
Forkhead Box Protein P3 (FOXP3) Represses ATF3 Transcriptional Activity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
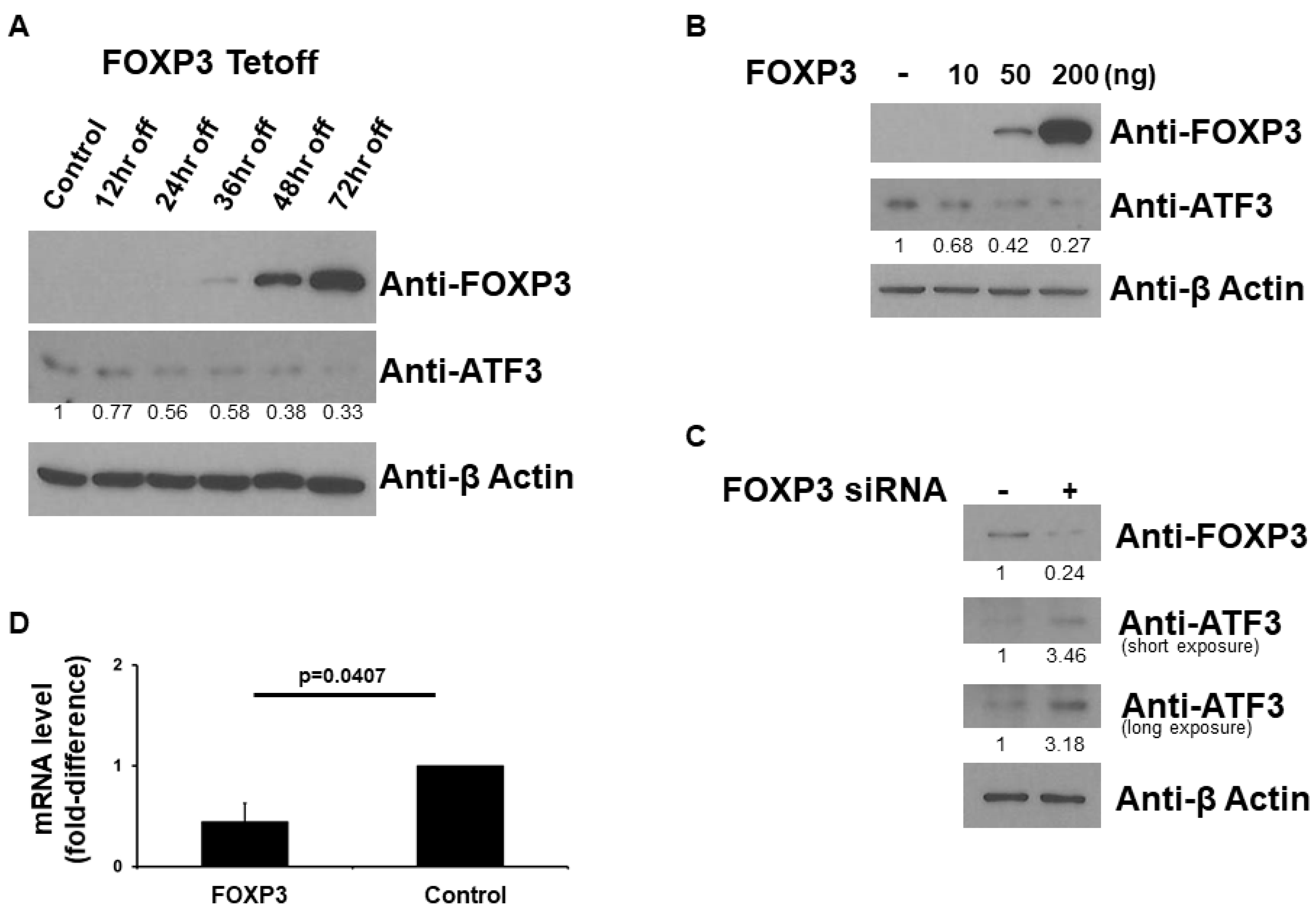
2.1. FOXP3 Decreases ATF3 Protein Level
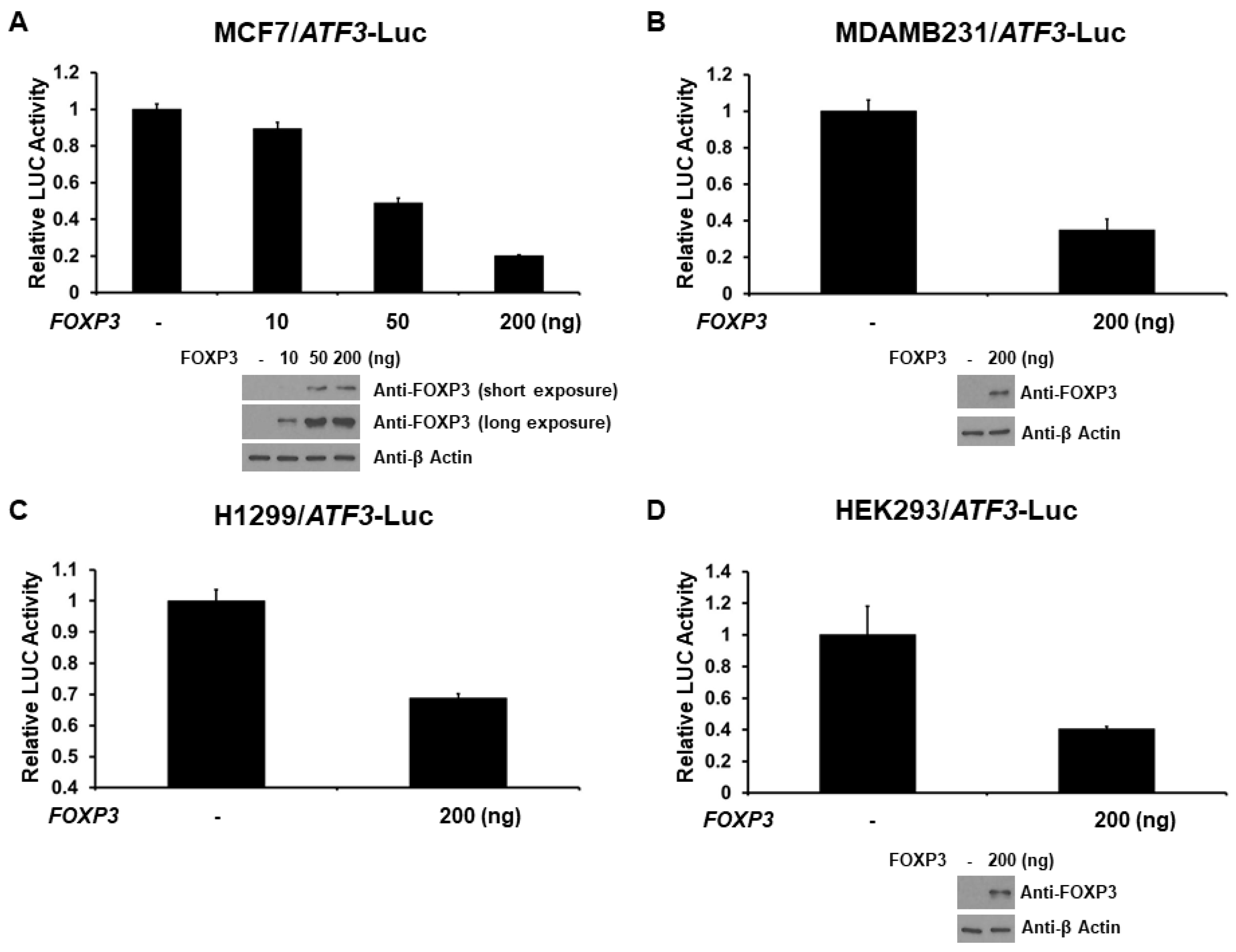
2.2. FOXP3 Is a Repressor of the ATF3 Promoter
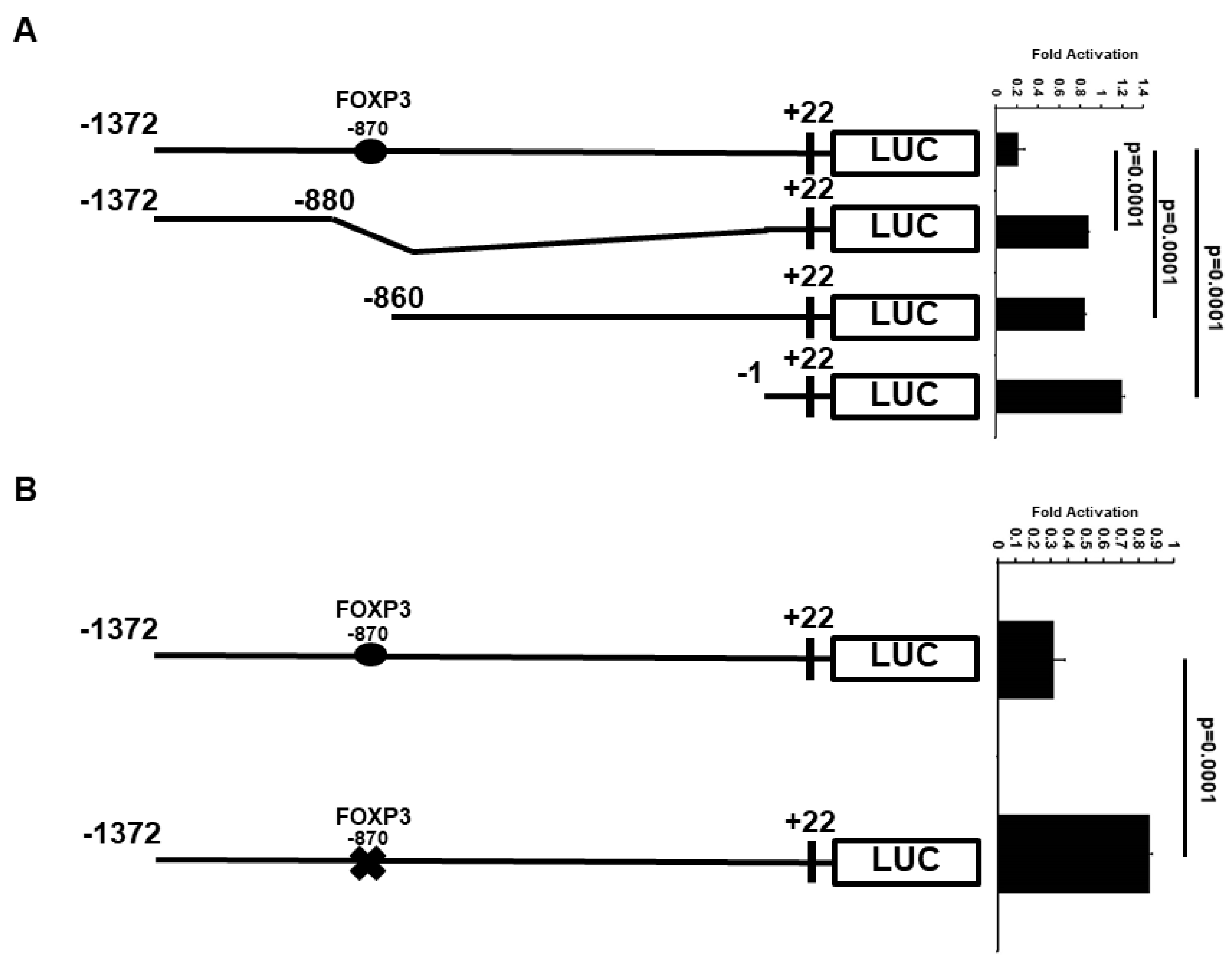
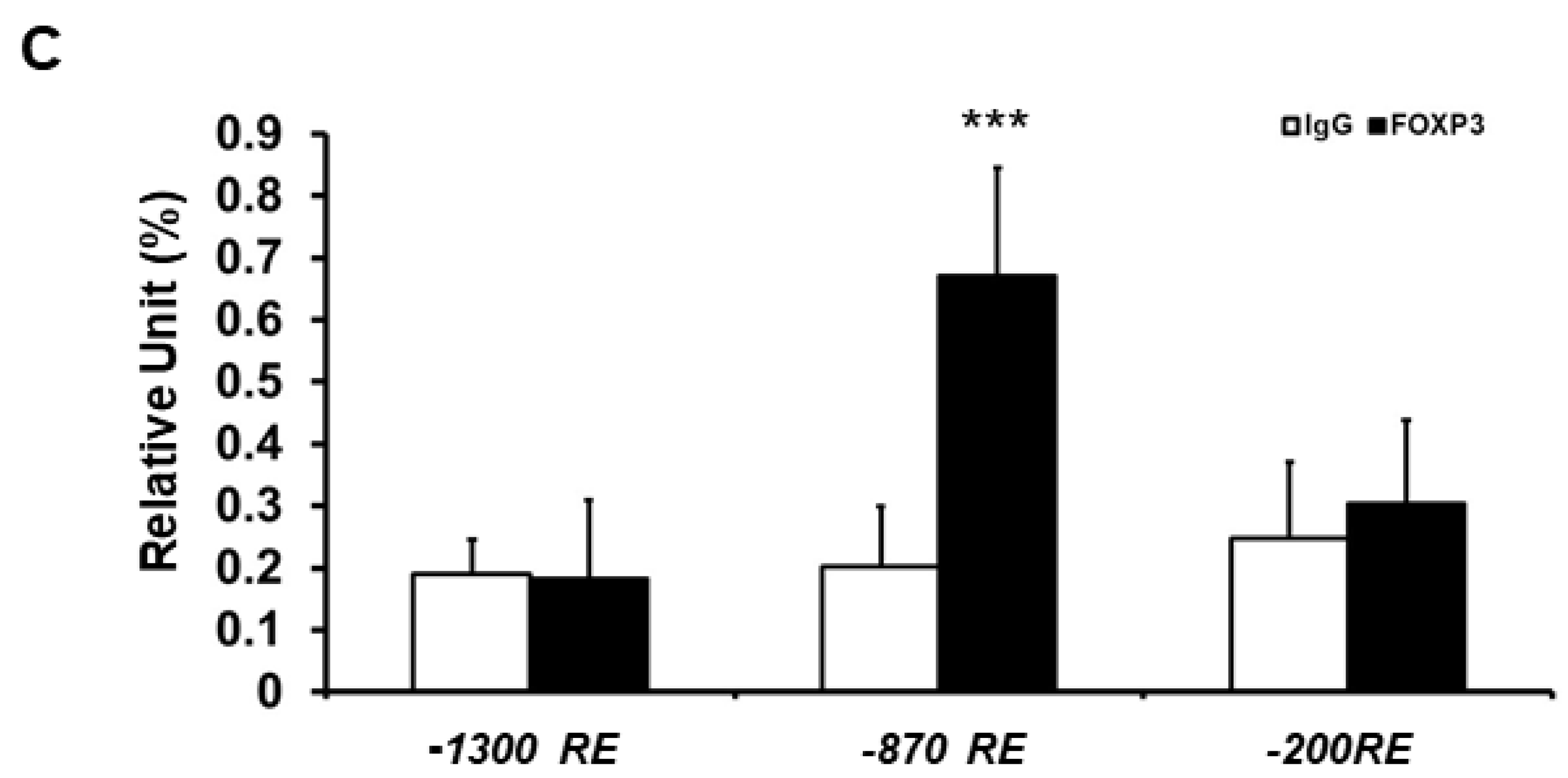
2.3. Minimal ATF3 Promoter Region Responsive to FOXP3 Repression
2.4. Phosphorylation at Tyr342 of FOXP3 Is Required for Full FOXP3-Mediated ATF3 Transcriptional Activity
3. Discussion
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. DNA Constructs
4.3. Cell Culture and Transfection
4.4. ATF3 Promoter Luciferase Reporter Assays
4.5. Western Blot Analysis
4.6. RT-PCR and Real-Time ChIP
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FOXP3 | Forkhead box protein P3 |
| ATF3 | Activating transcription factor 3 |
| SUMO | Small ubiquitin-like modifier |
References
- Chatila, T.A.; Blaeser, F.; Ho, N.; Lederman, H.M.; Voulgaropoulos, C.; Helms, C.; Bowcock, A.M. JM2, encoding a fork head-related protein, is mutated in X-linked autoimmunity-allergic disregulation syndrome. J. Clin. Investig. 2000, 106, R75–R81. [Google Scholar] [CrossRef]
- Bennett, C.L.; Christie, J.; Ramsdell, F.; Brunkow, M.E.; Ferguson, P.J.; Whitesell, L.; Kelly, T.E.; Saulsbury, F.T.; Chance, P.F.; Ochs, H.D. The immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) is caused by mutations of FOXP3. Nat. Genet. 2001, 27, 20–21. [Google Scholar] [CrossRef]
- Brunkow, M.E.; Jeffery, E.W.; Hjerrild, K.A.; Paeper, B.; Clark, L.B.; Yasayko, S.A.; Wilkinson, J.E.; Galas, D.; Ziegler, S.F.; Ramsdell, F. Disruption of a new forkhead/winged-helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat. Genet. 2001, 27, 68–73. [Google Scholar] [CrossRef]
