
Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease


{kind=link}
{kind=link}
Abstract
:1. Introduction
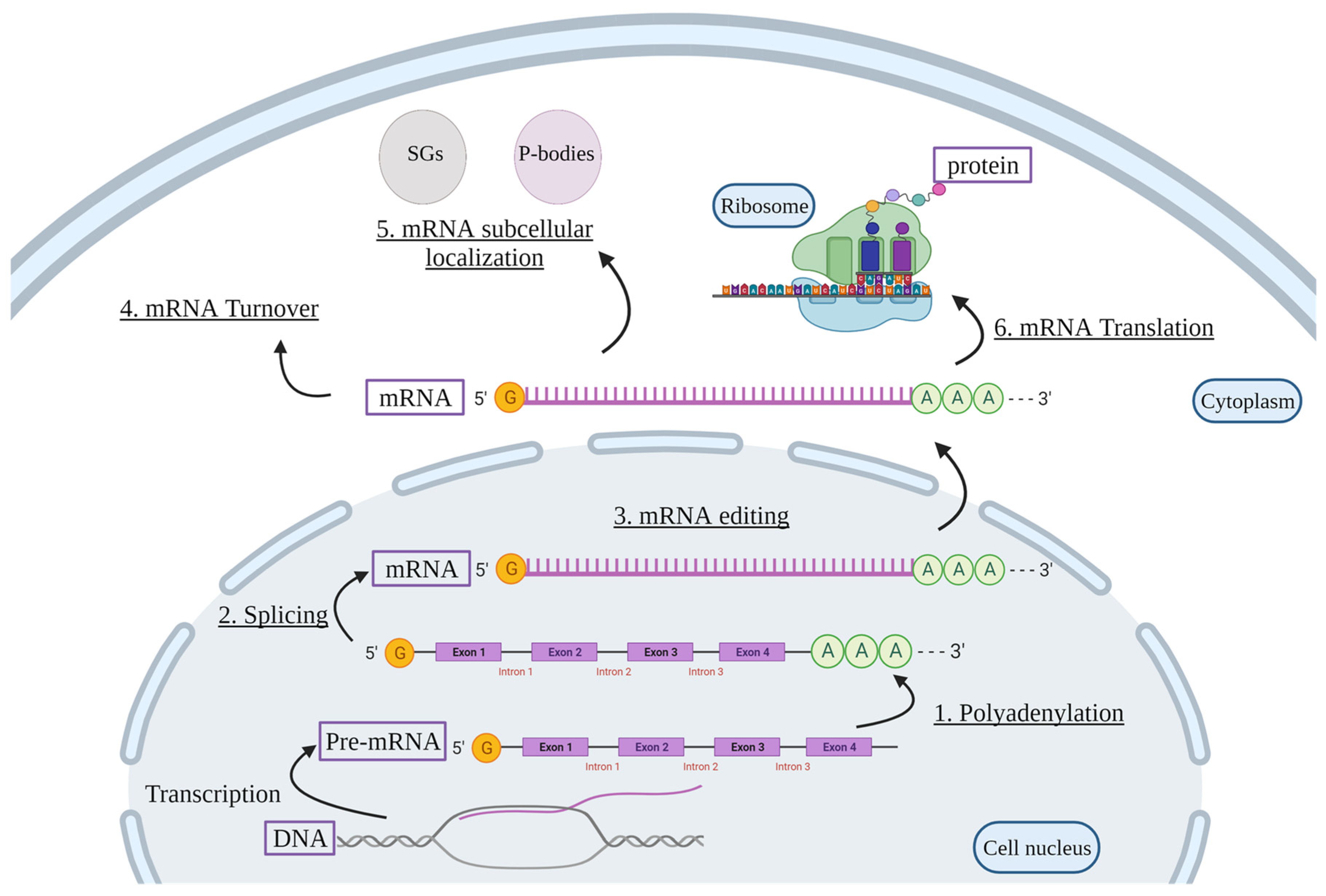
2. Post-Transcriptional Regulation of mRNAs
2.1. RNA Binding Proteins (RBPs)
2.1.1. Biological Functions of RBPs
Polyadenylation
Pre-mRNA Splicing
mRNA Editing
mRNA Turnover
mRNA Subcellular Localization
mRNA Translation
2.2. miRNAs
Biological Function of miRNAs
2.3. LncRNA
Biological Function of lncRNA
3. Post-Transcriptional Regulation in COPD
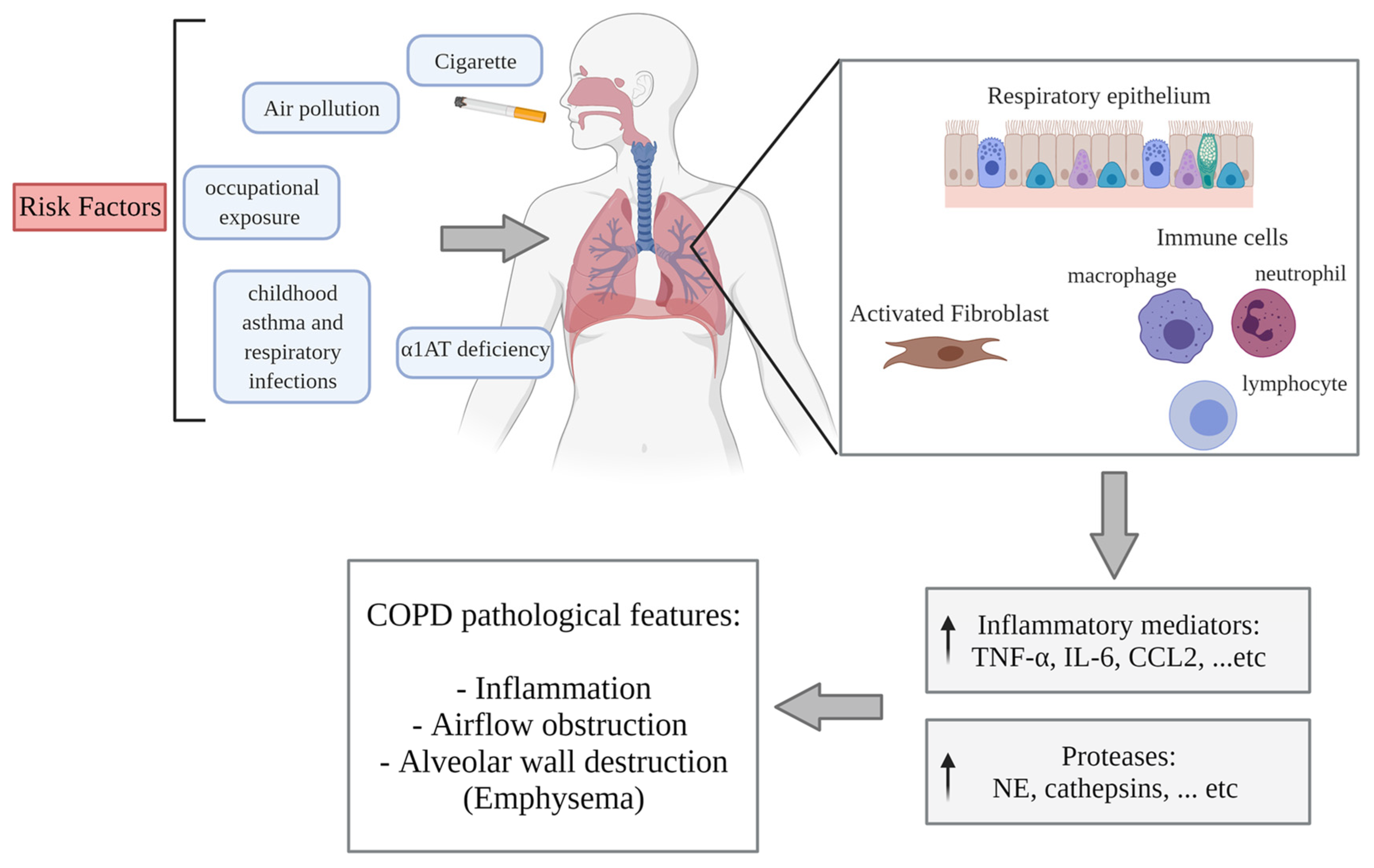
3.1. COPD Pathogenesis
3.2. RBPs
3.2.1. The Response of RBPs to CS
3.2.2. The Regulation of RBPs in COPD
RBPs in Inflammation
RBPs in Apoptosis and Protease Expression
3.2.3. Interplay of RBPs
3.3. miRNA
3.3.1. miRNA and Pathogenic Mechanisms of COPD
3.3.2. Interplay of RBPs with miRNAs
3.4. LncRNA in COPD
3.4.1. LncRNA and Pathogenic Mechanisms of COPD
3.4.2. Interplay of RBPs with lncRNA
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α1AT | Alpha 1-anti-trypsin |
| ADAR | Adenosine deaminases acting on RNA |
| AhR | Aryl hydrocarbon receptor |
| AUF1 | AU-binding factor 1 |
| ARE | AU-rich elements |
| AS | Alternative splicing |
| BAL | Bronchoalveolar lavage |
| CCL | C-C motif ligand |
| COPD | Chronic obstructive pulmonary disease |
| COX-2/PTGS2 | Cyclooxygenase-2 |
| CS | Cigarette smoke |
| CSE | Cigarette smoke extract |
| CXCL | C-X-C motif ligand |
| EMT | Epithelial-to-mesenchymal transition |
| GOLD | Global initiative for chronic obstructive lung disease |
| GR | Glucocorticoid receptors |
| hnRNP | Heterogeneous nuclear ribonucleoprotein |
| HOTAIR | HOX transcript antisense RNA |
| HuR | Hu antigen R |
| IL | Interleukin |
| IRP2 | Iron-responsive element binding protein 2 |
| LCPAT1 | Lung cancer progression–association transcript 1 |
| Linc-RoR | LincRNA regulator of reprogramming |
| LncRNA | Long non-coding RNAs |
| LPS | Lipopolysaccharide |
| MALAT1 | Metastasis-associated in lung adenocarcinoma transcript 1 |
| MEG3 | Maternally expressed gene 3 |
| MiRNA/MiR | MicroRNA |
| MMP | Matrix metalloproteinase |
| NE | Neutrophil Elastase |
| NF-κB | Nuclear Factor Kappa B |
| PABP | Poly(A)-binding protein |
| RBD | RNA-binding domain |
| RBM5 | RNA-binding motif protein 5 |
| RBP | RNA-binding protein |
| RISC | RNA-induced silencing complex |
| RNP | Ribonucleoprotein |
| SCAL1 | Smoke and cancer–associated lncRNA–1 |
| SGs | Stress granules |
| TIA-1 | T-cell Intracellular Antigen 1 |
| TNF-α | Tumor necrosis factor α |
| TTP | Tristetraprolin |
| UTR | Untranslated region |
| VEGF | Vascular endothelial growth factor |
References
- Orphanides, G.; Reinberg, D. A unified theory of gene expression. Cell 2002, 108, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Keene, J.D. RNA regulons: Coordination of post-transcriptional events. Nat. Rev. Genet. 2007, 8, 533–543. [Google Scholar] [CrossRef]
- He, R.Z.; Luo, D.X.; Mo, Y.Y. Emerging roles of lncRNAs in the post-transcriptional regulation in cancer. Genes Dis. 2019, 6, 6–15. [Google Scholar] [CrossRef]
- Corley, M.; Burns, M.C.; Yeo, G.W. How RNA-Binding Proteins Interact with RNA: Molecules and Mechanisms. Mol. Cell 2020, 78, 9–29. [Google Scholar] [CrossRef] [PubMed]
- Alles, J.; Fehlmann, T.; Fischer, U.; Backes, C.; Galata, V.; Minet, M.; Hart, M.; Abu-Halima, M.; Grasser, F.A.; Lenhof, H.P.; et al. An estimate of the total number of true human miRNAs. Nucleic Acids Res. 2019, 47, 3353–3364. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Zhang, L.; Guo, J.; Niu, Y.; Wu, Y.; Li, H.; Zhao, L.; Li, X.; Teng, X.; Sun, X.; et al. NONCODEV5: A comprehensive annotation database for long non-coding RNAs. Nucleic Acids Res. 2018, 46, D308–D314. [Google Scholar] [CrossRef] [PubMed]
- Hon, C.C.; Ramilowski, J.A.; Harshbarger, J.; Bertin, N.; Rackham, O.J.; Gough, J.; Denisenko, E.; Schmeier, S.; Poulsen, T.M.; Severin, J.; et al. An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 2017, 543, 199–204. [Google Scholar] [CrossRef] [Green Version]
- Tan, W.C.; Sin, D.D.; Bourbeau, J.; Hernandez, P.; Chapman, K.R.; Cowie, R.; FitzGerald, J.M.; Marciniuk, D.D.; Maltais, F.; Buist, A.S.; et al. Characteristics of COPD in never-smokers and ever-smokers in the general population: Results from the CanCOLD study. Thorax 2015, 70, 822–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, D.K.; Hirata, F.; Rishi, A.K.; Gairola, C.G. Cigarette smoke, inflammation, and lung injury: A mechanistic perspective. J. Toxicol. Environ. Health B Crit. Rev. 2009, 12, 45–64. [Google Scholar] [CrossRef] [PubMed]
- Stampfli, M.R.; Anderson, G.P. How cigarette smoke skews immune responses to promote infection, lung disease and cancer. Nat. Rev. Immunol. 2009, 9, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Martey, C.A.; Pollock, S.J.; Turner, C.K.; O’Reilly, K.M.; Baglole, C.J.; Phipps, R.P.; Sime, P.J. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: Implications for lung inflammation and cancer. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L981–L991. [Google Scholar] [CrossRef] [Green Version]
- Li, C.J.; Ning, W.; Matthay, M.A.; Feghali-Bostwick, C.A.; Choi, A.M. MAPK pathway mediates EGR-1-HSP70-dependent cigarette smoke-induced chemokine production. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 292, L1297–L1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmider, B.; Messier, E.M.; Chu, H.W.; Mason, R.J. Human alveolar epithelial cell injury induced by cigarette smoke. PLoS ONE 2011, 6, e26059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baglole, C.J.; Bushinsky, S.M.; Garcia, T.M.; Kode, A.; Rahman, I.; Sime, P.J.; Phipps, R.P. Differential induction of apoptosis by cigarette smoke extract in primary human lung fibroblast strains: Implications for emphysema. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 291, L19–L29. [Google Scholar] [CrossRef] [Green Version]
- Dreyfuss, G.; Kim, V.N.; Kataoka, N. Messenger-RNA-binding proteins and the messages they carry. Nat. Rev. Mol. Cell Biol. 2002, 3, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.G.; Dreyfuss, G. Conserved structures and diversity of functions of RNA-binding proteins. Science 1994, 265, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Glisovic, T.; Bachorik, J.L.; Yong, J.; Dreyfuss, G. RNA-binding proteins and post-transcriptional gene regulation. FEBS Lett. 2008, 582, 1977–1986. [Google Scholar] [CrossRef] [Green Version]
