
Atractylodin Suppresses TGF-β-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
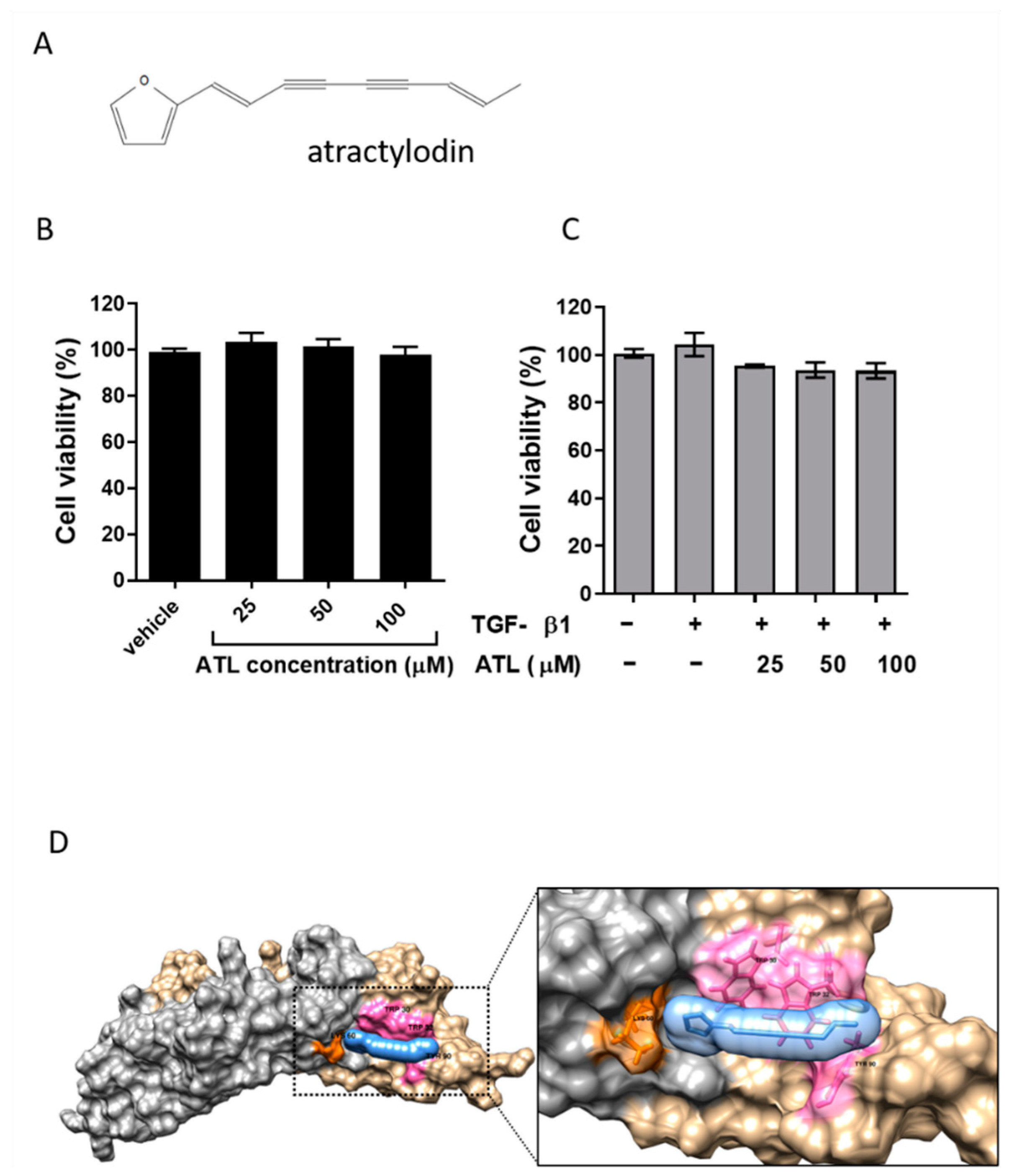
2.1. Effect of Atractylodin on Cell Viability of TGF-β1-Induced A549 Cells
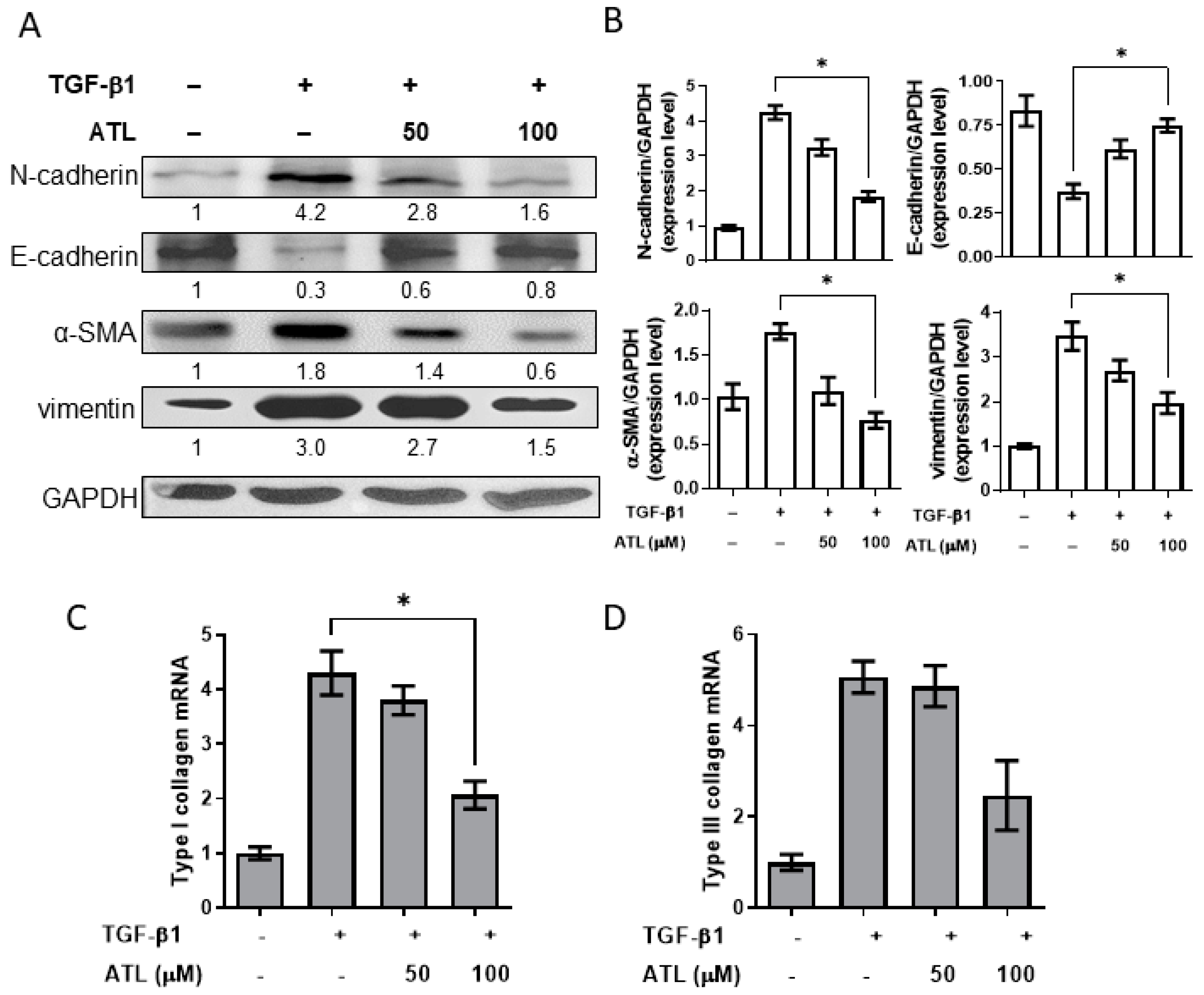
2.2. Atractylodin Alleviates TGF-β1-Induced EMT in A549 Cells
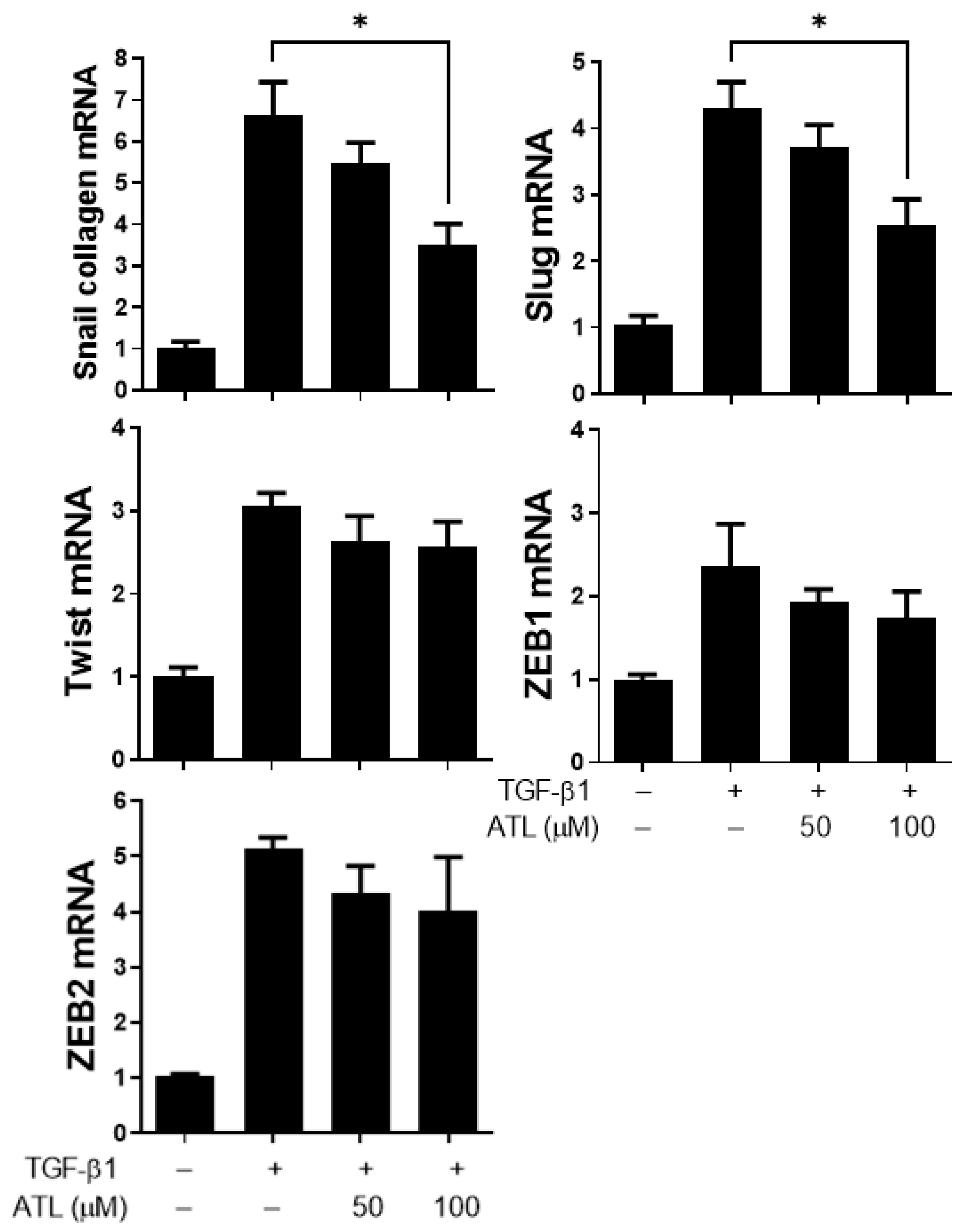
2.3. Atractylodin Inhibits EMT-Related Transcription Factor Expression in A549 Cells
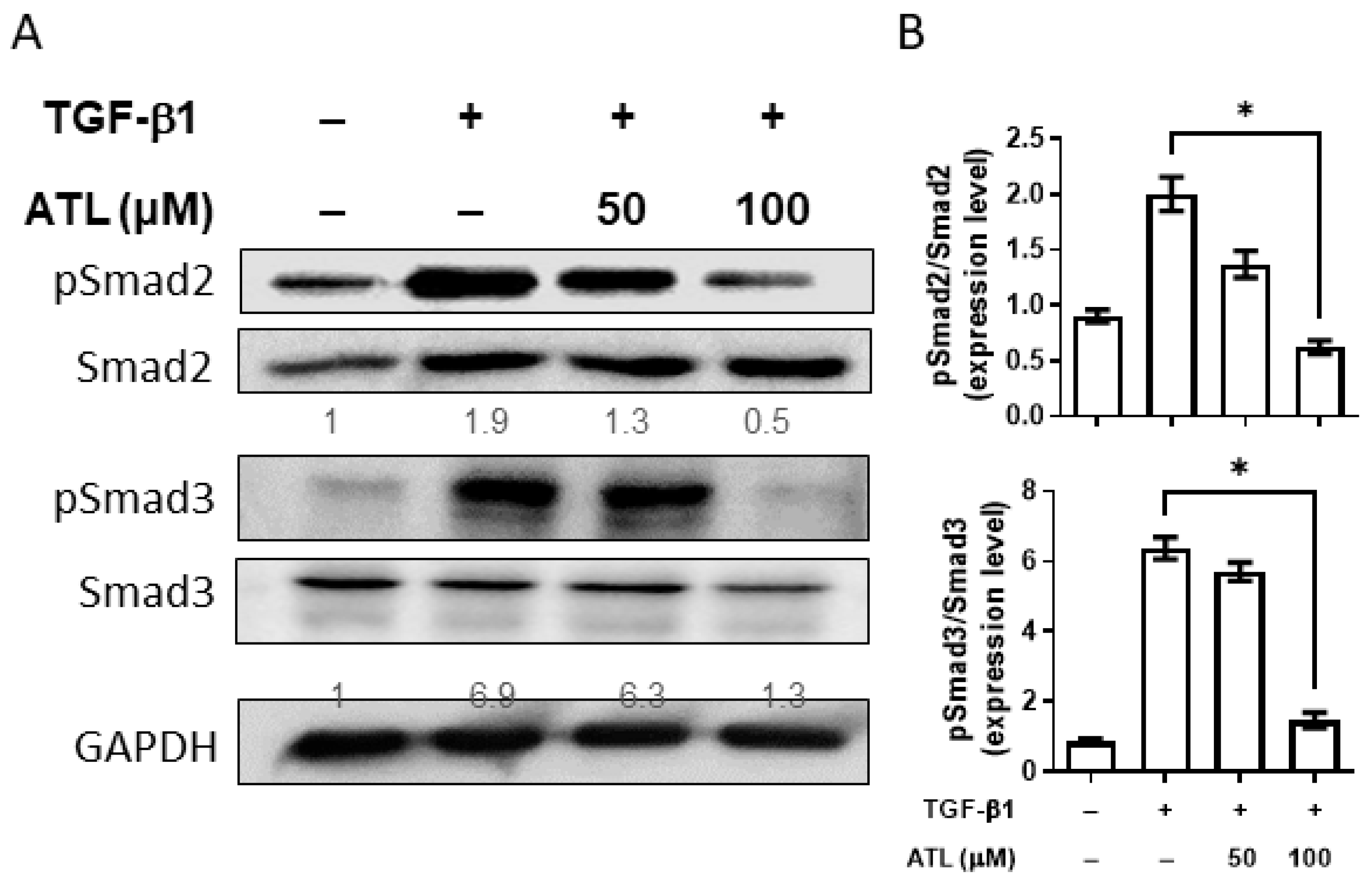
2.4. Atractylodin Reduces Smad-Dependent Pathway Activation in A549 Cells
2.5. Atractylodin Suppresses Smad-Independent Pathway Activation in A549 Cells
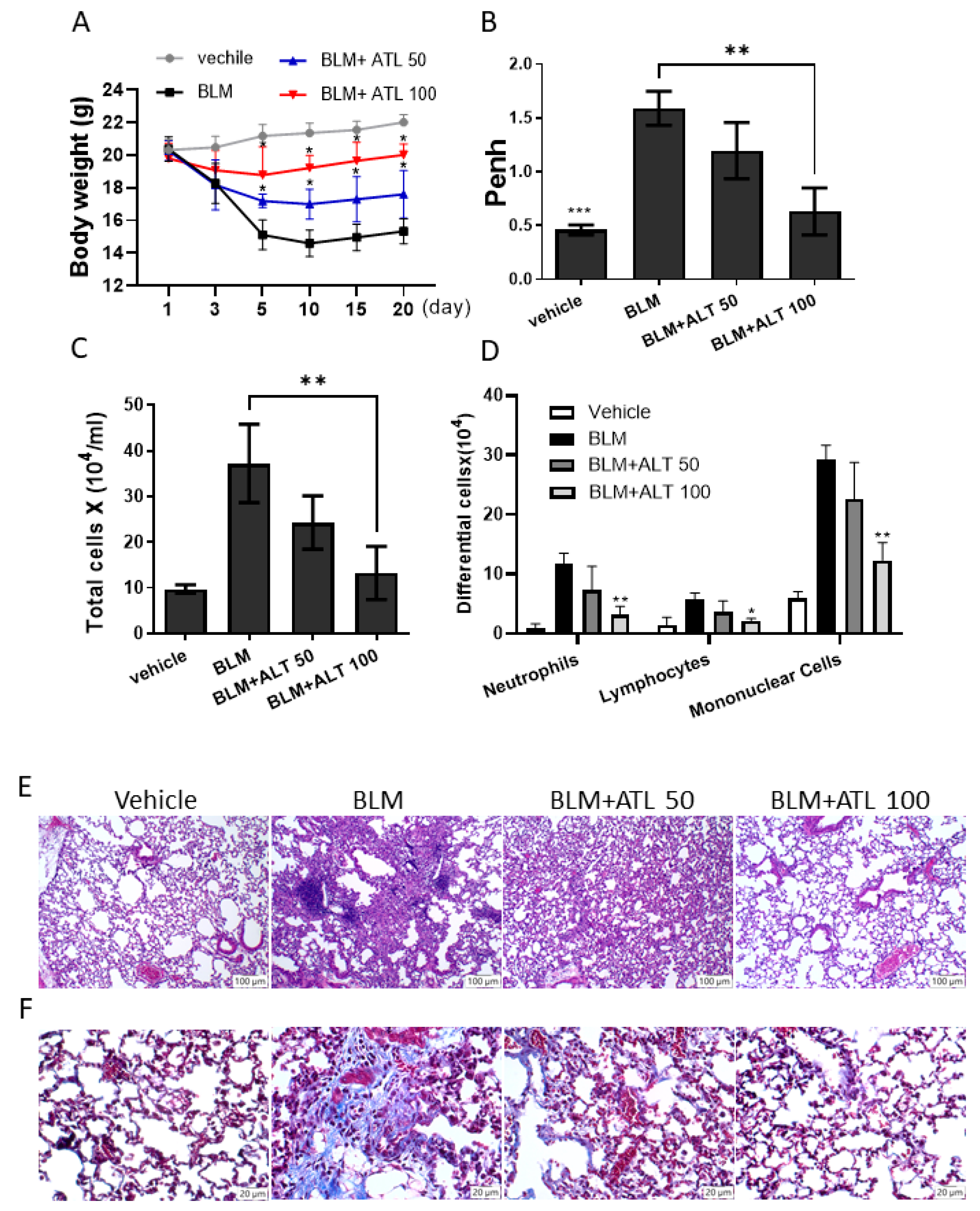
2.6. Atractylodin Decreases BLM-Induced Pulmonary Fibrosis in Mice
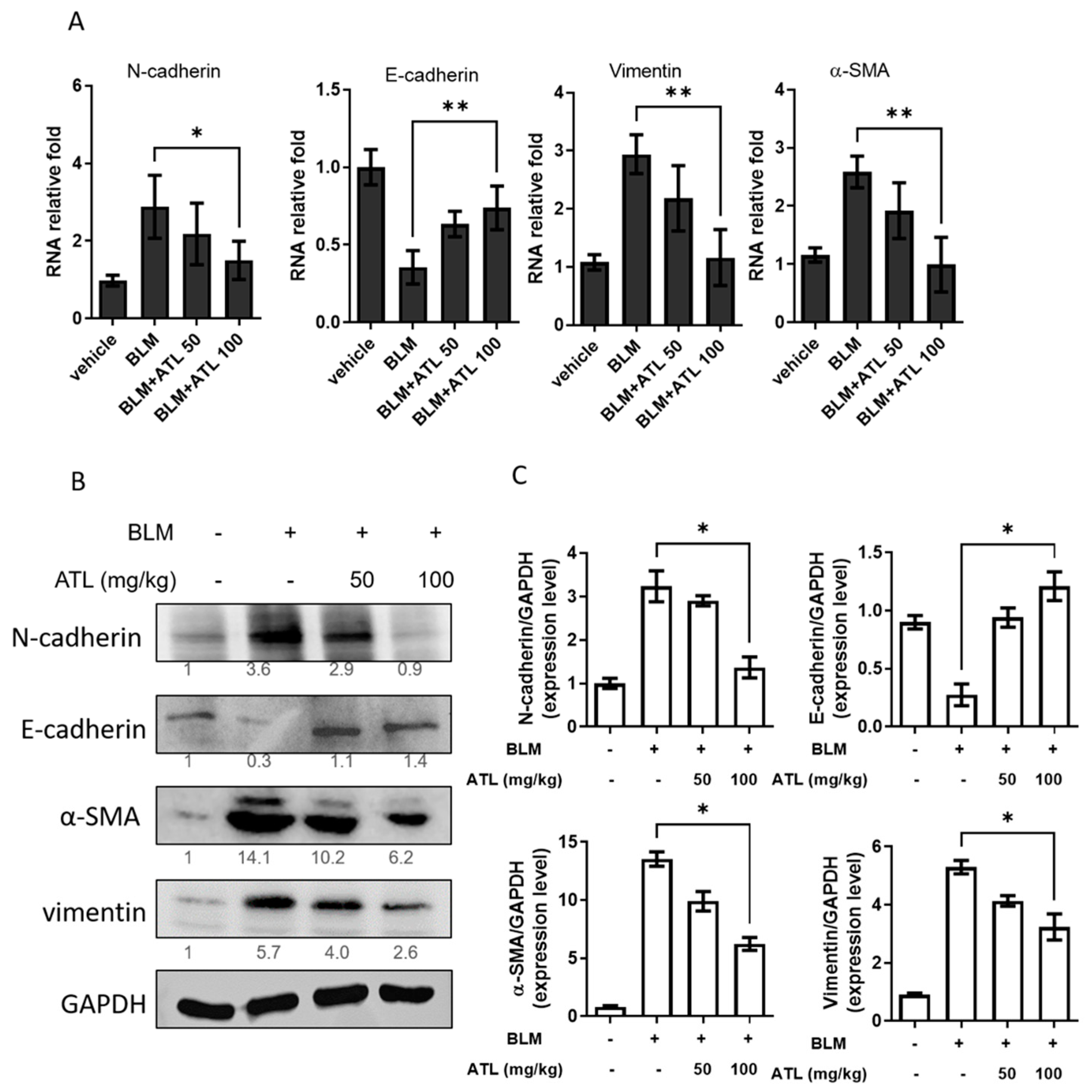
2.7. Atractylodin Down-Regulates BLM-Induced EMT in Mice Lung Tissues
3. Discussion
4. Material and Methods
4.1. Cell Viability Assay
4.2. Western Blot Assay
4.3. Quantitative Real-Time PCR
4.4. Animal Care and Experimental Procedures
4.5. Measurement of Airway Hyperresponsiveness
4.6. Histopathological and Immunohistochemical Examination
4.7. Molecular Docking
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Ley, B.; Collard, H.R.; King, T.E., Jr. Clinical course and prediction of survival in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 431–440. [Google Scholar] [CrossRef]