- Wildin, R.S.; Ramsdell, F.; Peake, J.; Faravelli, F.; Casanova, J.L.; Buist, N.; Levy-Lahad, E.; Mazzella, M.; Goulet, O.; Perroni, L.; et al. X-linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat. Genet. 2001, 27, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Khattri, R.; Cox, T.; Yasayko, S.A.; Ramsdell, F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat. Immunol. 2003, 4, 337–342. [Google Scholar] [CrossRef]
- Bennett, C.L.; Ochs, H.D. IPEX is a unique X-linked syndrome characterized by immune dysfunction, polyendocrinopathy, enteropathy, and a variety of autoimmune phenomena. Curr. Opin. Pediatr. 2001, 13, 533–538. [Google Scholar] [CrossRef]
- Martin, F.; Ladoire, S.; Mignot, G.; Apetoh, L.; Ghiringhelli, F. Human FOXP3 and cancer. Oncogene 2010, 29, 4121–4129. [Google Scholar] [CrossRef] [Green Version]
- Redpath, M.; Xu, B.; van Kempen, L.C.; Spatz, A. The dual role of the X-linked FoxP3 gene in human cancers. Mol. Oncol. 2011, 5, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, D.O.; Jasurda, J.S.; Egashira, N.; Ellsworth, B.S. The forkhead transcription factor, FOXP3, is required for normal pituitary gonadotropin expression in mice. Biol. Reprod. 2012, 86, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.L.; Ruan, L.W. Association between FOXP3 promoter polymorphisms and cancer risk: A meta-analysis. Oncol. Lett. 2014, 8, 2795–2799. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Wang, L.; Morrison, C.; Chang, X.; Zhang, H.; Li, W.; Liu, Y.; Wang, Y.; Liu, X.; Chan, M.W.; et al. FOXP3 is an X-linked breast cancer suppressor gene and an important repressor of the HER-2/ErbB2 oncogene. Cell 2007, 129, 1275–1286. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Katoh, H.; Wang, L.; Yu, X.; Du, Z.; Yan, X.; Zheng, P.; Liu, Y. FOXP3 regulates sensitivity of cancer cells to irradiation by transcriptional repression of BRCA1. Cancer Res. 2013, 73, 2170–2180. [Google Scholar] [CrossRef] [Green Version]