- Lunde, B.M.; Moore, C.; Varani, G. RNA-binding proteins: Modular design for efficient function. Nat. Rev. Mol. Cell Biol. 2007, 8, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Lorković, Z.J. RNA Binding Proteins; Landes Bioscience: Austin, TX, USA, 2012; p. 162. [Google Scholar]
- Auweter, S.D.; Oberstrass, F.C.; Allain, F.H. Sequence-specific binding of single-stranded RNA: Is there a code for recognition? Nucleic Acids Res. 2006, 34, 4943–4959. [Google Scholar] [CrossRef] [Green Version]
- Richter, J.D.; Zhao, X. The molecular biology of FMRP: New insights into fragile X syndrome. Nat. Rev. Neurosci. 2021, 22, 209–222. [Google Scholar] [CrossRef]
- Takagaki, Y.; MacDonald, C.C.; Shenk, T.; Manley, J.L. The human 64-kDa polyadenylylation factor contains a ribonucleoprotein-type RNA binding domain and unusual auxiliary motifs. Proc. Natl. Acad. Sci. USA 1992, 89, 1403–1407. [Google Scholar] [CrossRef] [Green Version]
- Shimberg, G.D.; Michalek, J.L.; Oluyadi, A.A.; Rodrigues, A.V.; Zucconi, B.E.; Neu, H.M.; Ghosh, S.; Sureschandra, K.; Wilson, G.M.; Stemmler, T.L.; et al. Cleavage and polyadenylation specificity factor 30: An RNA-binding zinc-finger protein with an unexpected 2Fe-2S cluster. Proc. Natl. Acad. Sci. USA 2016, 113, 4700–4705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neve, J.; Patel, R.; Wang, Z.; Louey, A.; Furger, A.M. Cleavage and polyadenylation: Ending the message expands gene regulation. RNA Biol. 2017, 14, 865–890. [Google Scholar] [CrossRef] [Green Version]
- Minvielle-Sebastia, L.; Keller, W. mRNA polyadenylation and its coupling to other RNA processing reactions and to transcription. Curr. Opin. Cell Biol. 1999, 11, 352–357. [Google Scholar] [CrossRef]
- Dettwiler, S.; Aringhieri, C.; Cardinale, S.; Keller, W.; Barabino, S.M. Distinct sequence motifs within the 68-kDa subunit of cleavage factor Im mediate RNA binding, protein-protein interactions, and subcellular localization. J. Biol. Chem. 2004, 279, 35788–35797. [Google Scholar] [CrossRef] [Green Version]
- Wahle, E. A novel poly(A)-binding protein acts as a specificity factor in the second phase of messenger RNA polyadenylation. Cell 1991, 66, 759–768. [Google Scholar] [CrossRef]
- Bienroth, S.; Keller, W.; Wahle, E. Assembly of a processive messenger RNA polyadenylation complex. EMBO J. 1993, 12, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zhou, H.L.; Hasman, R.A.; Lou, H. Hu proteins regulate polyadenylation by blocking sites containing U-rich sequences. J. Biol. Chem. 2007, 282, 2203–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staley, J.P.; Guthrie, C. Mechanical devices of the spliceosome: Motors, clocks, springs, and things. Cell 1998, 92, 315–326. [Google Scholar] [CrossRef] [Green Version]
- Zahler, A.M.; Lane, W.S.; Stolk, J.A.; Roth, M.B. SR proteins: A conserved family of pre-mRNA splicing factors. Genes Dev. 1992, 6, 837–847. [Google Scholar] [CrossRef] [Green Version]
- Will, C.L.; Luhrmann, R. Spliceosome structure and function. Cold Spring Harb. Perspect. Biol. 2011, 3, a003707. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.M.; Castle, J.; Garrett-Engele, P.; Kan, Z.; Loerch, P.M.; Armour, C.D.; Santos, R.; Schadt, E.E.; Stoughton, R.; Shoemaker, D.D. Genome-wide survey of human alternative pre-mRNA splicing with exon junction microarrays. Science 2003, 302, 2141–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Q.; Shai, O.; Lee, L.J.; Frey, B.J.; Blencowe, B.J. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 2008, 40, 1413–1415. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.T.; Sandberg, R.; Luo, S.; Khrebtukova, I.; Zhang, L.; Mayr, C.; Kingsmore, S.F.; Schroth, G.P.; Burge, C.B. Alternative isoform regulation in human tissue transcriptomes. Nature 2008, 456, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Burge, C.B. Splicing regulation: From a parts list of regulatory elements to an integrated splicing code. RNA 2008, 14, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Huelga, S.C.; Vu, A.Q.; Arnold, J.D.; Liang, T.Y.; Liu, P.P.; Yan, B.Y.; Donohue, J.P.; Shiue, L.; Hoon, S.; Brenner, S.; et al. Integrative genome-wide analysis reveals cooperative regulation of alternative splicing by hnRNP proteins. Cell Rep. 2012, 1, 167–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izquierdo, J.M. Hu antigen R (HuR) functions as an alternative pre-mRNA splicing regulator of Fas apoptosis-promoting receptor on exon definition. J. Biol. Chem. 2008, 283, 19077–19084. [Google Scholar] [CrossRef] [Green Version]
- Bass, B.L. RNA editing and hypermutation by adenosine deamination. Trends Biochem. Sci. 1997, 22, 157–162. [Google Scholar] [CrossRef]
- Schaub, M.; Keller, W. RNA editing by adenosine deaminases generates RNA and protein diversity. Biochimie 2002, 84, 791–803. [Google Scholar] [CrossRef]
- Valente, L.; Nishikura, K. ADAR gene family and A-to-I RNA editing: Diverse roles in posttranscriptional gene regulation. Prog. Nucleic Acid Res. Mol. Biol. 2005, 79, 299–338. [Google Scholar]
- Athanasiadis, A.; Rich, A.; Maas, S. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004, 2, e391. [Google Scholar] [CrossRef]
- Keller, W.; Wolf, J.; Gerber, A. Editing of messenger RNA precursors and of tRNAs by adenosine to inosine conversion. FEBS Lett. 1999, 452, 71–76. [Google Scholar] [CrossRef] [Green Version]
- Gerber, A.P.; Keller, W. RNA editing by base deamination: More enzymes, more targets, new mysteries. Trends Biochem. Sci. 2001, 26, 376–384. [Google Scholar] [CrossRef]
- Bass, B.L. RNA editing by adenosine deaminases that act on RNA. Annu. Rev. Biochem. 2002, 71, 817–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bass, B.L.; Nishikura, K.; Keller, W.; Seeburg, P.H.; Emeson, R.B.; O’Connell, M.A.; Samuel, C.E.; Herbert, A. A standardized nomenclature for adenosine deaminases that act on RNA. RNA 1997, 3, 947–949. [Google Scholar]
- Chen, C.X.; Cho, D.S.; Wang, Q.; Lai, F.; Carter, K.C.; Nishikura, K. A third member of the RNA-specific adenosine deaminase gene family, ADAR3, contains both single- and double-stranded RNA binding domains. RNA 2000, 6, 755–767. [Google Scholar] [CrossRef]
- Levanon, E.Y.; Eisenberg, E.; Yelin, R.; Nemzer, S.; Hallegger, M.; Shemesh, R.; Fligelman, Z.Y.; Shoshan, A.; Pollock, S.R.; Sztybel, D.; et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004, 22, 1001–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.D.; Kim, T.T.; Walsh, T.; Kobayashi, Y.; Matise, T.C.; Buyske, S.; Gabriel, A. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004, 14, 1719–1725. [Google Scholar] [CrossRef] [Green Version]
