
Bone Formation Ability and Cell Viability Enhancement of MC3T3-E1 Cells by Ferrostatin-1 a Ferroptosis Inhibitor of Cancer Cells

Abstract
:1. Introduction
2. Results
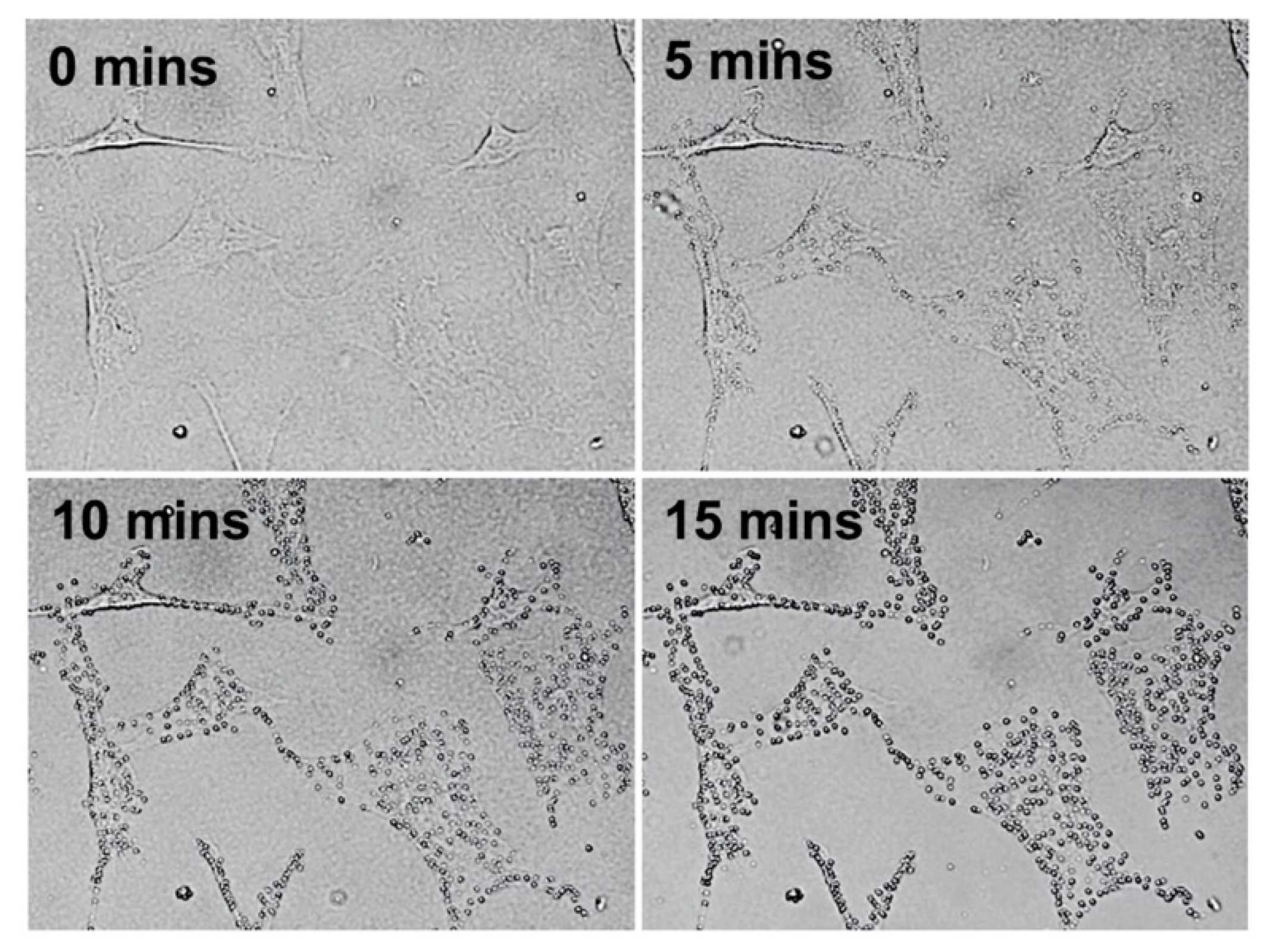
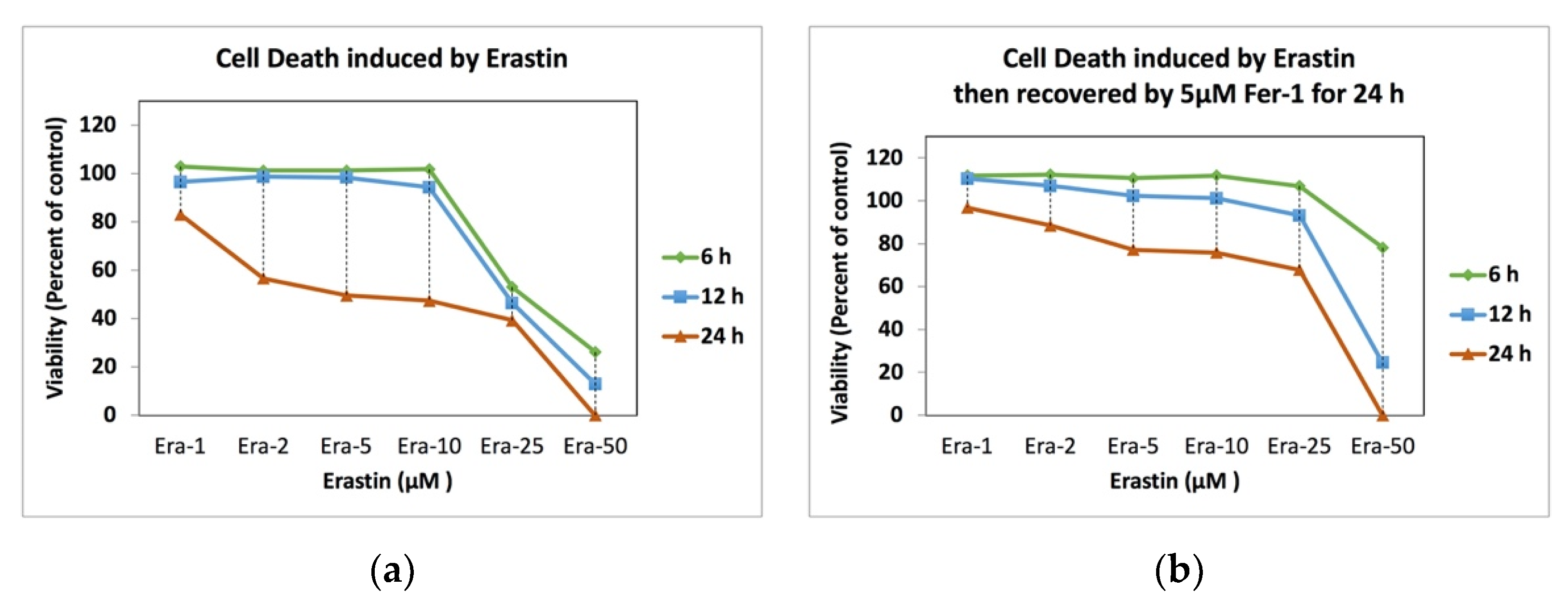
2.1. Erastin and Cell Observation
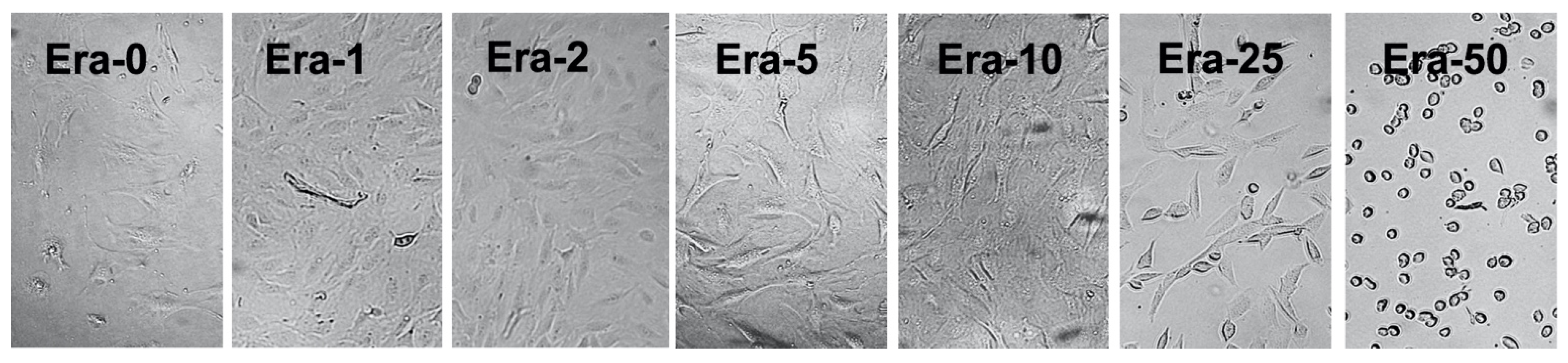
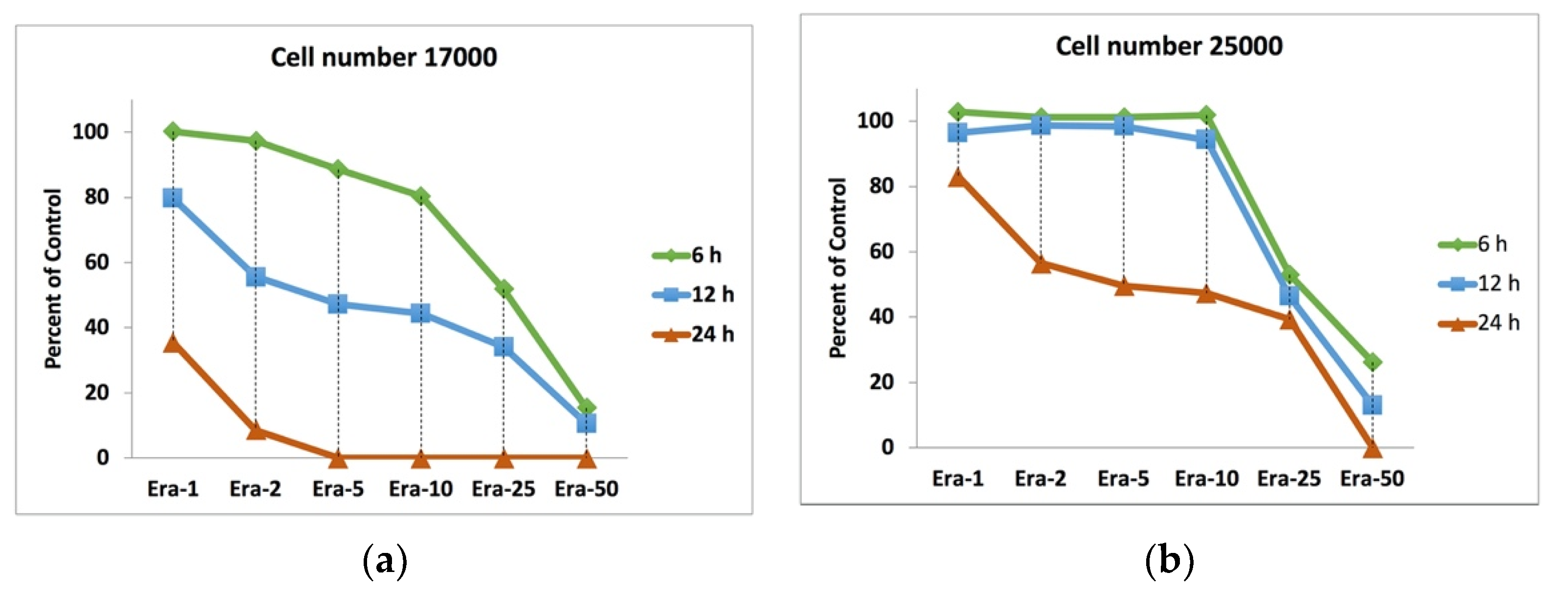
2.2. Erastin and Cell Viability
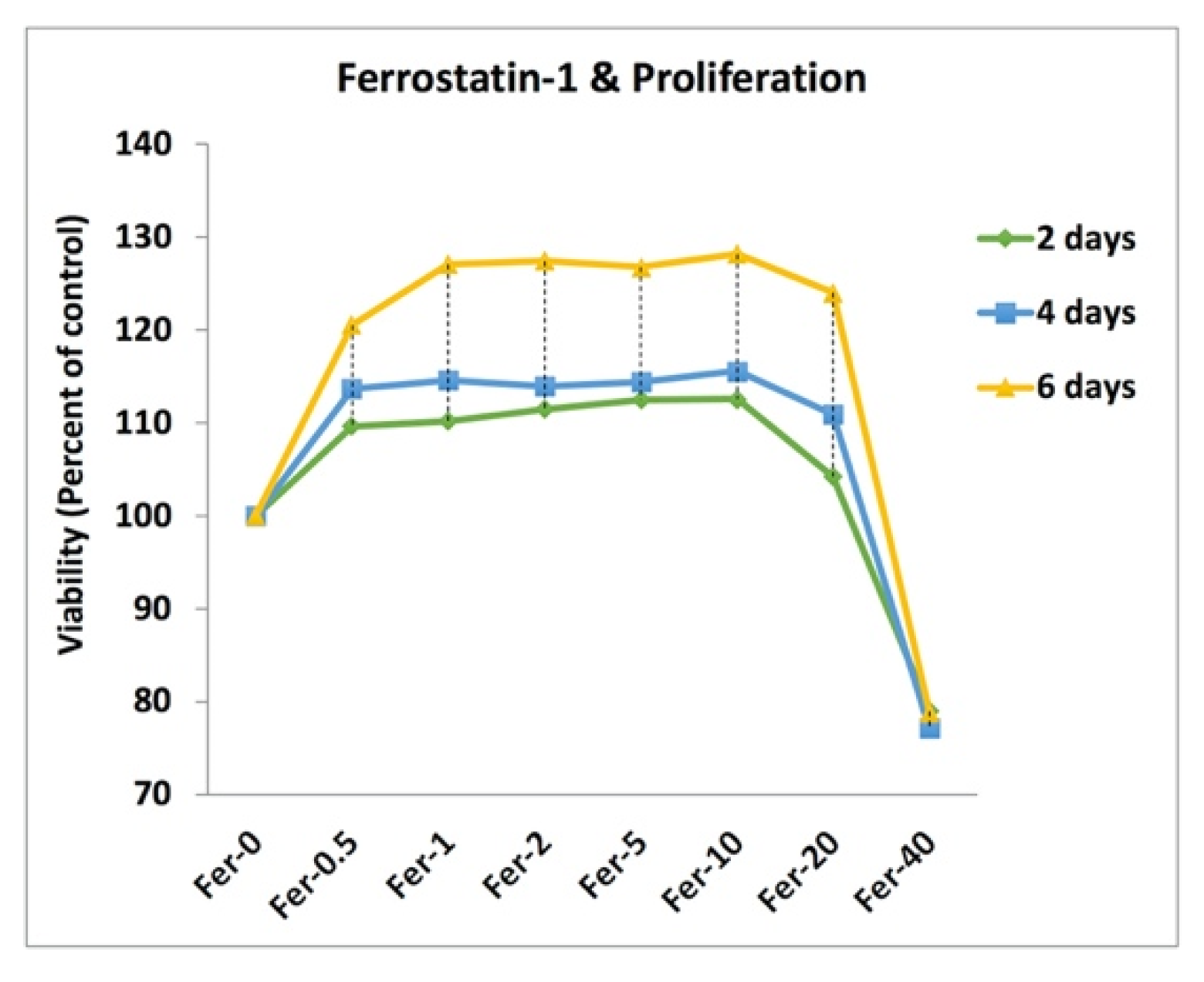
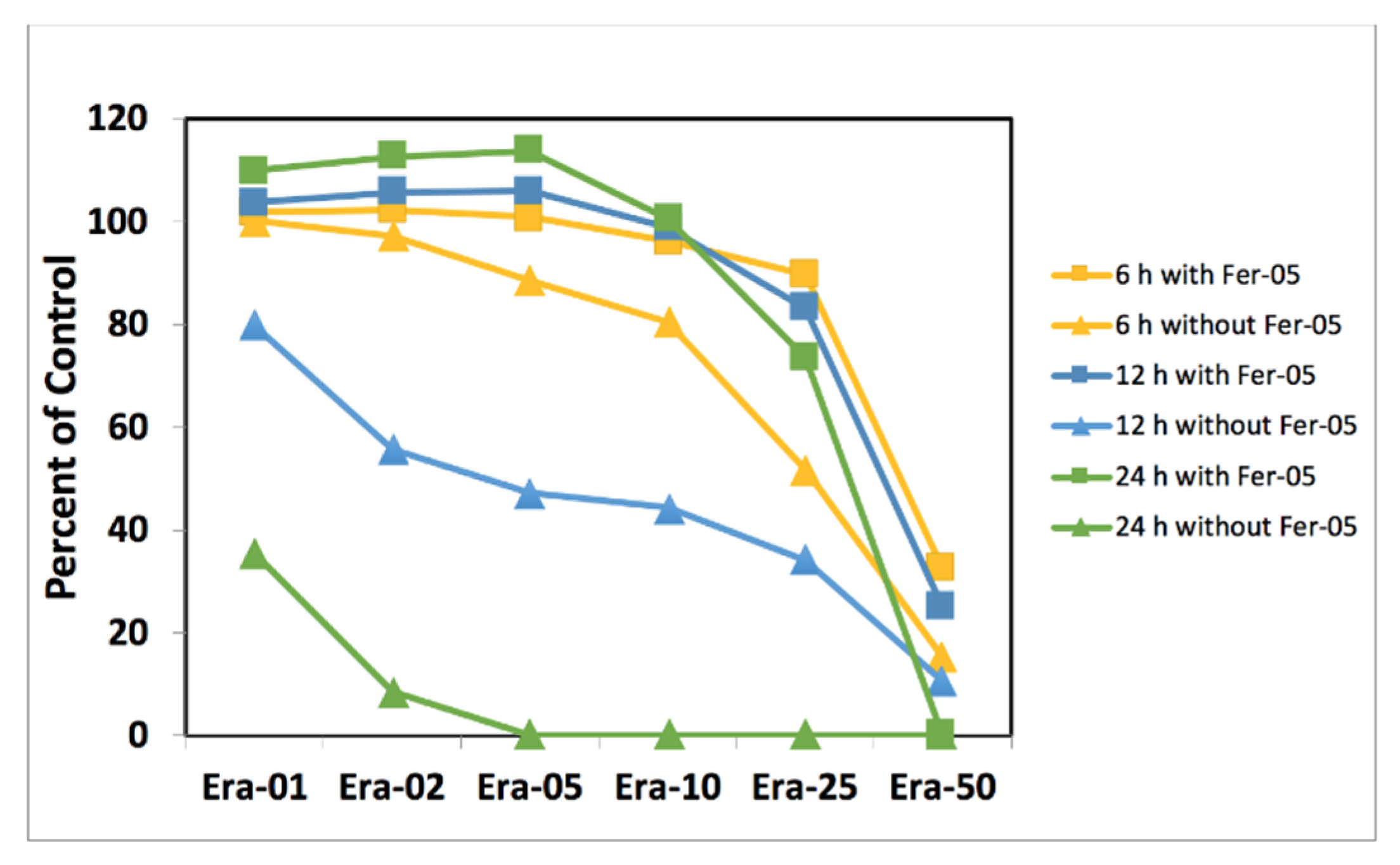
2.3. Cell Doping and Ferrostatin-1
2.4. Ferrostatin-1 and Ferroptosis Inhibition
2.5. Ferrostatin-1 and Ferroptosis Recovery
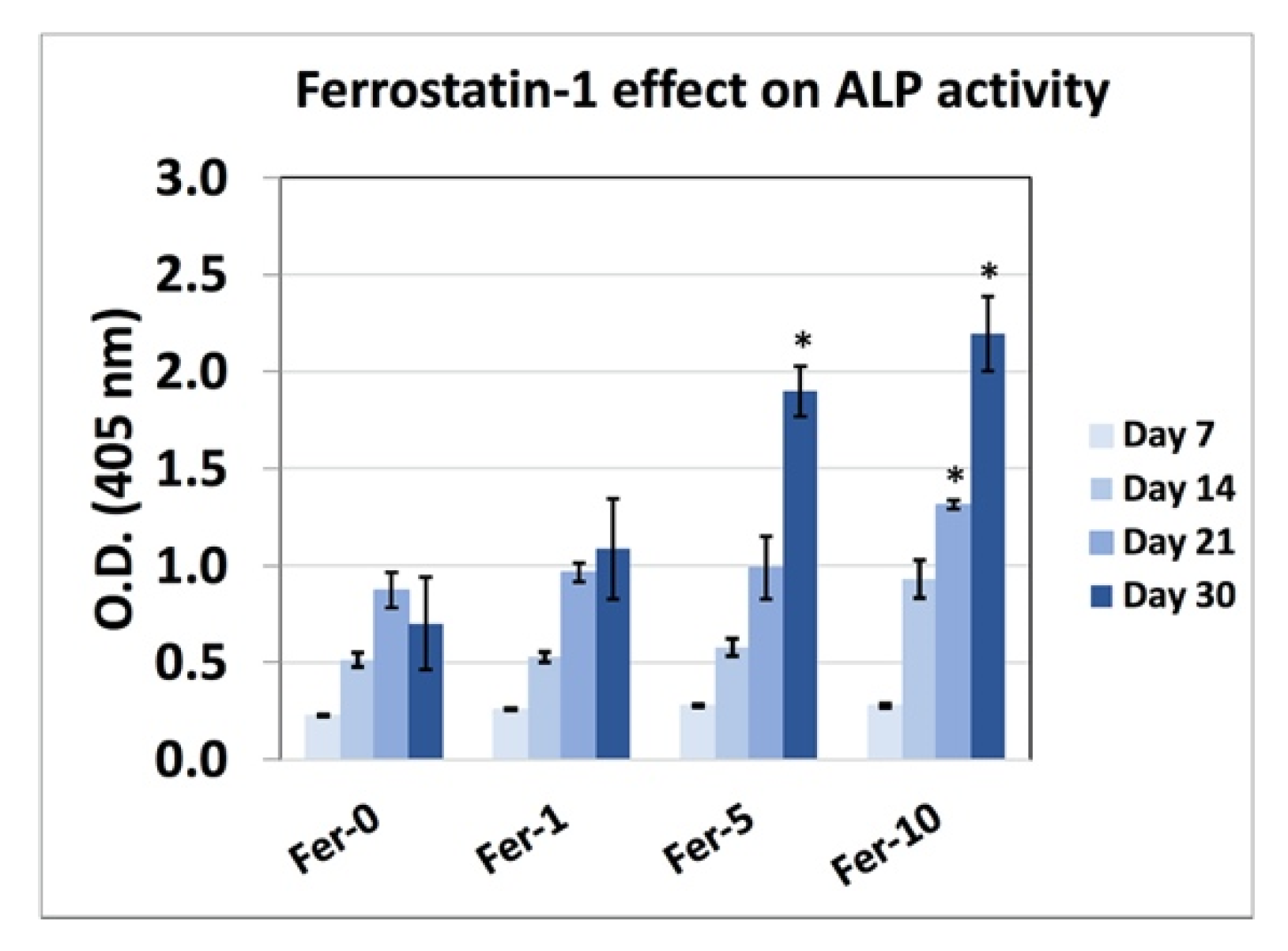
2.6. ALP Activity
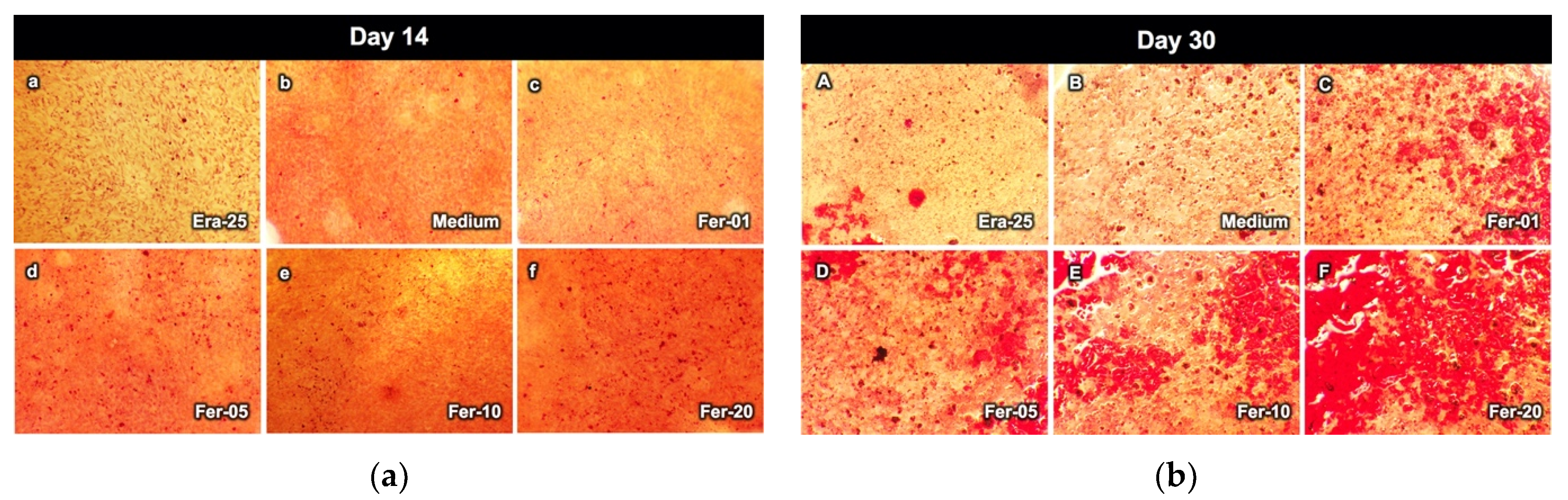
2.7. Alizarin Red Staining
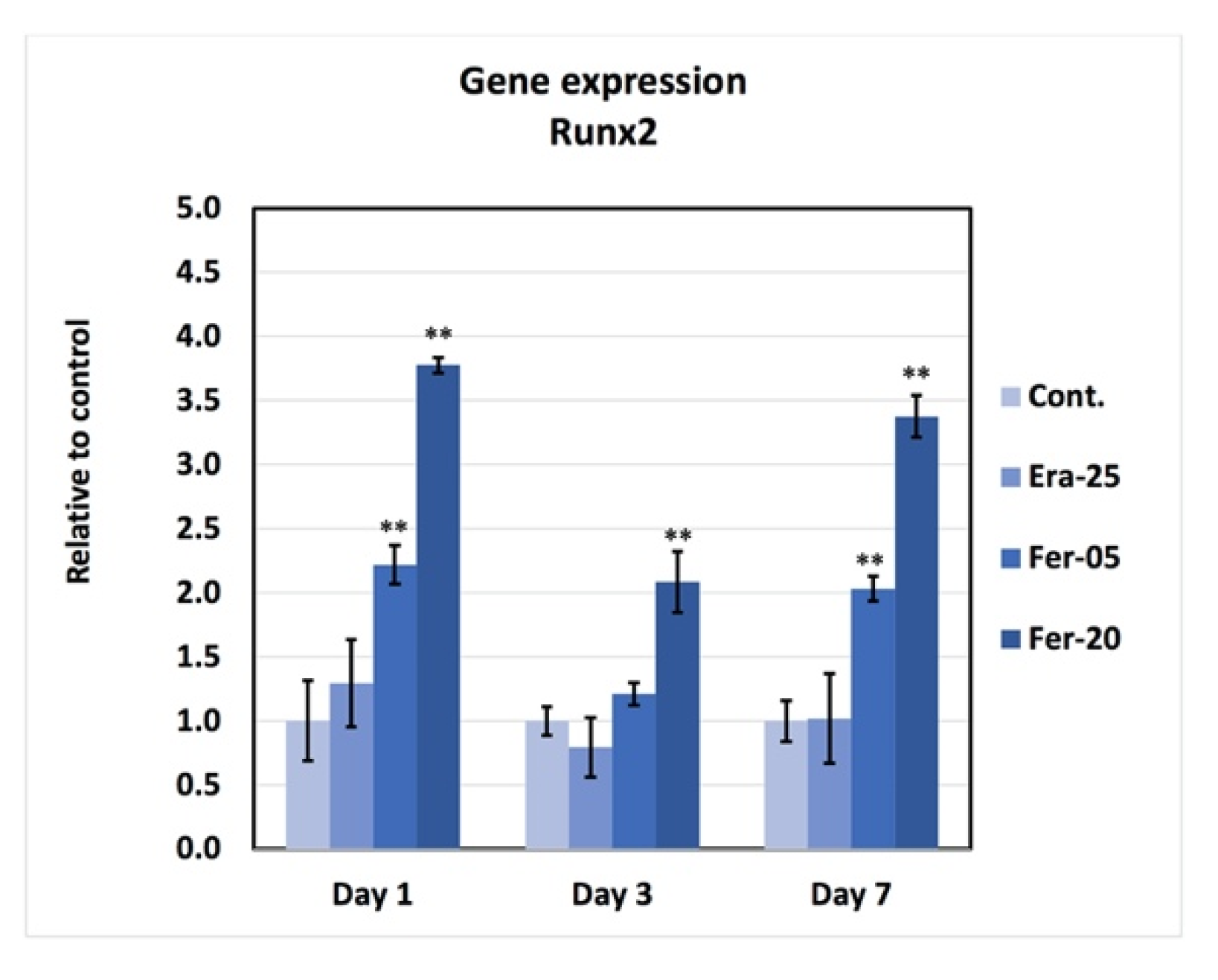
2.8. Gene Expression
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Proliferation and Observation
4.3. Erastin and Cell Viability
4.4. Viability Doping Effect of Ferrostatin-1
4.5. Ferrostatin-1 and Ferroptosis Inhibition
4.6. Cell Death Recovery
4.7. ALP Activity
4.8. Alizarin Red Staining
4.9. Gene Expression
