
The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders


Myocardial Pathophisiology Area, Centro Nacional de Investigaciones Cardiovasculares (CNIC), 28029 Madrid, Spain
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Int. J. Mol. Sci. 2021, 22(19), 10573; https://doi.org/10.3390/ijms221910573
Submission received: 8 September 2021
/
Revised: 27 September 2021
/
Accepted: 28 September 2021
/
Published: 30 September 2021
(This article belongs to the Special Issue PPARs as Key Mediators of Metabolic and Inflammatory Regulation)
Abstract
:Peroxisome proliferator-activated receptors (PPARs) are ligand-modulated nuclear receptors that play pivotal roles in nutrient sensing, metabolism, and lipid-related processes. Correct control of their target genes requires tight regulation of the expression of different PPAR isoforms in each tissue, and the dysregulation of PPAR-dependent transcriptional programs is linked to disorders, such as metabolic and immune diseases or cancer. Several PPAR regulators and PPAR-regulated factors are epigenetic effectors, including non-coding RNAs, epigenetic enzymes, histone modifiers, and DNA methyltransferases. In this review, we examine advances in PPARα and PPARγ-related epigenetic regulation in metabolic disorders, including obesity and diabetes, immune disorders, such as sclerosis and lupus, and a variety of cancers, providing new insights into the possible therapeutic exploitation of PPAR epigenetic modulation.
1. Introduction
1.1. Peroxisome Proliferator Activated Receptors
Peroxisome proliferator-activated receptors (PPARs) are a group of nuclear receptors (NRs) that act as ligand-activated transcription factors (TFs) [1]. Upon ligand binding, PPARs assemble with retinoid-X-receptors (RXRs), generating dimeric complexes that bind response elements in target genes to exert important regulatory functions [2]. PPARs are well known for their important functions in lipid and glucose homeostasis, nutrient sensing, inflammation, cellular differentiation, and development [3]. There are three PPAR isoforms: PPARα (NR1C1), PPARβ/δ (NR1C2), and PPARγ (NR1C3). The three PPAR isoforms are differentially expressed in distinct tissues and, more importantly, play different and contrasting roles upon ligand activation [4,5]. PPARα is expressed in tissues with high rates of fatty-acid catabolism, such as the liver, where it is mainly expressed. PPARα decreases glycolysis and lipogenesis, while enhancing glucose uptake, glycogen synthesis, and fatty acid oxidation. Although the PPARβ/δ isoform is expressed ubiquitously, its expression is prominent in the gastrointestinal tract and muscle, where it controls metabolism, glucose utilization, and lipid transport. PPARγ is mostly expressed in adipose tissue, where it promotes lipogenesis and adipocyte differentiation. It also improves insulin secretion by pancreatic β-cells, skeletal muscle sensitization to insulin, and gluconeogenesis in the liver.
Like other NRs, PPARs have a well-conserved structure. Between the N-terminal and C-terminal ends are a DNA binding domain (DBD), a flexible hinge, and a ligand-binding domain (LBD) [2]. The DBD includes a structure containing two zinc-fingers that recognize specific DNA sequences in the peroxisome proliferator response elements. These sequences consist of direct repeats of the hexanucleotide AGGTCA separated by a single nucleotide spacer [6]. The LBD contains 13 alpha helices and one four-stranded beta sheet and can interact with several ligands that activate or repress PPAR action [5,7]. Many natural and synthetic lipophilic acids are PPAR ligands, prominent among which are a wide variety of unsaturated fatty acids (docosahexaenoic and eicosapentanoic acids) and eicosanoids. Natural ligands include leukotriene B4 for PPARα and prostaglandin PGJ2 for PPARγ [5]. PPARα is also stimulated by a family of chemicals known as fibrates, such as fenofibrate and clofibrate. Similarly, PPARγ binds a group of synthetic molecules called thiazolidinediones (TZDs), including rosiglitazone and pioglitazone.
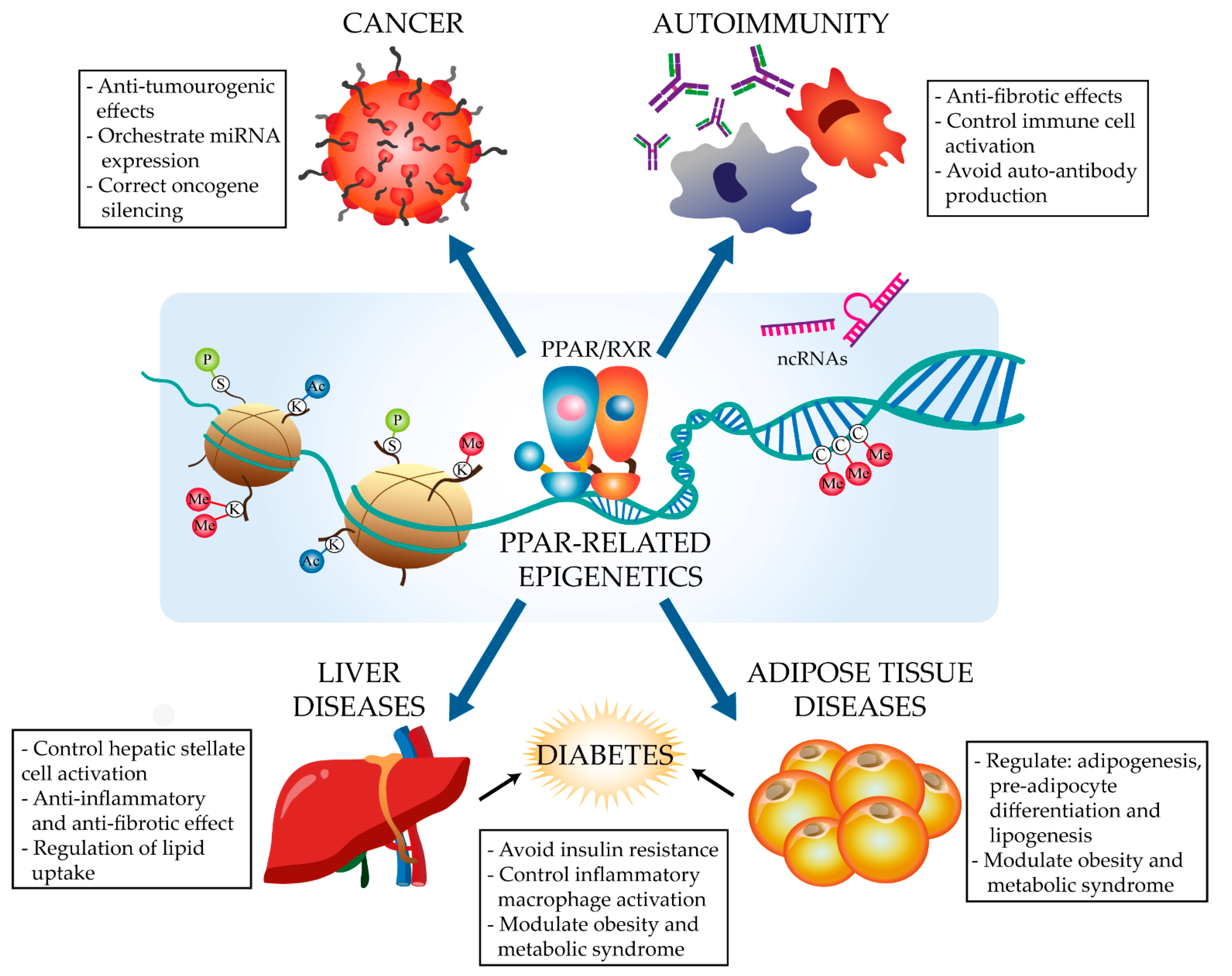
PPARs regulate energy metabolism and inflammation, exerting anti-fibrotic and anti-inflammatory effects in diverse conditions, including cancer, autoimmune diseases, liver steatosis, and type 2 diabetes (T2D) [8,9,10]. PPARs stimulate the expression of anti-inflammatory molecules and inhibit the production of extracellular matrix proteins and pro-inflammatory cytokines, as well as modulating the response and phenotype of immune cells such as macrophages and lymphocytes [10]. The activation of all three isoforms has been demonstrated to polarize macrophages to an anti-inflammatory M2 phenotype and to regulate CD4+ T cell survival and differentiation towards different Th and Treg lineages [11,12]. PPARγ has been demonstrated to act as a key transcription factor in alveolar macrophage and osteoclast identity and ontogeny [13]. While the PPAR pathways implicated in the control of these processes are well characterized [10], little is known about the epigenetic modulation of or by PPARα and PPARγ. Nevertheless, recent research has begun to identify features of the PPAR epigenome in different diseases (Figure 1). These advances, together with the amenability of PPARs to ligand-modulation and the increasing availability of synthetic ligands [5], are driving the study of the complex transcriptional and epigenetic regulation of PPARs in specific diseases. Here, we discuss recent advances focused on the PPARα- and PPARγ-related regulation of non-coding RNAs, histone modification, and DNA methylation in the context of cancer and metabolic and immune-related disorders, as well as the emerging therapeutic potential of these processes in these diseases.
1.2. Epigenetics
The term epigenetics was coined by Conrad Hal Waddington in 1942 to explain the link between genes and the environment. Epigenetics is the study of mechanisms of stable and heritable gene regulation that require no changes to DNA sequence and can be defined as the set of environmental influences that determine a phenotype [14]. The three main epigenetic mechanisms are DNA methylation, histone modification, and the binding of non-coding RNAs to regulatory elements. These mechanisms perpetually modulate gene expression states in order to ensure the correct cellular fate and state without altering the DNA sequence.
1.2.1. Major Epigenetic Modifications
DNA Methylation
DNA methylation, one of the most studied epigenetic modifications, is the addition of a methyl group (-CH3) to the fifth carbon atom of the cytosine ring, generating 5-methylcytosine (5meC). DNA methylation inhibits gene transcription [15] and is catalysed by the m5C DNA methyltransferase (DNMT) family of enzymes. These are classified into three groups, DNMT1, DNMT3a, and DNMT3b, which together establish and sustain the correct DNA methylation patterns. DNMTs, together with partners such as UHRF1 (ubiquitin like with PHD and ring finger domains 1), must be tightly regulated to avoid pathological outcomes, for instance the expression of oncogenes [16,17]. DNA methylation is a reversible epigenetic mark, and the removal of methyl groups is catalysed by the ten-eleven translocation (TET) methylcytosine dioxygenases [18].
Histone Modification
Histones are composed of the protein subunits H2A, H3, H3B, and H4 and act as cores around which DNA winds to form nucleosomes, the building blocks of chromatin. Histones can be modified by acetylation, methylation, ubiquitination, or phosphorylation. These specific modifications, or a combination of them, change the nucleosome conformation, thereby regulating access by the transcriptional machinery to the coding DNA [19,20,21]. The most well-studied histone modifications are acetylation and methylation [22]. Histone acetylation is the most frequent histone modification and is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). When histones are acetylated, the chromatin adopts a more relaxed and open conformation, allowing access to the gene transcription machinery. Histones can be methylated on lysines and arginines by histone methyltransferases (HMTs), with the reverse reaction catalysed by histone demethylases (HDMs) [23,24]. Histone methylation most often induces gene silencing by promoting the recruitment of DNMTs, followed by methyl-binding proteins and finally HDACs [25,26]. Nevertheless, histone methylation can promote the activity of positive transcriptional regulatory elements, such as de novo and pre-disposed enhancers or promoters [27,28,29].
Non-Coding RNAs
Non-coding RNAs (ncRNAs) are RNA molecules that do not translate into proteins but instead play important roles in gene expression regulation both transcriptionally, at the DNA level, and post-transcriptionally, at the mRNA level [30]. ncRNAs are a diverse group of molecules, and it is difficult to make general statements about their function and regulation [31]. The most widely studied ncRNAs in relation to epigenetics are micro RNAs (miRNAs) and long ncRNAs (lncRNAs). miRNAs are typically 18–24 nucleotides long and bind to complementary sequences in target mRNAs, marking them for degradation and thus preventing their translation into protein. lncRNAs, which can exceed 200 nucleotides, have a diverse interactome that includes DNA, proteins, peptides, mRNAs, and miRNAs, through which they regulate both transcription and translation [32].
The epigenome—the complete set of epigenetic marks—must be tightly regulated not only to sustain development and cell fate, but also to prevent pathogenic conditions that could otherwise arise at any moment during life [33]. The dynamic gene regulation afforded by epigenetics ensures a locally appropriate accessibility of chromatin to TFs and, therefore, the execution of a precise transcriptional program [34]. Given the importance of epigenetics for sustained gene transcription, all epigenetics programs within a cell need to work correctly in order to maintain cell function and phenotype, and to prevent possible inflammatory conditions derived from an altered epigenetic landscape [35]. Much recent research interest therefore focuses on the roles of specific epigenetic proteins, such as enzymes and TFs, in a range of biological processes, such as immune metabolism, inflammation, disease, and differentiation. Aberrant DNA and histone methylation, abnormal histone acetylation patterns, and altered ncRNA regulation have been linked to conditions, such as aging, neurological and metabolic disorders, allergies and other autoimmune diseases, and cancer [36,37,38,39,40,41,42,43,44,45,46,47]. Finding molecules able to modulate the epigenome would open up opportunities to specifically treat these conditions.
2. The PPARα and PPARγ Epigenetic Landscape in Disease
2.1. Cancer
PPARs have well known anti-tumourogenic effects [8]. PPARα activation can induce apoptosis and tumour cell death, preventing tumour expansion and inflammation. PPAR-related effects on tumour development have historically been linked to cell-cycle blockade genes such as p18, p21, and p27, leading to apoptosis through the inhibition of B-cell lymphoma 2 (Bcl-2) and reduced angiogenesis through the inhibition of vascular endothelial growth factor (VEGF) [48,49]. The implication of PPAR-related miRNAs and DNA modifications in tumour development has spurred interest in their potential as biomarkers and therapeutic targets. However, the evidence is disputed for some cancers and PPAR isoforms (Table 1).
