
Abscopal Effect and Drug-Induced Xenogenization: A Strategic Alliance in Cancer Treatment?

 , ,
, ,  and
and
Abstract
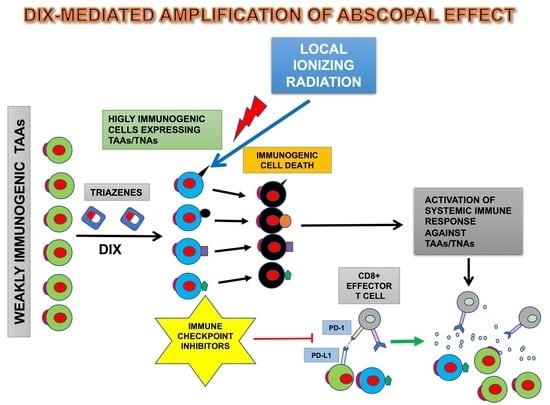
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Abscopal Effect
2.1. Preclinical and Clinical Investigations
2.2. Mechanism of RT-Induced Abscopal Effect and Immunogenic Consequences of Radiation
2.3. RT-Induced Immunosuppression
3. Drug-Induced Upregulation of Tumor Immunogenicity
3.1. Drug-Induced TAA Upregulation
3.1.1. Interferons
3.1.2. Epigenetic Drugs
3.1.3. Antitumor Agents
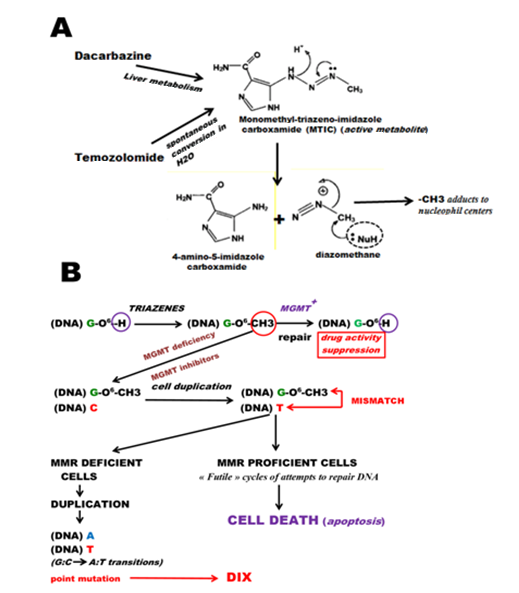
3.2. Drug-Induced Xenogenization (DIX)
3.2.1. Mechanism of Action of Triazenes
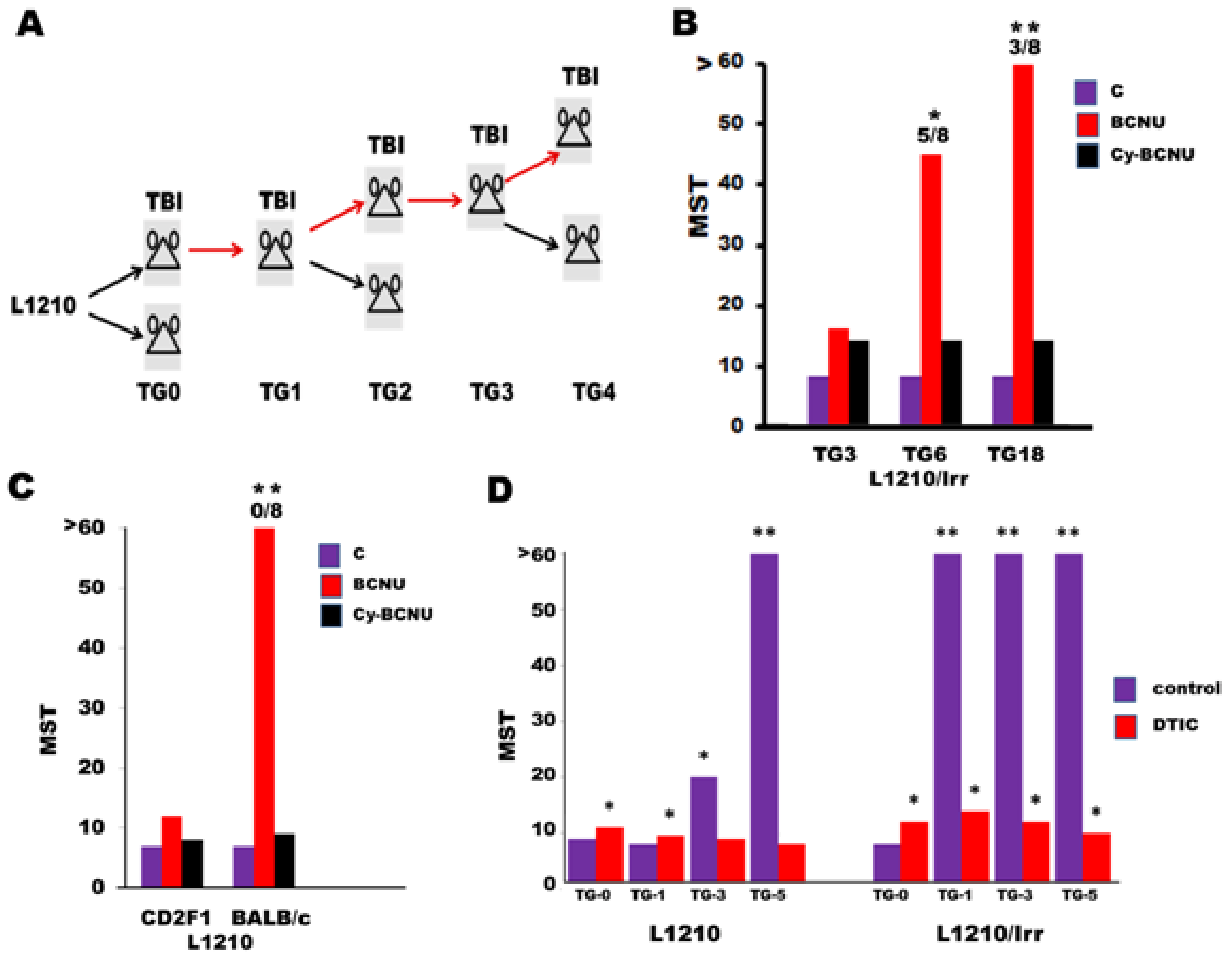
3.2.2. DIX and Ionizing Radiation
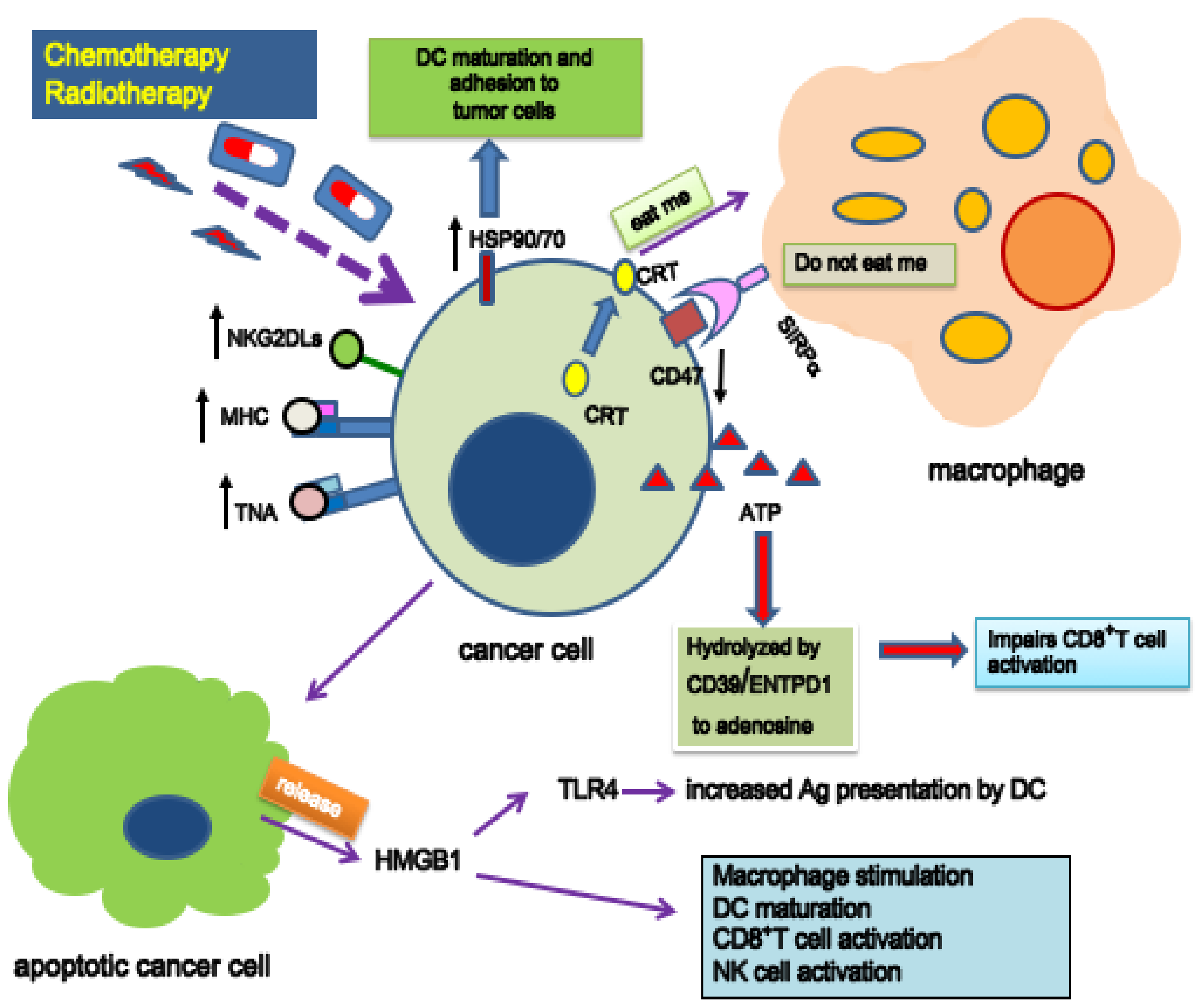
4. Immunogenic Cell Death
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kim, K.; Khang, D. Past, Present, and Future of Anticancer Nanomedicine. Int. J. Nanomed. 2020, 15, 5719–5743. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Ponziani, S.; Di Vittorio, G.; Pitari, G.; Cimini, A.M.; Ardini, M.; Gentile, R.; Iacobelli, S.; Sala, G.; Capone, E.; Flavell, D.J.; et al. Antibody-Drug Conjugates: The New Frontier of Chemotherapy. Int. J. Mol. Sci. 2020, 21, 5510. [Google Scholar] [CrossRef]
- Topatana, W.; Juengpanich, S.; Li, S.; Cao, J.; Hu, J.; Lee, J.; Suliyanto, K.; Ma, D.; Zhang, B.; Chen, M.; et al. Advances in synthetic lethality for cancer therapy: Cellular mechanism and clinical translation. J. Hematol. Oncol. 2020, 13, 118. [Google Scholar] [CrossRef]
- Yang, Y.; Li, X.; Wang, T.; Guo, Q.; Xi, T.; Zheng, L. Emerging agents that target signaling pathways in cancer stem cells. J. Hematol. Oncol. 2020, 13, 60. [Google Scholar] [CrossRef]
- Mole, R.H. Whole body irradiation; radiobiology or medicine? Br. J. Radiol. 1953, 26, 234–241. [Google Scholar] [CrossRef]
- Tochner, Z.; Slavin, S. Immune modulation by ionized irradiation. Curr. Opin. Immunol. 1988, 1, 261–268. [Google Scholar] [CrossRef]
- Desai, N.B.; Laine, A.M.; Timmerman, R.D. Stereotactic ablative body radiotherapy (SAbR) for oligometastatic cancer. Br. J. Radiol. 2017, 90, 20160500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalkidou, A.; Macmillan, T.; Grzeda, M.T.; Peacock, J.; Summers, J.; Eddy, S.; Coker, B.; Patrick, H.; Powell, H.; Berry, L.; et al. Stereotactic ablative body radiotherapy in patients with oligometastatic cancers: A prospective, registry-based, single-arm, observational, evaluation study. Lancet Oncol. 2021, 22, 98–106. [Google Scholar] [CrossRef]
- Palucka, A.K.; Coussens, L.M. The Basis of Oncoimmunology. Cell 2016, 164, 1233–1247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, S.; Li, D.; Zhu, X. Cancer immunotherapy: Pros, cons and beyond. Biomed. Pharmacol. 2020, 124, 109821. [Google Scholar] [CrossRef]
- Jia, Y.; Liu, L.; Shan, B. Future of immune checkpoint inhibitors: Focus on tumor immune microenvironment. Ann. Transl. Med. 2020, 8, 1095. [Google Scholar] [CrossRef]
- Londono, M.C.; Reig, M.; Group, R.M. Multidisciplinary Clinical Approach to Cancer Patients with Immune-Related Adverse Events Induced by Checkpoint Inhibitors. Cancers 2020, 12, 3446. [Google Scholar] [CrossRef]
- Perez-Ruiz, E.; Melero, I.; Kopecka, J.; Sarmento-Ribeiro, A.B.; Garcia-Aranda, M.; De Las Rivas, J. Cancer immunotherapy resistance based on immune checkpoints inhibitors: Targets, biomarkers, and remedies. Drug Resist. Updates 2020, 53, 100718. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.B.; Ha, S.J.; Kim, H.R. Clinical Insights Into Novel Immune Checkpoint Inhibitors. Front. Pharmacol. 2021, 12, 681320. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.J.; Darcy, P.K.; Trapani, J.A.; Kershaw, M.H.; Slaney, C.Y. Cross-talk between tumors at anatomically distinct sites. FEBS J. 2021, 288, 81–90. [Google Scholar] [CrossRef]
- Adjepong, D.; Malik, B.H. Radiation Therapy as a Modality to Create Abscopal Effects: Current and Future Practices. Cureus 2020, 12, e7054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demaria, S.; Formenti, S.C. The abscopal effect 67 years later: From a side story to center stage. Br. J. Radiol. 2020, 93, 20200042. [Google Scholar] [CrossRef] [PubMed]
- Franzese, O.; Torino, F.; Fuggetta, M.P.; Aquino, A.; Roselli, M.; Bonmassar, E.; Giuliani, A.; D’Atri, S. Tumor immunotherapy: Drug-induced neoantigens (xenogenization) and immune checkpoint inhibitors. Oncotarget 2017, 8, 41641–41669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden, E.B.; Marciscano, A.E.; Formenti, S.C. Radiation Therapy and the In Situ Vaccination Approach. Int. J. Radiat. Oncol. Biol. Phys. 2020, 108, 891–898. [Google Scholar] [CrossRef]
- Tesei, A.; Arienti, C.; Bossi, G.; Santi, S.; De Santis, I.; Bevilacqua, A.; Zanoni, M.; Pignatta, S.; Cortesi, M.; Zamagni, A.; et al. TP53 drives abscopal effect by secretion of senescence-associated molecular signals in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 2021, 40, 89. [Google Scholar] [CrossRef] [PubMed]
- Pouget, J.P.; Georgakilas, A.G.; Ravanat, J.L. Targeted and off-target (Bystander and Abscopal) effects of radiation therapy: Redox mechanisms and risk/benefit analysis. Antioxid. Redox Signal. 2018, 29, 1447–1487. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; Ten Hagen, T.L. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat. Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekpli, X.; Holme, J.A.; Sergent, O.; Lagadic-Gossmann, D. Role for membrane remodeling in cell death: Implication for health and disease. Toxicology 2013, 304, 141–157. [Google Scholar] [CrossRef] [PubMed]
- Korpela, E.; Liu, S.K. Endothelial perturbations and therapeutic strategies in normal tissue radiation damage. Radiat. Oncol. 2014, 9, 266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketteler, J.; Wittka, A.; Leonetti, D.; Roy, V.V.; Estephan, H.; Maier, P.; Reis, H.; Herskind, C.; Jendrossek, V.; Paris, F.; et al. Caveolin-1 regulates the ASMase/ceramide-mediated radiation response of endothelial cells in the context of tumor-stroma interactions. Cell Death Dis. 2020, 11, 228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, K.; He, C.; Guo, N.; Chan, C.; Ni, K.; Lan, G.; Tang, H.; Pelizzari, C.; Fu, Y.X.; Spiotto, M.T.; et al. Low-dose X-ray radiotherapy-radiodynamic therapy via nanoscale metal-organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng. 2018, 2, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Markovsky, E.; Budhu, S.; Samstein, R.M.; Li, H.; Russell, J.; Zhang, Z.; Drill, E.; Bodden, C.; Chen, Q.; Powell, S.N.; et al. An Antitumor Immune Response Is Evoked by Partial-Volume Single-Dose Radiation in 2 Murine Models. Int. J. Radiat. Oncol. Biol. Phys. 2019, 103, 697–708. [Google Scholar] [CrossRef]
- Mills, B.N.; Connolly, K.A.; Ye, J.; Murphy, J.D.; Uccello, T.P.; Han, B.J.; Zhao, T.; Drage, M.G.; Murthy, A.; Qiu, H.; et al. Stereotactic Body Radiation and Interleukin-12 Combination Therapy Eradicates Pancreatic Tumors by Repolarizing the Immune Microenvironment. Cell Rep. 2019, 29, 406–421. [Google Scholar] [CrossRef] [Green Version]
- Pomeranz Krummel, D.A.; Nasti, T.H.; Kaluzova, M.; Kallay, L.; Bhattacharya, D.; Melms, J.C.; Izar, B.; Xu, M.; Burnham, A.; Ahmed, T.; et al. Melanoma Cell Intrinsic GABAA Receptor Enhancement Potentiates Radiation and Immune Checkpoint Inhibitor Response by Promoting Direct and T Cell-Mediated Antitumor Activity. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109, 1040–1053. [Google Scholar] [CrossRef]
- Marconi, R.; Strolin, S.; Bossi, G.; Strigari, L. A meta-analysis of the abscopal effect in preclinical models: Is the biologically effective dose a relevant physical trigger? PLoS ONE 2017, 12, e0171559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, E.D.; Chang, J.Y. Time to abandon single-site irradiation for inducing abscopal effects. Nat. Rev. Clin. Oncol. 2019, 16, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H. Abscopal effect of stereotactic radiotherapy combined with anti-PD-1/PD-L1 immunotherapy: Mechanisms, clinical efficacy, and issues. Cancer Commun. 2020, 40, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Guha, C.; Schoenfeld, J.; Morris, Z.; Monjazeb, A.; Sikora, A.; Crittenden, M.; Shiao, S.; Khleif, S.; Gupta, S.; et al. Radiation dose and fraction in immunotherapy: One-size regimen does not fit all settings, so how does one choose? J. Immunother. Cancer 2021, 9, e002038. [Google Scholar] [CrossRef] [PubMed]
- Demaria, S.; Coleman, C.N.; Formenti, S.C. Radiotherapy: Changing the Game in Immunotherapy. Trends Cancer 2016, 2, 286–294. [Google Scholar] [CrossRef] [Green Version]
- Ngwa, W.; Irabor, O.C.; Schoenfeld, J.D.; Hesser, J.; Demaria, S.; Formenti, S.C. Using immunotherapy to boost the abscopal effect. Nat. Rev. Cancer 2018, 18, 313–322. [Google Scholar] [CrossRef]
- Rodriguez-Ruiz, M.E.; Vanpouille-Box, C.; Melero, I.; Formenti, S.C.; Demaria, S. Immunological Mechanisms Responsible for Radiation-Induced Abscopal Effect. Trends Immunol. 2018, 39, 644–655. [Google Scholar] [CrossRef]
- Lhuillier, C.; Rudqvist, N.P.; Elemento, O.; Formenti, S.C.; Demaria, S. Radiation therapy and anti-tumor immunity: Exposing immunogenic mutations to the immune system. Genome Med. 2019, 11, 40. [Google Scholar] [CrossRef] [Green Version]
- Houchens, D.P.; Bonmassar, E.; Gaston, M.R.; Kende, M.; Goldin, A. Drug-mediated immunogenic changes of virus-induced leukemia in vivo. Cancer Res. 1976, 36, 1347–1352. [Google Scholar]