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stockwell, B.R.; Jiang, X.; Gu, W. Emerging Mechanisms and Disease Relevance of Ferroptosis. Trends Cell Biol. 2020, 30, 478–490. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Taylor, R.; Cullen, S.P.; Martin, S. Apoptosis: Controlled demolition at the cellular level. Nat. Rev. Mol. Cell Biol. 2008, 9, 231–241. [Google Scholar] [CrossRef]
- Grootjans, S.; Berghe, T.V.; Vandenabeele, P. Initiation and execution mechanisms of necroptosis: An overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Wyllie, A.; Kerr, J.; Currie, A. Cell Death: The Significance of Apoptosis. Int. Rev. Cytol. 1980, 68, 251–306. [Google Scholar] [CrossRef]
- Thompson, C.B. Apoptosis in the Pathogenesis and Treatment of Disease. Science 1995, 267, 1456–1462. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Eleina, M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death Scott. NIH Public Access 2013, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Stockwell, B.R.; Angeli, J.P.F.; Bayir, H.; Bush, A.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [Green Version]
- Fleming, R.E.; Ponka, P. Iron Overload in Human Disease. N. Engl. J. Med. 2012, 366, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Meynard, D.; Babitt, J.L.; Lin, H.Y. The liver: Conductor of systemic iron balance. Blood 2014, 123, 168–176. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017, 66, 449–465. [Google Scholar] [CrossRef]
- Jeney, V. Clinical Impact and Cellular Mechanisms of Iron Overload-Associated Bone Loss. Front. Pharmacol. 2017, 8, 77. [Google Scholar] [CrossRef]
- Zilka, O.; Shah, R.; Li, B.; Angeli, J.P.F.; Griesser, M.; Conrad, M.; Pratt, D.A. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Central Sci. 2017, 3, 232–243. [Google Scholar] [CrossRef]
- Dolma, S.; Lessnick, S.L.; Hahn, W.C.; Stockwell, B.R. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003, 3, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Hou, W.; Song, X.; Yu, Y.; Huang, J.; Sun, X.; Kang, R.; Tang, D. Ferroptosis: Process and function. Cell Death Differ. 2016, 23, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Sang, W.; Zhong, Y.; Xue, S.; Yang, M.; Wang, C.; Lu, H.; Huan, R.; Mao, X.; Zhu, L.; et al. CoCrMo-Nanoparticles induced peri-implant osteolysis by promoting osteoblast ferroptosis via regulating Nrf2-ARE signalling pathway. Cell Prolif. 2021, e13142. [Google Scholar] [CrossRef]