2.1.1. Colorectal Cancer
Colorectal cancer (CRC) accounts for 7–10% of incident cancers and 3.2% of all cancer-related deaths worldwide, and the incidence is increasing in developed countries [50]. Several studies have shown that PPARγ plays a protective role in CRC and have described the pathways involved downstream of PPAR, opening up the possibility of using PPAR agonists to treat CRC [51]. However, less is known about the upstream pathways and epigenetic mechanisms involved in the action of PPARs in CRC, and research in this area is ongoing.
CRC is often associated with obesity, and the tissue hypoxia characteristic of obesity has been linked to altered expression of typical CRC miRNAs [52]. In 2017, Motawi et al. reported that PPARγ epigenetic regulation contributes to the CRC risk of obese patients [53], showing that obese CRC patients have upregulated expression of the miRNAs miR-27b, miR-130b, and miR-138. In line with the anti-tumourogenic role of PPARγ in CRC, the expression level of these miRNAs correlated negatively with PPARγ mRNA and protein expression, possibly as a result of direct targeting of PPARγ mRNA [53]. Motawi and coworker’s findings are strongly supported by several previous studies [54,55,56,57,58]. However, others reported downregulated expression of miR-27b and miR-138 in colonic cancer cells and tissues [59,60,61], although none of these studies discussed PPARγ. Interestingly, miR-506, which is frequently dysregulated in cancer, has been shown to inhibit PPARα expression in the hydroxicamptothecin-resistant colon cancer cell line SW1116 [62]. Moreover, targeted downregulation of PPAR signalling pathway by a set of miRNAs has been reported in CRC-derived liver metastasis [63]. Together, these findings suggest the therapeutic potential of targeting PPAR-interacting miRNAs in CRC.
There is also evidence for a role in CRC of PPAR-related DNA methylation. UHRF1 was demonstrated to foster Pparg promoter methylation and repressive histone modifications that suppress PPARγ expression in human-derived CRC cell lines [64]. These in vitro results are in step with studies in CRC patients reporting an association between increased methylation of Pparg [65] and PPARγ target genes [66] and decreased PPARγ expression [67]. Furthermore, hypermethylation of the Pparg promoter suppressed PPARγ expression and was associated with CRC regardless of patient body weight [53]. Interestingly, PPARα acts as a suppressor of colon carcinogenesis in mice and is downregulated in mouse colonic tumours. Mice lacking PPARα had increased expression of DNMT1 and protein arginine methyltransferase 6 (PRMT6), resulting in methylation of the tumour suppressor genes P21 and p27, respectively [68]. However, recent evidence indicates that PPARα, along with PPARδ, is overexpressed in human CRC [69]. The inconsistency between these studies could be explained by the significant differences in PPARα expression and function between mice and humans [70].
2.1.2. Liver Cancer
The most common type of primary liver cancer in humans is hepatocellular carcinoma (HCC), which is the third deadliest cancer in the world. A recent analysis of mouse and human single and bulk RNA-seq data revealed that PPARγ controls the expression of a set of antifibrotic miRNAs, including miR-30, miR-29c, and miR-338, that are important for the maintenance of low profibrotic protein levels during HCC-related liver fibrosis [71]. Conversely, other studies have reported that miRNA regulation of the PPAR pathway may contribute to HCC progression. For example, miR-27a inhibits the expression of PPARγ in hepatocarcinoma cells [72]. Interestingly, miR-27a also inhibits RXRα, possibly contributing to cell proliferation in rhabdomyosarcoma [73]. Given that PPAR forms obligate heterodimers with RXRs to regulate transcription, RXR-targeting miRNAs, like miR-27a and miR-34a [74], might also modify the binding capacity and activity of PPAR indirectly. One of the most differentially expressed miRNAs in human HCC samples is miR-9 [72,75], which has been shown to favour tumour growth and aggressiveness. Moreover, bioinformatic analysis identified putative miR-9 binding sites in the PPARα 3′UTR. However, it remains uncertain whether miR-9 contributes to the regulation of PPARα expression in HCC [75].
2.1.3. Other Cancers
PPARγ has been proposed as a therapeutic target in thyroid cancer [76], but although attempts have been made to correlate PPARγ expression with miR-27a, as yet there is no firm evidence linking miRNAs and PPARs in this type of cancer [77]. PPARs are also plausible therapeutic targets in lung cancer. In canine primary lung cancer cells, the Pparg promoter shows a significant loss of 5′-methylation. However, although PPARγ is highly expressed in canine non-small lung cancer cells, this change in the methylation pattern was unrelated to the observed changes in PPARγ protein expression [78]. PPARγ is also dysregulated in gingivo-buccal oral squamous cell carcinoma (OSCC-GB), with OSCC-GB patients showing significant differential methylation of the PPAR pathway genes Cd36, Cyp27a1, Olr1, and Pparg itself. The anti-cancer potential of targeting PPARs is highlighted by the finding that synthetic PPARγ ligands can reduce the incidence of carcinogen-induced tongue tumours [79]. However, current PPARγ ligands are cytotoxic. As an alternative, interest has emerged in the epigenetic action of DNA methyltransferase inhibitors (DNMTI), which is able to renew the transcription of key silenced genes in this cancer, including Pparg [79]. However, as yet, there have been no reports on the molecular mechanism underlying DNA methylation and PPARγ regulation in these tumours. In 1.25-dihydroxyvitamin D3-treated human prostate adenocarcinoma cells, expression of miR-17/92 correlated with PPARα downregulation. However, a direct effect of miR-17/92 on PPARα expression has not been demonstrated experimentally [80].

{kind=link}
Table 1.
PPAR epigenetics in different cancers.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Colorectal cancer | PPARα | miR-506 | PPARα expression inhibition in a hydroxicamptothecin resistant colon cancer cell line. | [62] |
| DNMT1 | Absence of PPARα caused P21 and P27 methylation by DNMT1. | [68] | ||
| PPARγ | miR-27b, miR-130b and miR-138 | Potential downregulation of PPARγ. | [53] | |
| UHRF1 | Epigenetic PPARγ inactivation in human-derived CRC cell lines. | [64] | ||
| Promoter hypermethylation | Hypermethylation of Pparg promoter caused PPARγ suppression. | [53] | ||
| Hepatocellular carcinoma | PPARα | miR-9 | Putative biding sites to PPARα 3’ UTR. | [75] |
| PPARγ | miR-30, miR-29c and miR-338 | Antifibrotic miRNAs regulated by PPARγ during HCC-related liver fibrosis. | [71] | |
| miR-27a | PPARγ inhibition in hepatocarcinoma cells. | [72] | ||
| Thyroid cancer | PPARγ | miR-27a | no relation obsrved yet. | [77] |
| Lung cancer | PPARγ | Promoter methylation | Significantly loss of 5′-methylation. | [78] |
| Gingivo-buccal oral squamous cell carcinoma | PPARγ | DNMTs | DNA methyltransferase inhibitors could renew PPARγ transcription. | [79] |
| Prostate cancer | PPARα | miR-17/92 | Possible direct PPARα targetting and dowregulation. | [80] |
Thus, although research is uncovering new PPAR epigenetics-related factors with potential for the treatment of different types of cancer, much of the evidence has been obtained in vitro or consists of observational data obtained from patient samples. Much further research is therefore needed before the field can contemplate moving to cancer clinical trials of therapies based on the modulation of PPAR epigenetics.
2.2. Immune Disorders
PPARs, especially PPARγ, contribute to the suppression of key pro-inflammatory genes such as NF-kB, INFγ, TNFα, TGFβ, and the interleukins IL-1a and IL-6 [1,10]. These actions are related to the key roles played by PPARs in autoimmune diseases, such as celiac disease [81] and lupus [82]. In sepsis patients and in LPS-treated THP-1 cells PPARγ has been shown to upregulate miR-142-3p. This miRNA targets the 3′-UTR of high mobility group box-1 (HMGB1), a protein with increased expression in many autoimmune diseases, and through miR-142-3p, PPARγ thus contributes to maintaining reduced HMGB1 expression [10,83]. Moreover, several studies have demonstrated PPAR-related regulation of histone and DNA modifications in asthma [84] and lupus [85]. PPARs thus regulate immune-related diseases and have the potential to serve as therapeutic targets in these diseases (Table 2).
2.2.1. Asthma
Asthma is an immune disorder characterized by hyper-responsiveness and inflammation of the airways and involving various immune cell types, such as Th2 lymphocytes or eosinophils and inflammatory cytokines. Asthma affects approximately 300 million people worldwide. Although several treatments are available, including corticoids, not all of them are effective and some can have adverse effects in some individuals. Luckily, accumulating evidence is starting to show that PPARs are not only involved in asthma pathogenesis, but could also serve as targets to reduce asthma symptoms [86].
A well-known cause of asthma is exposure to nicotine. Human primary lung fibroblasts from smokers and mouse primary lung fibroblasts from mice exposed to nicotine both show reduced PPARγ protein levels [87]. In the nicotine-exposed mice, treatment with the PPARγ pathway activator rosiglitazone restores the expression level of miR-98, a miRNA that negatively regulates the expression of airway remodelling proteins associated with collagen deposition and fibrosis [87]. Similarly, pioglitazone-mediated PPARγ activation in rats inhibits airway smooth muscle cell proliferation and remodelling by supressing the Smad-TGFβ1-miR-21 signalling pathway [88]. Human miR-21 is known to target phosphatase and tensin homolog deleted on chromosome ten (PTEN), thereby promoting airway smooth muscle cell proliferation [88]. However, the proposed beneficial role of PPARs in asthma was brought into question by the recent finding that IgE promotes airway inflammatory remodelling in asthma patients by activating the PPARγ pathway [89]. Moreover, there is currently a lack of specific mouse models for studying the implication of immune cells in asthma, thus impeding the identification of immune regulators linked to PPARs and associated miRNAs such as miR-98.
Research into the PPAR epigenetic regulatory network in asthma has also identified a group of lncRNAs in sputa from patients with eosinophilic asthma (the most common type of asthma) that appear to target and modulate PPAR target-gene mRNAs [90]. However, this report did not specify whether the effect was to increase or decrease PPAR pathway activity, and the samples came from a small pool of just six patients [90]. A study of the leukocyte methylome in asthma patients detected PPARα pathway enriched in differentially methylated regions [84], but the study design did not permit identification of the specific cell types affected.
The proposed anti-inflammatory actions of PPARs in asthma thus point pointing to the therapeutic potential of PPAR agonists in asthma-related disorders [86]. However, although evidence of PPAR-related epigenetic mechanisms in asthma is beginning to emerge, the roles of miRNAs, lncRNAs, and DNA methylation in these processes remains largely unknown.
2.2.2. Systemic Lupus Erythematosus
Systemic lupus erythematosus (SLE) is an autoimmune disease in which dysfunctional immune cells, such as antigen presenting cells, T cells, and B cells, lead to a multiple organ malfunction characteristic of each patient [91]. Among several advances in SLE research, PPARγ has emerged as a promising target, and the PPARγ agonists pioglitazone and rosiglitazone have yielded hopeful results in mouse models of the disease [82,92].
Monomethylation of the 20th lysine of histone 4 (H4K20) at the Pparg promoter has been demonstrated to increase the expression of the histone deacetylase HDAC9 [93]. Subsequent analysis of SLE patient samples and mouse models showed that histone modifications at the Pparg promoter influence cytokine and autoantibody production [94]. The authors showed that HDAC9 deletion in mouse CD4+ T cells increased H3K9ac and H3K18ac in the Pparg promoter, prompting a shift in T cell cytokine production towards a more anti-inflammatory class, accompanied by reduced anti-dsDNA autoantibody production by B cells, and therefore protection against proteinuria and renal disease [94].
In a very recent study of CD14+ monocytes from SLE patients, Liu Yu et al. reported the emergence of an immunosuppressive M2-phenotype upon TLR-induced epigenetic activation of PPARγ expression [85]. In these experiments, TLR2 activation with the synthetic ligand Pam3CSK4 triggered decreased expression and binding of the deacetylase Sirt1 to the Pparg promoter. ChIP-qPCR revealed that reduced Sirt1 binding leads to increased histone 3 acetylation in the Pparg promoter, with no changes in histone 4 acetylation, resulting in increased PPARγ protein expression and thus allowing the monocytic transition towards a M2 phenotype [85]. These findings are in line with increased Sirt1 expression in the CD4+ T cells of active SLE patients [95].
Taken together, these results highlight the importance of the epigenetic modulation of PPARγ in autoimmune diseases such as lupus, the protective role of TLR-Sirt1-PPARγ signalling in SLE, and the therapeutic potential of targeting this pathway and histone deactelyases in SLE.
2.2.3. Systemic Sclerosis (Scleroderma)
Systemic sclerosis (SSc), or scleroderma, is a rare and severe autoimmune disease featuring diffuse fibrosis and vascular abnormalities in organs, joints, and skin. Of SSc patients, 30% die within 10 years of diagnosis. One of the main challenges of SSc is the rapid worsening of the disease due to uncontrolled inflammation, collagen deposition, and dysregulation of fibroblast growth [96].
PPARγ expression is low in SSc lesions [97], and in SSc animal models, ligand activation of PPARγ reduces both TGFβ-dependent fibrogenesis and fibroblast hyperactivation [98]. In line with these findings, PPARγ has been shown to reduce Smad-dependent fibroblast activation and differentiation [99], and PPARγ activation blocks recruitment to DNA of the histone acetyl transferase p300 [100]. p300 is required for interaction with Smad3, activation of the pro-fibrogenic Smad3 pathway [101], and histone 4 hyperacetylation at the Col1a2 locus [100]. PPARγ activation thus leads to Smad3 pathway blockade and reduced collagen production, resulting in diminished inflammation and fibrosis [100,102]. Although no effective therapies have yet been devised for SSc [103], epigenetic-based strategies are being postulated as promising future SSc treatments [104,105,106]. The pharmacological modulation of PPARγ is one of the strategies being considered as a means of epigenetically reducing the fibrotic response in SSc patients.
Table 2.