- Schoenberg, D.R.; Maquat, L.E. Regulation of cytoplasmic mRNA decay. Nat. Rev. Genet. 2012, 13, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Blackshear, P.J. Tristetraprolin and other CCCH tandem zinc-finger proteins in the regulation of mRNA turnover. Biochem. Soc. Trans. 2002, 30 Pt 6, 945–952. [Google Scholar] [CrossRef]
- Carballo, E.; Lai, W.S.; Blackshear, P.J. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 1998, 281, 1001–1005. [Google Scholar] [CrossRef] [Green Version]
- Lai, W.S.; Carballo, E.; Strum, J.R.; Kennington, E.A.; Phillips, R.S.; Blackshear, P.J. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell Biol. 1999, 19, 4311–4323. [Google Scholar] [CrossRef] [Green Version]
- Kratochvill, F.; Machacek, C.; Vogl, C.; Ebner, F.; Sedlyarov, V.; Gruber, A.R.; Hartweger, H.; Vielnascher, R.; Karaghiosoff, M.; Rulicke, T.; et al. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Mol. Syst. Biol. 2011, 7, 560. [Google Scholar] [CrossRef] [PubMed]
- Sauer, I.; Schaljo, B.; Vogl, C.; Gattermeier, I.; Kolbe, T.; Muller, M.; Blackshear, P.J.; Kovarik, P. Interferons limit inflammatory responses by induction of tristetraprolin. Blood 2006, 107, 4790–4797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogilvie, R.L.; Abelson, M.; Hau, H.H.; Vlasova, I.; Blackshear, P.J.; Bohjanen, P.R. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J. Immunol. 2005, 174, 953–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, K.; Kedersha, N.; Shen, L.; Blackshear, P.J.; Anderson, P. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. USA 2004, 101, 2011–2016. [Google Scholar] [CrossRef] [Green Version]
- Winzen, R.; Thakur, B.K.; Dittrich-Breiholz, O.; Shah, M.; Redich, N.; Dhamija, S.; Kracht, M.; Holtmann, H. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol. Cell Biol. 2007, 27, 8388–8400. [Google Scholar] [CrossRef] [Green Version]
- Leppek, K.; Schott, J.; Reitter, S.; Poetz, F.; Hammond, M.C.; Stoecklin, G. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell 2013, 153, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Paschoud, S.; Dogar, A.M.; Kuntz, C.; Grisoni-Neupert, B.; Richman, L.; Kuhn, L.C. Destabilization of interleukin-6 mRNA requires a putative RNA stem-loop structure, an AU-rich element, and the RNA-binding protein AUF1. Mol. Cell Biol. 2006, 26, 8228–8241. [Google Scholar] [CrossRef] [Green Version]
- Gratacos, F.M.; Brewer, G. The role of AUF1 in regulated mRNA decay. Wiley Interdiscip. Rev. RNA 2010, 1, 457–473. [Google Scholar] [CrossRef] [Green Version]
- White, E.J.; Brewer, G.; Wilson, G.M. Post-transcriptional control of gene expression by AUF1: Mechanisms, physiological targets, and regulation. Biochim. Biophys. Acta 2013, 1829, 680–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Caldwell, M.C.; Lin, S.; Furneaux, H.; Gorospe, M. HuR regulates cyclin A and cyclin B1 mRNA stability during cell proliferation. EMBO J. 2000, 19, 2340–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa, A.; Cuadrado, A.; Fan, J.; Atasoy, U.; Muscat, G.E.; Munoz-Canoves, P.; Gorospe, M.; Munoz, A. Role of HuR in skeletal myogenesis through coordinate regulation of muscle differentiation genes. Mol. Cell Biol. 2003, 23, 4991–5004. [Google Scholar] [CrossRef] [Green Version]
- van der Giessen, K.; Di-Marco, S.; Clair, E.; Gallouzi, I.E. RNAi-mediated HuR depletion leads to the inhibition of muscle cell differentiation. J. Biol. Chem. 2003, 278, 47119–47128. [Google Scholar] [CrossRef] [Green Version]
- Dormoy-Raclet, V.; Menard, I.; Clair, E.; Kurban, G.; Mazroui, R.; Di Marco, S.; von Roretz, C.; Pause, A.; Gallouzi, I.E. The RNA-binding protein HuR promotes cell migration and cell invasion by stabilizing the beta-actin mRNA in a U-rich-element-dependent manner. Mol. Cell Biol. 2007, 27, 5365–5380. [Google Scholar] [CrossRef] [Green Version]
- Ishimaru, D.; Ramalingam, S.; Sengupta, T.K.; Bandyopadhyay, S.; Dellis, S.; Tholanikunnel, B.G.; Fernandes, D.J.; Spicer, E.K. Regulation of Bcl-2 expression by HuR in HL60 leukemia cells and A431 carcinoma cells. Mol. Cancer Res. 2009, 7, 1354–1366. [Google Scholar] [CrossRef] [Green Version]
- Doller, A.; Huwiler, A.; Muller, R.; Radeke, H.H.; Pfeilschifter, J.; Eberhardt, W. Protein kinase C alpha-dependent phosphorylation of the mRNA-stabilizing factor HuR: Implications for posttranscriptional regulation of cyclooxygenase-2. Mol. Biol. Cell 2007, 18, 2137–2148. [Google Scholar] [CrossRef]
- Fan, J.; Ishmael, F.T.; Fang, X.; Myers, A.; Cheadle, C.; Huang, S.K.; Atasoy, U.; Gorospe, M.; Stellato, C. Chemokine transcripts as targets of the RNA-binding protein HuR in human airway epithelium. J. Immunol. 2011, 186, 2482–2494. [Google Scholar] [CrossRef] [Green Version]
- Bai, D.; Gao, Q.; Li, C.; Ge, L.; Gao, Y.; Wang, H. A conserved TGFbeta1/HuR feedback circuit regulates the fibrogenic response in fibroblasts. Cell Signal. 2012, 24, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Adjibade, P.; Mazroui, R. Control of mRNA turnover: Implication of cytoplasmic RNA granules. Semin. Cell Dev. Biol. 2014, 34, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Mahboubi, H.; Stochaj, U. Cytoplasmic stress granules: Dynamic modulators of cell signaling and disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 884–895. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.Y.; Haqqi, T.M. Interleukin-1beta induced Stress Granules Sequester COX-2 mRNA and Regulates its Stability and Translation in Human OA Chondrocytes. Sci. Rep. 2016, 6, 27611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kedersha, N.; Ivanov, P.; Anderson, P. Stress granules and cell signaling: More than just a passing phase? Trends Biochem. Sci. 2013, 38, 494–506. [Google Scholar] [CrossRef] [Green Version]
- Jain, S.; Parker, R. The discovery and analysis of P Bodies. Adv. Exp. Med. Biol. 2013, 768, 23–43. [Google Scholar] [PubMed]
- Glasmacher, E.; Hoefig, K.P.; Vogel, K.U.; Rath, N.; Du, L.; Wolf, C.; Kremmer, E.; Wang, X.; Heissmeyer, V. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat. Immunol. 2010, 11, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Kahvejian, A.; Svitkin, Y.V.; Sukarieh, R.; M’Boutchou, M.N.; Sonenberg, N. Mammalian poly(A)-binding protein is a eukaryotic translation initiation factor, which acts via multiple mechanisms. Genes Dev. 2005, 19, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Dixon, D.A.; Balch, G.C.; Kedersha, N.; Anderson, P.; Zimmerman, G.A.; Beauchamp, R.D.; Prescott, S.M. Regulation of cyclooxygenase-2 expression by the translational silencer TIA-1. J. Exp. Med. 2003, 198, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Lal, A.; Kawai, T.; Yang, X.; Mazan-Mamczarz, K.; Gorospe, M. Antiapoptotic function of RNA-binding protein HuR effected through prothymosin alpha. EMBO J. 2005, 24, 1852–1862. [Google Scholar] [CrossRef]