- Cheresh, P.; Kim, S.-J.; Tulasiram, S.; Kamp, D.W. Oxidative stress and pulmonary fibrosis. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2013, 1832, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Schultz, G.S.; Wysocki, A. Interactions between extracellular matrix and growth factors in wound healing. Wound Repair Regen. 2009, 17, 153–162. [Google Scholar] [CrossRef]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [Green Version]
- Karimi-Shah, B.A.; Chowdhury, B.A. Forced Vital Capacity in Idiopathic Pulmonary Fibrosis—FDA Review of Pirfenidone and Nintedanib. N. Engl. J. Med. 2015, 372, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Milara, J.; Navarro, R.; Juan, G.; Peiró, T.; Serrano, A.; Ramón, M.; Morcillo, E.; Cortijo, J. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012, 67, 147–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, S.-L.; Yang, Y.; Liu, X.-L.; Xu, Q.-B. Emodin attenuates bleomycin-induced pulmonary fibrosis via anti-inflammatory and anti-oxidative activities in rats. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2018, 24, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frangogiannis, N.G. Transforming growth factor–β in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Xaubet, A.; Marin-Arguedas, A.; Lario, S.; Ancochea, J.; Morell, F.; Ruiz-Manzano, J.; Rodriguez-Becerra, E.; Rodriguez-Arias, J.M.; Inigo, P.; Sanz, S. Transforming growth factor-β1 gene polymorphisms are associated with disease progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2003, 168, 431–435. [Google Scholar] [CrossRef]