PPAR epigenetics in autoimmune diseases.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Asthma | PPARα | DNA methylation | Human white blood cells showed DNA methylation in several PPAR pathway. | [84] |
| PPARγ | miR-21 | The profibroti Smad-TGFβ1-miR-21c axis was supress upon PPARγ pioglitazone activation. | [87] | |
| miR-98 | This profibrotic miRNA was downregulated upon PPARγ rosiglitazone activation. | [80] | ||
| Not specified | set of lncRNAs | Modulation of PPAR signalling pathway in sputa from eosinophilic asthma patients. | [90] | |
| Systemic Lupus Erythematosus | PPARγ | H4K20me1 and HDAC9 | Decreased H3K9ac and H3K18ac in the Pparg promoter leading to pro-inflammatory T cell cytokines and B cell auto-antibodies. | [93,94] |
| PPARγ | Sirt1 | Reduced PPARγ expression due to H3 deacetylation, avoiding M2 monocytic transition. | [85] | |
| Systemic sclerosis | PPARγ | p300 | Ligand-activated PPARγ blocks histone acetylatransferase p300 avoiding Smad3 pathway activation and Col1a2 locus histone 4 hyperacetylation. | [99,100,101] |
2.3. Metabolism-Related Diseases
Metabolism-related diseases are a broad class of medical conditions, caused by both genetic and non-genetic defects, which lead to altered metabolic processes. These dysfunctions form a group of diseases that frequently derive from widespread nutritionally poor and unhealthy lifestyles [107]. Overnutrition or low-quality nutrition can lead to a wide range of symptoms converging in the pathologic condition called metabolic syndrome [108]. Some of these symptoms are high blood pressure, high levels of triglycerides, low high-density lipoprotein (HDL) concentrations, increased liver fat, non-alcoholic fatty liver disease (NAFLD), elevated amounts of visceral adipose tissue, insulin resistance and diabetes, high inflammatory state, and even cancer [107,109].
Much PPAR research in this area has focused on direct or indirect activation with natural or synthetic ligands [110]. For example, the important role of PPARs in glucose metabolism and effective insulin signaling prompted research into the use of PPARγ-activating TZDs as insulin-sensitizing drugs in T2D [111,112]. More recent approaches have sought to unravel the regulatory networks controlling PPAR expression and function. PPARs clearly play roles spanning many interconnected metabolic disorders. Given the profound effects of transcriptional and epigenetic modulation of PPARs in diverse diseases, new epigenetic targets may have promising therapeutic potential. Here, we focus on the underlying epigenetic mechanisms involving PPARs in three distinct but intimately related metabolic disorders: liver diseases, adipose tissue diseases, and T2D.
2.3.1. Liver Diseases
NAFLD includes a group of liver diseases unrelated to significant alcohol intake. Although the global prevalence and the development of these liver disorders are influenced by ethnicity and geographic origin, there is significant evidence linking NAFLD to poor dietary habits, obesity, adipose tissue dysregulation, and insulin resistance [113]. NAFLD progresses from diet-induced steatosis to a severe inflammatory state, resulting in hepatocyte damage and death that triggers the transdifferentiation of hepatic stellate cells (HSCs) into extracellular matrix-producing myofibroblast-like cells [114]. HSC activation is generally followed by a shift from adipogenesis to a fibrogenic state. This shift is accompanied by a downregulation in the expression of PPARs, which have an anti-inflammatory and protective action in the liver. The shift to fibrogenesis can lead to non-alcoholic steatohepatitis (NASH) and potentially to end-stage liver diseases such as hepatocellular carcinoma.
Several studies have explored epigenetic changes taking place during hepatic metabolic diseases and how they might regulate the expression of PPARs or modulate their binding to promoter and regulatory regions [115,116,117,118] (Table 3). Many epigenetic modifications take place during the progression of steatosis and inflammation and when HSC transdifferentiation begins. For example, many metabolic, proinflammatory, and fibrogenic pathways are regulated by miR-21. This miRNA, which is strongly overexpressed in NASH, represses PPARα expression by direct mRNA targeting and induces HSC activation [119]. Much research into the role PPARs in the hepatic response to dietary fat has focused on the balance between DNA methylation and demethylation and how this determines chromatin accessibility and subsequent changes in gene expression patterns. High dietary fat decreases the methylation of the Ppara promoter, resulting in increased PPARα protein expression and the consequent upregulation of carnitine palmitoyl transferase-1 and downregulation of fatty acid synthase, two important lipid metabolism-related enzymes [120,121]. These changes ensure adequate lipid metabolism in response to high dietary fat intake and reveal the important anti-inflammatory role of PPARα in liver diseases and the complex downstream network it controls.
In newborn and suckling mice, PPARα regulates increased liver DNA demethylation and an accompanying increase in the mRNA expression of β-oxidation-related genes [122]. The molecular mechanism underlying this process has not been thoroughly described. Nonetheless, this metabolic transition makes sense given the high dietary fat intake during suckling. A recent study of the livers of fetal and adult offspring of mice fed a high-fat diet during gestation revealed downregulation of the ten-eleven translocation (TET) enzymes TET1 and TET2, together with hypermethylation of Ppara and correspondingly lower levels of PPARα protein expression [131]. These findings suggest that dietary alterations during gestation and lactation could downregulate TET enzyme expression in offspring, favouring the hypermethylation of Ppara and decreased expression of its lipid metabolism-related target genes. However, further studies are needed to confirm this. TET enzymes require ascorbic acid as a cofactor, and ascorbic acid deficiency during the suckling period increases the hypermethylation of PPARα-dependent lipid metabolism genes such as fibroblast growth factor 21 (Fgf21) [132]. FGF21 is a mainly liver-secreted peptide hormone that stimulates adipocytes to take up glucose from the blood [133,134]. In adult mice, fasting-induced FGF21 signalling triggers further epigenetic modifications, such as phosphorylation of the histone demethylase Jumonji-D3 (JMJD3). Phosphorylated JMJD3 interacts directly with PPARα to upregulate the expression of autophagy-related genes [123]. Since this induced process is closely related to triglyceride hydrolysis and ketone body production, PPARα-dependent FGF21–JMJD3 autophagy signalling emerges as an important endocrine regulator and a potential therapeutic target in metabolic disorders [135,136,137].
Other histone modifying enzymes include protein arginine methyltransferase 5 (PRMT5), which regulates gene expression via the dimethylation of histone residues H4R3, H3R8, and H2R3. These methylation marks induce gene silencing through the recruitment of DNA methyltransferase 3a (DNMT3a). PRMT5 is abundant in the liver of fat-fed mice and is implicated in the development of hepatic steatosis [124]. Reduced or annulled PRMT5 expression triggers the overexpression of PPARα and an increased mitochondrial biogenesis [124]. Similarly, the methyltransferase PRMT6 has shown to be a repressor of PPARγ activity [128]. The repression of PPARs by PRMT activity thus presents a further possible target for the treatment of fatty liver.
Although PPARγ is more weakly expressed in the liver than PPARα, it is essential for liver function, and the DNA methylation status of the Pparg gene has been identified as a marker of liver disease progression. Analysis of the Pparg promoter in plasma cell-free DNA has identified differential DNA methylation patterns in specific CpGs that distinguish between mild and severe fibrosis in NAFLD patients [127]. This cell-free DNA is believed to originate in dying hepatocytes that release their genomic content to the systemic circulation, and thus could provide a noninvasive means of measuring liver status [116]. Taken together, these findings open up new prospective research directions and possibilities for the early diagnosis, screening, and treatment of NAFLD.
PPARγ modulates the expression of lipid uptake and metabolism genes and is a well characterized and important negative regulator of HSC transdifferentiation [125,138]. During this process, downregulation of miR-132 enhances the expression of methyl-CpG binding domain protein 2 (MeCP2), which binds to the 5’ region of Pparg, promoting H3K9 methylation and recruitment of the transcriptional repressor heterochromatin protein 1 (HP1α). MeCP2 additionally promotes expression of the H3K27 methyltransferase EZH2 (enhancer zeste homolog 2), generating a repression complex at the 3’ region of Pparg. Furthermore, MeCP2 induces the expression of the H3K4 methyltransferase ASH1 (absent small and homeotic disks protein 1), which opposes the action of PPARγ by positively regulating the expression of profibrogenic genes [139,140]. In line with these results, miR-132 was recently linked to human NAFLD [141], and strategies targeting MeCP2 and EZH2 have succeeded in decreasing fibrogenic markers characteristics [142,143]. Additionally, a novel mechanism was shown to promote hepatic lipogenesis through the lncRNA-H19/mi-130a/PPARγ axis [126], becoming a potential target to treat NAFLD.
Other miRNAs involved in PPARγ regulation include miR-29a, which is expressed upon rosiglitazone-induced PPARγ activation in a human HSC cell line and results in the inhibition of fibrosis-related genes [144]. Both miR-29a and miR-652 have been shown to contribute to the resolution of liver fibrosis by modulating the activity of CD4+ T cells and HSCs [145,146]. However, as yet, no relationship has been established between the prevention of HSC activation by miR-652 and PPARγ activity.
PPARγ is also involved in the regulation of adipogenic metabolism by certain demethylases that act as essential modulators of hepatic lipid homeostasis. For example, the H3K9-specific Jumonji demethylases JMJD1A and JMJD2B have been reported to bind to the Pparg promoter, and the loss of these enzymes resulted in an increase in the number of H3K9me2 marks in this region, leading to Pparg repression and higher levels of fibrosis markers [129]. Conversely, overexpression of these demethylases upregulated Pparg expression and increased lipid uptake and intracellular triglyceride accumulation, thus favouring adipogenesis and steatosis [130].
2.3.2. Adipose Tissue Diseases
Evidence accumulated over the past 20 years has established that adipose tissue is an endocrine organ involved in a wide array of metabolic and immune processes [147]. Defects in adipose tissue are typically related to obesity, diabetes and insulin resistance, cardiovascular diseases, cancer, longevity, and even fertility [148,149]. The main transcriptional modulators in adipose tissue are CCAAT/enhancer binding proteins (C/EBP) and PPARγ (specifically PPARγ2), which cooperate in fatty acid uptake and in preadipocyte differentiation to the mature adipocyte phenotype [150,151]. Given the important role of PPARγ in lipid homeostasis, there is intense interest in not only the transcriptional, but also the epigenetic regulation of PPARγ in the development and function of adipose tissue (Table 4 and Table 5).
The methylation status of the Pparg promoter undergoes characteristic changes during adipogenesis and obesity. Pparg promoter methylation correlates with low expression of PPARγ in preadipocytes of the mouse cell line 3T3-L1 [152], and preadipocyte differentiation to mature adipocytes is accompanied by progressive Pparg promoter demethylation as the expression of PPARγ protein increases, whereas obesity is associated with the reverse effect, with Pparg methylation increasing as PPARγ expression decreases [152].
Table 4.
PPARα epigenetics in adipose tissue diseases.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARα | Lsd1 | Targets PPARα to control beige adipocyte numbers | [153] |
| Bta-miR-199a-3p, -154c, -320a and -432 | Control lipid metabolism through PPARα | [154] | ||
| miR-519d | Suppresses PPARα protein translation in obese patients | [155] |
The expression and function of PPARγ in adipose tissue is determined by insertions of histone variants and histone modifications. A crucial protein in adipocyte differentiation is the complex formed by E1A-binding protein p400 and bromo-containing protein 8 (p400/Brd8). The p400/Brd8 complex can incorporate the histone variant H2A.Z, which preferentially locates within transcriptional regulatory sequences, into the promoter regions of PPARγ target genes [156]. In line with this finding, knockdown of Brd8 or H2A.Z results in cell arrest at the immature preadipocyte stage [156] because the PPARγ target genes involved in differentiation are incorrectly expressed. Histone modifications have been investigated in a genome-wide analysis in mouse and human adipocytes during adipogenesis, demonstrating enrichment of the H3K4me2/me3 and H3K27ac active histone marks in the promoters of Pparg1 and 2 [157]. Interestingly, a recent study showed that Pparg is repressed by the action of piperine, a major component of black pepper, resulting in the inhibition of various adipogenic genes [158]. In contrast, Pparg expression and lipogenesis are enhanced upon H3K4 methylation by the methyltransferases mixed-lineage leukemia proteins 3 and 4 (MLL3 and MLL4), which form a complex with ASC-2 and are recruited by C/EBPβ to the Pparg locus [159]. Another study reported that MLL4 induces H3K4me3 marks in the promoters of both C/EBPα and PPARγ through a process requiring the histone methylation regulator PTIP [160]. Moreover, MLL4 itself interacts with some adipogenic TFs, such as tonicity-responsive enhancer binding protein (TonEBP), enabling it to bind the Pparg promoter region, increase H3K9me2 marks, and thereby decrease PPARγ expression [161]. Another important methyltransferase in adipocyte differentiation is EZH2, which adds H3K27me marks to the promoter region of the histone deacetylase HDAC9c in adipose tissue, downregulating its expression [162]. Proposals to target EZH2–HDAC9c interaction for the treatment of age-associated osteoporosis and obesity are supported by the report that HDAC9c attenuates adipogenesis by interfering with PPARγ transcriptional activity [163]. Two other methyltransferases of interest are the H3K36 methyltransferase Nsd2 and the lysine methyltransferase 5 (KMT5A, also known as SETD8). Deletion of Nsd2 alters PPARγ target gene expression, adipogenesis, and adipose tissue function [164], whereas KMT5A, a PPARγ target gene expressed during adipocyte differentiation, boosts the expression of PPARγ and the levels of H4K20me marks in other PPARγ target genes in a positive feedback loop [93]. Research has also addressed the role of demethylases in PPARγ regulation in adipose tissue [165], with the histone demethylase JMJD2C reported to downregulate PPARγ transcriptional activation and decrease preadipocyte differentiation, and the H3K9-specific demethylase JHDM2A shown to facilitate the recruitment of PPARγ and RXRα while promoting brown adipogenesis [166,167,168].