- Kullmann, M.; Gopfert, U.; Siewe, B.; Hengst, L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5’UTR. Genes Dev. 2002, 16, 3087–3099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leandersson, K.; Riesbeck, K.; Andersson, T. Wnt-5a mRNA translation is suppressed by the Elav-like protein HuR in human breast epithelial cells. Nucleic Acids Res. 2006, 34, 3988–3999. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Jeon, K.; Lee, J.T.; Kim, S.; Kim, V.N. MicroRNA maturation: Stepwise processing and subcellular localization. EMBO J. 2002, 21, 4663–4670. [Google Scholar] [CrossRef] [Green Version]
- Tomari, Y.; Zamore, P.D. MicroRNA biogenesis: Drosha can’t cut it without a partner. Curr. Biol. 2005, 15, R61-4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Lee, Y.; Yeom, K.H.; Nam, J.W.; Heo, I.; Rhee, J.K.; Sohn, S.Y.; Cho, Y.; Zhang, B.T.; Kim, V.N. Molecular basis for the recognition of primary microRNAs by the Drosha-DGCR8 complex. Cell 2006, 125, 887–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamata, T.; Tomari, Y. Making RISC. Trends Biochem. Sci. 2010, 35, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Fabian, M.R.; Sonenberg, N.; Filipowicz, W. Regulation of mRNA translation and stability by microRNAs. Annu. Rev. Biochem. 2010, 79, 351–379. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Bernstein, E.; Kim, S.Y.; Carmell, M.A.; Murchison, E.P.; Alcorn, H.; Li, M.Z.; Mills, A.A.; Elledge, S.J.; Anderson, K.V.; Hannon, G.J. Dicer is essential for mouse development. Nat. Genet. 2003, 35, 215–217. [Google Scholar] [CrossRef]
- Wang, Y.; Medvid, R.; Melton, C.; Jaenisch, R.; Blelloch, R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat. Genet. 2007, 39, 380–385. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, A.; Vigorito, E.; Clare, S.; Warren, M.V.; Couttet, P.; Soond, D.R.; van Dongen, S.; Grocock, R.J.; Das, P.P.; Miska, E.A.; et al. Requirement of bic/microRNA-155 for normal immune function. Science 2007, 316, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Johnnidis, J.B.; Harris, M.H.; Wheeler, R.T.; Stehling-Sun, S.; Lam, M.H.; Kirak, O.; Brummelkamp, T.R.; Fleming, M.D.; Camargo, F.D. Regulation of progenitor cell proliferation and granulocyte function by microRNA-223. Nature 2008, 451, 1125–1129. [Google Scholar] [CrossRef]
- Ventura, A.; Young, A.G.; Winslow, M.M.; Lintault, L.; Meissner, A.; Erkeland, S.J.; Newman, J.; Bronson, R.T.; Crowley, D.; Stone, J.R.; et al. Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 2008, 132, 875–886. [Google Scholar] [CrossRef] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Jarroux, J.; Morillon, A.; Pinskaya, M. History, Discovery, and Classification of lncRNAs. Adv. Exp. Med. Biol 2017, 1008, 1–46. [Google Scholar] [PubMed]
- Dhanoa, J.K.; Sethi, R.S.; Verma, R.; Arora, J.S.; Mukhopadhyay, C.S. Long non-coding RNA: Its evolutionary relics and biological implications in mammals: A review. J. Anim. Sci. Technol. 2018, 60, 25. [Google Scholar] [CrossRef] [Green Version]
- Tsagakis, I.; Douka, K.; Birds, I.; Aspden, J.L. Long non-coding RNAs in development and disease: Conservation to mechanisms. J. Pathol. 2020, 250, 480–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.; Hao, Y.; Dong, X.; Gong, Q.; Chen, J.; Zhang, J.; Tian, W. Molecular mechanisms and function prediction of long noncoding RNA. Sci. World J. 2012, 2012, 541786. [Google Scholar] [CrossRef] [PubMed]
- Marchese, F.P.; Raimondi, I.; Huarte, M. The multidimensional mechanisms of long noncoding RNA function. Genome Biol. 2017, 18, 206. [Google Scholar] [CrossRef] [Green Version]
- Dykes, I.M.; Emanueli, C. Transcriptional and Post-transcriptional Gene Regulation by Long Non-coding RNA. Genom. Proteom. Bioinform. 2017, 15, 177–186. [Google Scholar] [CrossRef]
- Gil, N.; Ulitsky, I. Regulation of gene expression by cis-acting long non-coding RNAs. Nat. Rev. Genet. 2020, 21, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Xiong, Y.; Liao, W.; Meng, L.; Yang, S. Long noncoding RNA ATB participates in the development of renal cell carcinoma by downregulating p53 via binding to DNMT1. J. Cell Physiol. 2019, 234, 12910–12917. [Google Scholar] [CrossRef]
- Keren, H.; Lev-Maor, G.; Ast, G. Alternative splicing and evolution: Diversification, exon definition and function. Nat. Rev. Genet. 2010, 11, 345–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, W.; Zhu, W.; Dong, J.; Cheng, Y.; Yin, Z.; Shen, F. Mechanisms and Functions of Long Non-Coding RNAs at Multiple Regulatory Levels. Int. J. Mol. Sci. 2019, 20, 5573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kopp, F.; Chang, T.C.; Sataluri, A.; Chen, B.; Sivakumar, S.; Yu, H.; Xie, Y.; Mendell, J.T. Noncoding RNA NORAD Regulates Genomic Stability by Sequestering PUMILIO Proteins. Cell 2016, 164, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Vestbo, J.; Hurd, S.S.; Agusti, A.G.; Jones, P.W.; Vogelmeier, C.; Anzueto, A.; Barnes, P.J.; Fabbri, L.M.; Martinez, F.J.; Nishimura, M.; et al. Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 2013, 187, 347–365. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. The Top 10 Causes of Death. Available online: https://www.who.int/en/news-room/fact-sheets/detail/the-top-10-causes-of-death (accessed on 28 October 2021).
- Global Strategy for the Diagnosis; Management and Prevention of COPD. Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2020. Available online: https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=&ved=2ahUKEwiFyoOAqOrzAhVeQvEDHVvzCuYQjBB6BAgPEAE&url=https%3A%2F%2Fgoldcopd.org%2Fwp-content%2Fuploads%2F2020%2F03%2FGOLD-2020-POCKET-GUIDE-ver1.0_FINAL-WMV.pdf&usg=AOvVaw3jGwvv-lRohtDCg1_pHTST (accessed on 28 October 2021).
- Pauwels, R.A.; Rabe, K.F. Burden and clinical features of chronic obstructive pulmonary disease (COPD). Lancet 2004, 364, 613–620. [Google Scholar] [CrossRef]
- Decramer, M.; Janssens, W.; Miravitlles, M. Chronic obstructive pulmonary disease. Lancet 2012, 379, 1341–1351. [Google Scholar] [CrossRef]
- World Health Organization. Tobacco. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 28 October 2021).
- FDA. Chemicals in Cigarettes: From Plant to Product to Puff. Available online: https://www.fda.gov/tobacco-products/products-ingredients-components/chemicals-cigarettes-plant-product-puff (accessed on 28 October 2021).