- Fernandez, I.E.; Eickelberg, O. The impact of TGF-β on lung fibrosis: From targeting to biomarkers. Proc. Am. Thorac. Soc. 2012, 9, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, K. TGF-β signaling by Smad proteins. Cytokine Growth Factor Rev. 2000, 11, 15–22. [Google Scholar] [CrossRef]
- Ma, J.; Sanchez-Duffhues, G.; Goumans, M.-J.; ten Dijke, P. TGF-β-Induced Endothelial to Mesenchymal Transition in Disease and Tissue Engineering. Front. Cell Dev. Biol. 2020, 8, 260. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ma, L.; Huang, K.; Wei, Y.; Long, S.; Liu, Q.; Zhang, D.; Wu, S.; Wang, W.; Yang, G. Regorafenib-Attenuated, Bleomycin-Induced Pulmonary Fibrosis by Inhibiting the TGF-β1 Signaling Pathway. Int. J. Mol. Sci. 2021, 22, 1985. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Ribatti, D.; Lisi, S. SMADS-Mediate Molecular Mechanisms in Sjögren’s Syndrome. Int. J. Mol. Sci. 2021, 22, 3203. [Google Scholar] [CrossRef]
- Varga, J.; Abraham, D. Systemic sclerosis: A prototypic multisystem fibrotic disorder. J. Clin. Investig. 2007, 117, 557–567. [Google Scholar] [CrossRef]