Epigenetic analysis of the the Pparg gene has revealed increases in H3K9 and H3K27 acetylation marks, paralleling increased PPARγ expression during the differentiation from preadipocytes to mature adipocytes [169]. PPARγ expression is also increased upon the recruitment of C/EBP and the glucocorticoid receptor (GR) to the Pparg enhancer by a complex formed between RNA polymerase II transcription subunit 1 (MED1) and the histone acetyltransferase p300 [170]. Another study reported that the Pparg promoter and PPARγ target genes are bound by poly(ADP-Ribose)-Polymerase-1 (PARP1), which enhances their expression and thus acts as an adipogenic modulator [171]. However, in contrast with these results, p300 is known to interact with cyclin D1, which inhibits its acetyltransferase activity and thereby reduces Pparg expression [172]. These results provide evidence for a central role of PPARγ in the fine epigenetic regulation of adipocyte differentiation, development, and proliferation
Histone deacetylases regulated during adipogenesis include the fasting-induced NAD-dependent histone deacetylase sirtuin-1 (SIRT1). SIRT1 blocks PPARγ activity by docking with the NR co-repressor (NCoR) and the silencing mediator of retinoid and thyroid hormone receptors (SMRT). The resulting complex occupies PPAR binding sites, inhibiting the expression lipogenesis-related genes [173,174]. This finding has prompted interest in SIRT1 as a potential pharmacological target for obesity and obesity-related diseases [173,175]. Recent studies in mouse models of obesity have already demonstrated that HDAC inhibitors stimulate adipose tissue function and oxidative potential, improving the metabolic profile [176,177,178]. Additionally, epigenetic changes upon PPARγ-ligand binding have been studied in relation to their effects on adipogenesis. Rosiglitazone-induced PPARγ activation was found to require the methylcytosine dioxygenase TET2, which is important for demethylation. TET2 enhances the expression of PPARγ target genes and thus participates as an epigenetic regulator and a transcriptional modulator in adipocytes [179]. In 2017, Duteil and colleagues revealed that the lysine-specific demethylase 1 (Lsd1) targeted Ppara, maintaining the transcriptional program that sustains beige adipocyte homeostasis. PPARα pharmacological intervention could be used to fight obesity by preventing beige-to-white transition [153].
Research in the past few years has uncovered essential roles of ncRNAs in PPARγ regulation in adipose tissue. The levels of specific ncRNAs have been found to oscillate during adipogenesis and obesity, cell commitment, and adipocyte differentiation. For instance, in vitro studies showed that lncRNA U90926 inhibits Pparg promoter activity and therefore decreases its expression [180], whereas nuclear enriched abundant transcript 1 (NEAT1) regulates Pparg splicing [181], and the HOX antisense intergenic RNA (HOTAIR) enhances Pparg expression and adipocyte differentiation [182]. Another study in obese mice showed that lncRNA taurine upregulated gene1 (TUG1) diminished fatty acid accumulation, insulin intolerance, and inflammation by attenuating miR-204 and promoting GLUT4/PPARγ/AKT pathway [183].
Table 5.
PPARγ epigenetics in adipose tissue diseases.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Adipose tissue diseases | PPARγ | U90926 | Inhibition of Pparg transcription activity | [180] |
| NEAT1 | Regulation of Pparg splicing | [178] | ||
| HOTAIR | Increased expression of PPARγ | [182] | ||
| miR-155, miR-221 and miR-122 | Decreased expression of PPARγ in human bone-marrow-derived stromal cells | [184] | ||
| miR-540 | Decreased expression of PPARγ in adipose tissue-derived stromal cells | [185] | ||
| miR-27a/b, miR-31, miR-130/b, miR301a, miR-302a and miR-548d5p | Negative regulation of PPARγ and adipogenesis | [186,187] | ||
| miR-103, miR-143, miR-200a, miR-335 and miR-375 | Upregulation of Pparg | [187,188] | ||
| p400/Brd8 complex | Incorporation of the histone variant H2A.Z, which facilitates the expression of PPARγ target genes | [156] | ||
| MLL3 and MLL4 | Complex with ASC-2. Migration to the Pparg locus and methylation of H3K4, promoting enhanced Pparg expression | [159] | ||
| EZH2 | H3K27 methylation in the Hdac9c promoter. Enhanced adipogenesis | [162] | ||
| SETD8 (KMT5A) | Enhanced H4K20me marks in PPARγ target genes. | [93] | ||
| JMJD2C | Downregulation of PPARγ transcriptional activation | [166] | ||
| JHDM2A (JMJD1A) | Decreased H3K9me2 marks and facilitated recruitment of PPARγ, RXRα and PGC1α | [167,168] | ||
| Cyclin D1 | Interaction with p300 and HDACs to inhibit Pparg expression | [172] | ||
| SIRT1 | Blocked PPARγ mechanism of action | [173,174] | ||
| LncRNA TUG1 and miR-294 | Control fatty acid accumulation through GLUT4/PPARγ/AKT axis | [183] |
MiRNAs described to have an epigenetic effect on Pparg include miR-155, miR-221, and miR-122. These miRNAs are downregulated during adipogenesis in human bone-marrow-derived stromal cells, and their overexpression results in lower levels of PPARγ [184]. Moreover, bovine fat-enriched miRNAs, Bta-miR-199a-3p, -154c, -320a, and -432, targeted both Ppara and Pparg in order to control lipid metabolism [154]. Similarly, miR-540 acts as a negative regulator of adipogenesis in adipose tissue-derived stromal cells through binding to the 3′-UTR region of Pparg transcripts, blocking their expression [185]. Studies in the 3T3-L1 preadipocyte mouse cell line identified miR-27a/b, miR-31, miR-130/b, miR-301a, miR-302a, and miR-548d5p as negative regulators of Pparg expression and thereby inhibitors of adipogenesis [186,187]. In contrast, the expression of miR-103, miR-143, miR-200a, miR-335, and miR-375 accounts for the upregulation of Pparg under high-fat diet conditions [187,188,189]. Interestingly, miR-519d has been shown to be upregulated in obese patients and to suppress PPARα protein translation, resulting in an increased lipid accumulation during pre-adipocyte differentiation [155].
Together, these results demonstrate the importance and complexity of the epigenetic regulation of PPARs in the control of adipogenesis and adipocyte differentiation in homeostatic and pathological conditions. Since the mechanisms by which adipocytes acquire their specific identity are well known, the quest for new therapeutic applications appears to be very promising. Although some of studies cited here were carried out in human preadipocytes and human multipotent adipose-derived stem cells, most research has been performed in adipocytes from mouse models of obesity. Further research into the epigenetic control of PPARs in human studies is thus needed to move the field towards therapeutic applications in obesity and adipose tissue disorders.
2.3.3. Insulin Sensitivity and Resistance: Type 2 Diabetes
Diabetes is a metabolic disorder characterized by an inability to properly clear glucose from the blood. The most common form is T2D, in which two related features converge: insufficient insulin production by pancreatic β-cells and progressive insulin resistance [190]. T2D is intimately associated with obesity, inflammation, ageing, and steroid use, and over the past decades its incidence has worryingly increased in children [191,192,193]. Although research has traditionally focused on insulin signaling defects, some studies have emphasized the transcriptional and epigenetic basis of chronic inflammation in insulin resistance and T2D [194] (Table 6), and others have identified NRs, such as the glucocorticoid and vitamin D receptors, as common mediators of insulin resistance [195].
Although NRs require activating ligands, some researchers have concluded that post-translational modifications such as acetylation increase NR activity in the absence of external ligand [196]. Some histone deacetylases have been implicated in post-translational modifications of PPARs and their activity. High expression of the deacetylase HDAC3 correlated with high levels of proinflammatory markers and insulin resistance in peripheral blood mononuclear cells from T2D patients and hepatocytes from fat-fed E3 rats, which develop metabolic syndrome [197,198]. Inhibition of HDAC3 in adipocytes increased PPARγ acetylation and the expression of PPARγ target genes, including adipokines and adipocyte protein 2, resulting in decreased insulin resistance. These adipokines include adiponectin, which facilitates hepatic glucose output, and leptins, which are important regulators of feeding behaviour [196,199]. Adipose tissue-specific knockout of SIRT 1 triggers a hyperacetylated PPARγ state and enhanced PPARγ activity, leading to increased insulin sensitivity [175]. These results suggest that HDAC inhibitors have the potential to improve insulin sensitivity through a variety of actions. For example, HDAC inhibitors might release PPAR binding sites, as described for SIRT1, and promote maintenance of the acetylated state of PPARs and PPAR target genes. These inhibitors could also stimulate significant PPARγ activation. Recent studies have begun to explore the therapeutic potential of HDAC inhibitors in insulin resistance and obesity [200,201,202]. However, their application to human disease requires further research.
T2D is also closely related to immunity. During diabetes, adipose tissue macrophages (ATMs) are activated and shift to a pro-inflammatory phenotype, contributing to the propagation of the altered metabolic state by expressing the pro-inflammatory cytokines TNFα, IL-6, and MCP-1 [203,204]. Macrophage activation during T2D is in part mediated by epigenetic mechanisms [205]. The regulation of ATM alternative activation and insulin sensitivity correlate with PPARγ activation [206,207,208], and ATM alternative activation is held in check by DNA methylation at the Pparg promoter. DNA methylation blockade at the Pparg promoter boosts macrophage alternative activation, whereas DNA hypermethylation promotes inflammatory responses and insulin resistance [209]. In another study, DNMT3b downregulation in ATMs was found to promote an anti-inflammatory state and enhanced insulin sensitivity, revealing the contribution of DNMT3b-mediated methylation at the Pparg promoter to increased inflammatory conditions and insulin resistance [210]. Studies have also reported the contribution of other DNMTs to the epigenetic control of PPARγ target genes. For instance, hypermethylation of FGF21 by DNMT3a in human adipocytes decreased its expression and correlated with insulin resistance in patients [211]. In another study, methylation of the adiponectin promoter by DNMT1 reduced adiponectin expression in obese mice, and DNMT1 inhibition increased insulin sensitivity and ameliorated glucose intolerance [212]. DNMT inhibitors are thus able to lower DNA methylation that directly affects Pparg and PPARγ target genes, identifying these inhibitors as a promising potential treatment for T2D.
The adipogenesis inhibiting miRNA miR-27a has also been reported to promote insulin resistance [213], acting as a glucose metabolism mediator that regulates the PI3K–Akt–GLUT4 signalling pathway by targeting the 3’UTR region of Pparg transcripts, promoting insulin resistance [213]. MiR27a is also upregulated during obesity and induces ATM proinflammatory activation by targeting Pparg [214].
Table 6.
PPAR epigenetics in type 2 diabetes.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| Insulin sensitivity and resistance: Type 2 Diabetes | PPARγ | miR27-a | Target of Pparg transcripts, promoting insulin resistance. Induction of inflammatory ATM activation in obesity | [213,214] |
| HDAC3 | Decreased expression of PPARγ in E3 rat livers. Correlated with inflammation and insulin resistance | [196,197,198] | ||
| SIRT1 | Control of the PPARγ acetylation status and its activity | [175] | ||
| DNMT3b | Pparg promoter methylation. Increased inflammatory macrophage activation and insulin resistance | [209,210] | ||
| DNMT3a | Fgf21 hypermethylation in human adipocytes, insulin resistance | [211] | ||
| DNMT1 | Adiponectin promoter methylation in obese mice. Glucose intolerance | [212] |
Further epigenetic studies have focused on the PPAR coactivator 1α (PGC1α). This protein binds and modulates the activity of PPARγ and PPARα, thereby indirectly regulating the expression of PPAR target genes and functions [215,216]. Like PPARγ, PGC1α can be regulated by reversible acetylation. Its protein sequence contains 13 lysine acetylation sites, and acetylation/deacetylation of these sites depends on the cell energy state [217]. PGC1α can be activated by deacetylation mediated by SIRT1 [218,219]. This activation promotes the expression of PPAR target genes and increased expression of gluconeogenic genes [220]. In contrast, PGC1α is inactivated by acetylation by p300, SRC1/3, GCN5, or hepatic PCAF, producing the opposite effect [220]. Epigenetic changes thus not only control Pparg expression directly, but also regulate the availability and activity of obligate PPARγ coactivators. These studies increase the relevance of Pparg epigenetic modulation and underline the importance of continuing to develop new therapeutic approaches to apply these observations to the treatment of T2D.
3. Conclusions
Despite the importance of PPARs in the control of inflammation and lipid homeostasis in different disease contexts, efforts to decipher the diversity of PPAR-related epigenetic modulation are still at an early stage. This review provides a broad overview of PPAR biology and epigenetics in different diseases. PPARs have a complex and tightly regulated transcriptional network that when dysregulated can lead to disease conditions such as metabolic disorders, autoimmune diseases, or cancer. Although research in this area has characterized several factors of the PPAR regulatory network, the epigenetic effectors and regulators remain largely unknown. For instance, many studies discussed have established a correlation between PPAR and epigenetics in different diseases but have failed to establish a clear causal relationship. Nonetheless, the current evidence establishes that cancer-related, immune, and metabolic disorders have an epigenetic regulatory basis, in which PPARs act as central regulators of inflammation, fibrosis, immune responses, as well as lipid and glucose homeostasis. Some lines of research suggest a potential for therapeutic strategies based on PPAR epigenetics. For instance, HDAC and DNMT inhibitors could serve as therapies in PPAR-dependent inflammatory diseases such as obesity or cancer. Moreover, some PPAR network epigenetic effectors such as miRNAs could be used as early biomarkers of specific disorders. The PPAR epigenetic network is a fascinating emerging field of study that is beginning to identify promising targets for the treatment of cancer, immune, and metabolic disorders.
Funding
This research was funded by the Ministerio de Ciencia, Innovación y Universidades (MCNU) (SAF2017-90604-REDT-NurCaMeIn, RTI2018-095928-BI00) and the Comunidad de Madrid (MOIR-B2017/BMD-3684) to MR; the MCNU fellowships to JP (FPU17/01731) and to JM-M (PRE2019-087964). The CNIC is supported by the MCNU and the Pro CNIC Foundation.