- Sato, H.; Okonogi, N.; Nakano, T. Rationale of combination of anti-PD-1/PD-L1 antibody therapy and radiotherapy for cancer treatment. Int. J. Clin. Oncol. 2020, 25, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.; Bakhoum, S.F. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Kirchmair, A.; Sato, A.; Buque, A.; Rybstein, M.; Petroni, G.; Bloy, N.; Finotello, F.; Stafford, L.; Navarro Manzano, E.; et al. Mitochondrial DNA drives abscopal responses to radiation that are inhibited by autophagy. Nat. Immunol. 2020, 21, 1160–1171. [Google Scholar] [CrossRef] [PubMed]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Mukai, K.; Konno, H.; Akiba, T.; Uemura, T.; Waguri, S.; Kobayashi, T.; Barber, G.N.; Arai, H.; Taguchi, T. Activation of STING requires palmitoylation at the Golgi. Nat. Commun. 2016, 7, 11932. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Helgason, E.; Phung, Q.T.; Quan, C.L.; Iyer, R.S.; Lee, M.W.; Bowman, K.K.; Starovasnik, M.A.; Dueber, E.C. Molecular basis of Tank-binding kinase 1 activation by transautophosphorylation. Proc. Natl. Acad. Sci. USA 2012, 109, 9378–9383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferies, C.A. Regulating IRFs in IFN Driven Disease. Front. Immunol. 2019, 10, 325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marineau, A.; Khan, K.A.; Servant, M.J. Roles of GSK-3 and beta-Catenin in Antiviral Innate Immune Sensing of Nucleic Acids. Cells 2020, 9, 897. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.K.; Pan, D.; Bao, X.; Hu, M.; Li, F.; Li, C.Y. Endogenous Retrovirus Activation as a Key Mechanism of Anti-Tumor Immune Response in Radiotherapy. Radiat. Res. 2020, 193, 305–317. [Google Scholar] [CrossRef]
- Alcazer, V.; Bonaventura, P.; Depil, S. Human Endogenous Retroviruses (HERVs): Shaping the Innate Immune Response in Cancers. Cancers 2020, 12, 610. [Google Scholar] [CrossRef] [Green Version]
- Stok, J.E.; Vega Quiroz, M.E.; van der Veen, A.G. Self RNA Sensing by RIG-I-like Receptors in Viral Infection and Sterile Inflammation. J. Immunol. 2020, 205, 883–891. [Google Scholar] [CrossRef]
- Refolo, G.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Ciccosanti, F. Mitochondrial Interactome: A Focus on Antiviral Signaling Pathways. Front. Cell Dev. Biol. 2020, 8, 8. [Google Scholar] [CrossRef]
- Yamazaki, T.; Galluzzi, L. Mitochondrial control of innate immune signaling by irradiated cancer cells. Oncoimmunology 2020, 9, 1797292. [Google Scholar] [CrossRef]
- Abe, T.; Shapira, S.D. Negative Regulation of Cytosolic Sensing of DNA. Int. Rev. Cell Mol. Biol. 2019, 344, 91–115. [Google Scholar] [CrossRef]
- Zhou, R.; Zhang, Q.; Xu, P. TBK1, a central kinase in innate immune sensing of nucleic acids and beyond. Acta Biochim. Biophys. Sin. 2020, 52, 757–767. [Google Scholar] [CrossRef]
- Zhou, W.; Whiteley, A.T.; de Oliveira Mann, C.C.; Morehouse, B.R.; Nowak, R.P.; Fischer, E.S.; Gray, N.S.; Mekalanos, J.J.; Kranzusch, P.J. Structure of the Human cGAS-DNA Complex Reveals Enhanced Control of Immune Surveillance. Cell 2018, 174, 300–311. [Google Scholar] [CrossRef] [Green Version]
- Attermann, A.S.; Bjerregaard, A.M.; Saini, S.K.; Gronbaek, K.; Hadrup, S.R. Human endogenous retroviruses and their implication for immunotherapeutics of cancer. Ann. Oncol. 2018, 29, 2183–2191. [Google Scholar] [CrossRef] [PubMed]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, T.; Rodriguez, B.L.; Chen, L.; Corte, C.M.D.; Morikawa, N.; Fujimoto, J.; Cristea, S.; Nguyen, T.; Diao, L.; Li, L.; et al. Targeting DNA damage response promotes antitumor immunity through STING- mediated T-cell activation in small cell lung cancer. Cancer Discov. 2019, 9, 646–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Chen, J.; Hu, J.; Zhang, H.; Xu, F.; He, W.; Wang, X.; Li, M.; Lu, W.; Yeng, G.; et al. cGAS/STING axis mediates a topoisomerase II inhibitor-induced tumor immunogenicity. J. Clin. Investig. 2019, 130, 4850–4862. [Google Scholar] [CrossRef]
- Diamond, J.M.; Vanpouille-Box, S.; Spada, C.; Rudqvist, J.R.; Chapman, N.P.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcus, A.; Mao, A.J.; Lensink-Vasan, M.; Wang, L.; Vance, R.E.; Raulet, D.H. Tumor-derived cGAMP triggers a STING-mediated interferon response in non-tumor cells to activate the NK cell response. Immunity 2018, 49, 754–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donlon, N.E.; Power, R.; Hayes, C.; Reynolds, J.V.; Lysaght, J. Radiotherapy, immunotherapy, and the tumour microenvironment: Turning an immunosuppressive milieu into a therapeutic opportunity. Cancer Lett. 2021, 502, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, F.T. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell 2017, 31, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Kodet, O.; Němejcova, K.; Strnadová, K.; Havlínová, A.; Dundr, P.; Krajsová, I.; Štork, J.; Smetana, K., Jr.; Lukas, L. The Abscopal Effect in the Era of Checkpoint Inhibitors. Int. J. Mol. Sci. 2021, 22, 7204. [Google Scholar] [CrossRef]
- Parkes, E.E.; Walker, S.M.; Taggart, L.E.; McCabe, N.; Knight, L.A.; Wilkinson, R.; McCloskey, K.D.; Buckley, N.E.; Savage, K.I.; Salto-Tellez, M.; et al. Activation of STING-dependent innate immune signaling by S-Phase-Speci c DNA damage in breast cancer. J. Natl. Cancer Inst. 2016, 109, djw199. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Wu, Z.; Muluh, T.A.; Fu, S.; Wu, J. Effect of low-dose total-body radiotherapy on immune microenvironment. Transl. Oncol. 2021, 14, 101118. [Google Scholar] [CrossRef]
- Li, J.; Zeng, Z.; Wu, Q.; Chen, J.; Liu, X.; Zhang, J.; Luo, Y.; Sun, W.; Huang, Z.; Zhang, J.; et al. Immunological modulation of the Th1/Th2 shift by ionizing radiation in tumors. Int. J. Oncol. 2021, 59, 50. [Google Scholar] [CrossRef]
- Lugade, A.A.; Moran, J.P.; Gerber, S.A.; Rose, R.C.; Frelinger, J.G.; Lord, E.M. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J. Immunol. 2005, 174, 7516–7523. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.; Auh, S.L.; Wang, Y.; Burnette, B.; Wang, Y.; Meng, Y.; Beckett, M.; Sharma, R.; Chine, R.; Tu, T.; et al. Therapeutic effects of ablative radiation on local tumor require CD8+ T cells: Changing strategies for cancer treatment. Blood 2009, 114, 589–595. [Google Scholar] [CrossRef]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reits, E.A.; Hodge, J.W.; Herberts, C.A.; Groothuis, T.A.; Chakraborty, M.; Wansley, E.K.; Camphausen, K.; Luiten, R.M.; de Ru, A.H.; Neijssen, J.; et al. Radiation modulates the peptide repertoire, enhances MHC class I expression, and induces successful antitumor immunotherapy. J. Exp. Med. 2006, 203, 1259–1271. [Google Scholar] [CrossRef] [PubMed]
- Rudqvist, N.P.; Pilones, K.A.; Lhuillier, C.; Wennerberg, E.; Sidhom, J.W.; Emerson, R.O.; Robins, H.S.; Schneck, J.; Formenti, S.C.; Demaria, S. Radiotherapy and CTLA-4 Blockade Shape the TCR Repertoire of Tumor-Infiltrating T Cells. Cancer Immunol. Res. 2018, 6, 139–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruckert, M.; Flohr, A.S.; Hecht, M.; Gaipl, U.S. Radiotherapy and the immune system: More than just immune suppression. Stem Cells 2021, 39, 1155–1165. [Google Scholar] [CrossRef]
- Kohno, M.; Murakami, J.; Wu, L.; Chan, M.L.; Yun, Z.; Cho, B.C.J.; de Perrot, M. Foxp3(+) Regulatory T Cell Depletion after Nonablative Oligofractionated Irradiation Boosts the Abscopal Effects in Murine Malignant Mesothelioma. J. Immunol. 2020, 205, 2519–2531. [Google Scholar] [CrossRef]
- Piper, M.; Mueller, A.C.; Karam, S.D. The interplay between cancer associated fibroblasts and immune cells in the context of radiation therapy. Mol. Carcinog. 2020, 59, 754–765. [Google Scholar] [CrossRef] [PubMed]
- Formenti, S.C.; Demaria, S. Systemic effects of local radiotherapy. Lancet Oncol. 2009, 10, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, J.M.; Shinohara, E.; Summers, J.B.; Niermann, K.J.; Morimoto, A.; Brousal, J. The controversial abscopal effect. Cancer Treat. Rev. 2005, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Deng, L.; Chmura, S.; Burnette, B.; Liadis, N.; Darga, T.; Beckett, M.A.; Lingen, M.W.; Witt, M.; Weichselbaum, R.R.; et al. Radiation-induced equilibrium is a balance between tumor cell proliferation and T cell-mediated killing. J. Immunol. 2013, 190, 5874–5881. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; See, A.P.; Phallen, J.; Jackson, C.M.; Belcaid, Z.; Ruzevick, J.; Durham, N.; Meyer, C.; Harris, T.J.; Albesiano, E.; et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 343–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heylmann, D.; Ponath, V.; Kindler, T.; Kaina, B. Comparison of DNA repair and radiosensitivity of different blood cell populations. Sci. Rep. 2021, 11, 2478. [Google Scholar] [CrossRef]
- Deaglio, S.; Dwyer, K.M.; Gao, W.; Friedman, D.; Usheva, A.; Erat, A.; Chen, J.F.; Enjyoji, K.; Linden, J.; Oukka, M.; et al. Adenosine generation catalyzed by CD39 and CD73 expressed on regulatory T cells mediates immune suppression. J. Exp. Med. 2007, 204, 1257–1265. [Google Scholar] [CrossRef] [Green Version]
- De Leve, S.; Wirsdorfer, F.; Jendrossek, V. The CD73/Ado System-A New Player in RT Induced Adverse Late Effects. Cancers 2019, 11, 1578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aponte, P.M.; Caicedo, A. Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int. 2017, 2017, 5619472. [Google Scholar] [CrossRef] [PubMed]
- Feng, L.L.; Cai, Y.Q.; Zhu, M.C.; Xing, L.J.; Wang, X. The yin and yang functions of extracellular ATP and adenosine in tumor immunity. Cancer Cell Int. 2020, 20, 110. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Boeynaems, J.M. Purinergic signalling and immune cells. Purinergic Signal. 2014, 10, 529–564. [Google Scholar] [CrossRef] [Green Version]
- Buisseret, L.; Pommey, S.; Allard, B.; Garaud, S.; Bergeron, M.; Cousineau, I.; Ameye, L.; Bareche, Y.; Paesmans, M.; Crown, J.P.A.; et al. Clinical significance of CD73 in triple-negative breast cancer: Multiplex analysis of a phase III clinical trial. Ann. Oncol. 2018, 29, 1056–1062. [Google Scholar] [CrossRef] [Green Version]
- Jiang, T.; Xu, X.; Qiao, M.; Li, X.; Zhao, C.; Zhou, F.; Gao, G.; Wu, F.; Chen, X.; Su, C.; et al. Comprehensive evaluation of NT5E/CD73 expression and its prognostic significance in distinct types of cancers. BMC Cancer 2018, 18, 267. [Google Scholar] [CrossRef] [Green Version]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [Green Version]