- Zuo, T.; Liu, R.; Zhang, H.; Chang, X.; Liu, Y.; Wang, L.; Zheng, P.; Liu, Y. FOXP3 is a novel transcriptional repressor for the breast cancer oncogene SKP2. J. Clin. Investig. 2007, 117, 3765–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Xu, Y.; Hao, Q.; Wang, S.; Li, H.; Li, J.; Gao, Y.; Li, M.; Li, W.; Xue, X.; et al. FOXP3 suppresses breast cancer metastasis through downregulation of CD44. Int. J. Cancer 2015, 137, 1279–1290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Yi, B.; Wei, S.; Yang, W.H.; Hart, K.M.; Chauhan, P.; Zhang, W.; Mao, X.; Liu, X.; Liu, C.G.; et al. FOXP3-miR-146-NF-κB Axis and Therapy for Precancerous Lesions in Prostate. Cancer Res. 2015, 75, 1714–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, R.; Liu, C.; Chen, D.; Yang, W.H.; Liu, X.; Liu, C.G.; Dugas, C.M.; Tang, F.; Zheng, P.; Liu, Y.; et al. FOXP3 Controls an miR-146/NF-κB Negative Feedback Loop That Inhibits Apoptosis in Breast Cancer Cells. Cancer Res. 2015, 75, 1703–1713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Wang, Y.; Wang, M.; Li, Z.; Zhao, Z.; Wang, R.X.; Wu, R.; Yuan, Z.; Cui, R.; Jiao, K.; et al. MicroRNA-155, induced by FOXP3 through transcriptional repression of BRCA1, is associated with tumor initiation in human breast cancer. Oncotarget 2017, 8, 41451–41464. [Google Scholar] [CrossRef]
- Zhang, G.; Zhang, W.; Li, B.; Stringer-Reasor, E.; Chu, C.; Sun, L.; Bae, S.; Chen, D.; Wei, S.; Jiao, K.; et al. MicroRNA-200c and microRNA- 141 are regulated by a FOXP3-KAT2B axis and associated with tumor metastasis in breast cancer. Breast Cancer Res. 2017, 19, 73. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Yi, B.; Wei, S.; Rao, D.; He, Y.; Naik, G.; Bae, S.; Liu, X.M.; Yang, W.H.; Sonpavde, G.; et al. Loss of FOXP3 and TSC1 Accelerates Prostate Cancer Progression through Synergistic Transcriptional and Posttranslational Regulation of c-MYC. Cancer Res. 2019, 79, 1413–1425. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Liu, Y.; Li, M.Y.; Ng, C.S.H.; Yang, S.L.; Wang, S.; Zou, C.; Dong, Y.; Du, J.; Long, X.; et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/beta-catenin signaling pathway and EMT in non-small cell lung cancer. Mol. Cancer. 2017, 16, 124. [Google Scholar] [CrossRef]
- Qi, H.; Li, W.; Zhang, J.; Chen, J.; Peng, J.; Liu, Y.; Yang, S.; Du, J.; Long, X.; Ng, C.S.; et al. Glioma-associated oncogene homolog 1 stimulates FOXP3 to promote non-small cell lung cancer stemness. Am. J. Transl. Res. 2020, 12, 1839–1850. [Google Scholar] [PubMed]
- Masugi, Y.; Nishihara, R.; Yang, J.; Mima, K.; da Silva, A.; Shi, Y.; Inamura, K.; Cao, Y.; Song, M.; Nowak, J.A.; et al. Tumour CD274 (PD-L1) expression and T cells in colorectal cancer. Gut 2017, 66, 1463–1473. [Google Scholar] [CrossRef] [PubMed]
- Zaravinos, A.; Roufas, C.; Nagara, M.; de Lucas Moreno, B.; Oblovatskaya, M.; Efstathiades, C.; Dimopoulos, C.; Ayiomamitis, G.D. Cytolytic activity correlates with the mutational burden and deregulated expression of immune checkpoints in colorectal cancer. J. Exp. Clin. Cancer Res. 2019, 38, 364. [Google Scholar] [CrossRef] [Green Version]
- Hai, T.; Hartman, M.G. The molecular biology and nomenclature of the activating transcription factor/cAMP responsive element binding family of transcription factors: Activating transcription factor proteins and homeostasis. Gene 2001, 273, 1–11. [Google Scholar] [CrossRef]
- Hai, T.; Wolfgang, C.D.; Marsee, D.K.; Allen, A.E.; Sivaprasad, U. ATF3 and stress responses. Gene Expr. 1999, 7, 321–335. [Google Scholar]