- Cao, W.; Hu, C.; Wu, L.; Xu, L.; Jiang, W. Rosmarinic acid inhibits inflammation and angiogenesis of hepatocellular carcinoma by suppression of NF-κB signaling in H22 tumor-bearing mice. J. Pharmacol. Sci. 2016, 132, 131–137. [Google Scholar] [CrossRef] [Green Version]
- Chae, H.-S.; Kim, Y.-M.; Chin, Y.-W. Atractylodin inhibits interleukin-6 by blocking NPM-ALK activation and MAPKs in HMC-1. Molecules 2016, 21, 1169. [Google Scholar] [CrossRef]
- Yu, C.; Xiong, Y.; Chen, D.; Li, Y.; Xu, B.; Lin, Y.; Tang, Z.; Jiang, C.; Wang, L. Ameliorative effects of atractylodin on intestinal inflammation and co-occurring dysmotility in both constipation and diarrhea prominent rats. Korean J. Physiol. Pharmacol. Off. J. Korean Physiol. Soc. Korean Soc. Pharmacol. 2017, 21, 1026147. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Fan, K.; Wang, K.; Bian, C. Atractylodin attenuates lipopolysaccharide-induced acute lung injury by inhibiting NLRP3 inflammasome and TLR4 pathways. J. Pharmacol. Sci. 2018, 136, 203–211. [Google Scholar] [CrossRef]
- Tirino, V.; Camerlingo, R.; Bifulco, K.; Irollo, E.; Montella, R.; Paino, F.; Sessa, G.; Carriero, M.-V.; Normanno, N.; Rocco, G.; et al. TGF-beta1 exposure induces epithelial to mesenchymal transition both in CSCs and non-CSCs of the A549 cell line, leading to an increase of migration ability in the CD133+ A549 cell fraction. Cell Death Dis. 2013, 4, e620. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Francí, C.; Domínguez, D.; Monfar, M.; Baulida, J.; De Herreros, A.G. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- Bolós, V.; Peinado, H.; Pérez-Moreno, M.A.; Fraga, M.F.; Esteller, M.; Cano, A. The transcription factor Slug represses E-cadherin expression and induces epithelial to mesenchymal transitions: A comparison with Snail and E47 repressors. J. Cell Sci. 2003, 116, 499–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Park, S.Y.; Joo, C.-K. Transforming growth factor-β1 represses E-cadherin production via slug expression in lens epithelial cells. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2708–2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef] [Green Version]
- Krebs, A.M.; Mitschke, J.; Lasierra Losada, M.; Schmalhofer, O.; Boerries, M.; Busch, H.; Boettcher, M.; Mougiakakos, D.; Reichardt, W.; Bronsert, P.; et al. The EMT-activator Zeb1 is a key factor for cell plasticity and promotes metastasis in pancreatic cancer. Nat. Cell Biol. 2017, 19, 518–529. [Google Scholar] [CrossRef] [Green Version]
- ComijnJ, B.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates E-cadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Noble, P. Epithelial fibroblast triggering and interactions in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 123–129. [Google Scholar] [CrossRef]
- Roach, K.M.; Sutcliffe, A.; Matthews, L.; Elliott, G.; Newby, C.; Amrani, Y.; Bradding, P. A model of human lung fibrogenesis for the assessment of anti-fibrotic strategies in idiopathic pulmonary fibrosis. Sci. Rep. 2018, 8, 342. [Google Scholar] [CrossRef] [Green Version]
- Tanjore, H.; Xu, X.C.; Polosukhin, V.V.; Degryse, A.L.; Li, B.; Han, W.; Sherrill, T.P.; Plieth, D.; Neilson, E.G.; Blackwell, T.S. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. Am. J. Respir. Crit. Care Med. 2009, 180, 657–665. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Shen, Y.; Hong, J.; Xia, Q.; Zhou, F.; Liu, X. The contribution of TGF-β in Epithelial-Mesenchymal Transition (EMT): Down-regulation of E-cadherin via snail. Neoplasma 2015, 62, 1–15. [Google Scholar] [CrossRef]
- Bhatt, T.; Rizvi, A.; Batta, S.P.R.; Kataria, S.; Jamora, C. Signaling and mechanical roles of E-cadherin. Cell Commun. Adhes. 2013, 20, 189–199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, E.; Seo, J.; Yoon, H.; Cho, S. The Post-Translational Regulation of Epithelial–Mesenchymal Transition-Inducing Transcription Factors in Cancer Metastasis. Int. J. Mol. Sci. 2021, 22, 3591. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Chen, Y.-G. Regulation of TGF-β receptor activity. Cell Biosci. 2012, 2, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, R.; Dejeans, N.; Glorieux, C.; Creton, M.; Delaive, E.; Dieu, M.; Raes, M.; Levêque, P.; Gallez, B.; Depuydt, M.; et al. Hsp90 Is Cleaved by Reactive Oxygen Species at a Highly Conserved N-Terminal Amino Acid Motif. PLoS ONE 2012, 7, e40795. [Google Scholar]