- Sitkovsky, M.V. T regulatory cells: Hypoxia-adenosinergic suppression and re-direction of the immune response. Trends Immunol. 2009, 30, 102–108. [Google Scholar] [CrossRef]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [Green Version]
- Horenstein, A.L.; Chillemi, A.; Zaccarello, G.; Bruzzone, S.; Quarona, V.; Zito, A.; Serra, S.; Malavasi, F. A CD38/CD203a/CD73 ectoenzymatic pathway independent of CD39 drives a novel adenosinergic loop in human T lymphocytes. Oncoimmunology 2013, 2, e26246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wennerberg, E.; Spada, S.; Rudqvist, N.P.; Lhuillier, C.; Gruber, S.; Chen, Q.; Zhang, F.; Zhou, X.K.; Gross, S.S.; Formenti, S.C.; et al. CD73 Blockade Promotes Dendritic Cell Infiltration of Irradiated Tumors and Tumor Rejection. Cancer Immunol. Res. 2020, 8, 465–478. [Google Scholar] [CrossRef] [PubMed]
- De Leve, S.; Wirsdorfer, F.; Jendrossek, V. Targeting the Immunomodulatory CD73/Adenosine System to Improve the Therapeutic Gain of Radiotherapy. Front. Immunol. 2019, 10, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terp, M.G.; Olesen, K.A.; Arnspang, E.C.; Lund, R.R.; Lagerholm, B.C.; Ditzel, H.J.; Leth-Larsen, R. Anti-human CD73 monoclonal antibody inhibits metastasis formation in human breast cancer by inducing clustering and internalization of CD73 expressed on the surface of cancer cells. J. Immunol. 2013, 191, 4165–4173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Identifier: NCT02503774. Available online: https://clinicaltrials.gov/ (accessed on 5 August 2021).
- Tsukui, H.; Horie, H.; Koinuma, K.; Ohzawa, H.; Sakuma, Y.; Hosoya, Y.; Yamaguchi, H.; Yoshimura, K.; Lefor, A.K.; Sata, N.; et al. CD73 blockade enhances the local and abscopal effects of radiotherapy in a murine rectal cancer model. BMC Cancer 2020, 20, 411. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef] [Green Version]
- Kitabatake, K.; Kaji, Y.; Tsukimata, M. Involvement of CD73 and A2B receptor in radiation-induced DNA damage response and cell migration in human glioblastoma A 172 cells. Biol. Pharm. Bull. 2021; 44, 197–210. [Google Scholar] [CrossRef]
- Wirsdorfer, F.; Cappuccini, F.; Niazman, M.; de Leve, S.; Westendorf, A.M.; Ludemann, L.; Stuschke, M.; Jendrossek, V. Thorax irradiation triggers a local and systemic accumulation of immunosuppressive CD4+ FoxP3+ regulatory T cells. Radiat. Oncol. 2014, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Dovedi, S.J.; Cheadle, E.J.; Popple, A.L.; Poon, E.; Morrow, M.; Stewart, R.; Yusko, E.C.; Sanders, C.M.; Vignali, M.; Emerson, R.O.; et al. Fractionated radiation therapy stimulates antitumor immunity mediated by both resident and infiltrating polyclonal T-cell populations when combined with PD-1 blockade. Clin. Cancer Res. 2017, 23, 5514–5526. [Google Scholar] [CrossRef] [Green Version]
- Kachikwu, E.L.; Iwamoto, K.S.; Liao, Y.-P.; DeMarco, J.J.; Agazaryan, N.; Economou, J.S.; McBride, W.H.; Schaue, D. Radiation enhances regulatory T cell representation. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Klug, F.; Prakash, H.; Huber, P.E.; Seibel, T.; Bender, N.; Halama, N.; Pfirschke, C.; Voss, R.H.; Timke, C.; Umansky, L.; et al. Low-dose irradiation programs macrophage differentiation to an iNOS(+)/M1 phenotype that orchestrates effective T cell immunotherapy. Cancer Cell 2013, 24, 589–602. [Google Scholar] [CrossRef] [Green Version]
- Gao, H.; Dong, Z.; Gong, X.; Dong, J.; Zhang, Y.; Wei, W.; Wang, R.; Jin, S. Effects of various radiation doses on induced T-helper cell differentiation and related cytokine secretion. J. Radiat. Res. 2018, 59, 395–403. [Google Scholar] [CrossRef]
- Chandra, R.A.; Wilhite, T.J.; Balboni, T.A.; Alexander, B.M.; Spektor, A.; Ott, P.A.; Ng, A.K.; Hodi, F.S.; Schoenfeld, J.D. A systematic evaluation of abscopal responses following radiotherapy in patients with metastatic melanoma treated with ipilimumab. Oncoimmunology 2015, 4, e1046028. [Google Scholar] [CrossRef] [Green Version]
- Vanpouille-Box, C. Immune radiobiology. J. Transl. Med. 2021, 19, 25. [Google Scholar] [CrossRef]
- Keisari, Y.; Kelson, I. The Potentiation of Anti-Tumor Immunity by Tumor Abolition with Alpha Particles, Protons, or Carbon Ion Radiation and Its Enforcement by Combination with Immunoadjuvants or Inhibitors of Immune Suppressor Cells and Checkpoint Molecules. Cells 2021, 10, 228. [Google Scholar] [CrossRef]
- Zhao, X.; Shao, C. Radiotherapy-Mediated Immunomodulation and Anti-Tumor Abscopal Effect Combining Immune Checkpoint Blockade. Cancers 2020, 12, 2762. [Google Scholar] [CrossRef] [PubMed]
- Peres Lde, P.; da Luz, F.A.; Pultz Bdos, A.; Brigido, P.C.; de Araujo, R.A.; Goulart, L.R.; Silva, M.J. Peptide vaccines in breast cancer: The immunological basis for clinical response. Biotechnol. Adv. 2015, 33, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correale, P.; Aquino, A.; Giuliani, A.; Pellegrini, M.; Micheli, L.; Cusi, M.G.; Nencini, C.; Petrioli, R.; Prete, S.P.; De Vecchis, L.; et al. Treatment of colon and breast carcinoma cells with 5-fluorouracil enhances expression of carcinoembryonic antigen and susceptibility to HLA-A(*)02.01 restricted, CEA-peptide-specific cytotoxic T cells in vitro. Int. J. Cancer 2003, 104, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Franzese, O.; Aquino, A.; Fuggetta, M.P.; Roselli, M.; Bonmassar, E.; De Vecchis, L.; Torino, F. Drug-Induced Neoantigens: A New Horizon in Cancer Immunotherapy? Clin. Oncol. 2018, 3, 1411. [Google Scholar]
- Franzese, O.; Battaini, F.; Graziani, G.; Tentori, L.; Barbaccia, M.L.; Aquino, A.; Roselli, M.; Fuggetta, M.P.; Bonmassar, E.; Torino, F. Drug-induced xenogenization of tumors: A possible role in the immune control of malignant cell growth in the brain? Pharmacol. Res. 2018, 131, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Jennings, V.A.; Melcher, A.A.; Lise, S. The Immunogenic Potential of Recurrent Cancer Drug Resistance Mutations: An In Silico Study. Front. Immunol. 2020, 11, 524968. [Google Scholar] [CrossRef]
- Pettitt, S.J.; Frankum, J.; Punta, M.; Lise, S.; Alexander, J.; Chen, Y.; Yap, T.A.; Haider, S.; Tutt, A.N.J.; Lord, C.J. Clinical BRCA1/2 reversion analysis identifies hotspot mutations and predicted neoantigens associated with therapy resistance. Cancer Discov. 2020, 10, 1–14. [Google Scholar] [CrossRef]
- Anichini, A.; Perotti, V.E.; Sgambelluri, F.; Mortarini, R. Immune Escape Mechanisms in Non Small Cell Lung Cancer. Cancers 2020, 12, 3605. [Google Scholar] [CrossRef]
- Trinh, A.; Polyak, K. Tumor Neoantigens: When Too Much of a Good Thing Is Bad. Cancer Cell 2019, 36, 466–467. [Google Scholar] [CrossRef]
- Seledtsov, V.I.; Goncharov, A.G.; Seledtsova, G.V. Clinically feasible approaches to potentiating cancer cell-based immunotherapies. Hum. Vaccines Immunother. 2015, 11, 851–869. [Google Scholar] [CrossRef] [Green Version]
- Vanmeerbeek, I.; Sprooten, J.; De Ruysscher, D.; Tejpar, S.; Vandenberghe, P.; Fucikova, J.; Spisek, R.; Zitvogel, L.; Kroemer, G.; Galluzzi, L.; et al. Trial watch: Chemotherapy-induced immunogenic cell death in immuno-oncology. Oncoimmunology 2020, 9, 1703449. [Google Scholar] [CrossRef] [Green Version]
- Sharabi, A.; Haran-Ghera, N. Immune recovery after cyclophosphamide treatment in multiple myeloma: Implication for maintenance immunotherapy. Bone Marrow Res. 2011, 2011, 269519. [Google Scholar] [CrossRef] [Green Version]
- Emadi, A.; Jones, R.J.; Brodsky, R.A. Cyclophosphamide and cancer: Golden anniversary. Nat. Rev. Clin. Oncol. 2009, 6, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Heylmann, D.; Bauer, M.; Becker, H.; van Gool, S.; Bacher, N.; Steinbrink, K.; Kaina, B. Human CD4+CD25+ regulatory T cells are sensitive to low dose cyclophosphamide: Implications for the immune response. PLoS ONE 2013, 8, e83384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Attallah, A.M.; Needy, C.F.; Noguchi, P.D.; Elisberg, B.L. Enhancement of carcinoembryonic antigen expression by interferon. Int. J. Cancer 1979, 24, 49–52. [Google Scholar] [CrossRef] [PubMed]
- Guadagni, F.; Witt, P.L.; Robbins, P.F.; Schlom, J.; Greiner, J.W. Regulation of carcinoembryonic antigen expression in different human colorectal tumor cells by interferon-gamma. Cancer Res. 1990, 50, 6248–6255. [Google Scholar]
- Leon, J.A.; Mesa-Tejada, R.; Gutierrez, M.C.; Estabrook, A.; Greiner, J.W.; Schlom, J.; Fisher, P.B. Increased surface expression and shedding of tumor associated antigens by human breast carcinoma cells treated with recombinant human interferons or phorbol ester tumor promoters. Anticancer. Res. 1989, 9, 1639–1647. [Google Scholar]
- Greiner, J.W.; Guadagni, F.; Goldstein, D.; Smalley, R.V.; Borden, E.C.; Simpson, J.F.; Molinolo, A.; Schlom, J. Intraperitoneal administration of interferon-gamma to carcinoma patients enhances expression of tumor-associated glycoprotein-72 and carcinoembryonic antigen on malignant ascites cells. J. Clin. Oncol. 1992, 10, 735–746. [Google Scholar] [CrossRef]
- Greiner, J.W.; Hand, P.H.; Noguchi, P.; Fisher, P.B.; Pestka, S.; Schlom, J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant human leukocyte alpha-interferon treatment. Cancer Res. 1984, 44, 3208–3214. [Google Scholar]
- Shimada, S.; Ogawa, M.; Schlom, J.; Greiner, J.W. Comparison of the interferon-gamma-mediated regulation of tumor-associated antigens expressed by human gastric carcinoma cells. In Vivo 1993, 7, 1–8. [Google Scholar]
- Mobus, V.J.; Asphal, W.; Knapstein, P.G.; Kreienberg, R. Effects of interferon gamma on the proliferation and modulation of cell-surface structures of human ovarian carcinoma cell lines. J. Cancer Res. Clin. Oncol. 1993, 120, 27–34. [Google Scholar] [CrossRef]