- Henriksen, K.; Bollerslev, J.; Everts, V.; Karsdal, M.A. Osteoclast Activity and Subtypes as a Function of Physiology and Pathology—Implications for Future Treatments of Osteoporosis. Endocr. Rev. 2011, 32, 31–63. [Google Scholar] [CrossRef] [Green Version]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta (BBA)—Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [Green Version]
- Medeiros, D.M. Copper, iron, and selenium dietary deficiencies negatively impact skeletal integrity: A review. Exp. Biol. Med. 2016, 241, 1316–1322. [Google Scholar] [CrossRef] [Green Version]
- Tsay, J.; Yang, Z.; Ross, F.P.; Cunningham-Rundles, S.; Lin, H.; Coleman, R.; Mayer-Kuckuk, P.; Doty, S.B.; Grady, R.W.; Giardina, P.J.; et al. Bone loss caused by iron overload in a murine model: Importance of oxidative stress. Blood 2010, 116, 2582–2589. [Google Scholar] [CrossRef]
- Carraro, M.; Bernardi, P. Calcium and reactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium 2016, 60, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Maher, P.; Schubert, D. Requirement for cGMP in Nerve Cell Death Caused by Glutathione Depletion. J. Cell Biol. 1997, 139, 1317–1324. [Google Scholar] [CrossRef]