Acknowledgments
We thank Simon Bartlett for English editing, and María Piedad Menéndez Gutierrez for advice on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mirza, A.Z.; AlThagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The effect of regulating molecules on the structure of the PPAR-RXR complex. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2016, 1861, 1852–1863. [Google Scholar] [CrossRef] [PubMed]
- Feige, J.N.; Gelman, L.; Michalik, L.; Desvergne, B.; Wahli, W. From molecular action to physiological outputs: Peroxisome proliferator-activated receptors are nuclear receptors at the crossroads of key cellular functions. Prog. Lipid Res. 2006, 45, 120–159. [Google Scholar] [CrossRef] [PubMed]
- Hong, F.; Pan, S.; Guo, Y.; Xu, P.; Zhai, Y. PPARs as Nuclear Receptors for Nutrient and Energy Metabolism. Molecules 2019, 24, 2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grygiel-Gorniak, B. Peroxisome proliferator-activated receptors and their ligands: Nutritional and clinical implications—A review. Nutr. J. 2014, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.A.; Rusten, M.; AbuGhazaleh, R.D.; Wuertz, B.; Souksavong, V.; Escher, P.; Ondrey, F. Effects of PPAR-gamma agonists on oral cancer cell lines: Potential horizons for chemopreventives and adjunctive therapies. Head Neck 2020, 42, 2542–2554. [Google Scholar] [CrossRef] [PubMed]
- Zoete, V.; Grosdidier, A.; Michielin, O. Peroxisome proliferator-activated receptor structures: Ligand specificity, molecular switch and interactions with regulators. Biochim. Biophys. Acta 2007, 1771, 915–925. [Google Scholar] [CrossRef] [PubMed]
- Font-Diaz, J.; Jimenez-Panizo, A.; Caelles, C.; Vivanco, M.D.; Perez, P.; Aranda, A.; Estébanez-Perpina, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. Semin. Cancer Biol. 2021, 73, 58–75. [Google Scholar] [CrossRef]
- Fougerat, A.; Montagner, A.; Loiseau, N.; Guillou, H.; Wahli, W. Peroxisome Proliferator-Activated Receptors and Their Novel Ligands as Candidates for the Treatment of Non-Alcoholic Fatty Liver Disease. Cells 2020, 9, 1638. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.; Luo, S.; Zhan, Y.; Lu, Q. The roles of PPARgamma and its agonists in autoimmune diseases: A comprehensive review. J. Autoimmun. 2020, 113, 102510. [Google Scholar] [CrossRef]
- Nobs, S.P.; Natali, S.; Pohlmeier, L.; Okreglicka, K.; Schneider, C.; Kurrer, M.; Sallusto, F.; Kopf, M. PPARgamma in dendritic cells and T cells drives pathogenic type-2 effector responses in lung inflammation. J. Exp. Med. 2017, 214, 3015–3035. [Google Scholar] [CrossRef] [PubMed]
- Housley, W.J.; Adams, C.O.; Vang, A.G.; Brocke, S.; Nichols, F.C.; Lacombe, M.; Rajan, T.V.; Clark, R.B. Peroxisome Proliferator-Activated Receptor gamma Is Required for CD4+ T Cell-Mediated Lymphopenia-Associated Autoimmunity. J. Immunol. 2011, 187, 4161–4169. [Google Scholar] [CrossRef] [Green Version]
- Porcuna, J.; Menendez-Gutierrez, M.P.; Ricote, M. Molecular control of tissue-resident macrophage identity by nuclear receptors. Curr. Opin. Pharmacol. 2020, 53, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Tronick, E.; Hunter, R.G. Waddington, Dynamic Systems, and Epigenetics. Front. Behav. Neurosci. 2016, 10, 107. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2013, 38, 23–38. [Google Scholar] [CrossRef] [Green Version]
- Rajabi, H.; Tagde, A.; Alam, M.; Bouillez, A.; Pitroda, S.; Suzuki, Y.; Kufe, D. DNA methylation by DNMT1 and DNMT3b methyltransferases is driven by the MUC1-C oncoprotein in human carcinoma cells. Oncogene 2016, 35, 6439–6445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, Y.M.; Mahar, M.; Ewan, E.E.; Leahy, K.M.; Zhao, G.; Cavalli, V. Epigenetic regulator UHRF1 inactivates REST and growth suppressor gene expression via DNA methylation to promote axon regeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E12417–E12426. [Google Scholar] [CrossRef] [Green Version]
- Ginno, P.A.; Gaidatzis, D.; Feldmann, A.; Hoerner, L.; Imanci, D.; Burger, L.; Zilbermann, F.; Peters, A.; Edenhofer, F.; Smallwood, S.A.; et al. A genome-scale map of DNA methylation turnover identifies site-specific dependencies of DNMT and TET activity. Nat. Commun. 2020, 11, 2680. [Google Scholar] [CrossRef]
- Giaimo, B.D.; Ferrante, F.; Herchenrother, A.; Hake, S.B.; Borggrefe, T. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin 2019, 12, 37. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.L.; Grant, P.A. The Role of DNA Methylation and Histone Modifications in Transcriptional Regulation in Humans. Subcell Biochem. 2013, 61, 289–317. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Blythe, S.A.; Cha, S.W.; Tadjuidje, E.; Heasman, J.; Klein, P.S. beta-Catenin Primes Organizer Gene Expression by Recruiting a Histone H3 Arginine 8 Methyltransferase, Prmt2. Dev. Cell 2010, 19, 220–231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svobodova Kovarikova, A.; Legartova, S.; Krejci, J.; Bartova, E. H3K9me3 and H4K20me3 represent the epigenetic landscape for 53BP1 binding to DNA lesions. Aging (Albany NY) 2018, 10, 2585–2605. [Google Scholar] [CrossRef]
- Hervouet, E.; Peixoto, P.; Delage-Mourroux, R.; Boyer-Guittaut, M.; Cartron, P.-F. Specific or not specific recruitment of DNMTs for DNA methylation, an epigenetic dilemma. Clin. Epigenet. 2018, 10, 17. [Google Scholar] [CrossRef]
- Di Croce, L.; Raker, V.; Corsaro, M.; Fazi, F.; Fanelli, M.; Faretta, M.; Fuks, F.; Lo Coco, F.; Kouzarides, T.; Nervi, C.; et al. Methyltransferase Recruitment and DNA Hyermethylation of Target Promoters by an Oncogenic Transcription Factor. Science 2002, 295, 1079–1082. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, K.J.; Scelfo, A.; Jammula, S.; Cuomo, A.; Barozzi, I.; Stutzer, A.; Fischle, W.; Bonaldi, T.; Pasini, D. Polycomb-Dependent H3K27me1 and H3K27me2 Regulate Active Transcription and Enhancer Fidelity. Mol. Cell 2014, 53, 49–62. [Google Scholar] [CrossRef] [Green Version]
- Kaikkonen, M.U.; Spann, N.J.; Heinz, S.; Romanoski, C.E.; Allison, K.A.; Stender, J.D.; Chun, H.B.; Tough, D.F.; Prinjha, R.K.; Benner, C.; et al. Remodeling of the Enhancer Landscape during Macrophage Activation Is Coupled to Enhancer Transcription. Mol. Cell 2013, 51, 310–325. [Google Scholar] [CrossRef] [Green Version]
- Lara-Astiaso, D.; Weiner, A.; Lorenzo-Vivas, E.; Zaretsky, I.; Jaitin, D.A.; David, E.; Keren-Shaul, H.; Mildner, A.; Winter, D.; Jung, S.; et al. Immunogenetics. Chromatin state dynamics during blood formation. Science 2014, 345, 943–949. [Google Scholar] [CrossRef] [Green Version]
- Zhang, P.; Wu, W.; Chen, Q.; Chen, M. Non-Coding RNAs and their Integrated Networks. J. Integr. Bioinform. 2019, 16. [Google Scholar] [CrossRef]
- Frias-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Ghidini, M.; Salati, M.; Mirchev, M.B.; Valeri, N.; Hahne, J.C. MicroRNAs (miRNAs) and Long Non-Coding RNAs (lncRNAs) as New Tools for Cancer Therapy: First Steps from Bench to Bedside. Target. Oncol. 2020, 15, 261–278. [Google Scholar] [CrossRef]
- Zhang, W.; Qu, J.; Liu, G.-H.; Belmonte, J.C.I. The ageing epigenome and its rejuvenation. Nat. Rev. Mol. Cell Biol. 2020, 21, 137–150. [Google Scholar] [CrossRef]
- Zhao, Y.; Zheng, D.; Cvekl, A. Profiling of chromatin accessibility and identification of general cis-regulatory mechanisms that control two ocular lens differentiation pathways. Epigenet. Chromatin 2019, 12, 27. [Google Scholar] [CrossRef]
- Chen, S.; Yang, J.; Wei, Y.; Wei, X. Epigenetic regulation of macrophages: From homeostasis maintenance to host defense. Cell. Mol. Immunol. 2020, 17, 36–49. [Google Scholar] [CrossRef] [Green Version]
- Elnady, H.G.; Sherif, L.S.; Kholoussi, N.M.; Ali Azzam, M.; Foda, A.R.; Helwa, I.; Sabry, R.N.; Eissa, E.; Fahmy, R.F. Aberrant Expression of Immune-related MicroRNAs in Pediatric Patients with Asthma. Int. J. Mol. Cell Med. 2020, 9, 246–255. [Google Scholar]
- Neganova, M.E.; Klochkov, S.G.; Aleksandrova, Y.R.; Aliev, G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2020. [Google Scholar] [CrossRef]
- Islam, A.; Mohammad, E.; Khan, M.A. Aberration of the modulatory functions of intronic microRNA hsa-miR-933 on its host gene ATF2 results in type II diabetes mellitus and neurodegenerative disease development. Hum. Genom. 2020, 14, 34. [Google Scholar] [CrossRef]
- Yang, Y.; Wicks, J.; Haitchi, H.M.; Powell, R.M.; Manuyakorn, W.; Howarth, P.H.; Holgate, S.T.; Davies, D.E. Regulation of A Disintegrin and Metalloprotease-33Expression by Transforming Growth Factor-β. Am. J. Respir. Cell Mol. Biol. 2012, 46, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Harb, H.; Raedler, D.; Ballenberger, N.; Bock, A.; Kesper, D.A.; Renz, H.; Schaub, B. Childhood allergic asthma is associated with increased IL-13 and FOXP3 histone acetylation. J. Allergy Clin. Immunol. 2015, 136, 200–202. [Google Scholar] [CrossRef]
- Le, T.N.; Williams, S.R.; Alaimo, J.T.; Elsea, S.H. Genotype and phenotype correlation in 103 individuals with 2q37 deletion syndrome reveals incomplete penetrance and supports HDAC4 as the primary genetic contributor. Am. J. Med Genet. Part A 2019, 179, 782–791. [Google Scholar] [CrossRef]
- Wu, Y.; Cui, W.; Zhang, D.; Wu, W.; Yang, Z. The shortening of leukocyte telomere length relates to DNA hypermethylation of LINE-1 in type 2 diabetes mellitus. Oncotarget 2017, 8, 73964–73973. [Google Scholar] [CrossRef]
- Sun, Z.H.; Liu, Y.H.; Liu, J.D.; Xu, D.D.; Li, X.F.; Meng, X.M.; Ma, T.T.; Huang, C.; Li, J. MeCP2 Regulates PTCH1 Expression Through DNA Methylation in Rheumatoid Arthritis. Inflammation 2017, 40, 1497–1508. [Google Scholar] [CrossRef]
- Raveche, E.S.; Salerno, E.; Scaglione, B.J.; Manohar, V.; Abbasi, F.; Lin, Y.C.; Fredrickson, T.; Landgraf, P.; Ramachandra, S.; Huppi, K.; et al. Abnormal microRNA-16 locus with synteny to human 13q14 linked to CLL in NZB mice. Blood 2007, 109, 5079–5086. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Bidzhekov, K.; Noels, H.; Shagdarsuren, E.; Gan, L.; Denecke, B.; Hristov, M.; Koppel, T.; Jahantigh, M.N.; Lutgens, E.; et al. Delivery of MicroRNA-126 by Apoptotic Bodies Induces CXCL12-Dependent Vascular Protection. Sci. Signal. 2009, 2, ra81. [Google Scholar] [CrossRef]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Dovey, O.M.; Foster, C.T.; Conte, N.; Edwards, S.A.; Edwards, J.M.; Singh, R.; Vassiliou, G.; Bradley, A.; Cowley, S.M. Histone deacetylase 1 and 2 are essential for normal T-cell development and genomic stability in mice. Blood 2013, 121, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Hu, L.; Li, X.; Geng, J.; Dai, M.; Bai, X. MicroRNA-130b promotes lung cancer progression via PPARγ/VEGF-A/BCL-2-mediated suppression of apoptosis. J. Exp. Clin. Cancer Res. 2016, 35, 105. [Google Scholar] [CrossRef] [Green Version]
- Koga, H.; Sakisaka, S.; Harada, M.; Takagi, T.; Hanada, S.; Taniguchi, E.; Kawaguchi, T.; Sasatomi, K.; Kimura, R.; Hashimoto, O.; et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology 2001, 33, 1087–1097. [Google Scholar] [CrossRef]
- Rawla, P.; Sunkara, T.; Barsouk, A. Epidemiology of colorectal cancer: Incidence, mortality, survival, and risk factors. Prz. Gastroenterol. 2019, 14, 89–103. [Google Scholar] [CrossRef]
- Park, J.-I.; Kwak, J.-Y. The Role of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer. PPAR Res. 2012, 2012, 876418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.; Baldassarre, A.; Masotti, A.; Calura, E.; Martini, P.; Vari, R.; Scazzocchio, B.; Gessani, S.; Del Corno, M. Integrated Transcriptome Analysis of Human Visceral Adipocytes Unravels Dysregulated microRNA-Long Non-coding RNA-mRNA Networks in Obesity and Colorectal Cancer. Front. Oncol. 2020, 10, 1089. [Google Scholar] [CrossRef]