- Ozzello, L.; Derosa, C.; Habif, D.; Cantell, K.; Pestka, S. Up-regulation of a tumor-associated antigen (tag-72) by interferon-alpha and interferon-gamma in patients with cutaneous breast-cancer recurrences. Int. J. Oncol. 1995, 6, 985–991. [Google Scholar] [CrossRef]
- Colombatti, M.; Bisconti, M.; Lorenzi, P.; Stevanoni, G.; Dipasquale, B.; Gerosa, M.; Tridente, G. Human glioma cell lines: Tumour associated antigens distribution and sensitivity to antibody-toxin or ligand-toxin conjugates. A preliminary report. Acta Neurochir. Suppl. 1988, 43, 121–125. [Google Scholar] [CrossRef]
- Colombatti, M.; Dipasquale, B.; Del-l’Arciprete, L.; Gerosa, M.; Tridente, G. Heterogeneity and modulation of tumor-associated antigens in human glioblastoma cell lines. J. Neurosurg. 1989, 71, 388–397. [Google Scholar] [CrossRef] [PubMed]
- Tran, R.; Hand, P.H.; Greiner, J.W.; Pestka, S.; Schlom, J. Enhancement of surface antigen expression on human breast carcinoma cells by recombinant human interferons. J. Interferon. Res. 1988, 8, 75–88. [Google Scholar] [CrossRef]
- Flieger, D.; Hoff, A.S.; Sauerbruch, T.; Schmidt-Wolf, I.G. Influence of cytokines, monoclonal antibodies and chemotherapeutic drugs on epithelial cell adhesion molecule (EpCAM) and LewisY antigen expression. Clin. Exp. Immunol. 2001, 123, 9–14. [Google Scholar] [CrossRef]
- Weidanz, J.A.; Nguyen, T.; Woodburn, T.; Neethling, F.A.; Chiriva-Internati, M.; Hildebrand, W.H.; Lustgarten, J. Levels of specific peptide-HLA class I complex predicts tumor cell susceptibility to CTL killing. J. Immunol. 2006, 177, 5088–5097. [Google Scholar] [CrossRef] [Green Version]
- Dunn, I.S.; Haggerty, T.J.; Kono, M.; Durda, P.J.; Butera, D.; Macdonald, D.B.; Benson, E.M.; Rose, L.B.; Kurnick, J.T. Enhancement of human melanoma antigen expression by IFN-beta. J. Immunol. 2007, 179, 2134–2142. [Google Scholar] [CrossRef] [Green Version]
- Bao, L.; Dunham, K.; Lucas, K. MAGE-A1, MAGE-A3, and NY-ESO-1 can be upregulated on neuroblastoma cells to facilitate cytotoxic T lymphocyte-mediated tumor cell killing. Cancer Immunol. Immunother. 2011, 60, 1299–1307. [Google Scholar] [CrossRef]
- Grabbe, S.; Bruvers, S.; Beissert, S.; Granstein, R.D. Interferon-gamma inhibits tumor antigen presentation by epidermal antigen-presenting cells. J. Leukoc. Biol. 1994, 55, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Barrero, M.J. Epigenetic Strategies to Boost Cancer Immunotherapies. Int. J. Mol. Sci. 2017, 18, 1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroesen, M.; Gielen, P.; Brok, I.C.; Armandari, I.; Hoogerbrugge, P.M.; Adema, G.J. HDAC inhibitors and immunotherapy; a double edged sword? Oncotarget 2014, 5, 6558–6572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, D.; Qiu, Y.; Jiao, Y.; Qiu, Z.; Liu, D. Small Molecules Targeting HATs, HDACs, and BRDs in Cancer Therapy. Front. Oncol. 2020, 10, 560487. [Google Scholar] [CrossRef]
- Milazzo, G.; Mercatelli, D.; Di Muzio, G.; Triboli, L.; De Rosa, P.; Perini, G.; Giorgi, F.M. Histone Deacetylases (HDACs): Evolution, Specificity, Role in Transcriptional Complexes, and Pharmacological Actionability. Genes 2020, 11, 556. [Google Scholar] [CrossRef] [PubMed]
- Adams, G.E.; Chandru, A.; Cowley, S.M. Co-repressor, co-activator and general transcription factor: The many faces of the Sin3 histone deacetylase (HDAC) complex. Biochem. J. 2018, 475, 3921–3932. [Google Scholar] [CrossRef] [PubMed]
- Villagra, A.; Sotomayor, E.M.; Seto, E. Histone deacetylases and the immunological network: Implications in cancer and inflammation. Oncogene 2010, 29, 157–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, R.; Kikuchi, J.; Wada, T.; Ozawa, K.; Kano, Y.; Furukawa, Y. HDAC inhibitors augment cytotoxic activity of rituximab by upregulating CD20 expression on lymphoma cells. Leukemia 2010, 24, 1760–1768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Jost, E.; Belohlavek, C.; Nagel, G.; Fritz, G.; Kaina, B. Intrinsic anticancer drug resistance of malignant melanoma cells is abrogated by IFN-beta and valproic acid. Cancer Res. 2011, 71, 4150–4160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Pan, X.; Zhang, W.; Guo, H.; Cheng, S.; He, Q.; Yang, B.; Ding, L. Epigenetic strategies synergize with PD-L1/PD-1 targeted cancer immunotherapies to enhance antitumor responses. Acta Pharmacol. Sin. B 2020, 10, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Ebelt, N.D.; Zuniga, E.; Johnson, B.L.; Diamond, D.J.; Manuel, E.R. 5-Azacytidine Potentiates Anti-tumor Immunity in a Model of Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 11, 538. [Google Scholar] [CrossRef] [Green Version]
- Shiozawa, M.; Chang, C.H.; Huang, Y.C.; Chen, Y.C.; Chi, M.S.; Hao, H.C.; Chang, Y.C.; Takeda, S.; Chi, K.H.; Wang, Y.S. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018, 19, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodge, J.W.; Garnett, C.T.; Farsaci, B.; Palena, C.; Tsang, K.Y.; Ferrone, S.; Gameiro, S.R. Chemotherapy-induced immunogenic modulation of tumor cells enhances killing by cytotoxic T lymphocytes and is distinct from immunogenic cell death. Int. J. Cancer 2013, 133, 624–636. [Google Scholar] [CrossRef]
- Aquino, A.; Formica, V.; Prete, S.P.; Correale, P.P.; Massara, M.C.; Turriziani, M.; De Vecchis, L.; Bonmassar, E. Drug-induced increase of carcinoembryonic antigen expression in cancer cells. Pharmacol. Res. 2004, 49, 383–396. [Google Scholar] [CrossRef]
- Aquino, A.; Prete, S.P.; Greiner, J.W.; Giuliani, A.; Graziani, G.; Turriziani, M.; De Filippi, R.; Masci, G.; Bonmassar, E.; De Vecchis, L. Effect of the combined treatment with 5-fluorouracil, gamma-interferon or folinic acid on carcinoembryonic antigen expression in colon cancer cells. Clin. Cancer Res. 1998, 4, 2473–2481. [Google Scholar]
- Aquino, A.; Prete, S.P.; Guadagni, F.; Greiner, J.W.; Giuliani, A.; Orlando, L.; Masci, G.; De Santis, S.; Bonmassar, E.; Graziani, G. Effect of 5-fluorouracil on carcinoembryonic antigen expression and shedding at clonal level in colon cancer cells. Anticancer Res. 2000, 20, 3475–3484. [Google Scholar]
- Cappelletti, D.; Cardillo, A.; Bonanno, E.; Prete, S.P.; Cucchiara, G.; Turriziani, M.; Greiner, J.W.; Cottarelli, A.; Breda, E.; Aquino, A.; et al. Drug-induced modulation of carcinoembryonic antigen (CEA) expression in neoplastic cells from a patient with rectal cancer. J. Exp. Clin. Cancer Res. 2000, 19, 467–469. [Google Scholar]
- Correale, P.; Cusi, M.G.; Del Vecchio, M.T.; Aquino, A.; Prete, S.P.; Tsang, K.Y.; Micheli, L.; Nencini, C.; La Placa, M.; Montagnani, F.; et al. Dendritic cell-mediated cross-presentation of antigens derived from colon carcinoma cells exposed to a highly cytotoxic multidrug regimen with gemcitabine, oxaliplatin, 5-fluorouracil, and leucovorin, elicits a powerful human antigen-specific CTL response with antitumor activity in vitro. J. Immunol. 2005, 175, 820–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Filippi, R.; Prete, S.P.; Giuliani, A.; Silvi, E.; Yamaue, H.; Nieroda, C.A.; Greiner, J.W.; De Vecchis, L.; Bonmassar, E. Differential effects of recombinant interferon-alpha and 5-fluorouracil against colon cancer cells or against peripheral blood mononuclear cells. Anticancer Res. 1994, 14, 1767–1773. [Google Scholar]
- Ohtsukasa, S.; Okabe, S.; Yamashita, H.; Iwai, T.; Sugihara, K. Increased expression of CEA and MHC class I in colorectal cancer cell lines exposed to chemotherapy drugs. J. Cancer Res. Clin. Oncol. 2003, 129, 719–726. [Google Scholar] [CrossRef]
- Prete, S.P.; Aquino, A.; Masci, G.; Orlando, L.; Giuliani, A.; De Santis, S.; De Vecchis, L.; De Filippi, R.; Greiner, J.W.; Bonmassar, E.; et al. Drug-induced changes of carcinoembryonic antigen expression in human cancer cells: Effect of 5-fluorouracil. J. Pharmacol. Exp. Ther. 1996, 279, 1574–1581. [Google Scholar] [PubMed]
- Prete, S.P.; Rossi, L.; Correale, P.P.; Turriziani, M.; Baier, S.; Tamburrelli, G.; De Vecchis, L.; Bonmassar, E.; Aquino, A. Combined effects of protein kinase inhibitors and 5-fluorouracil on CEA expression in human colon cancer cells. Pharmacol. Res. 2005, 52, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Prete, S.P.; Turriziani, M.; Massara, M.C.; De Rossi, A.; Correale, P.; De Vecchis, L.; Torino, F.; Bonmassar, L.; Aquino, A. Combined effects of 5-fluorouracil, folinic acid and oxaliplatin on the expression of carcinoembryonic antigen in human colon cancer cells: Pharmacological basis to develop an active antitumor immunochemotherapy. J. Exp. Clin. Cancer Res. 2008, 27, 5. [Google Scholar] [CrossRef] [Green Version]
- Aquino, A.; Prete, S.P.; Balduzzi, A.; Fossile, E.; Formica, V.; Torino, F.; Bonmassar, L.; Di Giacomo, A.; Cappelletti, D.; Cardillo, A.; et al. A novel method for monitoring response to chemotherapy based on the detection of circulating cancer cells: A case report. J. Chemother. 2002, 14, 412–416. [Google Scholar] [CrossRef]
- Bonmassar, L.; Fossile, E.; Scoppola, A.; Graziani, G.; Prete, S.P.; Formica, V.; Cappelletti, D.; De Vecchis, L.; Cardillo, A.; Concolino, F.; et al. Detection of circulating tumor cells is improved by drug-induced antigen up-regulation: Preclinical and clinical studies. Anticancer Res. 2010, 30, 4721–4730. [Google Scholar]
- Yang, S.; Haluska, F.G. Treatment of melanoma with 5-fluorouracil or dacarbazine in vitro sensitizes cells to antigen-specific CTL lysis through perforin/granzyme- and Fas-mediated pathways. J. Immunol. 2004, 172, 4599–4608. [Google Scholar] [CrossRef] [Green Version]
- Contessa, A.R.; Bonmassar, A.; Giampietri, A.; Circolo, A.; Goldin, A.; Fioretti, M.C. In vitro generation of a highly immunogenic subline of L1210 leukemia following exposure to 5-(3,3’-dimethyl-1-triazeno)imidazole-4-carboxamide. Cancer Res. 1981, 41, 2476–2482. [Google Scholar] [PubMed]
- Koido, S.; Kan, S.; Yoshida, K.; Yoshizaki, S.; Takakura, K.; Namiki, Y.; Tsukinaga, S.; Odahara, S.; Kajihara, M.; Okamoto, M.; et al. Immunogenic modulation of cholangiocarcinoma cells by chemoimmunotherapy. Anticancer Res. 2014, 34, 6353–6361. [Google Scholar] [PubMed]