- Fowles, J.; Dybing, E. Application of toxicological risk assessment principles to the chemical constituents of cigarette smoke. Tob. Control. 2003, 12, 424–430. [Google Scholar] [CrossRef] [Green Version]
- Athanazio, R. Airway disease: Similarities and differences between asthma, COPD and bronchiectasis. Clinics 2012, 67, 1335–1343. [Google Scholar] [CrossRef]
- King, P.T. Inflammation in chronic obstructive pulmonary disease and its role in cardiovascular disease and lung cancer. Clin. Transl. Med. 2015, 4, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J. Allergy Clin. Immunol 2016, 138, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, S.; Mahler, D.A.; Marcus, P.; Owen, C.A.; Yawn, B.; Rennard, S. Inflammation in COPD: Implications for management. Am. J. Med. 2012, 125, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, L.E.; Barnes, P.J. Chemokine receptors as therapeutic targets in chronic obstructive pulmonary disease. Trends Pharm. Sci. 2006, 27, 546–553. [Google Scholar] [CrossRef]
- Frankenberger, M.; Eder, C.; Hofer, T.P.; Heimbeck, I.; Skokann, K.; Kassner, G.; Weber, N.; Moller, W.; Ziegler-Heitbrock, L. Chemokine expression by small sputum macrophages in COPD. Mol. Med. 2011, 17, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, J.A.; Zago, M.; Nair, P.; Li, P.Z.; Bourbeau, J.; Tan, W.C.; Hamid, Q.; Eidelman, D.H.; Benedetti, A.L.; Baglole, C.J. Decreased expression of the NF-kappaB family member RelB in lung fibroblasts from Smokers with and without COPD potentiates cigarette smoke-induced COX-2 expression. Respir. Res. 2015, 16, 54. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Chen, P.; Hanaoka, M.; Droma, Y.; Kubo, K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with the severity of airflow limitation in stable COPD. Respirology 2008, 13, 1014–1021. [Google Scholar]
- Bagdonas, E.; Raudoniute, J.; Bruzauskaite, I.; Aldonyte, R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int. J. Chron. Obs. Pulm. Dis. 2015, 10, 995–1013. [Google Scholar]
- Owen, C.A. Roles for proteinases in the pathogenesis of chronic obstructive pulmonary disease. Int. J. Chron. Obs. Pulm. Dis. 2008, 3, 253–268. [Google Scholar] [CrossRef] [Green Version]
- Barnes, P.J.; Shapiro, S.D.; Pauwels, R.A. Chronic obstructive pulmonary disease: Molecular and cellular mechanisms. Eur. Respir. J. 2003, 22, 672–688. [Google Scholar] [CrossRef]
- Ahn, K.S.; Aggarwal, B.B. Transcription factor NF-kappaB: A sensor for smoke and stress signals. Ann. N. Y. Acad. Sci. 2005, 1056, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Zago, M.; Sheridan, J.A.; Nair, P.; Rico de Souza, A.; Gallouzi, I.E.; Rousseau, S.; Di Marco, S.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. Aryl Hydrocarbon Receptor-Dependent Retention of Nuclear HuR Suppresses Cigarette Smoke-Induced Cyclooxygenase-2 Expression Independent of DNA-Binding. PLoS ONE 2013, 8, e74953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srikantan, S.; Gorospe, M. HuR function in disease. Front. Biosci. 2012, 17, 189–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hudy, M.H.; Proud, D. Cigarette smoke enhances human rhinovirus-induced CXCL8 production via HuR-mediated mRNA stabilization in human airway epithelial cells. Respir. Res. 2013, 14, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, S.N.; Garcha, D.S.; Mackay, A.J.; Patel, A.R.; Singh, R.; Sapsford, R.J.; Donaldson, G.C.; Wedzicha, J.A. Human rhinovirus infection during naturally occurring COPD exacerbations. Eur. Respir. J. 2014, 44, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Owuor, N.; Nalamala, N.; Gimenes, J.A., Jr.; Sajjan, U.S. Rhinovirus and COPD airway epithelium. Pulm. Crit. Care Med. 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Angeloni, D. Molecular analysis of deletions in human chromosome 3p21 and the role of resident cancer genes in disease. Brief. Funct. Genom. Proteomic 2007, 6, 19–39. [Google Scholar] [CrossRef]
- Timmer, T.; Terpstra, P.; van den Berg, A.; Veldhuis, P.M.; Ter Elst, A.; Voutsinas, G.; Hulsbeek, M.M.; Draaijers, T.G.; Looman, M.W.; Kok, K.; et al. A comparison of genomic structures and expression patterns of two closely related flanking genes in a critical lung cancer region at 3p21.3. Eur. J. Hum. Genet. 1999, 7, 478–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamsai, D.; Watkins, D.N.; O’Connor, A.E.; Merriner, D.J.; Gursoy, S.; Bird, A.D.; Kumar, B.; Miller, A.; Cole, T.J.; Jenkins, B.J.; et al. In vivo evidence that RBM5 is a tumour suppressor in the lung. Sci. Rep. 2017, 7, 16323. [Google Scholar] [CrossRef] [Green Version]
- Hao, Y.Q.; Su, Z.Z.; Lv, X.J.; Li, P.; Gao, P.; Wang, C.; Bai, Y.; Zhang, J. RNA-binding motif protein 5 negatively regulates the activity of Wnt/beta-catenin signaling in cigarette smoke-induced alveolar epithelial injury. Oncol. Rep. 2015, 33, 2438–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlier, F.M.; Dupasquier, S.; Ambroise, J.; Detry, B.; Lecocq, M.; Bietry-Claudet, C.; Boukala, Y.; Gala, J.L.; Bouzin, C.; Verleden, S.E.; et al. Canonical WNT pathway is activated in the airway epithelium in chronic obstructive pulmonary disease. EBioMedicine 2020, 61, 103034. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Peiro, T.; Serrano, A.; Cortijo, J. Epithelial to mesenchymal transition is increased in patients with COPD and induced by cigarette smoke. Thorax 2013, 68, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, S.; Srisuma, S.; Demeo, D.L.; Shapiro, S.D.; Bueno, R.; Silverman, E.K.; Reilly, J.J.; Mariani, T.J. Molecular biomarkers for quantitative and discrete COPD phenotypes. Am. J. Respir. Cell Mol. Biol. 2009, 40, 359–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeMeo, D.L.; Mariani, T.; Bhattacharya, S.; Srisuma, S.; Lange, C.; Litonjua, A.; Bueno, R.; Pillai, S.G.; Lomas, D.A.; Sparrow, D.; et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am. J. Hum. Genet. 2009, 85, 493–502. [Google Scholar] [CrossRef] [Green Version]
- Qiu, W.; Cho, M.H.; Riley, J.H.; Anderson, W.H.; Singh, D.; Bakke, P.; Gulsvik, A.; Litonjua, A.A.; Lomas, D.A.; Crapo, J.D.; et al. Genetics of sputum gene expression in chronic obstructive pulmonary disease. PLoS ONE 2011, 6, e24395. [Google Scholar] [CrossRef] [PubMed]
- Hardin, M.; Zielinski, J.; Wan, E.S.; Hersh, C.P.; Castaldi, P.J.; Schwinder, E.; Hawrylkiewicz, I.; Sliwinski, P.; Cho, M.H.; Silverman, E.K. CHRNA3/5, IREB2, and ADCY2 are associated with severe chronic obstructive pulmonary disease in Poland. Am. J. Respir. Cell Mol. Biol. 2012, 47, 203–208. [Google Scholar] [CrossRef] [Green Version]
- Cloonan, S.M.; Glass, K.; Laucho-Contreras, M.E.; Bhashyam, A.R.; Cervo, M.; Pabon, M.A.; Konrad, C.; Polverino, F.; Siempos, I.I.; Perez, E.; et al. Mitochondrial iron chelation ameliorates cigarette smoke-induced bronchitis and emphysema in mice. Nat. Med. 2016, 22, 163–174. [Google Scholar] [CrossRef]
- Ghio, A.J.; Hilborn, E.D.; Stonehuerner, J.G.; Dailey, L.A.; Carter, J.D.; Richards, J.H.; Crissman, K.M.; Foronjy, R.F.; Uyeminami, D.L.; Pinkerton, K.E. Particulate matter in cigarette smoke alters iron homeostasis to produce a biological effect. Am. J. Respir. Crit. Care Med. 2008, 178, 1130–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philippot, Q.; Deslee, G.; Adair-Kirk, T.L.; Woods, J.C.; Byers, D.; Conradi, S.; Dury, S.; Perotin, J.M.; Lebargy, F.; Cassan, C.; et al. Increased iron sequestration in alveolar macrophages in chronic obstructive pulmonary disease. PLoS ONE 2014, 9, e96285. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Gu, X.; Wu, N.; Zhang, P.; Liu, Y.; Jiang, S. Human antigen R enhances the epithelial-mesenchymal transition via regulation of ZEB-1 in the human airway epithelium. Respir. Res. 2018, 19, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, L.; Col, J.D.; Casolari, P.; Memoli, D.; Conti, V.; Vatrella, A.; Vonakis, B.M.; Papi, A.; Caramori, G.; Stellato, C. Differential expression of RNA-binding proteins in bronchial epithelium of stable COPD patients. Int. J. Chron. Obs. Pulm. Dis. 2018, 13, 3173–3190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricciardi, L.; Giurato, G.; Memoli, D.; Pietrafesa, M.; Dal Col, J.; Salvato, I.; Nigro, A.; Vatrella, A.; Caramori, G.; Casolaro, V.; et al. Posttranscriptional Gene Regulatory Networks in Chronic Airway Inflammatory Diseases: In silico Mapping of RNA-Binding Protein Expression in Airway Epithelium. Front. Immunol. 2020, 11, 579889. [Google Scholar] [CrossRef] [PubMed]
