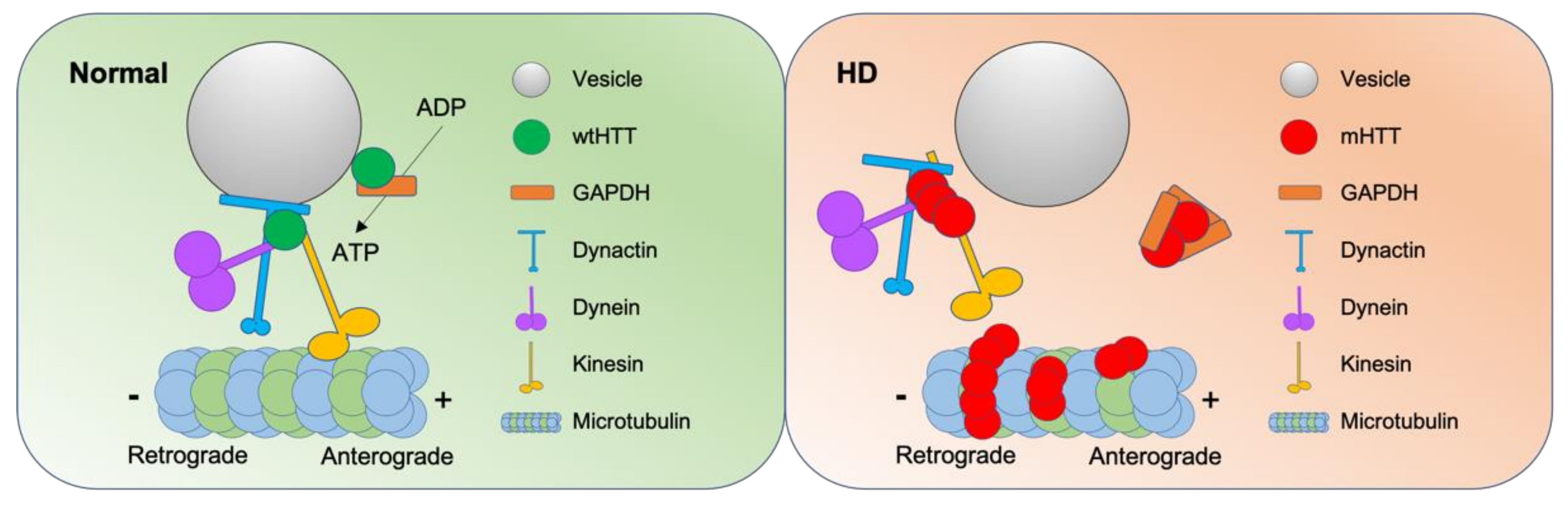{kind=link}
{kind=link}
Table 1.
Non-cell autonomous cell death pathway in HD related to astrocytes.
| HD Pathology | Specimen | Brain Region/Cell Type | Experimental Method | Reference |
|---|---|---|---|---|
| Less EAAT2 mRNA | Postmortem | Cingulate cortex | RNA sequencing | [78] |
| Neostriatum | In situ hybridization | [79] | ||
| Less EAAT2 protein | Postmortem | Striatum | Immunohistochemistry | [34] |
| Striatum, cortex | Western blot | [80] | ||
| Mouse; R6/2 | Striatum, cortex, hippocampus, midbrain | Quantitative proteomics | [81] | |
| Less glutamate uptake | Postmortem | Prefrontal cortex | Glutamate uptake assay | [84] |
| Cell; astrocyte | differentiated from Q77 monkey iPSC | Glutamate uptake assay | [82] | |
| Mouse; Q175 | Single corticostriatal synapse | Imaging assay with glutamate sensor | [83] | |
| Less Kir4.1 mRNA | Postmortem | cingulate cortex | RNA sequencing | [78] |
| Less Kir4.1 protein Higher extracellular K+More excitable | Mouse; R6/2 | Striatal MSN and astrocyte | qPCR, IHC, Western blot, Virus microinjection, Electrophysiology | [86] |
| Altered Ca2+ signal | Mouse; R6/2 | Striatal astrocyte | Virus microinjectionElectrophysiology | [87] |
| More excitotoxicity | Cell; neuron and astrocyte | Co-culture of HD neurons and astrocytes from human iPSC | Cell count after glutamate exposure | [89] |
| Co-culture of wild type neurons with mHTT infected glia from rat primary culture | [90] |
Table 2.