- Botta, C.; Bestoso, E.; Apollinari, S.; Cusi, M.G.; Pastina, P.; Abbruzzese, A.; Sperlongano, P.; Misso, G.; Caraglia, M.; Tassone, P.; et al. Immune-modulating effects of the newest cetuximab-based chemoimmunotherapy regimen in advanced colorectal cancer patients. J. Immunother. 2012, 35, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Botta, C.; Martino, E.C.; Ulivieri, C.; Battaglia, G.; Carfagno, T.; Rossetti, M.G.; Fioravanti, A.; Guidelli, G.M.; Cheleschi, S.; et al. Phase Ib study of poly-epitope peptide vaccination to thymidylate synthase (TSPP) and GOLFIG chemo-immunotherapy for treatment of metastatic colorectal cancer patients. Oncoimmunology 2016, 5, e1101205. [Google Scholar] [CrossRef] [Green Version]
- Correale, P.; Cusi, M.G.; Tsang, K.Y.; Del Vecchio, M.T.; Marsili, S.; Placa, M.L.; Intrivici, C.; Aquino, A.; Micheli, L.; Nencini, C.; et al. Chemo-immunotherapy of metastatic colorectal carcinoma with gemcitabine plus FOLFOX 4 followed by subcutaneous granulocyte macrophage colony-stimulating factor and interleukin-2 induces strong immunologic and antitumor activity in metastatic colon cancer patients. J. Clin. Oncol. 2005, 23, 8950–8958. [Google Scholar] [CrossRef] [Green Version]
- Correale, P.; Del Vecchio, M.T.; Di Genova, G.; Savellini, G.G.; La Placa, M.; Terrosi, C.; Vestri, M.; Urso, R.; Lemonnier, F.; Aquino, A.; et al. 5-fluorouracil-based chemotherapy enhances the antitumor activity of a thymidylate synthase-directed polyepitopic peptide vaccine. J. Natl. Cancer. Inst. 2005, 97, 1437–1445. [Google Scholar] [CrossRef]
- Correale, P.; Del Vecchio, M.T.; La Placa, M.; Montagnani, F.; Di Genova, G.; Savellini, G.G.; Terrosi, C.; Mannucci, S.; Giorgi, G.; Francini, G.; et al. Chemotherapeutic drugs may be used to enhance the killing efficacy of human tumor antigen peptide-specific CTLs. J. Immunother. 2008, 31, 132–147. [Google Scholar] [CrossRef]
- Correale, P.; Sabatino, M.; Cusi, M.G.; Micheli, L.; Nencini, C.; Pozzessere, D.; Petrioli, R.; Aquino, A.; De Vecchis, L.; Turriziani, M.; et al. In vitro generation of cytotoxic T lymphocytes against HLA-A2.1-restricted peptides derived from human thymidylate synthase. J. Chemother. 2001, 13, 519–526. [Google Scholar] [CrossRef]
- Cusi, M.G.; Botta, C.; Pastina, P.; Rossetti, M.G.; Dreassi, E.; Guidelli, G.M.; Fioravanti, A.; Martino, E.C.; Gandolfo, C.; Pagliuchi, M.; et al. Phase I trial of thymidylate synthase poly-epitope peptide (TSPP) vaccine in advanced cancer patients. Cancer Immunol. Immunother. 2015, 64, 1159–1173. [Google Scholar] [CrossRef]
- Galaine, J.; Turco, C.; Vauchy, C.; Royer, B.; Mercier-Letondal, P.; Queiroz, L.; Loyon, R.; Mouget, V.; Boidot, R.; Laheurte, C.; et al. CD4 T cells target colorectal cancer antigens upregulated by oxaliplatin. Int. J. Cancer 2019, 145, 3112–3125. [Google Scholar] [CrossRef] [PubMed]
- Gravett, A.M.; Dalgleish, A.G.; Copier, J. In vitro culture with gemcitabine augments death receptor and NKG2D ligand expression on tumour cells. Sci. Rep. 2019, 9, 1544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Galat, V.; Galat, Y.; Lee, Y.K.A.; Wainwright, D.; Wu, J. NK cell-based cancer immunotherapy: From basic biology to clinical development. J. Hematol. Oncol. 2021, 14, 7. [Google Scholar] [CrossRef] [PubMed]
- Bonmassar, E.; Bonmassar, A.; Vadlamudi, S.; Goldin, A. Immunological alteration of leukemic cells in vivo after treatment with an antitumor drug. Proc. Natl. Acad. Sci. USA 1970, 66, 1089–1095. [Google Scholar] [CrossRef] [Green Version]
- Bethesda. Dacarbazine. In Drugs and Lactation Database (LactMed); National Library of Medicine: Bethesda, MD, USA, 2006. [Google Scholar]
- Nardelli, B.; Contessa, A.R.; Romani, L.; Sava, G.; Nisi, C.; Fioretti, M.C. Immunogenic changes of murine lymphoma cells following in vitro treatment with aryl-triazene derivatives. Cancer Immunol. Immunother. 1984, 16, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Bonmassar, A.; Frati, L.; Fioretti, M.C.; Romani, L.; Giampietri, A.; Goldin, A. Changes of the immunogenic properties of K36 lymphoma treated in vivo with 5(3,3-dimethyl-1-triazeno) imidazole-4-carboxamide (DTIC). Eur. J. Cancer 1979, 15, 933–939. [Google Scholar] [CrossRef]
- Bonmassar, E.; Bonmassar, A.; Vadlamudi, S.; Goldin, A. Antigenic changes of L1210 leukemia in mice treated with 5-(3,3-dimethyl-1-triazeno)imidazole-4-carboxamide. Cancer Res. 1972, 32, 1446–1450. [Google Scholar]
- Bonmassar, E.; Testorelli, C.; Franco, P.; Goldin, A.; Cudkowicz, G. Changes of the immunogenic properties of a radiation-induced mouse lymphoma following treatment with antitumor drugs. Cancer Res. 1975, 35, 1957–1962. [Google Scholar]
- Fioretti, M.C.; Romani, L.; Taramelli, D.; Goldin, A. Antigenic properties of lymphoma sublines derived from a drug-treated immunogenic L5178Y leukemia. Transplantation 1978, 26, 449–451. [Google Scholar] [PubMed]
- Romani, L.; Grohmann, U.; Puccetti, P.; Rossi, M.A.; Fioretti, M.C. Cell-mediated immunity to chemically xenogenized tumors. V. Failure of novel antigens to increase the frequency of tumor-specific cytotoxic T cells. Int. J. Immunopharmacol. 1990, 12, 743–749. [Google Scholar] [CrossRef]
- Puccetti, P.; Romani, L.; Fioretti, M.C. Chemical xenogenization of experimental tumors. Cancer Metastasis Rev. 1987, 6, 93–111. [Google Scholar] [CrossRef]
- Boon, T.; Kellermann, O. Rejection by syngeneic mice of cell variants obtained by mutagenesis of a malignant teratocarcinoma cell line. Proc. Natl. Acad. Sci. USA 1977, 74, 272–275. [Google Scholar] [CrossRef] [Green Version]
- Nicolin, A.; Vadlamudi, S.; Goldin, A. Antigenicity of L1210 leukemic sublines induced by drugs. Cancer Res. 1972, 32, 653–657. [Google Scholar]
- Giampietri, A.; Fioretti, M.C.; Goldin, A.; Bonmassar, E. Drug-mediated antigenic changes in murine leukemia cells: Antagonistic effects of quinacrine, an antimutagenic compound. J. Natl. Cancer Inst. 1980, 64, 297–301. [Google Scholar] [CrossRef]
- Vecchi, A.; Fioretti, M.C.; Mantovani, A.; Barzi, A.; Spreafico, F. The immunodepressive and hematotoxic activity of imidazole-4-carboxamide,5-(3,3-dimethyl-1-triazeno) in mice. Transplantation 1976, 22, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Puccetti, P.; Fuschiotti, P.; Dominici, P.; Borri-Voltattorni, C.; Romani, L.; Fioretti, M.C. DNA methylating activity in murine lymphoma cells xenogenized by triazene derivatives. Int. J. Cancer 1987, 39, 769–773. [Google Scholar] [CrossRef]
- Kobayashi, H.; Kodama, T.; Shirai, T.; Kaji, H.; Hosokawa, M.; Sendo, F.; Saito, H.; Takeichi, N. Artificial regression of rat tumors infected with Friend virus (xenogenization): An effect produced by acquired antigen. J. Med. Sci. 1969, 44, 133–134. [Google Scholar]
- Johnson, H.G.; Bach, M.K. Apparent antimutagenic activity of quinacrine hydrochloride in Detroit-98 human sternal marrow cells grown in culture. Cancer Res. 1969, 29, 1367–1370. [Google Scholar] [PubMed]
- Fioretti, M.C.; Bianchi, R.; Romani, L.; Bonmassar, E. Drug-induced immunogenic changes of murine leukemia cells: Dissociation of onset of resistance and emergence of novel immunogenicity. J. Natl. Cancer. Inst. 1983, 71, 1247–1251. [Google Scholar]
- Grohmann, U.; Puccetti, P.; Belladonna, M.L.; Fallarino, F.; Bianchi, R.; Binaglia, L.; Sagakuchi, K.; Mage, M.G.; Appella, E.; Fioretti, M.C. Multiple point mutations in an endogenous retroviral gene confer high immunogenicity on a drug-treated murine tumor. J. Immunol. 1995, 154, 4630–4641. [Google Scholar]
- Marchesi, F.; Turriziani, M.; Tortorelli, G.; Avvisati, G.; Torino, F.; De Vecchis, L. Triazene compounds: Mechanism of action and related DNA repair systems. Pharmacol. Res. 2007, 56, 275–287. [Google Scholar] [CrossRef]
- Loveless, A. Possible relevance of O-6 alkylation of deoxyguanosine to the mutagenicity and carcinogenicity of nitrosamines and nitrosamides. Nature 1969, 223, 206–207. [Google Scholar] [CrossRef] [PubMed]
- Margison, G.P.; Povey, A.C.; Kaina, B.; Santibanez Koref, M.F. Variability and regulation of O6-alkylguanine-DNA alkyltransferase. Carcinogenesis 2003, 24, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. Rev. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef]
- Wiewrodt, D.; Nagel, G.; Dreimuller, N.; Hundsberger, T.; Perneczky, A.; Kaina, B. MGMT in primary and recurrent human glioblastomas after radiation and chemotherapy and comparison with p53 status and clinical outcome. Int. J. Cancer 2008, 122, 1391–1399. [Google Scholar] [CrossRef]
- Christmann, M.; Nagel, G.; Horn, S.; Krahn, U.; Wiewrodt, D.; Sommer, C.; Kaina, B. MGMT activity, promoter methylation and immunohistochemistry of pretreatment and recurrent malignant gliomas: A comparative study on astrocytoma and glioblastoma. Int. J. Cancer 2010, 127, 2106–2118. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.C.; Gorlia, T.; Hamou, M.F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [Green Version]
- Caporali, S.; Falcinelli, S.; Starace, G.; Russo, M.T.; Bonmassar, E.; Jiricny, J.; D’Atri, S. DNA damage induced by temozolomide signals to both ATM and ATR: Role of the mismatch repair system. Mol. Pharmacol. 2004, 66, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Kaina, B.; Ziouta, A.; Ochs, K.; Coquerelle, T. Chromosomal instability, reproductive cell death and apoptosis induced by O6-methylguanine in Mex-, Mex+ and methylation-tolerant mismatch repair compromised cells: Facts and models. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 1997, 381, 227–241. [Google Scholar] [CrossRef]
- Knizhnik, A.V.; Roos, W.P.; Nikolova, T.; Quiros, S.; Tomaszowski, K.H.; Christmann, M.; Kaina, B. Survival and death strategies in glioma cells: Autophagy, senescence and apoptosis triggered by a single type of temozolomide-induced DNA damage. PLoS ONE 2013, 8, e55665. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Kaina, B. Are There Thresholds in Glioblastoma Cell Death Responses Triggered by Temozolomide? Int. J. Mol. Sci. 2019, 20, 1562. [Google Scholar] [CrossRef] [Green Version]