- Motawi, T.K.; Shaker, O.G.; Ismail, M.F.; Sayed, N.H. Peroxisome Proliferator-Activated Receptor Gamma in Obesity and Colorectal Cancer: The Role of Epigenetics. Sci. Rep. 2017, 7, 10714. [Google Scholar] [CrossRef]
- Baffa, R.; Fassan, M.; Volinia, S.; O’Hara, B.; Liu, C.G.; Palazzo, J.P.; Gardiman, M.; Rugge, M.; Gomella, L.G.; Croce, C.M.; et al. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. J. Pathol. 2009, 219, 214–221. [Google Scholar] [CrossRef]
- Colangelo, T.; Fucci, A.; Votino, C.; Sabatino, L.; Pancione, M.; Laudanna, C.; Binaschi, M.; Bigioni, M.; Maggi, C.A.; Parente, D.; et al. MicroRNA-130b Promotes Tumor Development and Is Associated with Poor Prognosis in Colorectal Cancer. Neoplasia 2013, 15, 1086–1099. [Google Scholar] [CrossRef]
- Karbiener, M.; Fischer, C.; Nowitsch, S.; Opriessnig, P.; Papak, C.; Ailhaud, G.; Dani, C.; Amri, E.Z.; Scheideler, M. microRNA miR-27b impairs human adipocyte differentiation and targets PPARγ. Biochem. Biophys. Res. Commun. 2009, 390, 247–251. [Google Scholar] [CrossRef]
- Lin, Q.; Gao, Z.; Alarcon, R.M.; Ye, J.; Yun, Z. A role of miR-27 in the regulation of adipogenesis. FEBS J. 2009, 276, 2348–2358. [Google Scholar] [CrossRef]
- Yi, R.; Li, Y.; Wang, F.; Gu, J.; Isaji, T.; Li, J.; Qi, R.; Zhu, X.; Zhao, Y. Transforming growth factor (TGF) β1 acted through miR-130b to increase integrin α5 to promote migration of colorectal cancer cells. Tumor Biol. 2016, 37, 10763–10773. [Google Scholar] [CrossRef]
- Matsuyama, R.; Okuzaki, D.; Okada, M.; Oneyama, C. Micro RNA -27b suppresses tumor progression by regulating ARFGEF 1 and focal adhesion signaling. Cancer Sci. 2016, 107, 28–35. [Google Scholar] [CrossRef]
- Qin, Y.Z.; Xie, X.C.; Liu, H.Z.; Lai, H.; Qiu, H.; Ge, L.Y. Screening and preliminary validation of miRNAs with the regulation of hTERT in colorectal cancer. Oncol. Rep. 2015, 33, 2728–2736. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Yu, H.; Yi, S.; Peng, X.; Su, P.; Xiao, Z.; Liu, R.; Tang, A.; Li, X.; Liu, F.; et al. The tumor suppressor miR-138-5p targets PD-L1 in colorectal cancer. Oncotarget 2016, 7, 45370–45384. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.L.; Zhang, C.P.; Nie, F.; Xu, X.T.; Zhu, M.M.; Xiao, S.D.; Ran, Z.H. MicroRNA 506 regulates expression of PPAR alpha in hydroxycamptothecin-resistant human colon cancer cells. FEBS Lett. 2011, 585, 3560–3568. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Li, H.; Sun, L.; Shen, S.; Zhou, Q.; Yuan, Y.; Xing, C. Epigenetic Alternations of MicroRNAs and DNA Methylation Contribute to Liver Metastasis of Colorectal Cancer. Dig. Dis. Sci. 2019, 64, 1523–1534. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, L.; Fucci, A.; Pancione, M.; Carafa, V.; Nebbioso, A.; Pistore, C.; Babbio, F.; Votino, C.; Laudanna, C.; Ceccarelli, M.; et al. UHRF1 coordinates peroxisome proliferator activated receptor gamma (PPARG) epigenetic silencing and mediates colorectal cancer progression. Oncogene 2012, 31, 5061–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.B.; Lee, E.J.; Jung, E.H.; Chun, H.K.; Chang, D.K.; Song, S.Y.; Park, J.; Kim, D.H. Aberrant Methylation of APC, MGMT, RASSF2A, and Wif-1 Genes in Plasma as a Biomarker for Early Detection of Colorectal Cancer. Clin. Cancer Res. 2009, 15, 6185–6191. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Chu, F.H.; Xu, W.R.; Sun, J.Q.; Sun, X.; Ma, X.M.; Yu, M.W.; Yang, G.W.; Wang, X.M. Identification of regulatory role of DNA methylation in colon cancer gene expression via systematic bioinformatics analysis. Medicine 2017, 96, e8487. [Google Scholar] [CrossRef] [PubMed]
- Pancione, M.; Sabatino, L.; Fucci, A.; Carafa, V.; Nebbioso, A.; Forte, N.; Febbraro, A.; Parente, D.; Ambrosino, C.; Normanno, N.; et al. Epigenetic Silencing of Peroxisome Proliferator-Activated Receptor γ Is a Biomarker for Colorectal Cancer Progression and Adverse Patients’ Outcome. PLoS ONE 2010, 5, e14229. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARα Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Yaghoubizadeh, M.; Pishkar, L.; Basati, G. Aberrant Expression of Peroxisome Proliferator-Activated Receptors in Colorectal Cancer and Their Association with Cancer Progression and Prognosis. Gastrointest. Tumors 2020, 7, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Nagano, T.; Shah, Y.; Cheung, C.; Ito, S.; Gonzalez, F.J. The PPARα-Humanized Mouse: A Model to Investigate Species Differences in Liver Toxicity Mediated by PPARα. Toxicol. Sci. 2008, 101, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.; Bitter, C.; Winkler, S.; Weichenhan, D.; Thavamani, A.; Hengstler, J.G.; Borkham-Kamphorst, E.; Kohlbacher, O.; Plass, C.; Geffers, R.; et al. Identification of Pparγ-modulated miRNA hubs that target the fibrotic tumor microenvironment. Proc. Natl. Acad. Sci. USA 2020, 117, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, J.; Fei, B.-Y.; Shao, D.; Pan, Y.; Mo, Z.-H.; Sun, B.-Z.; Zhang, D.; Zheng, X.; Zhang, M.; et al. MiR-27a Promotes Hepatocellular Carcinoma Cell Proliferation Through Suppression of its Target Gene Peroxisome Proliferator-activated Receptor γ. Chin. Med. J. 2015, 128, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Tombolan, L.; Zampini, M.; Casara, S.; Boldrin, E.; Zin, A.; Bisogno, G.; Rosolen, A.; De Pitta, C.; Lanfranchi, G. MicroRNA-27a Contributes to Rhabdomyosarcoma Cell Proliferation by Suppressing RARA and RXRA. PLoS ONE 2015, 10, e0125171. [Google Scholar] [CrossRef]
- Oda, Y.; Nakajima, M.; Tsuneyama, K.; Takamiya, M.; Aoki, Y.; Fukami, T.; Yokoi, T. Retinoid X receptor α in human liver is regulated by miR-34a. Biochem. Pharmacol. 2014, 90, 179–187. [Google Scholar] [CrossRef] [Green Version]
- Cui, M.; Xiao, Z.; Wang, Y.; Zheng, M.; Song, T.; Cai, X.; Sun, B.; Ye, L.; Zhang, X. Long Noncoding RNA HULC Modulates Abnormal Lipid Metabolism in Hepatoma Cells through an miR-9–Mediated RXRA Signaling Pathway. Cancer Res. 2015, 75, 846–857. [Google Scholar] [CrossRef] [Green Version]
- Catalano, M.G.; Poli, R.; Pugliese, M.; Fortunati, N.; Boccuzzi, G. Emerging molecular therapies of advanced thyroid cancer. Mol. Asp. Med. 2010, 31, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Toraih, E.A.; Fawzy, M.S.; Abushouk, A.I.; Shaheen, S.; Hobani, Y.H.; Alruwetei, A.M.; Mansouri, O.A.; Kandil, E.; Badran, D.I. Prognostic value of the miRNA-27a and PPAR/RXRα signaling axis in patients with thyroid carcinoma. Epigenomics 2020, 12, 1825–1843. [Google Scholar] [CrossRef]
- Herrera, C.L.; Kim, D.Y.; Kumar, S.R.; Bryan, J.N. Peroxisome proliferator activated receptor γ protein expression is asymmetrically distributed in primary lung tumor and metastatic to lung osteosarcoma samples and does not correlate with gene methylation. BMC Veter-Res. 2015, 11, 230. [Google Scholar] [CrossRef] [Green Version]
- Das, D.; Ghosh, S.; Maitra, A.; Biswas, N.K.; Panda, C.K.; Roy, B.; Sarin, R.; Majumder, P.P. Epigenomic dysregulation-mediated alterations of key biological pathways and tumor immune evasion are hallmarks of gingivo-buccal oral cancer. Clin. Epigenet. 2019, 11, 178. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-L.W.; Welsh, J.; Tenniswood, M. 1,25-Dihydroxyvitamin D3 modulates lipid metabolism in prostate cancer cells through miRNA mediated regulation of PPARA. J. Steroid Biochem. Mol. Biol. 2013, 136, 247–251. [Google Scholar] [CrossRef]
- Mohan, M.; Okeoma, C.M.; Sestak, K. Dietary Gluten and Neurodegeneration: A Case for Preclinical Studies. Int. J. Mol. Sci. 2020, 21, 5407. [Google Scholar] [CrossRef] [PubMed]
- Aprahamian, T.; Bonegio, R.G.; Weitzner, Z.; Gharakhanian, R.; Rifkin, I.R. Peroxisome proliferator-activated receptor gamma agonists in the prevention and treatment of murine systemic lupus erythematosus. Immunology 2014, 142, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Luo, G.; Li, X.; Chen, J.; Wu, J.; Peng, Y. PPARγ inhibits HMGB1 expression through upregulation of miR-142-3p in vitro and in vivo. Cell. Signal. 2016, 28, 158–164. [Google Scholar] [CrossRef]
- Jeong, A.; Imboden, M.; Ghantous, A.; Novoloaca, A.; Carsin, A.-E.; Kogevinas, M.; Schindler, C.; Lovison, G.; Herceg, Z.; Cuenin, C.; et al. DNA Methylation in Inflammatory Pathways Modifies the Association between BMI and Adult-Onset Non-Atopic Asthma. Int. J. Environ. Res. Public Health 2019, 16, 600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Luo, S.; Zhan, Y.; Wang, J.; Zhao, R.; Li, Y.; Zeng, J.; Lu, Q. Increased Expression of PPAR-γ Modulates Monocytes Into a M2-Like Phenotype in SLE Patients: An Implicative Protective Mechanism and Potential Therapeutic Strategy of Systemic Lupus Erythematosus. Front. Immunol. 2021, 11, 579372. [Google Scholar] [CrossRef]
- Banno, A.; Reddy, A.; Lakshmi, S.; Reddy, A.R.C. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl. Recept. Res. 2018, 5, 101306. [Google Scholar] [CrossRef] [Green Version]
- Wongtrakool, C.; Ko, J.; Jang, A.J.; Grooms, K.; Chang, S.; Sylber, C.; Kosmider, B.; Bahmed, K.; Blackburn, M.R.; Sutliff, R.L.; et al. MicroRNA-98 reduces nerve growth factor expression in nicotine-induced airway remodeling. J. Biol. Chem. 2020, 295, 18051–18064. [Google Scholar] [CrossRef]
- Liu, L.; Pan, Y.; Zhai, C.; Zhu, Y.; Ke, R.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; et al. Activation of peroxisome proliferation–activated receptor-γ inhibits transforming growth factor-β1-induced airway smooth muscle cell proliferation by suppressing Smad–miR-21 signaling. J. Cell. Physiol. 2018, 234, 669–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fang, L.; Wang, X.; Sun, Q.; Papakonstantinou, E.; S’Ng, C.; Tamm, M.; Stolz, D.; Roth, M. IgE Downregulates PTEN through MicroRNA-21-5p and Stimulates Airway Smooth Muscle Cell Remodeling. Int. J. Mol. Sci. 2019, 20, 875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.-J.; Mao, D.; Gao, W.; Hu, H. Peripheral whole blood lncRNA expression analysis in patients with eosinophilic asthma. Medicine 2018, 97, e9817. [Google Scholar] [CrossRef]
- Moulton, V.R.; Suárez-Fueyo, A.; Meidan, E.; Li, H.; Mizui, M.; Tsokos, G.C. Pathogenesis of Human Systemic Lupus Erythematosus: A Cellular Perspective. Trends Mol. Med. 2017, 23, 615–635. [Google Scholar] [CrossRef]
- Rőszer, T.; Menendez-Gutierrez, M.P.; Lefterova, M.I.; Alameda, D.; Nunez, V.; Lazar, M.A.; Fischer, T.; Ricote, M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J. Immunol. 2011, 186, 621–631. [Google Scholar] [CrossRef]
- Wakabayashi, K.-I.; Okamura, M.; Tsutsumi, S.; Nishikawa, N.S.; Tanaka, T.; Sakakibara, I.; Kitakami, J.-I.; Ihara, S.; Hashimoto, Y.; Hamakubo, T.; et al. The Peroxisome Proliferator-Activated Receptor γ/Retinoid X Receptor α Heterodimer Targets the Histone Modification Enzyme PR-Set7/Setd8 Gene and Regulates Adipogenesis through a Positive Feedback Loop. Mol. Cell. Biol. 2009, 29, 3544–3555. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Cao, Q.; Reilly, C.; Young, N.; Garcia, B.A.; Mishra, N. Histone Deacetylase 9 Deficiency Protects against Effector T Cell-mediated Systemic Autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [Green Version]
- Hu, N.; Qiu, X.; Luo, Y.; Yuan, J.; Li, Y.; Lei, W.; Zhang, G.; Zhou, Y.; Su, Y.; Lu, Q. Abnormal histone modification patterns in lupus CD4+ T cells. J. Rheumatol. 2008, 35, 804–810. [Google Scholar]
- Xu, X.; Ramanujam, M.; Visvanathan, S.; Assassi, S.; Liu, Z.; Li, L. Transcriptional insights into pathogenesis of cutaneous systemic sclerosis using pathway driven meta-analysis assisted by machine learning methods. PLoS ONE 2020, 15, e0242863. [Google Scholar] [CrossRef]