- Hogg, J.C.; Timens, W. The pathology of chronic obstructive pulmonary disease. Annu. Rev. Pathol. 2009, 4, 435–459. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.M.; Williams, A.; Yauk, C.L.; Vincent, R. Overexpression of tumor necrosis factor-alpha in the lungs alters immune response, matrix remodeling, and repair and maintenance pathways. Am. J. Pathol. 2012, 180, 1413–1430. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.A.; Carballo, E.; Lee, D.M.; Lai, W.S.; Thompson, M.J.; Patel, D.D.; Schenkman, D.I.; Gilkeson, G.S.; Broxmeyer, H.E.; Haynes, B.F.; et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 1996, 4, 445–454. [Google Scholar] [CrossRef] [Green Version]
- Prabhala, P.; Ammit, A.J. Tristetraprolin and its role in regulation of airway inflammation. Mol. Pharm. 2015, 87, 629–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mino, T.; Takeuchi, O. Post-transcriptional regulation of cytokine mRNA controls the initiation and resolution of inflammation. Biotechnol. Genet. Eng. Rev. 2013, 29, 49–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smoak, K.; Cidlowski, J.A. Glucocorticoids regulate tristetraprolin synthesis and posttranscriptionally regulate tumor necrosis factor alpha inflammatory signaling. Mol. Cell Biol. 2006, 26, 9126–9135. [Google Scholar] [CrossRef] [Green Version]
- Ishmael, F.T.; Fang, X.; Galdiero, M.R.; Atasoy, U.; Rigby, W.F.; Gorospe, M.; Cheadle, C.; Stellato, C. Role of the RNA-binding protein tristetraprolin in glucocorticoid-mediated gene regulation. J. Immunol. 2008, 180, 8342–8353. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Y.; Sadri, N.; Schneider, R.J. Endotoxic shock in AUF1 knockout mice mediated by failure to degrade proinflammatory cytokine mRNAs. Genes Dev. 2006, 20, 3174–3184. [Google Scholar] [CrossRef] [Green Version]
- Sadri, N.; Schneider, R.J. Auf1/Hnrnpd-deficient mice develop pruritic inflammatory skin disease. J. Investig. Derm. 2009, 129, 657–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.L.; Wait, R.; Mahtani, K.R.; Sully, G.; Clark, A.R.; Saklatvala, J. The 3’ untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA-stabilizing factor HuR. Mol. Cell Biol. 2001, 21, 721–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atasoy, U.; Curry, S.L.; Lopez de Silanes, I.; Shyu, A.B.; Casolaro, V.; Gorospe, M.; Stellato, C. Regulation of eotaxin gene expression by TNF-alpha and IL-4 through mRNA stabilization: Involvement of the RNA-binding protein HuR. J. Immunol. 2003, 171, 4369–4378. [Google Scholar] [CrossRef] [Green Version]
- Bradford, E.; Jacobson, S.; Varasteh, J.; Comellas, A.P.; Woodruff, P.; O’Neal, W.; DeMeo, D.L.; Li, X.; Kim, V.; Cho, M.; et al. The value of blood cytokines and chemokines in assessing COPD. Respir. Res. 2017, 18, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morissette, M.C.; Parent, J.; Milot, J. Alveolar epithelial and endothelial cell apoptosis in emphysema: What we know and what we need to know. Int. J. Chron. Obs. Pulm. Dis. 2009, 4, 19–31. [Google Scholar]
- Aoshiba, K.; Yokohori, N.; Nagai, A. Alveolar wall apoptosis causes lung destruction and emphysematous changes. Am. J. Respir. Cell Mol. Biol. 2003, 28, 555–562. [Google Scholar] [CrossRef]
- Kasahara, Y.; Tuder, R.M.; Taraseviciene-Stewart, L.; Le Cras, T.D.; Abman, S.; Hirth, P.K.; Waltenberger, J.; Voelkel, N.F. Inhibition of VEGF receptors causes lung cell apoptosis and emphysema. J. Clin. Investig. 2000, 106, 1311–1319. [Google Scholar] [CrossRef] [Green Version]
- Kasahara, Y.; Tuder, R.M.; Cool, C.D.; Lynch, D.A.; Flores, S.C.; Voelkel, N.F. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am. J. Respir. Crit. Care Med. 2001, 163 Pt 1, 737–744. [Google Scholar] [CrossRef] [Green Version]
- Kanazawa, H.; Tochino, Y.; Asai, K.; Hirata, K. Simultaneous assessment of hepatocyte growth factor and vascular endothelial growth factor in epithelial lining fluid from patients with COPD. Chest 2014, 146, 1159–1165. [Google Scholar] [CrossRef] [PubMed]
- Essafi-Benkhadir, K.; Onesto, C.; Stebe, E.; Moroni, C.; Pages, G. Tristetraprolin inhibits Ras-dependent tumor vascularization by inducing vascular endothelial growth factor mRNA degradation. Mol. Biol. Cell 2007, 18, 4648–4658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, S.C.; Claffey, K.P. Regulation of human vascular endothelial growth factor mRNA stability in hypoxia by heterogeneous nuclear ribonucleoprotein L. J. Biol. Chem. 1999, 274, 1359–1365. [Google Scholar] [CrossRef] [Green Version]
- Heiner, M.; Hui, J.; Schreiner, S.; Hung, L.H.; Bindereif, A. HnRNP L-mediated regulation of mammalian alternative splicing by interference with splice site recognition. RNA Biol. 2010, 7, 56–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaudreau, M.C.; Grapton, D.; Helness, A.; Vadnais, C.; Fraszczak, J.; Shooshtarizadeh, P.; Wilhelm, B.; Robert, F.; Heyd, F.; Moroy, T. Heterogeneous Nuclear Ribonucleoprotein L is required for the survival and functional integrity of murine hematopoietic stem cells. Sci. Rep. 2016, 6, 27379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenderdos, K.; Condliffe, A. The neutrophil in chronic obstructive pulmonary disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 531–539. [Google Scholar] [CrossRef]
- Jasper, A.E.; McIver, W.J.; Sapey, E.; Walton, G.M. Understanding the role of neutrophils in chronic inflammatory airway disease. F1000Research 2019, 8. [Google Scholar] [CrossRef]
- Kapellos, T.S.; Bassler, K.; Aschenbrenner, A.C.; Fujii, W.; Schultze, J.L. Dysregulated Functions of Lung Macrophage Populations in COPD. J. Immunol Res. 2018, 2018, 2349045. [Google Scholar] [CrossRef] [Green Version]
- Van Tubergen, E.A.; Banerjee, R.; Liu, M.; Vander Broek, R.; Light, E.; Kuo, S.; Feinberg, S.E.; Willis, A.L.; Wolf, G.; Carey, T.; et al. Inactivation or loss of TTP promotes invasion in head and neck cancer via transcript stabilization and secretion of MMP9, MMP2, and IL-6. Clin. Cancer Res. 2013, 19, 1169–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huwiler, A.; Akool, E.S.; Aschrafi, A.; Hamada, F.M.; Pfeilschifter, J.; Eberhardt, W. ATP potentiates interleukin-1 beta-induced MMP-9 expression in mesangial cells via recruitment of the ELAV protein HuR. J. Biol. Chem. 2003, 278, 51758–51769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akool, E.S.; Kleinert, H.; Hamada, F.M.; Abdelwahab, M.H.; Forstermann, U.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol. Cell Biol. 2003, 23, 4901–4916. [Google Scholar] [CrossRef] [Green Version]
- Krishnamurthy, P.; Rajasingh, J.; Lambers, E.; Qin, G.; Losordo, D.W.; Kishore, R. IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ. Res. 2009, 104, e9–e18. [Google Scholar] [CrossRef]
- Wang, I.X.; So, E.; Devlin, J.L.; Zhao, Y.; Wu, M.; Cheung, V.G. ADAR regulates RNA editing, transcript stability, and gene expression. Cell Rep. 2013, 5, 849–860. [Google Scholar] [CrossRef] [Green Version]
- Stellos, K.; Gatsiou, A.; Stamatelopoulos, K.; Perisic Matic, L.; John, D.; Lunella, F.F.; Jae, N.; Rossbach, O.; Amrhein, C.; Sigala, F.; et al. Adenosine-to-inosine RNA editing controls cathepsin S expression in atherosclerosis by enabling HuR-mediated post-transcriptional regulation. Nat. Med. 2016, 22, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.; Nath, S.; Lora, A.; Samaha, G.; Elgamal, Z.; Kaiser, R.; Taggart, C.; Weldon, S.; Geraghty, P. Cathepsin S: Investigating an old player in lung disease pathogenesis, comorbidities, and potential therapeutics. Respir. Res. 2020, 21, 111. [Google Scholar] [CrossRef] [PubMed]
- Andrault, P.M.; Schamberger, A.C.; Chazeirat, T.; Sizaret, D.; Renault, J.; Staab-Weijnitz, C.A.; Hennen, E.; Petit-Courty, A.; Wartenberg, M.; Saidi, A.; et al. Cigarette smoke induces overexpression of active human cathepsin S in lungs from current smokers with or without COPD. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 317, L625–L638. [Google Scholar] [CrossRef] [PubMed]