- Yin, X.; Dewille, J.W.; Hai, T. A potential dichotomous role of ATF3, an adaptive-response gene, in cancer development. Oncogene 2008, 27, 2118–2127. [Google Scholar] [CrossRef] [Green Version]
- Pelzer, A.E.; Bektic, J.; Haag, P.; Berger, A.P.; Pycha, A.; Schafer, G.; Rogatsch, H.; Horninger, W.; Bartsch, G.; Klocker, H. The expression of transcription factor activating transcription factor 3 in the human prostate and its regulation by androgen in prostate cancer. J. Urol. 2006, 175, 1517–1522. [Google Scholar] [CrossRef]
- Lu, D.; Wolfgang, C.D.; Hai, T. Activating transcription factor 3, a stress-inducible gene, suppresses Ras-stimulated tumorigenesis. J. Biol. Chem. 2006, 281, 10473–10481. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Li, X.; Guo, B. KLF6 induces apoptosis in prostate cancer cells through up-regulation of ATF3. J. Biol. Chem. 2008, 283, 29795–29801. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Jin, S.; Amundson, S.A.; Tong, T.; Fan, W.; Zhao, H.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 2002, 21, 7488–7496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Li, Y.; Jadhav, K.; Pan, X.; Zhu, Y.; Hu, S.; Chen, S.; Chen, L.; Tang, Y.; Wang, H.H.; et al. Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism. Nat. Metab. 2021, 3, 59–74. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Brennan, V.C.; Gutierrez, N.M.; Wang, X.; Wang, L.; Yang, W.H. SUMOylation of ATF3 alters its transcriptional activity on regulation of TP53 gene. J. Cell Biochem. 2013, 114, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.M.; Yang, W.H. Loss of SUMOylation on ATF3 inhibits proliferation of prostate cancer cells by modulating CCND1/2 activity. Int. J. Mol. Sci. 2013, 14, 8367–8380. [Google Scholar] [CrossRef]
