1. Introduction
Bisphenols have been in commercial use as plasticizers for over 70 years. They are reported to be estrogenic mimics that may interfere with hormonal homeostasis. One very prevalent bisphenol, bisphenol A (BPA), is used for manufacturing polysulfones and polycarbonate plastics, epoxy resins, and thermal paper. BPA is considered an endocrine and metabolic disruptor, able to interfere with important physiological systems, such as insulin-glucagon signaling [
1,
2,
3,
4]. Comparatively, BPA has one of the highest production volumes of any chemical worldwide, with global production estimated at 7.7 million metric tons in 2015, and it is expected to reach 10.6 million metric tons by 2022 [
5]. Mammals are exposed to BPA daily through several routes, such as the consumption of food and drink, drugs, air born inhalation, and contact materials, such as various plastics, medical devices, and store receipts [
6,
7,
8]. However, the main exposure route of BPA is through diet as many food packages contain BPA, allowing it to leach into the food and be ingested [
6,
9,
10,
11,
12]. Due to its pervasiveness in the environment, BPA has been detected in the urine and sera in 90% of the people sampled, as well as in the amniotic fluid, placenta, and breast milk of women [
7,
13,
14,
15,
16,
17,
18]. It has become increasingly clear that BPA can bioaccumulate in the food chain. In fact, in a study in Africa, BPA reached very high concentrations in food (940 ng/g), biological fluids (209 ng/mL), consumer and PCPs (3.6 μg/g), and semisolids (154 μg/g) [
19].
Considering the prevalence of BPA in the biome and its suspected disruption of human physiology, many groups have used various model organisms, including mice or human cell lines, in an attempt to determine how BPA interacts with different biological signaling systems [
20]. Some of these groups have performed a microarray analysis after BPA exposure and deposited this information in public data banks [
21,
22]. However, most of the published datasets are relatively small, and meta-analysis studies that attempt to integrate existing microarray datasets regarding exposure to BPA are currently lacking. There is an opportunity to combine the existing datasets to improve the accuracy of the identified genes and pathways involved in BPA exposure.
It is somewhat surprising that most of the literature on data mining and chemometric data calculations refers to the exposure-instrumental/biological testing loop while less attention has been paid to exposure-gene expression correlations treatment with advanced environmetrics. The impact of BPA on cardiometabolic factors [
23] has shown a positive correlation between patients’ BPA concentrations and diabetes (87%), overweight (28%), obesity (85%), elevated waist circumference (100%), cardiovascular diseases (80%), and hypertension (66%) in cross-sectional studies. Unfortunately, none of these studies can confirm if BPA can be proven as a risk factor of the observed anomalies or if these elevated BPA concentrations result from the already pre-disordered status of the given patient. The problem is of increasing importance as BPA (at environmentally relevant concentrations) has been confirmed to affect pre-implanted embryos and has been detected in samples of serum and follicular fluid collected from women and the umbilical cord at ca. 1–2 ng/mL levels [
11]. For this reason, it is warranted to pay more attention to studies on the exposure-gene expression loop to unveil these interrelationships.
The amount of functional genomics data in the form of expression profiles from various experimental designs and model organisms are increasing rapidly, with over 1000 new submissions yearly to the ArrayExpress repository (
https://www.ebi.ac.uk/arrayexpress/, accessed on 10 February 2020) [
24]. Currently, the largest repositories of public functional genomics data are ArrayExpresses and NCBI Geo (
https://www.ncbi.nlm.nih.gov/geo/, accessed on 10 February 2020) [
25], which, in January 2020, contained 72,578 and 97,273 unique experiments, respectively; although 59,374 were found in both databases and, hence, redundant [
26]. Currently, the majority of these are in the form of microarray data, although, since 2018, the number of RNASeq experiments submitted to ArrayExpress is higher than the number of microarray submissions [
24]. Utilizing these databanks for novel large-scale analysis poses challenges due to the diversity of the technical platforms used to generate the data, resulting in differences in file formats, signal levels, and data variance, as well as differences in experimental design. Although several attempts have been made to simplify data retrieval and data selection, such as the All Of gene Expression (AOE) web portal [
26] and Biostudies database, which is now becoming the successor of ArrayExpress [
24], the challenges of between-experiments normalization and adjusting for the differences in experimental design remain. These difficulties in combining and analysing functional genomics data from various sources necessitate innovative and more powerful methods to utilize these data for novel analyses.