Therapeutic targets for HD.
| Target | Strategy | Mode of Action | Disease Model | Clinical Trial & NCTno. | References |
|---|---|---|---|---|---|
| mHTT gene | CRISPR/Cas9 | Excise mHTT DNA selectively | Cell iPS | [174] | |
| Mouse BacHD | [175] | ||||
| Mouse HD140Q | [176] | ||||
| Mouse R6/2 | [177] | ||||
| KRAB-ZFPs | Inhibition of translation or transcriptdegradation | Mouse R6/1,2 | [178,179] | ||
| shRNA | Mouse R6/2 | [180] | |||
| Mouse N171-82Q | [181,182] | ||||
| siRNA | Mouse HTT injected | [183] | |||
| Mouse Hdh-150Q | [184] | ||||
| miRNA | Mouse HD140Q | Phase I/II [235], NCT04120493 | [185] | ||
| Antisense nucleotide | Bind to HTT mRNA | Mouse BACHD | Phase II, NCT02519036 | [236,237] | |
| Transcriptional dysregulation | Phenylbutyrate | Inactivate histone deacetylase | Mouse N171-82Q | Phase II, NCT00212316 | [202] |
| Sodium butyrate | Mouse R6/2 | [203] | |||
| HDACi 4b | Mouse N171–82Q | [204,205,206] | |||
| HDACi LBH589 | Transgenic Rodent HD Models | [207] | |||
| Tubastatin A | Cell primary neuron | [208] | |||
| CKD-504 | Phase I, NCT03713892 | ||||
| Mithramycin | Increase H3K9 | Mouse R6/2 | [209] | ||
| mHTT aggregation | Cystamine | Suppress mHTT crosslinking | Mouse R6/2 | [238,239] | |
| Congo red | Bind and inhibit polyglutamineoligomerization | Mouse R6/2 | [240] | ||
| ChrysamineG, Direct fast yellow | [210] | ||||
| Trehalose | [211] | ||||
| mHTT fragmentation | Minocycline | Inhibit caspase | Mouse R6/2 | Phase III, NCT00277355 | [241] |
| Z-VAD-FMK | Cell X57 | [216] | |||
| mHTT lowering | Blood transfusion | Remove circulating mHTT | Mouse zQ175 | [217] | |
| mHTT post-modification | Insulin, exendin-4 | Increase mHTT Phosphorylation | Cell SH-SY5Y | [242] | |
| GM1 | Mouse YAC128 | [243] | |||
| RCAN1-1L | Cell ST14A | [244] | |||
| SGK | Cell primary neuron | [245] | |||
| Transactivation | KD3010 | Increased PPARδ transactivation | Mouse pCAGGS-loxP-STOP-loxP | [246] | |
| Mitochondrial dysfunction | Creatine | Inactivate mitochondrial permeability transition | Mouse R6/2 | Phase II, NCT00026988 | [218] |
| Coenzyme Q10 | Enhance electron transport | Phase II, NCT00920699 | [219,220] | ||
| PGC-1α | Upregulate mitochondrial gene | [222] | |||
| Metabolism | rhIGF-1 | increase glucose uptake | Mouse YAC128 Mouse R6/2 | [223] | |
| Autophagy | Niclosamide | Inhibit mTOR | Cell HEK293, N2a | [225] | |
| MAP4343, 17βE2, Isoquercitrin | Regulate stress response | C.elegans HD mutants | [228] | ||
| Apoptosis | Z-VAD-FMK, Z-DEVD-FMK, Z-LEHD-FMK | Inhibit caspase | Cell primary neuron | [227] | |
| PG3d | Cell COS-7 | [229] | |||
| Lithium chloride | Rat QA injected | [230] | |||
| Excitotoxicity | Memantine | Inhibit NMDA receptor | Phase IV, NCT00652457; Phase II, NCT00652457 | ||
| Necrostatin-1 | Inhibit RIP1 kinase | Mouse R6/2 | [233,247] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, C.; Yousefian-Jazi, A.; Choi, S.-H.; Chang, I.; Lee, J.; Ryu, H. Non-Cell Autonomous and Epigenetic Mechanisms of Huntington’s Disease. Int. J. Mol. Sci. 2021, 22, 12499. https://doi.org/10.3390/ijms222212499
AMA Style
Kim C, Yousefian-Jazi A, Choi S-H, Chang I, Lee J, Ryu H. Non-Cell Autonomous and Epigenetic Mechanisms of Huntington’s Disease. International Journal of Molecular Sciences. 2021; 22(22):12499. https://doi.org/10.3390/ijms222212499
Chicago/Turabian StyleKim, Chaebin, Ali Yousefian-Jazi, Seung-Hye Choi, Inyoung Chang, Junghee Lee, and Hoon Ryu. 2021. "Non-Cell Autonomous and Epigenetic Mechanisms of Huntington’s Disease" International Journal of Molecular Sciences 22, no. 22: 12499. https://doi.org/10.3390/ijms222212499
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.