- Wang, C.M.; Yang, W.H.; Liu, R.; Wang, L.; Yang, W.H. FOXP3 Activates SUMO-Conjugating UBC9 Gene in MCF7 Breast Cancer Cells. Int. J. Mol. Sci. 2018, 19, 2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spitz, F.; Furlong, E.E. Transcription factors: From enhancer binding to developmental control. Nat. Rev. Genet. 2012, 13, 613–626. [Google Scholar] [CrossRef]
- Francois, M.; Donovan, P.; Fontaine, F. Modulating transcription factor activity: Interfering with protein-protein interaction networks. Semin. Cell Dev. Biol. 2020, 99, 12–19. [Google Scholar] [CrossRef]
- Karagianni, P.; Talianidis, I. Transcription factor networks regulating hepatic fatty acid metabolism. Biochim. Biophys. Acta 2015, 1851, 2–8. [Google Scholar] [CrossRef]
- Daly, M.E. Transcription factor defects causing platelet disorders. Blood Rev. 2017, 31, 1–10. [Google Scholar] [CrossRef]
- Bushweller, J.H. Targeting transcription factors in cancer—From undruggable to reality. Nat. Rev. Cancer 2019, 19, 611–624. [Google Scholar] [CrossRef]
- Lee, S.M.; Gao, B.; Fang, D. FoxP3 maintains Treg unresponsiveness by selectively inhibiting the promoter DNA-binding activity of AP-1. Blood 2008, 111, 3599–3606. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.E.; Shin, J.S.; Moon, J.H.; Hong, S.W.; Jung, D.J.; Kim, J.H.; Hwang, I.Y.; Shin, Y.J.; Gong, E.Y.; Lee, D.H.; et al. Foxp3 is a key downstream regulator of p53-mediated cellular senescence. Oncogene 2017, 36, 219–230. [Google Scholar] [CrossRef]
- Yan, C.; Boyd, D.D. ATF3 regulates the stability of p53: A link to cancer. Cell Cycle 2006, 5, 926–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Gao, C.; Kawauchi, J.; Hashimoto, Y.; Tsuchida, N.; Kitajima, S. Transcriptional activation of the human stress-inducible transcriptional repressor ATF3 gene promoter by p53. Biochem. Biophys. Res. Commun. 2002, 297, 1302–1310. [Google Scholar] [CrossRef]