- Tomicic, M.T.; Christmann, M.; Kaina, B. Topotecan triggers apoptosis in p53-deficient cells by forcing degradation of XIAP and survivin thereby activating caspase-3-mediated Bid cleavage. J. Pharmacol. Exp. Ther. 2010, 332, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotzer, M.A.; Eggert, A.; Zuzak, T.J.; Janss, A.J.; Marwaha, S.; Wiewrodt, B.R.; Ikegaki, N.; Brodeur, G.M.; Phillips, P.C. Resistance to TRAIL-induced apoptosis in primitive neuroectodermal brain tumor cells correlates with a loss of caspase-8 expression. Oncogene 2000, 19, 4604–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonmassar, L.; Marchesi, F.; Pascale, E.; Franzese, O.; Margison, G.P.; Bianchi, A.; D’Atri, S.; Bernardini, S.; Lattuada, D.; Bonmassar, E.; et al. Triazene compounds in the treatment of acute myeloid leukemia: A short review and a case report. Curr. Med. Chem. 2013, 20, 2389–2401. [Google Scholar] [CrossRef]
- Yu, W.; Zhang, L.; Wei, Q.; Shao, A. O(6)-Methylguanine-DNA Methyltransferase (MGMT): Challenges and New Opportunities in Glioma Chemotherapy. Front. Oncol. 2019, 9, 1547. [Google Scholar] [CrossRef] [Green Version]
- Graziani, G.; Faraoni, I.; Grohmann, U.; Bianchi, R.; Binaglia, L.; Margison, G.P.; Watson, A.J.; Orlando, L.; Bonmassar, E.; D’Atri, S. O6-alkylguanine-DNA alkyltransferase attenuates triazene-induced cytotoxicity and tumor cell immunogenicity in murine L1210 leukemia. Cancer Res. 1995, 55, 6231–6236. [Google Scholar]
- Hu, Y.H.; Jiao, B.H.; Wang, C.Y.; Wu, J.L. Refulation of temozolomide resistance in glioma cells via the RIP2/NF-kB/MGMT pathway CNSW. Neurosci. Ther. 2021, 27, 552–563. [Google Scholar] [CrossRef]
- Bianchi, R.; Citti, L.; Beghetti, R.; Romani, L.; D’Incalci, M.; Puccetti, P.; Fioretti, M.C. O6-methylguanine-DNA methyltransferase activity and induction of novel immunogenicity in murine tumor cells treated with methylating agents. Cancer Chemother. Pharmacol. 1992, 29, 277–282. [Google Scholar] [CrossRef]
- Dumenco, L.L.; Allay, E.; Norton, K.; Gerson, S.L. The prevention of thymic lymphomas in transgenic mice by human O6-alkylguanine-DNA alkyltransferase. Science 1993, 259, 219–222. [Google Scholar] [CrossRef]
- Khan, O.; Middleton, M.R. The therapeutic potential of O6-alkylguanine DNA alkyltransferase inhibitors. Expert Opin. Investig. Drugs 2007, 16, 1573–1584. [Google Scholar] [CrossRef]
- Liu, L.; Gerson, S.L. Targeted modulation of MGMT: Clinical implications. Clin. Cancer Res. 2006, 12, 328–331. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Zhao, L.; Zhong, R.; Peng, Y. The specific role of O(6)-methylguanine-DNA methyltransferase inhibitors in cancer chemotherapy. Future Med. Chem. 2018, 10, 1971–1996. [Google Scholar] [CrossRef]
- D’Atri, S.; Graziani, G.; Lacal, P.M.; Nistico, V.; Gilberti, S.; Faraoni, I.; Watson, A.J.; Bonmassar, E.; Margison, G.P. Attenuation of O(6)-methylguanine-DNA methyltransferase activity and mRNA levels by cisplatin and temozolomide in jurkat cells. J. Pharmacol. Exp. Ther. 2000, 294, 664–671. [Google Scholar]
- Kong, X.T.; Nguyen, N.T.; Choi, Y.J.; Zhang, G.; Nguyen, H.N.; Filka, E.; Green, S.; Yong, W.H.; Liau, L.M.; Green, R.M.; et al. Phase 2 Study of Bortezomib Combined With Temozolomide and Regional Radiation Therapy for Upfront Treatment of Patients With Newly Diagnosed Glioblastoma Multiforme: Safety and Efficacy Assessment. Int. J. Radiat. Oncol. Biol. Phys. 2018, 100, 1195–1203. [Google Scholar] [CrossRef]
- Vlachostergios, P.J.; Hatzidaki, E.; Befani, C.D.; Liakos, P.; Papandreou, C.N. Bortezomib overcomes MGMT-related resistance of glioblastoma cell lines to temozolomide in a schedule-dependent manner. Investig. New Drugs 2013, 31, 1169–1181. [Google Scholar] [CrossRef]
- Rahman, M.A.; Gras Navarro, A.; Brekke, J.; Engelsen, A.; Bindesboll, C.; Sarowar, S.; Bahador, M.; Bifulco, E.; Goplen, D.; Waha, A.; et al. Bortezomib administered prior to temozolomide depletes MGMT, chemosensitizes glioblastoma with unmethylated MGMT promoter and prolongs animal survival. Br. J. Cancer 2019, 121, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Li, X.; Gao, F.; de Groot, J.F.; Koul, D.; Yung, W.K.A. PARP mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro-oncol. 2021, 23, 920–931. [Google Scholar] [CrossRef]
- Pegg, A.E.; Boosalis, M.; Samson, L.; Moschel, R.C.; Byers, T.L.; Swenn, K.; Dolan, M.E. Mechanism of inactivation of human O6-alkylguanine-DNA alkyltransferase by O6-benzylguanine. Biochemistry 1993, 32, 11998–12006. [Google Scholar] [CrossRef]
- Zhang, J.; Stevens, M.F.; Bradshaw, T.D. Temozolomide: Mechanisms of action, repair and resistance. Curr. Mol. Pharmacol. 2012, 5, 102–114. [Google Scholar] [CrossRef]
- Barvaux, V.A.; Lorigan, P.; Ranson, M.; Gillum, A.M.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. Sensitization of a human ovarian cancer cell line to temozolomide by simultaneous attenuation of the Bcl-2 antiapoptotic protein and DNA repair by O6-alkylguanine-DNA alkyltransferase. Mol. Cancer Ther. 2004, 3, 1215–1220. [Google Scholar]
- Clemons, M.; Kelly, J.; Watson, A.J.; Howell, A.; McElhinney, R.S.; McMurry, T.B.; Margison, G.P. O6-(4-bromothenyl)guanine reverses temozolomide resistance in human breast tumour MCF-7 cells and xenografts. Br. J. Cancer 2005, 93, 1152–1156. [Google Scholar] [CrossRef] [Green Version]
- Kaina, B.; Margison, G.P.; Christmann, M. Targeting O-methylguanine-DNA methyltransferase with specific inhibitors as a strategy in cancer therapy. Cell. Mol. Life Sci. 2010, 67, 3663–3681. [Google Scholar] [CrossRef]
- Caporaso, P.; Turriziani, M.; Venditti, A.; Marchesi, F.; Buccisano, F.; Tirindelli, M.C.; Alvino, E.; Garbin, A.; Tortorelli, G.; Toppo, L.; et al. Novel role of triazenes in haematological malignancies: Pilot study of Temozolomide, Lomeguatrib and IL-2 in the chemo-immunotherapy of acute leukaemia. DNA Repair 2007, 6, 1179–1186. [Google Scholar] [CrossRef]
- Turriziani, M.; Caporaso, P.; Bonmassar, L.; Buccisano, F.; Amadori, S.; Venditti, A.; Cantonetti, M.; D’Atri, S.; Bonmassar, E. O6-(4-bromothenyl)guanine (PaTrin-2), a novel inhibitor of O6-alkylguanine DNA alkyl-transferase, increases the inhibitory activity of temozolomide against human acute leukaemia cells in vitro. Pharmacol. Res. 2006, 53, 317–323. [Google Scholar] [CrossRef]
- Ranson, M.; Hersey, P.; Thompson, D.; Beith, J.; McArthur, G.A.; Haydon, A.; Davis, I.D.; Kefford, R.F.; Mortimer, P.; Harris, P.A.; et al. Randomized trial of the combination of lomeguatrib and temozolomide compared with temozolomide alone in chemotherapy naive patients with metastatic cutaneous melanoma. J. Clin. Oncol. 2007, 25, 2540–2545. [Google Scholar] [CrossRef] [Green Version]
- Tawbi, H.A.; Villaruz, L.; Tarhini, A.; Moschos, S.; Sulecki, M.; Viverette, F.; Shipe-Spotloe, J.; Radkowski, R.; Kirkwood, J.M. Inhibition of DNA repair with MGMT pseudosubstrates: Phase I study of lomeguatrib in combination with dacarbazine in patients with advanced melanoma and other solid tumours. Br. J. Cancer 2011, 105, 773–777. [Google Scholar] [CrossRef]
- Watson, A.J.; Middleton, M.R.; McGown, G.; Thorncroft, M.; Ranson, M.; Hersey, P.; McArthur, G.; Davis, I.D.; Thomson, D.; Beith, J.; et al. O(6)-methylguanine-DNA methyltransferase depletion and DNA damage in patients with melanoma treated with temozolomide alone or with lomeguatrib. Br. J. Cancer 2009, 100, 1250–1256. [Google Scholar] [CrossRef]
- Khan, O.A.; Ranson, M.; Michael, M.; Olver, I.; Levitt, N.C.; Mortimer, P.; Watson, A.J.; Margison, G.P.; Midgley, R.; Middleton, M.R. A phase II trial of lomeguatrib and temozolomide in metastatic colorectal cancer. Br. J. Cancer 2008, 98, 1614–1618. [Google Scholar] [CrossRef]
- Sabharwal, A.; Waters, R.; Danson, S.; Clamp, A.; Lorigan, P.; Thatcher, N.; Margison, G.P.; Middleton, M.R. Predicting the myelotoxicity of chemotherapy: The use of pretreatment O6-methylguanine-DNA methyltransferase determination in peripheral blood mononuclear cells. Melanoma Res. 2011, 21, 502–508. [Google Scholar] [CrossRef]
- Koch, D.; Hundsberger, T.; Boor, S.; Kaina, B. Local intracerebral administration of O(6)-benzylguanine combined with systemic chemotherapy with temozolomide of a patient suffering from a recurrent glioblastoma. J. Neuro-oncol. 2007, 82, 85–89. [Google Scholar] [CrossRef]
- Maier, P.; Spier, I.; Laufs, S.; Veldwijk, M.R.; Fruehauf, S.; Wenz, F.; Zeller, W.J. Chemoprotection of human hematopoietic stem cells by simultaneous lentiviral overexpression of multidrug resistance 1 and O(6)-methylguanine-DNA methyltransferase(P140K). Gene Ther. 2010, 17, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Tolcher, A.W.; Gerson, S.L.; Denis, L.; Geyer, C.; Hammond, L.A.; Patnaik, A.; Goetz, A.D.; Schwartz, G.; Edwards, T.; Reyderman, L.; et al. Marked inactivation of O6-alkylguanine-DNA alkyltransferase activity with protracted temozolomide schedules. Br. J. Cancer 2003, 88, 1004–1011. [Google Scholar] [CrossRef]
- Omuro, A.; Chan, T.A.; Abrey, L.E.; Khasraw, M.; Reiner, A.S.; Kaley, T.J.; Deangelis, L.M.; Lassman, A.B.; Nolan, C.P.; Gavrilovic, I.T.; et al. Phase II trial of continuous low-dose temozolomide for patients with recurrent malignant glioma. Neuro-oncol. 2013, 15, 242–250. [Google Scholar] [CrossRef]
- Kaina, B.; Fritz, G.; Coquerelle, T. Contribution of O6-alkylguanine and N-alkylpurines to the formation of sister chromatid exchanges, chromosomal aberrations, and gene mutations: New insights gained from studies of genetically engineered mammalian cell lines. Environ. Mol. Mutagen. 1993, 22, 283–292. [Google Scholar] [CrossRef]
- Kaina, B.; Christmann, M. DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair 2019, 78, 128–141. [Google Scholar] [CrossRef]