- Wei, J.; Ghosh, A.K.; Sargent, J.L.; Komura, K.; Wu, M.; Huang, Q.-Q.; Jain, M.; Whitfield, M.L.; Feghali-Bostwick, C.; Varga, J. PPARγ Downregulation by TGFß in Fibroblast and Impaired Expression and Function in Systemic Sclerosis: A Novel Mechanism for Progressive Fibrogenesis. PLoS ONE 2010, 5, e13778. [Google Scholar] [CrossRef] [Green Version]
- Kohno, S.; Endo, H.; Hashimoto, A.; Hayashi, I.; Murakami, Y.; Kitasato, H.; Kojima, F.; Kawai, S.; Kondo, H. Inhibition of skin sclerosis by 15deoxy Δ12,14-prostaglandin J2 and retrovirally transfected prostaglandin D synthase in a mouse model of bleomycin-induced scleroderma. Biomed. Pharmacother. 2006, 60, 18–25. [Google Scholar] [CrossRef]
- Nuwormegbe, S.A.; Sohn, J.H.; Kim, S.W. A PPAR-Gamma Agonist Rosiglitazone Suppresses Fibrotic Response in Human Pterygium Fibroblasts by Modulating the p38 MAPK Pathway. Investig. Opthalmol. Vis. Sci. 2017, 58, 5217–5226. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Bhattacharyya, S.; Wei, J.; Kim, S.; Barak, Y.; Mori, Y.; Varga, J. Peroxisome proliferator-activated receptor-γ abrogates Smad-dependent collagen stimulation by targeting the p300 transcriptional coactivator. FASEB J. 2009, 23, 2968–2977. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Xia, Y.; Lin, X.; Feng, X.-H.; Wang, Y. Smad3 signaling activates bone marrow-derived fibroblasts in renal fibrosis. Lab. Investig. 2014, 94, 545–556. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Bhattacharyya, S.; Lakos, G.; Chen, S.-J.; Mori, Y.; Varga, J. Disruption of transforming growth factor ? Signaling and profibrotic responses in normal skin fibroblasts by peroxisome proliferator-activated receptor? Arthritis Rheum. 2004, 50, 1305–1318. [Google Scholar] [CrossRef]
- Connolly, M.K. Systemic sclerosis (scleroderma): Remaining challenges. Ann. Transl. Med. 2021, 9, 438. [Google Scholar] [CrossRef]
- Ding, W.; Pu, W.; Wang, L.; Jiang, S.; Zhou, X.; Tu, W.; Yu, L.; Zhang, J.; Guo, S.; Liu, Q.; et al. Genome-Wide DNA Methylation Analysis in Systemic Sclerosis Reveals Hypomethylation of IFN-Associated Genes in CD4+ and CD8+ T Cells. J. Investig. Dermatol. 2018, 138, 1069–1077. [Google Scholar] [CrossRef] [Green Version]
- Hemmatazad, H.; Rodrigues, H.M.; Maurer, B.; Brentano, F.; Pileckyte, M.; Distler, J.H.W.; Gay, R.E.; Michel, B.A.; Gay, S.; Huber, L.C.; et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. 2009, 60, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Huber, L.C.; Distler, J.H.W.; Moritz, F.; Hemmatazad, H.; Hauser, T.; Michel, B.A.; Gay, R.E.; Matucci-Cerinic, M.; Gay, S.; Distler, O.; et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007, 56, 2755–2764. [Google Scholar] [CrossRef]
- Saklayen, M.G. The Global Epidemic of the Metabolic Syndrome. Curr. Hypertens. Rep. 2018, 20, 12. [Google Scholar] [CrossRef] [Green Version]
- Sherling, D.H.; Perumareddi, P.; Hennekens, C.H. Metabolic Syndrome. J. Cardiovasc. Pharmacol. Ther. 2017, 22, 365–367. [Google Scholar] [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef]
- Hong, F.; Xu, P.; Zhai, Y. The Opportunities and Challenges of Peroxisome Proliferator-Activated Receptors Ligands in Clinical Drug Discovery and Development. Int. J. Mol. Sci. 2018, 19, 2189. [Google Scholar] [CrossRef] [Green Version]
- Bansal, G.; Thanikachalam, P.V.; Maurya, R.K.; Chawla, P.; Ramamurthy, S. An overview on medicinal perspective of thiazolidine-2,4-dione: A remarkable scaffold in the treatment of type 2 diabetes. J. Adv. Res. 2020, 23, 163–205. [Google Scholar] [CrossRef] [PubMed]
- Hevener, A.L.; Olefsky, J.M.; Reichart, D.; Nguyen, M.T.; Bandyopadyhay, G.; Leung, H.Y.; Watt, M.J.; Benner, C.; Febbraio, M.A.; Nguyen, A.K.; et al. Macrophage PPAR gamma is required for normal skeletal muscle and hepatic insulin sensitivity and full antidiabetic effects of thiazolidinediones. J. Clin. Investig. 2007, 117, 1658–1669. [Google Scholar] [CrossRef]
- Brunt, E.M.; Wong, V.W.-S.; Nobili, V.; Day, C.P.; Sookoian, S.; Maher, J.J.; Bugianesi, E.; Sirlin, C.B.; Neuschwander-Tetri, B.A.; Rinella, M.E. Nonalcoholic fatty liver disease. Nat. Rev. Dis. Prim. 2015, 1, 15080. [Google Scholar] [CrossRef]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef]
- Hajri, T.; Zaiou, M.; Fungwe, T.; Ouguerram, K.; Besong, S. Epigenetic Regulation of Peroxisome Proliferator-Activated Receptor Gamma Mediates High-Fat Diet-Induced Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 1355. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Mann, J. Epigenetics and Liver Fibrosis. Cell. Mol. Gastroenterol. Hepatol. 2017, 4, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Claveria-Cabello, A.; Colyn, L.; Arechederra, M.; Urman, J.M.; Berasain, C.; Avila, M.A.; Fernandez-Barrena, M.G. Epigenetics in Liver Fibrosis: Could HDACs Be a Therapeutic Target? Cells 2020, 9, 2321. [Google Scholar] [CrossRef]
- Sodum, N.; Kumar, G.; Bojja, S.L.; Kumar, N.; Rao, C.M. Epigenetics in NAFLD/NASH: Targets and therapy. Pharmacol. Res. 2021, 167, 105484. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Rodrigues, C.; Castro, R.E. Modulation of liver steatosis by miR-21/PPARα. Cell Death Discov. 2018, 4, 9. [Google Scholar] [CrossRef]
- Moody, L.; Xu, G.B.; Chen, H.; Pan, Y.-X. Epigenetic regulation of carnitine palmitoyltransferase 1 (Cpt1a) by high fat diet. Biochim. Biophys. Acta (BBA)-Bioenerg. 2018, 1862, 141–152. [Google Scholar] [CrossRef]
- Wang, L.; Chen, L.; Tan, Y.; Wei, J.; Chang, Y.; Jin, T.; Zhu, H. Betaine supplement alleviates hepatic triglyceride accumulation of apolipoprotein E deficient mice via reducing methylation of peroxisomal proliferator-activated receptor alpha promoter. Lipids Health Dis. 2013, 12, 34. [Google Scholar] [CrossRef] [Green Version]
- Ehara, T.; Kamei, Y.; Yuan, X.; Takahashi, M.; Kanai, S.; Tamura, E.; Tsujimoto, K.; Tamiya, T.; Nakagawa, Y.; Shimano, H.; et al. Ligand-Activated PPARα-Dependent DNA Demethylation Regulates the Fatty Acid β-Oxidation Genes in the Postnatal Liver. Diabetes 2014, 64, 775–784. [Google Scholar] [CrossRef] [Green Version]
- Byun, S.; Seok, S.; Kim, Y.-C.; Zhang, Y.; Yau, P.; Iwamori, N.; Xu, H.E.; Ma, J.; Kemper, B.; Kemper, J.K. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat. Commun. 2020, 11, 807. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Liu, J.; Zhang, X.-O.; Sibley, K.; Najjar, S.M.; Lee, M.M.; Wu, Q. Inhibition of protein arginine methyltransferase 5 enhances hepatic mitochondrial biogenesis. J. Biol. Chem. 2018, 293, 10884–10894. [Google Scholar] [CrossRef] [Green Version]
- Mann, J.; Chu, D.C.; Maxwell, A.; Oakley, F.; Zhu, N.; Tsukamoto, H.; Mann, D.A. MeCP2 Controls an Epigenetic Pathway That Promotes Myofibroblast Transdifferentiation and Fibrosis. Gastroenterology 2010, 138, 705–714.e4. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Tang, T.; Wang, G.-D.; Liu, B. LncRNA-H19 promotes hepatic lipogenesis by directly regulating miR-130a/PPARγ axis in non-alcoholic fatty liver disease. Biosci. Rep. 2019, 39, BSR20181722. [Google Scholar] [CrossRef] [Green Version]
- Hardy, T.; Zeybel, M.; Day, C.P.; Dipper, C.; Masson, S.; McPherson, S.; Henderson, E.; Tiniakos, D.; White, S.; French, J.; et al. Plasma DNA methylation: A potential biomarker for stratification of liver fibrosis in non-alcoholic fatty liver disease. Gut 2016, 66, 1321–1328. [Google Scholar] [CrossRef]
- Hwang, J.W.; So, Y.; Bae, G.; Kim, S.; Kim, Y.K. Protein arginine methyltransferase 6 suppresses adipogenic differentiation by repressing peroxisome proliferator-activated receptor γ activity. Int. J. Mol. Med. 2019, 43, 2462–2470. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, S.; Zhao, Y.; Lin, C.; Zhong, F.; Jin, L.; He, F.; Wang, H. Histone H3K9 demethylase JMJD1A modulates hepatic stellate cells activation and liver fibrosis by epigenetically regulating peroxisome proliferator-activated receptor γ. FASEB J. 2015, 29, 1830–1841. [Google Scholar] [CrossRef]
- Kim, J.-H.; Jung, D.Y.; Nagappan, A.; Jung, M.H. Histone H3K9 demethylase JMJD2B induces hepatic steatosis through upregulation of PPARγ2. Sci. Rep. 2018, 8, 13734. [Google Scholar] [CrossRef]
- Pang, H.; Ling, D.; Cheng, Y.; Akbar, R.; Jin, L.; Ren, J.; Wu, H.; Chen, B.; Zhou, Y.; Zhu, H.; et al. Gestational high-fat diet impaired demethylation of Ppar α and induced obesity of offspring. J. Cell. Mol. Med. 2021, 25, 5404–5416. [Google Scholar] [CrossRef] [PubMed]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T.; et al. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 21228. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [Green Version]
- Jimenez, V.; Jambrina, C.; Casana, E.; Sacristan, V.; Muñoz, S.; Darriba, S.; Rodó, J.; Mallol, C.; Garcia, M.; León, X.; et al. FGF21 gene therapy as treatment for obesity and insulin resistance. EMBO Mol. Med. 2018, 10, e8791. [Google Scholar] [CrossRef]
- Longo, R.; Peri, C.; Cricrì, D.; Coppi, L.; Caruso, D.; Mitro, N.; De Fabiani, E.; Crestani, M. Ketogenic Diet: A New Light Shining on Old but Gold Biochemistry. Nutrients 2019, 11, 2497. [Google Scholar] [CrossRef] [Green Version]
- Seok, S.; Kim, Y.-C.; Byun, S.; Choi, S.; Xiao, Z.; Iwamori, N.; Zhang, Y.; Wang, C.; Ma, J.; Ge, K.; et al. Fasting-induced JMJD3 histone demethylase epigenetically activates mitochondrial fatty acid β-oxidation. J. Clin. Investig. 2018, 128, 3144–3159. [Google Scholar] [CrossRef]
- Yuan, X.; Tsujimoto, K.; Hashimoto, K.; Kawahori, K.; Hanzawa, N.; Hamaguchi, M.; Seki, T.; Nawa, M.; Ehara, T.; Kitamura, Y.; et al. Epigenetic modulation of Fgf21 in the perinatal mouse liver ameliorates diet-induced obesity in adulthood. Nat. Commun. 2018, 9, 636. [Google Scholar] [CrossRef]
- Hazra, S.; Miyahara, T.; Rippe, R.A.; Tsukamoto, H. PPAR Gamma and Hepatic Stellate Cells. Comp. Hepatol. 2004, 3 (Suppl. 1), S7. [Google Scholar] [CrossRef] [Green Version]
- Perugorria, M.J.; Wilson, C.L.; Zeybel, M.; Walsh, M.; Amin, S.; Robinson, S.; White, S.A.; Burt, A.D.; Oakley, F.; Tsukamoto, H.; et al. Histone methyltransferase ASH1 orchestrates fibrogenic gene transcription during myofibroblast transdifferentiation. Hepatology 2012, 56, 1129–1139. [Google Scholar] [CrossRef]
- Byrd, K.N.; Shearn, A. ASH1, a Drosophila trithorax group protein, is required for methylation of lysine 4 residues on histone H3. Proc. Natl. Acad. Sci. USA 2003, 100, 11535–11540. [Google Scholar] [CrossRef] [Green Version]
- Zong, Y.; Yan, J.; Jin, L.; Xu, B.; He, Z.; Zhang, R.; Hu, C.; Jia, W. Relationship between circulating miR-132 and non-alcoholic fatty liver disease in a Chinese population. Hereditas 2020, 157, 22. [Google Scholar] [CrossRef] [PubMed]
- Lau-Corona, D.; Bae, W.K.; Hennighausen, L.; Waxman, D.J. Sex-biased genetic programs in liver metabolism and liver fibrosis are controlled by EZH1 and EZH2. PLoS Genet. 2020, 16, e1008796. [Google Scholar] [CrossRef]
- Moran-Salvador, E.; Garcia-Macia, M.; Sivaharan, A.; Sabater, L.; Zaki, M.Y.; Oakley, F.; Knox, A.; Page, A.; Luli, S.; Mann, J.; et al. Fibrogenic Activity of MECP2 Is Regulated by Phosphorylation in Hepatic Stellate Cells. Gastroenterology 2019, 157, 1398–1412.e9. [Google Scholar] [CrossRef] [Green Version]
- Jing, F.; Geng, Y.; Xu, X.-Y.; Xu, H.-Y.; Shi, J.-S.; Xu, Z.-H. MicroRNA29a Reverts the Activated Hepatic Stellate Cells in the Regression of Hepatic Fibrosis through Regulation of ATPase H+ Transporting V1 Subunit C1. Int. J. Mol. Sci. 2019, 20, 796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xuan, J.; Guo, S.-L.; Huang, A.; Xu, H.-B.; Shao, M.; Yang, Y.; Wen, W. MiR-29a and miR-652 Attenuate Liver Fibrosis by Inhibiting the Differentiation of CD4+ T Cells. Cell Struct. Funct. 2017, 42, 95–103. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, Y.; Itami, S.; Kuroda, M.; Yoshizato, K.; Kawada, N.; Murakami, Y. MiR-29a Assists in Preventing the Activation of Human Stellate Cells and Promotes Recovery From Liver Fibrosis in Mice. Mol. Ther. 2016, 24, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Galic, S.; Oakhill, J.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Khandekar, M.J.; Cohen, P.; Spiegelman, B.M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 2011, 11, 886–895. [Google Scholar] [CrossRef] [PubMed]