- Dou, L.; Yang, F.; Xu, L.; Zou, Q. A comprehensive review of the imbalance classification of protein post-translational modifications. Brief. Bioinform. 2021, 8, bbab089. [Google Scholar] [CrossRef]
- Keenan, E.K.; Zachman, D.K.; Hirschey, M.D. Discovering the landscape of protein modifications. Mol. Cell 2021, 81, 1868–1878. [Google Scholar] [CrossRef]
- Deng, G.; Song, X.; Fujimoto, S.; Piccirillo, C.A.; Nagai, Y.; Greene, M.I. Foxp3 Post-translational Modifications and Treg Suppressive Activity. Front. Immunol. 2019, 10, 2486. [Google Scholar] [CrossRef] [Green Version]
- Nakahira, K.; Morita, A.; Kim, N.S.; Yanagihara, I. Phosphorylation of FOXP3 by LCK downregulates MMP9 expression and represses cell invasion. PLoS ONE 2013, 8, e77099. [Google Scholar] [CrossRef] [Green Version]
- Mailer, R.K.; Joly, A.L.; Liu, S.; Elias, S.; Tegner, J.; Andersson, J. IL-1beta promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci. Rep. 2015, 5, 14674. [Google Scholar] [CrossRef] [Green Version]
- Joly, A.L.; Seitz, C.; Liu, S.; Kuznetsov, N.V.; Gertow, K.; Westerberg, L.S.; Paulsson-Berne, G.; Hansson, G.K.; Andersson, J. Alternative Splicing of FOXP3 Controls Regulatory T Cell Effector Functions and Is Associated with Human Atherosclerotic Plaque Stability. Circ. Res. 2018, 122, 1385–1394. [Google Scholar] [CrossRef]
- Mailer, R.K. IPEX as a Consequence of Alternatively Spliced FOXP3. Front. Pediatr. 2020, 8, 594375. [Google Scholar] [CrossRef] [PubMed]
- Joly, A.L.; Andersson, J. Alternative splicing, FOXP3 and cardiovascular disease. Aging 2019, 11, 1905–1906. [Google Scholar] [CrossRef] [PubMed]