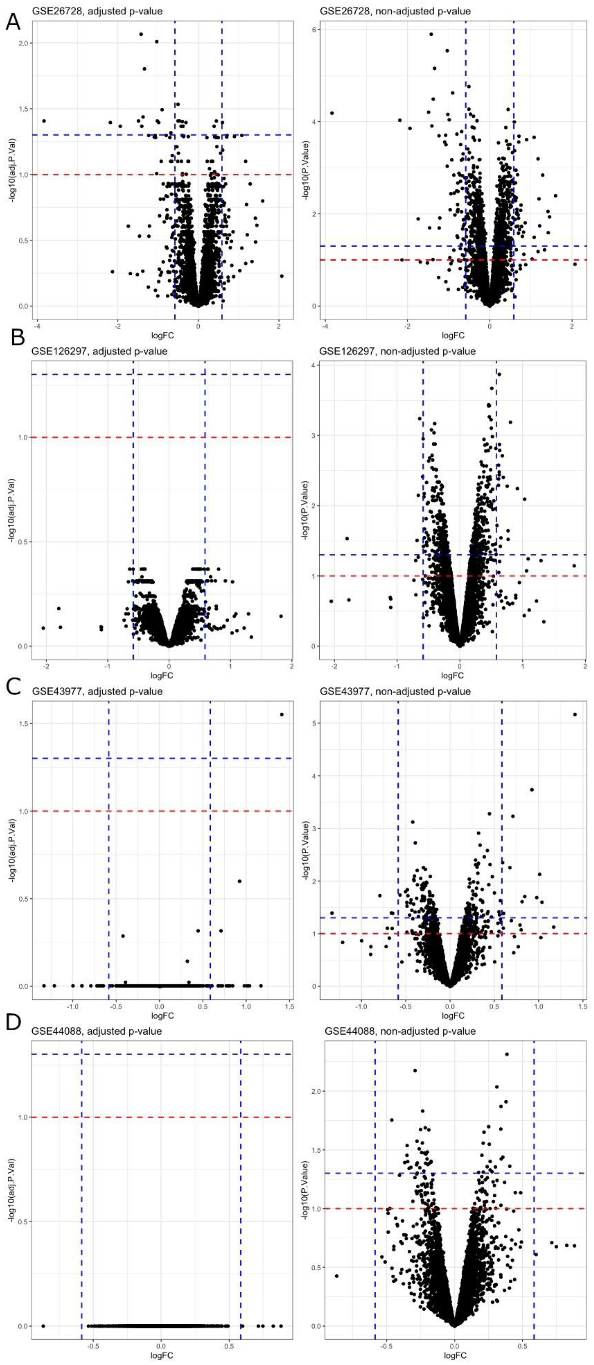
In this manuscript, we studied the gene expression changes from four available microarray datasets of mice under the influence of BPA exposure. The standard approach in analysing gene expression changes is to perform differential expression analysis with statistical tests for differences in intensity [
27]. We performed this traditional differential gene expression analysis of individual GEO datasets. However, this method suffers from various issues, such as uncertainty in
p-value choice to select the right set of “important” genes in terms of biological effects and the necessity of dealing with the problem of multiple comparisons. In contrast, machine learning methods, especially feature selection methods, are widely used today in gene expression analysis, providing the ability to select the right set of “important” genes in terms of the quality of the prediction model [
27,
28]. In this study, we focused on applying machine learning methods in terms of feature selection (FS), revealing key genes influenced by BPA exposure.