- Wigington, C.P.; Jung, J.; Rye, E.A.; Belauret, S.L.; Philpot, A.M.; Feng, Y.; Santangelo, P.J.; Corbett, A.H. Post-transcriptional regulation of programmed cell death 4 (PDCD4) mRNA by the RNA-binding proteins human antigen R (HuR) and T-cell intracellular antigen 1 (TIA1). J. Biol. Chem. 2015, 290, 3468–3487. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, X.; Liu, Q.; Zhang, S.; Hu, W. Translation repression via modulation of the cytoplasmic poly(A)-binding protein in the inflammatory response. Elife 2017, 6, e27786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004, 64, 3753–3756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.; Liu, Z.; Li, S.; Yang, C.; Xue, R.; Xi, Y.; Wang, L.; Wang, S.; He, Q.; Huang, J.; et al. Down-regulation of c-Met and Bcl2 by microRNA-206, activates apoptosis, and inhibits tumor cell proliferation, migration and colony formation. Oncotarget 2015, 6, 25533–25574. [Google Scholar] [CrossRef] [Green Version]
- Ke, Y.; Zhao, W.; Xiong, J.; Cao, R. Downregulation of miR-16 promotes growth and motility by targeting HDGF in non-small cell lung cancer cells. FEBS Lett. 2013, 587, 3153–3157. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xiao, Q.; Wang, X.; Wang, Z.; Hu, J.; Zhang, Z.; Gong, Z.; Chen, S. miR-16 regulates proliferation and invasion of lung cancer cells via the ERK/MAPK signaling pathway by targeted inhibition of MAPK kinase 1 (MEK1). J. Int. Med. Res. 2019, 47, 5194–5204. [Google Scholar] [CrossRef] [Green Version]
- Kao, S.C.; Fulham, M.; Wong, K.; Cooper, W.; Brahmbhatt, H.; MacDiarmid, J.; Pattison, S.; Sagong, J.O.; Huynh, Y.; Leslie, F.; et al. A Significant Metabolic and Radiological Response after a Novel Targeted MicroRNA-based Treatment Approach in Malignant Pleural Mesothelioma. Am. J. Respir. Crit. Care Med. 2015, 191, 1467–1469. [Google Scholar] [CrossRef]
- van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Schembri, F.; Sridhar, S.; Perdomo, C.; Gustafson, A.M.; Zhang, X.; Ergun, A.; Lu, J.; Liu, G.; Zhang, X.; Bowers, J.; et al. MicroRNAs as modulators of smoking-induced gene expression changes in human airway epithelium. Proc. Natl. Acad. Sci. USA 2009, 106, 2319–2324. [Google Scholar] [CrossRef] [Green Version]
- Graff, J.W.; Powers, L.S.; Dickson, A.M.; Kim, J.; Reisetter, A.C.; Hassan, I.H.; Kremens, K.; Gross, T.J.; Wilson, M.E.; Monick, M.M. Cigarette smoking decreases global microRNA expression in human alveolar macrophages. PLoS ONE 2012, 7, e44066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izzotti, A.; Calin, G.A.; Arrigo, P.; Steele, V.E.; Croce, C.M.; De Flora, S. Downregulation of microRNA expression in the lungs of rats exposed to cigarette smoke. FASEB J. 2009, 23, 806–812. [Google Scholar] [CrossRef]
- Izzotti, A.; Larghero, P.; Longobardi, M.; Cartiglia, C.; Camoirano, A.; Steele, V.E.; De Flora, S. Dose-responsiveness and persistence of microRNA expression alterations induced by cigarette smoke in mouse lung. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2011, 717, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hassan, F.; Nuovo, G.J.; Crawford, M.; Boyaka, P.N.; Kirkby, S.; Nana-Sinkam, S.P.; Cormet-Boyaka, E. MiR-101 and miR-144 regulate the expression of the CFTR chloride channel in the lung. PLoS ONE 2012, 7, e50837. [Google Scholar] [CrossRef] [Green Version]
- Rogers, S.; de Souza, A.R.; Zago, M.; Iu, M.; Guerrina, N.; Gomez, A.; Matthews, J.; Baglole, C.J. Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci. Rep. 2017, 7, 40539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halappanavar, S.; Nikota, J.; Wu, D.; Williams, A.; Yauk, C.L.; Stampfli, M. IL-1 receptor regulates microRNA-135b expression in a negative feedback mechanism during cigarette smoke-induced inflammation. J. Immunol. 2013, 190, 3679–3686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leuenberger, C.; Schuoler, C.; Bye, H.; Mignan, C.; Rechsteiner, T.; Hillinger, S.; Opitz, I.; Marsland, B.; Faiz, A.; Hiemstra, P.S.; et al. MicroRNA-223 controls the expression of histone deacetylase 2: A novel axis in COPD. J. Mol. Med. 2016, 94, 725–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpi, S.; Polini, B.; Nieri, D.; Dubbini, N.; Celi, A.; Nieri, P.; Neri, T. Expression Analysis of Muscle-Specific miRNAs in Plasma-Derived Extracellular Vesicles from Patients with Chronic Obstructive Pulmonary Disease. Diagnostics 2020, 10, 502. [Google Scholar] [CrossRef] [PubMed]
- Van Pottelberge, G.R.; Mestdagh, P.; Bracke, K.R.; Thas, O.; van Durme, Y.M.; Joos, G.F.; Vandesompele, J.; Brusselle, G.G. MicroRNA expression in induced sputum of smokers and patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2011, 183, 898–906. [Google Scholar] [CrossRef]
- Chatila, W.M.; Criner, G.J.; Hancock, W.W.; Akimova, T.; Moldover, B.; Chang, J.K.; Cornwell, W.; Santerre, M.; Rogers, T.J. Blunted expression of miR-199a-5p in regulatory T cells of patients with chronic obstructive pulmonary disease compared to unaffected smokers. Clin. Exp. Immunol. 2014, 177, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, S.; Bogaard, H.J.; Gomez-Arroyo, J.; Alhussaini, A.; Kraskauskas, D.; Cool, C.D.; Voelkel, N.F. MicroRNA-199a-5p is associated with hypoxia-inducible factor-1alpha expression in lungs from patients with COPD. Chest 2012, 142, 663–672. [Google Scholar] [CrossRef] [Green Version]
- Conickx, G.; Mestdagh, P.; Avila Cobos, F.; Verhamme, F.M.; Maes, T.; Vanaudenaerde, B.M.; Seys, L.J.; Lahousse, L.; Kim, R.Y.; Hsu, A.C.; et al. MicroRNA Profiling Reveals a Role for MicroRNA-218-5p in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Am. J. Respir. Crit. Care Med. 2017, 195, 43–56. [Google Scholar] [CrossRef]
- Du, Y.; Ding, Y.; Chen, X.; Mei, Z.; Ding, H.; Wu, Y.; Jie, Z. MicroRNA-181c inhibits cigarette smoke-induced chronic obstructive pulmonary disease by regulating CCN1 expression. Respir. Res. 2017, 18, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.B.; Li, Z.H.; Gao, S. Circulating miR-146a/b correlates with inflammatory cytokines in COPD and could predict the risk of acute exacerbation COPD. Medicine 2018, 97, e9820. [Google Scholar] [CrossRef]
- Zago, M.; Rico de Souza, A.; Hecht, E.; Rousseau, S.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. The NF-kappaB family member RelB regulates microRNA miR-146a to suppress cigarette smoke-induced COX-2 protein expression in lung fibroblasts. Toxicol. Lett. 2014, 226, 107–116. [Google Scholar] [CrossRef]
- Sato, T.; Liu, X.; Nelson, A.; Nakanishi, M.; Kanaji, N.; Wang, X.; Kim, M.; Li, Y.; Sun, J.; Michalski, J.; et al. Reduced miR-146a increases prostaglandin E(2)in chronic obstructive pulmonary disease fibroblasts. Am. J. Respir. Crit. Care Med. 2010, 182, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Christenson, S.A.; Brandsma, C.A.; Campbell, J.D.; Knight, D.A.; Pechkovsky, D.V.; Hogg, J.C.; Timens, W.; Postma, D.S.; Lenburg, M.; Spira, A. miR-638 regulates gene expression networks associated with emphysematous lung destruction. Genome Med. 2013, 5, 114. [Google Scholar] [CrossRef] [Green Version]
- Savarimuthu Francis, S.M.; Davidson, M.R.; Tan, M.E.; Wright, C.M.; Clarke, B.E.; Duhig, E.E.; Bowman, R.V.; Hayward, N.K.; Fong, K.M.; Yang, I.A. MicroRNA-34c is associated with emphysema severity and modulates SERPINE1 expression. BMC Genom. 2014, 15, 88. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ding, L.; Hu, Q.; Xia, J.; Sun, J.; Wang, X.; Xiong, H.; Gurbani, D.; Li, L.; Liu, Y.; et al. MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol. Cancer 2017, 16, 141. [Google Scholar] [CrossRef]
- Wheeler, M.A.; Clark, I.C.; Tjon, E.C.; Li, Z.; Zandee, S.E.J.; Couturier, C.P.; Watson, B.R.; Scalisi, G.; Alkwai, S.; Rothhammer, V.; et al. MAFG-driven astrocytes promote CNS inflammation. Nature 2020, 578, 593–599. [Google Scholar] [CrossRef]
- Demedts, I.K.; Bracke, K.R.; Van Pottelberge, G.; Testelmans, D.; Verleden, G.M.; Vermassen, F.E.; Joos, G.F.; Brusselle, G.G. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2007, 175, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Andriani, F.; Majorini, M.T.; Mano, M.; Landoni, E.; Miceli, R.; Facchinetti, F.; Mensah, M.; Fontanella, E.; Dugo, M.; Giacca, M.; et al. MiR-16 regulates the pro-tumorigenic potential of lung fibroblasts through the inhibition of HGF production in an FGFR-1- and MEK1-dependent manner. J. Hematol. Oncol. 2018, 11, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, L.; Sevinc, K.; Papazoglou, I.M.; Tildy, B.; Detillieux, K.; Halayko, A.J.; Chung, K.F.; Perry, M.M. Airway smooth muscle inflammation is regulated by microRNA-145 in COPD. FEBS Lett. 2016, 590, 1324–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Sun, Y.; Rao, Q.; Xu, H.; Li, L.; Chang, C. Androgen receptor (AR) suppresses miRNA-145 to promote renal cell carcinoma (RCC) progression independent of VHL status. Oncotarget 2015, 6, 31203–31215. [Google Scholar] [CrossRef] [Green Version]