- Anczuków, O.; Akerman, M.; Cléry, A.; Wu, J.; Shen, C.; Shirole, N.H.; Raimer, A.; Sun, S.; Jensen, M.A.; Hua, Y.; et al. SRSF1-Regulated Alternative Splicing in Breast Cancer. Mol. Cell 2015, 60, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Sheng, J.; Zhao, Q.; Zhao, J.; Zhang, W.; Sun, Y.; Qin, P.; Lv, Y.; Bai, L.; Yang, Q.; Chen, L.; et al. SRSF1 modulates PTPMT1 alternative splicing to regulate lung cancer cell radioresistance. EBioMedicine 2018, 38, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbagallo, D.; Caponnetto, A.; Cirnigliaro, M.; Brex, D.; Barbagallo, C.; D’Angeli, F.; Morrone, A.; Caltabiano, R.; Barbagallo, G.M.; Ragusa, M.; et al. CircSMARCA5 Inhibits Migration of Glioblastoma Multiforme Cells by Regulating a Molecular Axis Involving Splicing Factors SRSF1/SRSF3/PTB. Int. J. Mol. Sci. 2018, 19, 480. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wang, R.; Li, X.; Yu, L.; Hua, D.; Sun, C.; Shi, C.; Luo, W.; Rao, C.; Jiang, Z.; et al. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. J. Clin. Investig. 2019, 129, 676–693. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-M.; Yang, W.H.; Cardoso, L.; Gutierrez, N.; Yang, R.H.; Yang, W.-H. Forkhead Box Protein P3 (FOXP3) Represses ATF3 Transcriptional Activity. Int. J. Mol. Sci. 2021, 22, 11400. https://doi.org/10.3390/ijms222111400
Wang C-M, Yang WH, Cardoso L, Gutierrez N, Yang RH, Yang W-H. Forkhead Box Protein P3 (FOXP3) Represses ATF3 Transcriptional Activity. International Journal of Molecular Sciences. 2021; 22(21):11400. https://doi.org/10.3390/ijms222111400
Chicago/Turabian StyleWang, Chiung-Min, William Harry Yang, Leticia Cardoso, Ninoska Gutierrez, Richard Henry Yang, and Wei-Hsiung Yang. 2021. "Forkhead Box Protein P3 (FOXP3) Represses ATF3 Transcriptional Activity" International Journal of Molecular Sciences 22, no. 21: 11400. https://doi.org/10.3390/ijms222111400