- Maher, P.; van Leyen, K.; Dey, P.N.; Honrath, B.; Dolga, A.; Methner, A. The role of Ca2+ in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium 2018, 70, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Tan, S.; Sagara, Y.; Liu, Y.; Maher, P.; Schubert, D. The Regulation of Reactive Oxygen Species Production during Programmed Cell Death. J. Cell Biol. 1998, 141, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Boonrungsiman, S.; Gentleman, E.; Carzaniga, R.; Evans, N.; McComb, D.W.; Porter, A.E.; Stevens, M.M. The role of intracellular calcium phosphate in osteoblast-mediated bone apatite formation. Proc. Natl. Acad. Sci. USA 2012, 109, 14170–14175. [Google Scholar] [CrossRef] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.; Gao, Y.-H.; Inada, M.; et al. Targeted Disruption of Cbfa1 Results in a Complete Lack of Bone Formation owing to Maturational Arrest of Osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-S.; Kim, H.-J.; Li, Q.; Chi, X.-Z.; Ueta, C.; Komori, T.; Wozney, J.M.; Kim, E.-G.; Choi, J.-Y.; Ryoo, H.-M.; et al. Runx2 Is a Common Target of Transforming Growth Factor β1 and Bone Morphogenetic Protein 2, and Cooperation between Runx2 and Smad5 Induces Osteoblast-Specific Gene Expression in the Pluripotent Mesenchymal Precursor Cell Line C2C12. Mol. Cell. Biol. 2000, 20, 8783–8792. [Google Scholar] [CrossRef] [Green Version]
- Xiao, W.; Beibei, F.; Guangsi, S.; Yu, J.; Wen, Z.; Xi, H.; Youjia, X. Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass. J. Endocrinol. 2015, 226, 121–134. [Google Scholar] [CrossRef]
- Jiang, Y.; Yan, Y.; Wang, X.; Zhu, G.; Xu, Y.-J. Hepcidin inhibition on the effect of osteogenesis in zebrafish. Biochem. Biophys. Res. Commun. 2016, 476, 1–6. [Google Scholar] [CrossRef]
- Zarjou, A.; Jeney, V.; Arosio, P.; Poli, M.; Zavaczki, E.; Balla, G.; Balla, J. Ferritin ferroxidase activity: A potent inhibitor of osteogenesis. J. Bone Miner. Res. 2010, 25, 164–172. [Google Scholar] [CrossRef]
- Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Carrasco, S.; Cannata-Ortiz, P.; Sanchez-Niño, M.D.; Ortega, M.R.; Egido, J.; Linkermann, A.; Ortiz, A.; et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid–Induced AKI. J. Am. Soc. Nephrol. 2016, 28, 218–229. [Google Scholar] [CrossRef]