- Jin, L.; Guo, S.; Zhang, X.; Mo, Y.; Ke, S.; Duan, C. Optimal treatment strategy for adult patients with newly diagnosed glioblastoma: A systematic review and network meta-analysis. Neurosurg. Rev. 2020, 44, 1943–1955. [Google Scholar] [CrossRef]
- Guadagni, F.; Roselli, M.; Fuggetta, M.P.; Perno, C.F.; Goldin, A.; Giuliani, A. Increased immunogenicity of murine lymphoma cells following exposure to gamma rays in vivo. Chemioterapia 1984, 3, 358–364. [Google Scholar]
- Bonmassar, E.; Cudkowicz, G.; Vadlamudi, S.; Goldin, A. Influence of tumor-host differences at a single histocompatibility locus (H-1) on the antileukemic effect of 1,3-bis(2-chloroethyl)-1-nitrosourea (NSC 409962). Cancer Res. 1970, 30, 2538–2542. [Google Scholar]
- Riccardi, C.; Bartocci, A.; Puccetti, P.; Spreafico, F.; Bonmassar, E.; Goldin, A. Combined effects of antineoplastic agents and anti-lymphoma allograft reaction. Eur. J. Cancer 1980, 16, 23–33. [Google Scholar] [CrossRef]
- Wang, W.; Thomas, R.; Sizova, O.; Su, D.M. Thymic Function Associated With Cancer Development, Relapse, and Antitumor Immunity—A Mini-Review. Front. Immunol. 2020, 11, 773. [Google Scholar] [CrossRef]
- Haynes, L.; Maue, A.C. Effects of aging on T cell function. Curr. Opin. Immunol. 2009, 21, 414–417. [Google Scholar] [CrossRef]
- Ahmed, A.; Tait, S.W.G. Targeting immunogenic cell death in cancer. Mol. Oncol. 2020, 14, 2994–3006. [Google Scholar] [CrossRef]
- Sato, H.; Demaria, S.; Ohno, T. The role of radiotherapy in the age of immunotherapy. Jpn. J. Clin. Oncol. 2021, 51, 513–522. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Warren, S.; Adjemian, S.; Agostinis, P.; Martinez, A.B.; Chan, T.A.; Coukos, G.; Demaria, S.; Deutsch, E.; et al. Consensus guidelines for the definition, detection and interpretation of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000337. [Google Scholar] [CrossRef] [Green Version]
- Van Schaik, T.A.; Chen, K.S.; Shah, K. Therapy-Induced Tumor Cell Death: Friend or Foe of Immunotherapy? Front. Oncol. 2021, 11, 678562. [Google Scholar] [CrossRef]
- Feola, S.; Chiaro, J.; Martins, B.; Cerullo, V. Uncovering the Tumor Antigen Landscape: What to Know about the Discovery Process. Cancers 2020, 12, 1660. [Google Scholar] [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020, 11, 1013. [Google Scholar] [CrossRef]
- Solari, J.I.G.; Filippi-Chiela, E.; Pilar, E.S.; Nunes, V.; Gonzalez, E.A.; Figueiro, F.; Andrade, C.F.; Klamt, F. Damage-associated molecular patterns (DAMPs) related to immunogenic cell death are differentially triggered by clinically relevant chemotherapeutics in lung adenocarcinoma cells. BMC Cancer 2020, 20, 474. [Google Scholar] [CrossRef]
- Obeid, M.; Tesniere, A.; Ghiringhelli, F.; Fimia, G.M.; Apetoh, L.; Perfettini, J.L.; Castedo, M.; Mignot, G.; Panaretakis, T.; Casares, N.; et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat. Med. 2007, 13, 54–61. [Google Scholar] [CrossRef]
- Fucikova, J.; Spisek, R.; Kroemer, G.; Galluzzi, L. Calreticulin and cancer. Cell Res. 2021, 31, 5–16. [Google Scholar] [CrossRef]
- Catalan, R.; Orozco-Morales, M.; Hernandez-Pedro, N.Y.; Guijosa, A.; Colin-Gonzalez, A.L.; Avila-Moreno, F.; Arrieta, O. CD47-SIRPalpha Axis as a Biomarker and Therapeutic Target in Cancer: Current Perspectives and Future Challenges in Nonsmall Cell Lung Cancer. J. Immunol. Res. 2020, 2020, 9435030. [Google Scholar] [CrossRef]
- Lamberti, M.J.; Nigro, A.; Mentucci, F.M.; Rumie Vittar, N.B.; Casolaro, V.; Dal Col, J. Dendritic Cells and Immunogenic Cancer Cell Death: A Combination for Improving Antitumor Immunity. Pharmaceutics 2020, 12, 256. [Google Scholar] [CrossRef] [Green Version]
- Cerwenka, A.; Kopitz, J.; Schirmacher, P.; Roth, W.; Gdynia, G. HMGB1: The metabolic weapon in the arsenal of NK cells. Mol. Cell. Oncol. 2016, 3, e1175538. [Google Scholar] [CrossRef] [Green Version]
- Minute, L.; Teijeira, A.; Sanchez-Paulete, A.R.; Ochoa, M.C.; Alvarez, M.; Otano, I.; Etxeberrria, I.; Bolaños, E.; Azpilikueta, A.; Garasa, S.; et al. Cellular cytotoxicity is a form of immunogenic cell death. J. Immunother. Cancer 2020, 8, e000325. [Google Scholar] [CrossRef]
- Wu, L.; Yang, L. The function and mechanism of HMGB1 in lung cancer and its potential therapeutic implications. Oncol. Lett. 2018, 15, 6799–6805. [Google Scholar] [CrossRef]
- Ho, W.S.; Wang, H.; Maggio, D.; Kovach, J.S.; Zhang, Q.; Song, Q.; Marincola, F.M.; Heiss, J.D.; Gilbert, M.R.; Lu, R.; et al. Pharmacologic inhibition of protein phosphatase-2A achieves durable immune-mediated antitumor activity when combined with PD-1 blockade. Nat. Commun. 2018, 9, 2126. [Google Scholar] [CrossRef] [Green Version]
- Opzoomer, J.W.; Sosnowska, D.; Anstee, J.E.; Spicer, J.F.; Arnold, J.N. Cytotoxic Chemotherapy as an Immune Stimulus: A Molecular Perspective on Turning Up the Immunological Heat on Cancer. Front. Immunol. 2019, 10, 1654. [Google Scholar] [CrossRef] [Green Version]
- Pfirschke, C.; Engblom, C.; Rickelt, S.; Cortez-Retamozo, V.; Garris, C.; Pucci, F.; Yamazaki, T.; Poirier-Colame, V.; Newton, A.; Redouane, Y.; et al. Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 2016, 44, 343–354. [Google Scholar] [CrossRef] [Green Version]
- Borrie, A.E.; Vareki, S.M. T Lymphocyte-Based Cancer Immunotherapeutics. Int. Rev. Cell Mol. Biol. 2018, 341, 201–276. [Google Scholar] [CrossRef]
- D’Amico, L.; Menzel, U.; Prummer, M.; Muller, P.; Buchi, M.; Kashyap, A.; Haessler, U.; Yermanos, A.; Gebleux, R.; Briendl, M.; et al. A novel anti-HER2 anthracycline-based antibody-drug conjugate induces adaptive anti-tumor immunity and potentiates PD-1 blockade in breast cancer. J. Immunother. Cancer 2019, 7, 16. [Google Scholar] [CrossRef]
- Wakita, D.; Iwai, T.; Harada, S.; Suzuki, M.; Yamamoto, K.; Sugimoto, M. Cisplatin Augments Antitumor T-Cell Responses Leading to a Potent Therapeutic Effect in Combination With PD-L1 Blockade. Anticancer Res. 2019, 39, 1749–1760. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Bastian, I.N.; Long, M.D.; Dow, M.; Li, W.; Liu, T.; Ngu, R.K.; Antonucci, L.; Huang, J.Y.; Phung, Q.T.; et al. Activation of NF-kappaB and p300/CBP potentiates cancer chemoimmunotherapy through induction of MHC-I antigen presentation. Proc. Natl. Acad. Sci. USA 2021, 118, e2025840118. [Google Scholar] [CrossRef]
- Gasser, S.; Orsulic, S.; Brown, E.J.; Raulet, D.H. The DNA damage pathway regulates innate immune system ligands of the NKG2D receptor. Nature 2005, 436, 1186–1190. [Google Scholar] [CrossRef] [Green Version]
- Ghiringhelli, F.; Menard, C.; Puig, P.E.; Ladoire, S.; Roux, S.; Martin, F.; Solary, E.; Le Cesne, A.; Zitvogel, L.; Chauffert, B. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol. Immunother. 2007, 56, 641–648. [Google Scholar] [CrossRef]
- Khallouf, H.; Marten, A.; Serba, S.; Teichgraber, V.; Buchler, M.W.; Jager, D.; Schmidt, J. 5-Fluorouracil and interferon-alpha immunochemotherapy enhances immunogenicity of murine pancreatic cancer through upregulation of NKG2D ligands and MHC class I. J. Immunother. 2012, 35, 245–253. [Google Scholar] [CrossRef]
- Moschella, F.; Torelli, G.F.; Valentini, M.; Urbani, F.; Buccione, C.; Petrucci, M.T.; Natalino, F.; Belardelli, F.; Foa, R.; Proietti, E. Cyclophosphamide induces a type I interferon-associated sterile inflammatory response signature in cancer patients’ blood cells: Implications for cancer chemoimmunotherapy. Clin. Cancer Res. 2013, 19, 4249–4261. [Google Scholar] [CrossRef] [Green Version]
- Moschella, F.; Valentini, M.; Arico, E.; Macchia, I.; Sestili, P.; D’Urso, M.T.; Alessandri, C.; Belardelli, F.; Proietti, E. Unraveling cancer chemoimmunotherapy mechanisms by gene and protein expression profiling of responses to cyclophosphamide. Cancer Res. 2011, 71, 3528–3539. [Google Scholar] [CrossRef] [Green Version]
- Senovilla, L.; Aranda, F.; Galluzzi, L.; Kroemer, G. Impact of myeloid cells on the efficacy of anticancer chemotherapy. Curr. Opin. Immunol. 2014, 30, 24–31. [Google Scholar] [CrossRef]
- Zitvogel, L.; Apetoh, L.; Ghiringhelli, F.; Kroemer, G. Immunological aspects of cancer chemotherapy. Nat. Rev. Immunol. 2008, 8, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Briegert, M.; Kaina, B. Human monocytes, but not dendritic cells derived from them, are defective in base excision repair and hypersensitive to methylating agents. Cancer Res. 2007, 67, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Goldstein, M.; Heylmann, D.; Kaina, B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS ONE 2012, 7, e39956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, M.; Goldstein, M.; Christmann, M.; Becker, H.; Heylmann, D.; Kaina, B. Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. USA 2011, 108, 21105–21110. [Google Scholar] [CrossRef] [Green Version]
- Bailly, C.; Thuru, X.; Quesnel, B. Combined cytotoxic chemotherapy and immunotherapy of cancer: Modern times. NAR Cancer 2020, 2, zcaa002. [Google Scholar] [CrossRef] [Green Version]
- Derakhshani, A.; Hashemzadeh, S.; Asadzadeh, Z.; Shadbad, M.A.; Rasibonab, F.; Safarpour, H.; Jafarlou, V.; Solimando, A.G.; Racanelli, V.; Singh, P.K.; et al. Cytotoxic T-Lymphocyte Antigen-4 in Colorectal Cancer: Another Therapeutic Side of Capecitabine. Cancers 2021, 13, 2414. [Google Scholar] [CrossRef]
- Cioccoloni, G.; Aquino, A.; Notarnicola, M.; Caruso, M.G.; Bonmassar, E.; Zonfrillo, M.; Caporali, S.; Faraoni, I.; Villiva, C.; Fuggetta, M.P.; et al. Fatty acid synthase inhibitor orlistat impairs cell growth and down-regulates PD-L1 expression of a human T-cell leukemia line. J. Chemother. 2020, 32, 30–40. [Google Scholar] [CrossRef]
- Vacchelli, E.; Eggermont, A.; Fridman, W.H.; Galon, J.; Tartour, E.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Adoptive cell transfer for anticancer immunotherapy. Oncoimmunology 2013, 2, e24238. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Waxman, D.J. Medium dose intermittent cyclophosphamide induces immunogenic cell death and cancer cell autonomous type I interferon production in glioma models. Cancer Lett. 2020, 470, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Hotchkiss, K.M.; Sampson, J.H. Temozolomide treatment outcomes and immunotherapy efficacy in brain tumor. J. Neuro-oncol. 2021, 151, 55–62. [Google Scholar] [CrossRef]