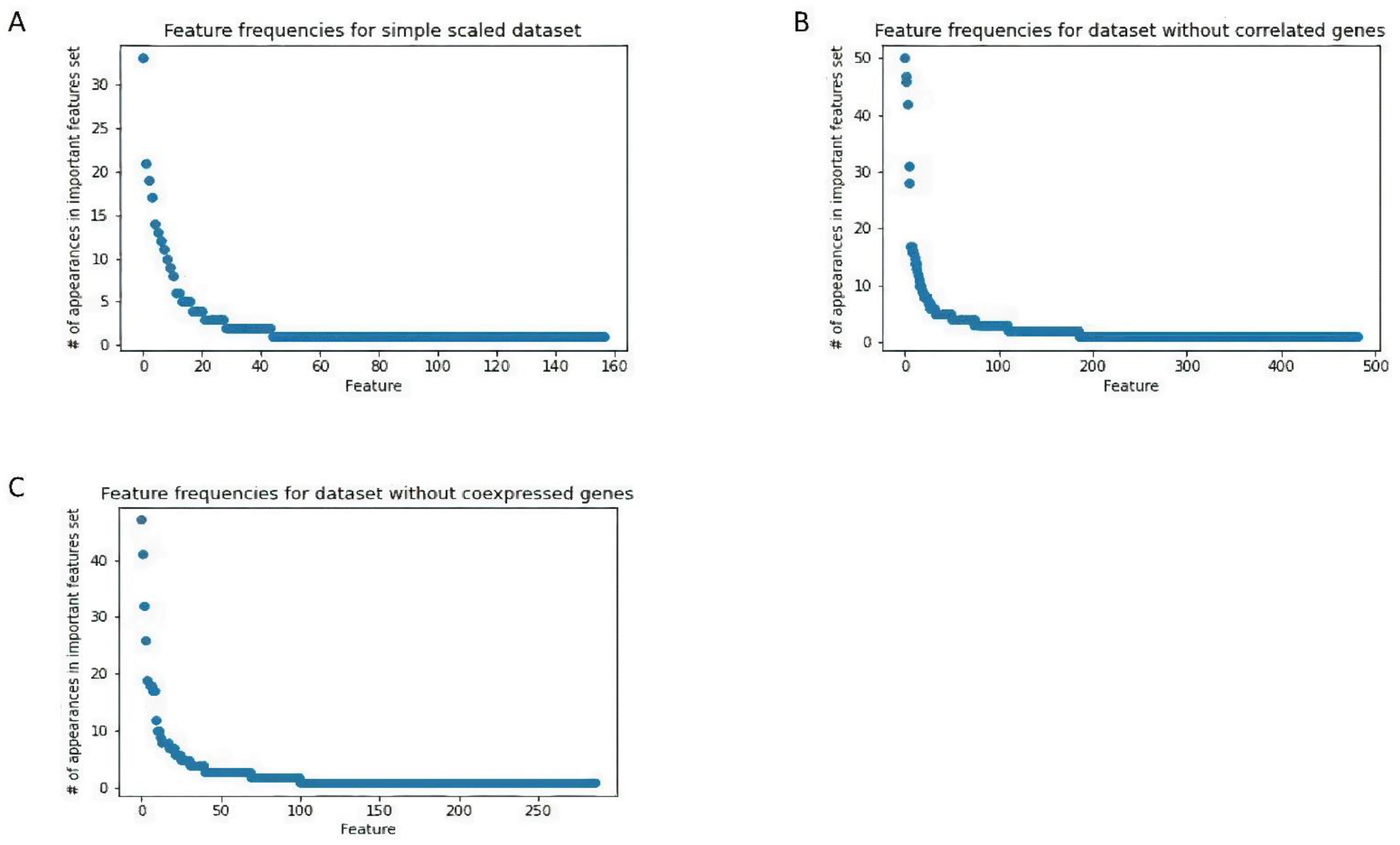
We constructed three joint datasets following three different correlation-based pre-processing approaches, namely using all of the common genes through four GEO datasets, uncorrelated, and no co-expressed genes, respectively. By applying machine learning methods to these joint datasets, we identified genes whose expression was significantly changed in all of the BPA microanalysis data tested. We went on to determine that a subset of these genes is involved in the regulation of cell survival and apoptosis. Our results highlight the benefit of combining existing datasets for integrated analysis for a specific topic when individual datasets are limited in size, in our case when studying the effects of BPA.
3. Discussion
Bisphenols are important pollutants that significantly infiltrated the biome. Their potential to disrupt physiology has led several groups to perform microarray analysis using biological material from mice after BPA intervention. The large datasets created by these works have been deposited in public data banks [
21,
22], but no other analysis has been performed. Using machine learning, we mined a subset of these microarray datasets and were able to define not only a method for performing a meta-analysis of these large datasets but also produce pathways conserved across different BPA interventions within a species.