- Bellaye, P.-S.; Wheildon, N.; Shimbori, C.; Upagupta, C.; Kolb, M. Pathogenesis of Pulmonary Fibrosis—The Role of Extracellular Heat Shock Protein-90 (HSP90) in Myofibroblast Differentiation and Persistence. QJM Int. J. Med. 2016, 109 (Suppl. 1), S1. [Google Scholar]
- Sontake, V.; Wang, Y.; Kasam, R.K.; Sinner, D.; Reddy, G.B.; Naren, A.P.; McCormack, F.X.; White, E.S.; Jegga, A.G.; Madala, S.K. Hsp90 regulation of fibroblast activation in pulmonary fibrosis. JCI Insight 2017, 2, e91454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sibinska, Z.; Tian, X.; Korfei, M.; Kojonazarov, B.; Kolb, J.S.; Klepetko, W.; Kosanovic, D.; Wygrecka, M.; Ghofrani, H.A.; Weissmann, N.; et al. Amplified canonical transforming growth factor-β signaling via heat shock protein 90 in pulmonary fibrosis. Eur. Respir. J. 2017, 49, 1501941. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, X.; Huang, W.; Ge, X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed. Pharmacother. 2021, 135, 111067. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Lin, X.; Feng, X.F. Critical regulation of TGFbeta signaling by Hsp90. Proc. Natl. Acad. Sci. USA 2008, 105, 9244–9249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.C.; Yang, C.C.; Lin, C.H.; Hsia, T.C.; Chao, W.C.; Lin, C.C. Atractylodin ameliorates ovalbumin-induced asthma in a mouse model and exerts immunomodulatory effects on Th2 immunity and dendritic cell function. Mol. Med. Rep. 2020, 22, 4909–4918. [Google Scholar] [CrossRef] [PubMed]