- Zitvogel, L.; Tesniere, A.; Kroemer, G. Cancer despite immunosurveillance: Immunoselection and immunosubversion. Nat. Rev. Immunol. 2006, 6, 715–727. [Google Scholar] [CrossRef]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E.; et al. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef]
- Xiang, Y.; Chen, L.; Li, L.; Huang, Y. Restoration and Enhancement of Immunogenic Cell Death of Cisplatin by Coadministration with Digoxin and Conjugation to HPMA Copolymer. ACS Appl. Mater. Interfaces 2020, 12, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Mignot, G.; Chalmin, F.; Ladoire, S.; Bruchard, M.; Chevriaux, A.; Martin, F.; Apetoh, L.; Rebe, C.; Ghiringhelli, F. 5-Fluorouracil selectively kills tumor-associated myeloid-derived suppressor cells resulting in enhanced T cell-dependent antitumor immunity. Cancer Res. 2010, 70, 3052–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emile, J.F.; Julie, C.; Le Malicot, K.; Lepage, C.; Tabernero, J.; Mini, E.; Folprecht, G.; Van Laethem, J.L.; Dimet, S.; Boulagnon-Rombi, C.; et al. Prospective validation of a lymphocyte infiltration prognostic test in stage III colon cancer patients treated with adjuvant FOLFOX. Eur. J. Cancer 2017, 82, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Denkert, C.; von Minckwitz, G.; Brase, J.C.; Sinn, B.V.; Gade, S.; Kronenwett, R.; Pfitzner, B.M.; Salat, C.; Loi, S.; Schmitt, W.D.; et al. Tumor-infiltrating lymphocytes and response to neoadjuvant chemotherapy with or without carboplatin in human epidermal growth factor receptor 2-positive and triple-negative primary breast cancers. J. Clin. Oncol. 2015, 33, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, H.; Bruchard, M.; Berger, H.; Derangere, V.; Odoul, L.; Euvrard, R.; Ladoire, S.; Chalmin, F.; Vegran, F.; Rebe, C.; et al. Bleomycin exerts ambivalent antitumor immune effect by triggering both immunogenic cell death and proliferation of regulatory T cells. PLoS ONE 2013, 8, e65181. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Kepp, O.; Kroemer, G. Immunogenic cell death in radiation therapy. Oncoimmunology 2013, 2, e26536. [Google Scholar] [CrossRef] [Green Version]
- Mothersill, C.; Seymour, C.B. Radiation-induced bystander effects--implications for cancer. Nat. Rev. Cancer 2004, 4, 158–164. [Google Scholar] [CrossRef]
- Daguenet, E.; Louati, S.; Wozny, A.S.; Vial, N.; Gras, M.; Guy, J.B.; Vallard, A.; Rodriguez-Lafrasse, C.; Magne, N. Radiation-induced bystander and abscopal effects: Important lessons from preclinical models. Br. J. Cancer 2020, 123, 339–348. [Google Scholar] [CrossRef]
- Okwan-Duodu, D.; Pollack, B.P.; Lawson, D.; Khan, M.K. Role of radiation therapy as immune activator in the era of modern immunotherapy for metastatic malignant melanoma. Am. J. Clin. Oncol. 2015, 38, 119–125. [Google Scholar] [CrossRef]
- Golden, E.B.; Apetoh, L. Radiotherapy and immunogenic cell death. Semin. Radiat. Oncol. 2015, 25, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; Kepp, O.; Ghiringhelli, F.; Apetoh, L.; Aymeric, L.; Locher, C.; Tesniere, A.; Martins, I.; Ly, A.; Haynes, N.M.; et al. Chemotherapy and radiotherapy: Cryptic anticancer vaccines. Semin. Immunol. 2010, 22, 113–124. [Google Scholar] [CrossRef]
- Gupta, G.; Borglum, K.; Chen, H. Immunogenic Cell Death: A Step Ahead of Autophagy in Cancer Therapy. J. Cancer Immunol. 2021, 3, 47–59. [Google Scholar] [CrossRef]
- Falasca, L.; Torino, F.; Marconi, M.; Costantini, M.; Pompeo, V.; Sentinelli, S.; De Salvo, L.; Patrizio, M.; Padula, C.; Gallucci, M.; et al. AMBRA1 and SQSTM1 expression pattern in prostate cancer. Apoptosis 2015, 20, 1577–1586. [Google Scholar] [CrossRef] [Green Version]
- White, E.; Lattime, E.C.; Guo, J.Y. Autophagy regulates stress responses, metabolism, and anticancer immunity. Trends Cancer 2021, 8, 778–789. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.; Chargari, C.; Galluzzi, L.; Kroemer, G. Optimising efficacy and reducing toxicity of anticancer radioimmunotherapy. Lancet Oncol. 2019, 20, e452–e463. [Google Scholar] [CrossRef]
- Galluzzi, L.; Humeau, J.; Buque, A.; Zitvogel, L.; Kroemer, G. Immunostimulation with chemotherapy in the era of immune checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2020, 17, 725–741. [Google Scholar] [CrossRef] [PubMed]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Hwang, W.L.; Pike, L.R.G.; Royce, T.J.; Mahal, B.A.; Loeffler, J.S. Safety of combining radiotherapy with immune-checkpoint inhibition. Nat. Rev. Clin. Oncol. 2018, 15, 477–494. [Google Scholar] [CrossRef]
- Tang, J.; Yu, J.X.; Hubbard-Lucey, V.M.; Neftelinov, S.T.; Hodge, J.P.; Lin, Y. Trial watch: The clinical trial landscape for PD1/PDL1 immune checkpoint inhibitors. Nat. Rev. Drug Discov. 2018, 17, 854–855. [Google Scholar] [CrossRef]
- Vacchelli, E.; Aranda, F.; Eggermont, A.; Galon, J.; Sautes-Fridman, C.; Cremer, I.; Zitvogel, L.; Kroemer, G.; Galluzzi, L. Trial Watch: Chemotherapy with immunogenic cell death inducers. Oncoimmunology 2014, 3, e27878. [Google Scholar] [CrossRef] [Green Version]
- Baracco, E.E.; Stoll, G.; Van Endert, P.; Zitvogel, L.; Vacchelli, E.; Kroemer, G. Contribution of annexin A1 to anticancer immunosurveillance. Oncoimmunology 2019, 8, e1647760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schcolnik-Cabrera, A.; Oldak, B.; Juarez, M.; Cruz-Rivera, M.; Flisser, A.; Mendlovic, F. Calreticulin in phagocytosis and cancer: Opposite roles in immune response outcomes. Apoptosis 2019, 24, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wang, G.; Chen, Y.; Wang, H.; Hua, Y.; Cai, Z. Immunogenic cell death in cancer therapy: Present and emerging inducers. J. Cell. Mol. Med. 2019, 23, 4854–4865. [Google Scholar] [CrossRef]
- Lu, Q.; Chen, X.; Wang, S.; Lu, Y.; Yang, C.; Jiang, G. Potential New Cancer Immunotherapy: Anti-CD47-SIRPalpha Antibodies. OncoTargets Ther. 2020, 13, 9323–9331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, Y.; Gao, J.; Fu, Y.; Hua, P.; Jing, Y.; Cai, M.; Wang, H.; Tong, T. The role of CD47-SIRPalpha immune checkpoint in tumor immune evasion and innate immunotherapy. Life Sci. 2021, 273, 119150. [Google Scholar] [CrossRef] [PubMed]
- Zingoni, A.; Vulpis, E.; Loconte, L.; Santoni, A. NKG2D Ligand Shedding in Response to Stress: Role of ADAM10. Front. Immunol. 2020, 11, 447. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Xin, J.; Wang, J.; Yao, C.; Zhang, Z. Role of NKG2D and its ligands in cancer immunotherapy. Am. J. Cancer Res. 2019, 9, 2064–2078. [Google Scholar]
- Martins, I.; Kepp, O.; Galluzzi, L.; Senovilla, L.; Schlemmer, F.; Adjemian, S.; Menger, L.; Michaud, M.; Zitvogel, L.; Kroemer, G. Surface-exposed calreticulin in the interaction between dying cells and phagocytes. Ann. N. Y. Acad. Sci. 2010, 1209, 77–82. [Google Scholar] [CrossRef]
- Grombacher, T.; Mitra, S.; Kaina, B. Induction of the alkyltransferase (MGMT) gene by DNA damaging agents and the glucocorticoid dexamethasone and comparison with the response of base excision repair genes. Carcinogenesis 1996, 17, 2329–2336. [Google Scholar] [CrossRef] [Green Version]
- Aasland, D.; Reich, T.R.; Tomicic, M.T.; Switzeny, O.J.; Kaina, B.; Christmann, M. Repair gene O6 -methylguanine-DNA methyltransferase is controlled by SP1 and up-regulated by glucocorticoids, but not by temozolomide and radiation. J. Neurochem. 2018, 144, 139–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Wu, X. Study and analysis of antitumor resistance mechanism of PD1/PD-L1 immune checkpoint blocker. Cancer Med. 2020, 9, 8086–8121. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Bartok, O.; Patkar, S.; Eli, G.B.; Cohen, S.; Litchfield, K.; Levy, R.; Jimenez-Sanchez, A.; Trabish, S.; Lee, J.S.; et al. UVB-Induced Tumor Heterogeneity Diminishes Immune Response in Melanoma. Cell 2019, 179, 219–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAbee, J.H.; Rath, B.H.; Valdez, K.; Young, D.L.; Wu, X.; Shankavaram, U.T.; Camphausen, K.; Tofilon, P.J. Radiation Drives the Evolution of Orthotopic Xenografts Initiated from Glioblastoma Stem-like Cells. Cancer Res. 2019, 79, 6032–6043. [Google Scholar] [CrossRef] [Green Version]
- Cudkowicz, G.; Bennett, M. Peculiar immunobiology of bone marrow allografts. I. Graft rejection by irradiated responder mice. J. Exp. Med. 1971, 134, 83–102. [Google Scholar] [CrossRef]
- Riccardi, C.; Fioretti, M.C.; Giampietri, A.; Puccetti, P.; Goldin, A.; Bonmassar, E. Growth inhibition of normal or drug-treated lymphoma cells in lethally irradiated mice. J. Natl. Cancer Inst. 1978, 60, 1083–1090. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franzese, O.; Torino, F.; Giannetti, E.; Cioccoloni, G.; Aquino, A.; Faraoni, I.; Fuggetta, M.P.; De Vecchis, L.; Giuliani, A.; Kaina, B.; et al. Abscopal Effect and Drug-Induced Xenogenization: A Strategic Alliance in Cancer Treatment? Int. J. Mol. Sci. 2021, 22, 10672. https://doi.org/10.3390/ijms221910672
Franzese O, Torino F, Giannetti E, Cioccoloni G, Aquino A, Faraoni I, Fuggetta MP, De Vecchis L, Giuliani A, Kaina B, et al. Abscopal Effect and Drug-Induced Xenogenization: A Strategic Alliance in Cancer Treatment? International Journal of Molecular Sciences. 2021; 22(19):10672. https://doi.org/10.3390/ijms221910672
Chicago/Turabian StyleFranzese, Ornella, Francesco Torino, Elisa Giannetti, Giorgia Cioccoloni, Angelo Aquino, Isabella Faraoni, Maria Pia Fuggetta, Liana De Vecchis, Anna Giuliani, Bernd Kaina, and et al. 2021. "Abscopal Effect and Drug-Induced Xenogenization: A Strategic Alliance in Cancer Treatment?" International Journal of Molecular Sciences 22, no. 19: 10672. https://doi.org/10.3390/ijms221910672