Our research confirms the importance of combining datasets in a meta-analysis but also highlights the importance of different pre-processing steps before applying machine learning methods, especially for small datasets. In this study, we focused on various correlation-based gene averaging processes. We showed that the usage of these strategies leads to different, but, in general, related, solutions. This suggests that co-expression-based pre-processing produces a dataset modification that promotes a solution candidate. Using hard voting, the final result was aggregated from the results of running three models on dataset modifications. The strategies to improve this process might include a deeper investigation of the differences in outcomes between the models, as well as more sensitive aggregation.
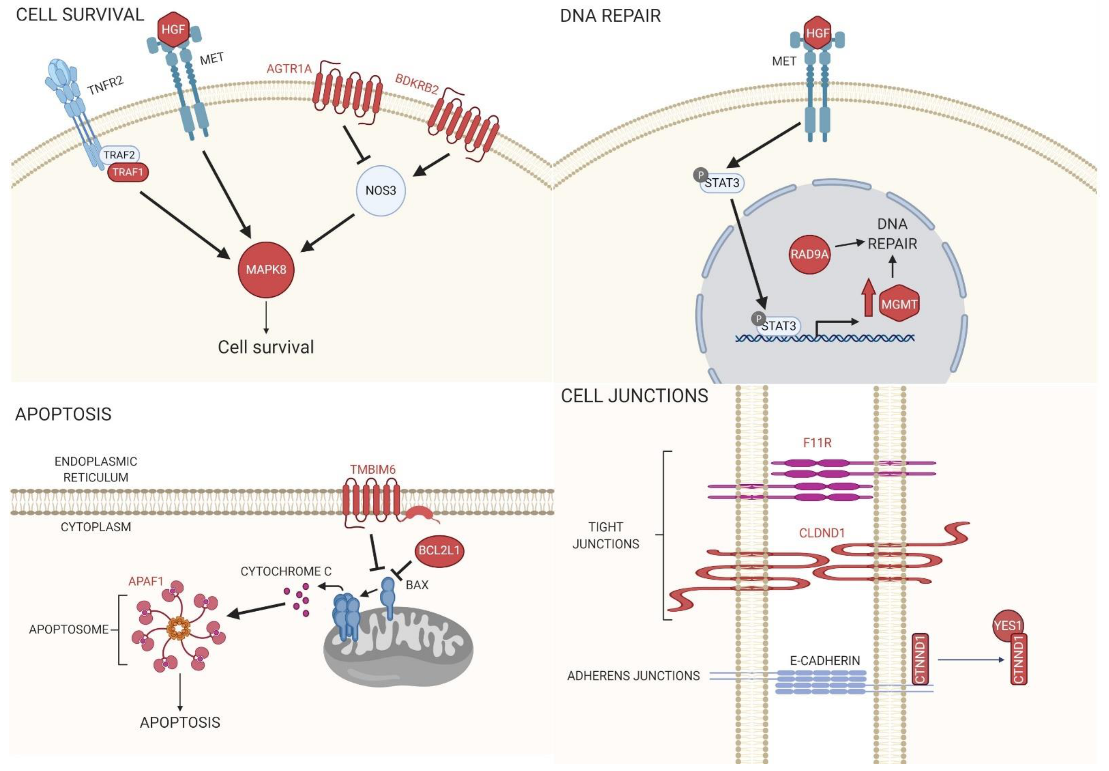
BPA exposure has been shown to disrupt mitochondria integrity, leading to elevated ROS levels and apoptosis rates in human granulosa and HT-22 cells, which are derived from the mouse brain [
30,
31]. The BPA-inhibited proliferation of neural progenitor cells and rat embryonic midbrain cells through the suppression of the JNK signaling pathway has also been reported [
32,
33]. Our gene ontology analysis using DAVID indicated that a significant group of the top 30 transcripts recovered from machine learning models were linked to the regulation of apoptosis (see
Table 8). Furthermore, by using DAVID and STRING, it became evident that many of the genes encoding these transcripts categorized as being involved in the regulation of apoptosis were, in fact, cell survival pathways that converged on Mitogen-Activated Protein Kinase 8 (Mapk8, also known as Jnk1) (
Figure 3). The protein of one of the transcripts recovered, Hepatocyte growth factor (Hgf), activates MET Proto-Oncogene, Receptor Tyrosine Kinase (Met), which not only regulates Mapk8 activation but also leads to increased transcriptional expression of another gene in our dataset,
O-6-Methylguanine-DNA Methyltransferase (
Mgmt) [
34]. Mgmt and Rad9a proteins, whose transcript levels were also shown to be affected by BPA, are both involved in repairing damaged DNA [
35]. Furthermore, in mouse macrophages, it was determined that BPA-induced mitochondrial disruption reduced BCL2 protein expression, which led to caspase-dependent apoptosis [
36]. In our study, one of the top 30 genes was
Bcl2l1, whose protein is known to inhibit Bax-induced apoptosis (
Figure 3). Furthermore, the protein of another gene recovered in the study,
Apaf1, acts downstream of Bax, and, along with Caspase-9, forms an apoptosome to induce apoptosis (
Figure 3) [
37]. Interestingly, we also showed that
Tmbim6 transcript levels are affected by BPA, and
Tmbim6 was shown to inhibit Bax-induced apoptosis (
Figure 3) [
38].