- Liu, Q.; Wang, K. The induction of ferroptosis by impairing STAT3/Nrf2/GPx4 signaling enhances the sensitivity of osteosarcoma cells to cisplatin. Cell Biol. Int. 2019, 43, 1245–1256. [Google Scholar] [CrossRef]
- Komori, T. Regulation of Osteoblast Differentiation by Runx2. In Osteoimmunology; Springer: Boston, MA, USA, 2009; Volume 658, pp. 43–49. [Google Scholar] [CrossRef]
- Ducy, P.; Zhang, R.; Geoffroy, V.; Ridall, A.L.; Karsenty, G. Osf2/Cbfa1: A Transcriptional Activator of Osteoblast Differentiation. Cell 1997, 89, 747–754. [Google Scholar] [CrossRef] [Green Version]
- Morabito, N.; Gaudio, A.; Lasco, A.; Atteritano, M.; Pizzoleo, M.A.; Cincotta, M.; La Rosa, M.; Guarino, R.; Meo, A.; Frisina, N. Osteoprotegerin and RANKL in the Pathogenesis of Thalassemia-Induced Osteoporosis: New Pieces of the Puzzle. J. Bone Miner. Res. 2004, 19, 722–727. [Google Scholar] [CrossRef]
- Hou, J.-M.; Xue, Y.; Lin, Q.-M. Bovine lactoferrin improves bone mass and microstructure in ovariectomized rats via OPG/RANKL/RANK pathway. Acta Pharmacol. Sin. 2012, 33, 1277–1284. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequences |
|---|---|
| RUNX2 |
5′-CCGCACGACAACCGCACCAT-3′ and 5′-CGCTCCGGCCCACAAATCTC-3′ |
| GAPDH |
5′-ACCACAGTCCATGCCATCAC-3′ and 5′-TCCACCACCCTGTTGCTGTA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valanezhad, A.; Odatsu, T.; Abe, S.; Watanabe, I. Bone Formation Ability and Cell Viability Enhancement of MC3T3-E1 Cells by Ferrostatin-1 a Ferroptosis Inhibitor of Cancer Cells. Int. J. Mol. Sci. 2021, 22, 12259. https://doi.org/10.3390/ijms222212259
Valanezhad A, Odatsu T, Abe S, Watanabe I. Bone Formation Ability and Cell Viability Enhancement of MC3T3-E1 Cells by Ferrostatin-1 a Ferroptosis Inhibitor of Cancer Cells. International Journal of Molecular Sciences. 2021; 22(22):12259. https://doi.org/10.3390/ijms222212259
Chicago/Turabian StyleValanezhad, Alireza, Tetsurou Odatsu, Shigeaki Abe, and Ikuya Watanabe. 2021. "Bone Formation Ability and Cell Viability Enhancement of MC3T3-E1 Cells by Ferrostatin-1 a Ferroptosis Inhibitor of Cancer Cells" International Journal of Molecular Sciences 22, no. 22: 12259. https://doi.org/10.3390/ijms222212259