- Lyu, Z.; Ji, X.; Chen, G.; An, B. Atractylodin ameliorates lipopolysaccharide and d-galactosamine-induced acute liver failure via the suppression of inflammation and oxidative stress. Int. Immunopharmacol. 2019, 72, 348–357. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chang, K.-W.; Zhang, X.; Lin, S.-C.; Lin, Y.-C.; Li, C.-H.; Akhrymuk, I.; Lin, S.-H.; Lin, C.-C. Atractylodin Suppresses TGF-β-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. Int. J. Mol. Sci. 2021, 22, 11152. https://doi.org/10.3390/ijms222011152
Chang K-W, Zhang X, Lin S-C, Lin Y-C, Li C-H, Akhrymuk I, Lin S-H, Lin C-C. Atractylodin Suppresses TGF-β-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice. International Journal of Molecular Sciences. 2021; 22(20):11152. https://doi.org/10.3390/ijms222011152
Chicago/Turabian StyleChang, Kai-Wei, Xiang Zhang, Shih-Chao Lin, Yu-Chao Lin, Chia-Hsiang Li, Ivan Akhrymuk, Sheng-Hao Lin, and Chi-Chien Lin. 2021. "Atractylodin Suppresses TGF-β-Mediated Epithelial-Mesenchymal Transition in Alveolar Epithelial Cells and Attenuates Bleomycin-Induced Pulmonary Fibrosis in Mice" International Journal of Molecular Sciences 22, no. 20: 11152. https://doi.org/10.3390/ijms222011152