Three of the top 30 genes,
F11 Receptor (
F11r, also known as
Jam-1),
Claudin Domain Containing 1 (
Cldnd1), and
Catenin Delta 1 (
Ctnnd1), are associated with either tight or adherens junctions (see
Table 8 and
Figure 3). In a recent study, the reproductive toxicity of BPA was investigated. Male CD-1 mice were orally administrated BPA, and the results showed that this exposure was sufficient to induce disorders in spermatogenesis, including damaging the tight junctions between Sertoli cells [
39]. Another study examined the effect of BPA in female rats on the expression levels of tight junction (TJ) transcripts in the uterus during early pregnancy. This study found profound alterations in the TJ gene transcript levels of uterine epithelial cells when the rats were exposed to BPA, which led to changes in fluid and ion transport across the epithelium, blocking the receptivity of the uterus to blastocyst implantation [
40]. In fact, this study saw profound effects on claudin transcript levels, such as
Cldnd1, including low expression levels or even the loss of expression.
Interestingly, our study recovered transcripts for two receptors that are in pathways regulated by Angiotensin I Converting Enzyme (ACE),
Angiotensin II Receptor Type 1a (
Agtr1a), and
Bradykinin Receptor B2 (
Bdkrb2) (see
Table 8 and
Figure 3). In both rat cardiac cells and human endothelial cell lines, it was shown that BPA was proangiogenic, including the upregulation of Nitric Oxide Synthase 3 [
41,
42,
43]. In another report, it was discovered in rat striatum that the inhibition of ACE was able to alleviate the ROS-inducing effects of a BPA + 1-methyl-4-phenylpyridinium ion (MPP(+)) mixture [
44]. Interestingly, both
Agtr1a and
Bdkrb2 signal upstream of
Nos3, where
Agtr1a leads to
Nos3 inhibition and
Bdkrb2 leads to activation (
Figure 3).
In terms of computational methods, in this paper, we suggest using a new cross-validation-based greedy feature selection algorithm with three different preprocessing strategies. Using this approach, one has the flexibility to incorporate different machine learning models and stopping criteria into the feature selection procedure depending on the properties of the data. We also provided gene importance analysis based on the frequencies of the genes’ appearances in the feature lists from 100 runs of the proposed algorithm. For small datasets, this process is more stable than using feature selection techniques based on a single run.
Our results highlight the value of integrating data from multiple datasets for co-analysis, revealing new biological knowledge. However, a key limitation of our study is still a lack of publicly available microarray data after BPA exposure, which restricts our investigation to the baseline machine learning methods. This is also an important constraint for analyzing the differences between the results from datasets without correlated and without co-expressed genes. We used co-expression analysis with the WGCNA package for each GEO dataset, but it should be carefully used for datasets with less than 15 samples [
45]. This means that a pre-processing method should be attentively chosen based on the available data.
In summary, we developed a new approach for the meta-analyses of microarray data, which could be very useful for analyzing other datasets relating to any environmental pollutants. The pathways that we have identified align well with the previous evidence for the molecular actions of BPA and prompt further studies into pathways that relate to the regulation of cell survival, DNA repair, apoptosis, and cellular junctions.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}