- Agra Andrieu, N.; Motino, O.; Mayoral, R.; Llorente Izquierdo, C.; Fernandez-Alvarez, A.; Bosca, L.; Casado, M.; Martin-Sanz, P. Cyclooxygenase-2 is a target of microRNA-16 in human hepatoma cells. PLoS ONE 2012, 7, e50935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahos, R.; Bozinovski, S. Role of alveolar macrophages in chronic obstructive pulmonary disease. Front. Immunol. 2014, 5, 435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woodruff, P.G.; Koth, L.L.; Yang, Y.H.; Rodriguez, M.W.; Favoreto, S.; Dolganov, G.M.; Paquet, A.C.; Erle, D.J. A distinctive alveolar macrophage activation state induced by cigarette smoking. Am. J. Respir. Crit. Care Med. 2005, 172, 1383–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, J.; Yang, F.; Wang, Y.; Wang, Y.; Xue, G.; Mei, Q.; Wang, F.; Sun, S. MicroRNA-23a antisense enhances 5-fluorouracil chemosensitivity through APAF-1/caspase-9 apoptotic pathway in colorectal cancer cells. J. Cell Biochem. 2014, 115, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheng, X.; Wan, Y.; Chen, T.; Zhou, Q.; Wang, Z.; Zhu, H. MicroRNA-421 Inhibits Apoptosis by Downregulating Caspase-3 in Human Colorectal Cancer. Cancer Manag. Res. 2020, 12, 7579–7587. [Google Scholar] [CrossRef] [PubMed]
- Damico, R.; Simms, T.; Kim, B.S.; Tekeste, Z.; Amankwan, H.; Damarla, M.; Hassoun, P.M. p53 mediates cigarette smoke-induced apoptosis of pulmonary endothelial cells: Inhibitory effects of macrophage migration inhibitor factor. Am. J. Respir. Cell Mol. Biol. 2011, 44, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N.; Reddy, M.A.; Natarajan, R. Distinct roles of heterogeneous nuclear ribonuclear protein K and microRNA-16 in cyclooxygenase-2 RNA stability induced by S100b, a ligand of the receptor for advanced glycation end products. J. Biol. Chem. 2008, 283, 36221–36233. [Google Scholar] [CrossRef] [Green Version]
- Young, L.E.; Moore, A.E.; Sokol, L.; Meisner-Kober, N.; Dixon, D.A. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol. Cancer Res. 2012, 10, 167–180. [Google Scholar] [CrossRef] [Green Version]
- Mubaid, S.; Ma, J.F.; Omer, A.; Ashour, K.; Lian, X.J.; Sanchez, B.J.; Robinson, S.; Cammas, A.; Dormoy-Raclet, V.; Di Marco, S.; et al. HuR counteracts miR-330 to promote STAT3 translation during inflammation-induced muscle wasting. Proc. Natl. Acad. Sci. USA 2019, 116, 17261–17270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef] [Green Version]
- Kedde, M.; van Kouwenhove, M.; Zwart, W.; Oude Vrielink, J.A.; Elkon, R.; Agami, R. A Pumilio-induced RNA structure switch in p27-3’ UTR controls miR-221 and miR-222 accessibility. Nat. Cell Biol. 2010, 12, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, D.; Li, D.; Zheng, Z.; Xu, J.; Liang, X.; Zhang, C.; Wang, S.; Wang, J.; Lu, W. Long non-coding RNA expression patterns in lung tissues of chronic cigarette smoke induced COPD mouse model. Sci Rep. 2018, 8, 7609. [Google Scholar] [CrossRef] [PubMed]
- Bi, H.; Zhou, J.; Wu, D.; Gao, W.; Li, L.; Yu, L.; Liu, F.; Huang, M.; Adcock, I.M.; Barnes, P.J.; et al. Microarray analysis of long non-coding RNAs in COPD lung tissue. Inflamm. Res. 2015, 64, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Shen, Z.; Guo, J.; Sun, S. Screening of long non-coding RNA and TUG1 inhibits proliferation with TGF-beta induction in patients with COPD. Int. J. Chron. Obs. Pulm. Dis 2016, 11, 2951–2964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thai, P.; Statt, S.; Chen, C.H.; Liang, E.; Campbell, C.; Wu, R. Characterization of a novel long noncoding RNA, SCAL1, induced by cigarette smoke and elevated in lung cancer cell lines. Am. J. Respir. Cell Mol. Biol. 2013, 49, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Lin, H.; Yu, W.; Zhang, F.; Wang, R.; Yu, H.; Qian, B. LncRNA LCPAT1 is involved in DNA damage induced by CSE. Biochem. Biophys. Res. Commun. 2019, 508, 512–515. [Google Scholar] [CrossRef]
- Lu, L.; Luo, F.; Liu, Y.; Liu, X.; Shi, L.; Lu, X.; Liu, Q. Posttranscriptional silencing of the lncRNA MALAT1 by miR-217 inhibits the epithelial-mesenchymal transition via enhancer of zeste homolog 2 in the malignant transformation of HBE cells induced by cigarette smoke extract. Toxicol. Appl. Pharm. 2015, 289, 276–285. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, F.; Xu, Y.; Wang, B.; Zhao, Y.; Xu, W.; Shi, L.; Lu, X.; Liu, Q. Epithelial-mesenchymal transition and cancer stem cells, mediated by a long non-coding RNA, HOTAIR, are involved in cell malignant transformation induced by cigarette smoke extract. Toxicol. Appl. Pharm. 2015, 282, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.J.; Huang, H.B.; Shen, H.B.; Chen, W.; Yang, Z.H. Role of long non-coding RNA MALAT1 in chronic obstructive pulmonary disease. Exp. Med. 2020, 20, 2691–2697. [Google Scholar] [CrossRef] [PubMed]
- Lei, Z.; Guo, H.; Zou, S.; Jiang, J.; Kui, Y.; Song, J. Long non-coding RNA maternally expressed gene regulates cigarette smoke extract induced lung inflammation and human bronchial epithelial apoptosis via miR-149-3p. Exp. Med. 2021, 21, 60. [Google Scholar] [CrossRef]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett. 2016, 590, 2884–2895. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Liu, J.; Li, W.; Liu, G.; Li, Z. LncRNA-HOTAIR promotes TNF-alpha production in cardiomyocytes of LPS-induced sepsis mice by activating NF-kappaB pathway. Biochem. Biophys. Res. Commun. 2016, 471, 240–246. [Google Scholar] [CrossRef]
- Liu, J.; Huang, G.Q.; Ke, Z.P. Silence of long intergenic noncoding RNA HOTAIR ameliorates oxidative stress and inflammation response in ox-LDL-treated human macrophages by upregulating miR-330-5p. J. Cell Physiol. 2019, 234, 5134–5142. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Komiya, M.; Matsumura, K.; Negishi, L.; Suda, S.; Okuno, M.; Yokota, N.; Osada, T.; Nagashima, T.; Hiyoshi, M.; et al. MYU, a Target lncRNA for Wnt/c-Myc Signaling, Mediates Induction of CDK6 to Promote Cell Cycle Progression. Cell Rep. 2016, 16, 2554–2564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Liu, X.; Xie, M.; Liu, M.; Ye, M.; Li, M.; Chen, X.M.; Li, X.; Zhou, R. The NF-kappaB-Responsive Long Noncoding RNA FIRRE Regulates Posttranscriptional Regulation of Inflammatory Gene Expression through Interacting with hnRNPU. J. Immunol. 2017, 199, 3571–3582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Zhang, A.; Ho, T.T.; Zhang, Z.; Zhou, N.; Ding, X.; Zhang, X.; Xu, M.; Mo, Y.Y. Linc-RoR promotes c-Myc expression through hnRNP I and AUF1. Nucleic Acids Res. 2016, 44, 3059–3069. [Google Scholar] [CrossRef] [PubMed]
- Meisner, N.C.; Hintersteiner, M.; Mueller, K.; Bauer, R.; Seifert, J.M.; Naegeli, H.U.; Ottl, J.; Oberer, L.; Guenat, C.; Moss, S.; et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat. Chem. Biol. 2007, 3, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.F.; Preet, R.; Aguado, A.; Vishwakarma, V.; Stevens, L.E.; Vyas, A.; Padhye, S.; Xu, L.; Weir, S.J.; Anant, S.; et al. Impact of HuR inhibition by the small molecule MS-444 on colorectal cancer cell tumorigenesis. Oncotarget 2016, 7, 74043–74058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trang, P.; Medina, P.P.; Wiggins, J.F.; Ruffino, L.; Kelnar, K.; Omotola, M.; Homer, R.; Brown, D.; Bader, A.G.; Weidhaas, J.B.; et al. Regression of murine lung tumors by the let-7 microRNA. Oncogene 2010, 29, 1580–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aloufi, N.; Alluli, A.; Eidelman, D.H.; Baglole, C.J. Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease. Int. J. Mol. Sci. 2021, 22, 11963. https://doi.org/10.3390/ijms222111963
Aloufi N, Alluli A, Eidelman DH, Baglole CJ. Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease. International Journal of Molecular Sciences. 2021; 22(21):11963. https://doi.org/10.3390/ijms222111963
Chicago/Turabian StyleAloufi, Noof, Aeshah Alluli, David H. Eidelman, and Carolyn J. Baglole. 2021. "Aberrant Post-Transcriptional Regulation of Protein Expression in the Development of Chronic Obstructive Pulmonary Disease" International Journal of Molecular Sciences 22, no. 21: 11963. https://doi.org/10.3390/ijms222111963