- Heydari, H.; Ghiasi, R.; Ghaderpour, S.; Keyhanmanesh, R. The Mechanisms Involved in Obesity-Induced Male Infertility. Curr. Diabetes Rev. 2021, 17, 259–267. [Google Scholar] [CrossRef]
- Siersbaek, R.; Nielsen, R.; Mandrup, S. PPARγ in adipocyte differentiation and metabolism—Novel insights from genome-wide studies. FEBS Lett. 2010, 584, 3242–3249. [Google Scholar] [CrossRef] [Green Version]
- Lefterova, M.I.; Zhang, Y.; Steger, D.J.; Schupp, M.; Schug, J.; Cristancho, A.; Feng, D.; Zhuo, D.; Stoeckert, J.C.J.; Liu, X.S.; et al. PPAR and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 2008, 22, 2941–2952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiki, K.; Kano, F.; Shiota, K.; Murata, M. Expression of the peroxisome proliferator activated receptor γ gene is repressed by DNA methylation in visceral adipose tissue of mouse models of diabetes. BMC Biol. 2009, 7, 38. [Google Scholar] [CrossRef] [Green Version]
- Duteil, D.; Tosic, M.; Willmann, D.; Georgiadi, A.; Kanouni, T.; Schüle, R. Lsd1 prevents age-programed loss of beige adipocytes. Proc. Natl. Acad. Sci. USA 2017, 114, 5265–5270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Zhang, B.; Lan, X.; Zhang, C.; Lei, C.; Chen, H. Comparative Transcriptome Analysis Reveals Significant Differences in MicroRNA Expression and Their Target Genes between Adipose and Muscular Tissues in Cattle. PLoS ONE 2014, 9, e102142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, R.; Nardelli, C.; Pilone, V.; Buonomo, T.; Liguori, R.; Castanò, I.; Buono, P.; Masone, S.; Persico, G.; Forestieri, P.; et al. miR-519d Overexpression Is Associated With Human Obesity. Obesity 2010, 18, 2170–2176. [Google Scholar] [CrossRef] [PubMed]
- Couture, J.-P.; Nolet, G.; Beaulieu, E.; Blouin, R.; Gévry, N. The p400/Brd8 Chromatin Remodeling Complex Promotes Adipogenesis by Incorporating Histone Variant H2A.Z at PPARγ Target Genes. Endocrinology 2012, 153, 5796–5808. [Google Scholar] [CrossRef] [Green Version]
- Mikkelsen, T.S.; Xu, Z.; Zhang, X.; Wang, L.; Gimble, J.M.; Lander, E.S.; Rosen, E.D. Comparative Epigenomic Analysis of Murine and Human Adipogenesis. Cell 2010, 143, 156–169. [Google Scholar] [CrossRef] [Green Version]
- Park, U.; Hwang, J.; Youn, H.; Kim, E.; Um, S. Piperine inhibits adipocyte differentiation via dynamic regulation of histone modifications. Phytother. Res. 2019, 33, 2429–2439. [Google Scholar] [CrossRef]
- Lee, J.-E.; Wang, C.; Xu, S.; Cho, Y.-W.; Wang, L.; Feng, X.; Baldridge, A.; Sartorelli, V.; Zhuang, L.; Peng, W.; et al. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. Elife 2013, 2, e01503. [Google Scholar] [CrossRef]
- Cho, Y.-W.; Hong, S.; Jin, Q.; Wang, L.; Lee, J.-E.; Gavrilova, O.; Ge, K. Histone Methylation Regulator PTIP Is Required for PPARγ and C/EBPα Expression and Adipogenesis. Cell Metab. 2009, 10, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Lee, H.H.; Ye, B.J.; Lee-Kwon, W.; Choi, S.Y.; Kwon, H.M. TonEBP suppresses adipogenesis and insulin sensitivity by blocking epigenetic transition of PPARγ2. Sci. Rep. 2015, 5, 10937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Chung, C.-C.; Liu, Y.-C.; Yeh, S.-P.; Hsu, J.L.; Hung, M.-C.; Su, H.-L.; Li, L.-Y. Enhancer of Zeste Homolog 2 and Histone Deacetylase 9c Regulate Age-Dependent Mesenchymal Stem Cell Differentiation into Osteoblasts and Adipocytes. Stem Cells 2016, 34, 2183–2193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.-H.; Yeh, F.-L.; Yeh, S.-P.; Ma, H.-T.; Hung, S.-C.; Hung, M.-C.; Li, L.-Y. Myocyte Enhancer Factor-2 Interacting Transcriptional Repressor (MITR) Is a Switch That Promotes Osteogenesis and Inhibits Adipogenesis of Mesenchymal Stem Cells by Inactivating Peroxisome Proliferator-activated Receptor γ-2. J. Biol. Chem. 2011, 286, 10671–10680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, L.; Jang, Y.; Park, Y.-K.; Lee, J.-E.; Jain, S.; Froimchuk, E.; Broun, A.; Liu, C.; Gavrilova, O.; Ge, K. Depletion of Nsd2-mediated histone H3K36 methylation impairs adipose tissue development and function. Nat. Commun. 2018, 9, 1796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nic-Can, G.I.; Rodas-Junco, B.A.; Carrillo-Cocom, L.M.; Zepeda-Pedreguera, A.; Peñaloza-Cuevas, R.; Aguilar-Ayala, F.J.; Rojas-Herrera, R.A. Epigenetic Regulation of Adipogenic Differentiation by Histone Lysine Demethylation. Int. J. Mol. Sci. 2019, 20, 3918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizcano, F.; Romero, C.; Vargas, D. Regulation of adipogenesis by nucelar receptor PPARγ is modulated by the histone demethylase JMJD2C. Genet. Mol. Biol. 2010, 34, 19–24. [Google Scholar] [CrossRef]
- Tateishi, K.; Okada, Y.; Kallin, E.M.; Zhang, Y. Role of Jhdm2a in regulating metabolic gene expression and obesity resistance. Nature 2009, 458, 757–761. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Tachibana, M.; Magoori, K.; Kudo, H.; Tanaka, T.; Okamura, M.; Naito, M.; Kodama, T.; Shinkai, Y.; Sakai, J. Obesity and metabolic syndrome in histone demethylase JHDM2a-deficient mice. Genes Cells 2009, 14, 991–1001. [Google Scholar] [CrossRef]
- Lee, J.-E.; Ge, K. Transcriptional and epigenetic regulation of PPARγ expression during adipogenesis. Cell Biosci. 2014, 4, 29. [Google Scholar] [CrossRef] [Green Version]
- Steger, D.J.; Grant, G.; Schupp, M.; Tomaru, T.; Lefterova, M.I.; Schug, J.; Manduchi, E.; Stoeckert, C.J.; Lazar, M.A. Propagation of adipogenic signals through an epigenomic transition state. Genes Dev. 2010, 24, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Erener, S.; Hesse, M.; Kostadinova, R.; Hottiger, M.O. Poly(ADP-Ribose)Polymerase-1 (PARP1) Controls Adipogenic Gene Expression and Adipocyte Function. Mol. Endocrinol. 2012, 26, 79–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, M.; Rao, M.; Bouras, T.; Wang, C.; Wu, K.; Zhang, X.; Li, Z.; Yao, T.-P.; Pestell, R.G. Cyclin D1 Inhibits Peroxisome Proliferator-activated Receptor γ-mediated Adipogenesis through Histone Deacetylase Recruitment. J. Biol. Chem. 2005, 280, 16934–16941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Zhang, F.; Yan, T.; Liu, Z.; Wang, C.; Ge, X.; Zhai, Q. C/EBPα regulates SIRT1 expression during adipogenesis. Cell Res. 2010, 20, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef]
- Ferrari, A.; Fiorino, E.; Longo, R.; Barilla, S.; Mitro, N.; Cermenati, G.; Giudici, M.; Caruso, D.; Mai, A.; Guerrini, U.; et al. Attenuation of diet-induced obesity and induction of white fat browning with a chemical inhibitor of histone deacetylases. Int. J. Obes. 2016, 41, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Aguilar, E.C.; Silva, J.; Navia-Pelaez, J.M.; Leonel, A.J.; Lopes, L.G.; Menezes-Garcia, Z.; Ferreira, A.; Capettini, L.; Teixeira, L.G.; Lemos, V.S.; et al. Sodium butyrate modulates adipocyte expansion, adipogenesis, and insulin receptor signaling by upregulation of PPAR-γ in obese Apo E knockout mice. Nutrition 2017, 47, 75–82. [Google Scholar] [CrossRef]
- Yoon, G.-E.; Jung, J.K.; Lee, Y.-H.; Jang, B.-C.; Kim, J.I. Histone deacetylase inhibitor CG200745 ameliorates high-fat diet-induced hypertension via inhibition of angiotensin II production. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 393, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Bian, F.; Ma, X.; Villivalam, S.D.; You, D.; Choy, L.R.; Paladugu, A.; Fung, S.; Kang, S. TET2 facilitates PPARγ agonist–mediated gene regulation and insulin sensitization in adipocytes. Metabolism 2018, 89, 39–47. [Google Scholar] [CrossRef]
- Chen, J.; Liu, Y.; Lu, S.; Yin, L.; Zong, C.; Cui, S.; Qin, D.; Yang, Y.; Guan, Q.; Li, X.; et al. The role and possible mechanism of lncRNA U90926 in modulating 3T3-L1 preadipocyte differentiation. Int. J. Obes. 2016, 41, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Cooper, D.R.; Carter, G.; Li, P.; Patel, R.; Watson, J.E.; Patel, N.A. Long Non-Coding RNA NEAT1 Associates with SRp40 to Temporally Regulate PPARγ2 Splicing during Adipogenesis in 3T3-L1 Cells. Genes 2014, 5, 1050–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Divoux, A.; Karastergiou, K.; Xie, H.; Guo, W.; Perera, R.J.; Fried, S.K.; Smith, S.R. Identification of a novel lncRNA in gluteal adipose tissue and evidence for its positive effect on preadipocyte differentiation. Obesity 2014, 22, 1781–1785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Gu, M.; Ma, Y.; Peng, Y. LncRNA TUG1 reduces inflammation and enhances insulin sensitivity in white adipose tissue by regulating miR-204/SIRT1 axis in obesity mice. Mol. Cell. Biochem. 2020, 475, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Skårn, M.; Namløs, H.M.; Noordhuis, P.; Wang, M.-Y.; Meza-Zepeda, L.A.; Myklebost, O. Adipocyte Differentiation of Human Bone Marrow-Derived Stromal Cells Is Modulated by MicroRNA-155, MicroRNA-221, and MicroRNA-222. Stem Cells Dev. 2012, 21, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, Y.; Zhang, S.; Ye, L.; Cui, J.; Sun, Q.; Li, K.; Wu, H.; Liu, L. MiR-540 as a Novel Adipogenic Inhibitor Impairs Adipogenesis Via Suppression of PPARγ. J. Cell. Biochem. 2015, 116, 969–976. [Google Scholar] [CrossRef]
- Li, H.; Xue, M.; Xu, J.; Qin, X. MiR-301a is involved in adipocyte dysfunction during obesity-related inflammation via suppression of PPARgamma. Pharmazie 2016, 71, 84–88. [Google Scholar]
- Huang, Q.; Ma, C.; Chen, L.; Luo, D.; Chen, R.; Liang, F. Mechanistic Insights into the Interaction between Transcription Factors and Epigenetic Modifications and the Contribution to the Development of Obesity. Front. Endocrinol. 2018, 9, 370. [Google Scholar] [CrossRef] [Green Version]
- McGregor, R.A.; Choi, M.S. microRNAs in the Regulation of Adipogenesis and Obesity. Curr. Mol. Med. 2011, 11, 304–316. [Google Scholar] [CrossRef]
- Xie, H.; Lim, B.; Lodish, H.F. MicroRNAs Induced During Adipogenesis that Accelerate Fat Cell Development Are Downregulated in Obesity. Diabetes 2009, 58, 1050–1057. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.; Martin, S. Environmental/lifestyle factors in the pathogenesis and prevention of type 2 diabetes. BMC Med. 2017, 15, 131. [Google Scholar] [CrossRef]
- Ojo, O. Dietary Intake and Type 2 Diabetes. Nutrients 2019, 11, 2177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, R.J. Type-2 diabetes mellitus and cardiovascular disease. Future Cardiol. 2018, 14, 491–509. [Google Scholar] [CrossRef]
- Ogurtsova, K.; da Rocha Fernandes, J.D.; Huang, Y.; Linnenkamp, U.; Guariguata, L.; Cho, N.; Cavan, D.; Shaw, J.; Makaroff, L. IDF Diabetes Atlas: Global estimates for the prevalence of diabetes for 2015 and 2040. Diabetes Res. Clin. Pract. 2017, 128, 40–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naidoo, V.; Naidoo, M.; Ghai, M. Cell- and tissue-specific epigenetic changes associated with chronic inflammation in insulin resistance and type 2 diabetes mellitus. Scand. J. Immunol. 2018, 88, e12723. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.; Zhou, Y.; Evertts, A.G.; Xu, S.; Griffin, M.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell. Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Ye, X.; Guo, W.; Lu, H.; Gao, Z. Inhibition of HDAC3 promotes ligand-independent PPARγ activation by protein acetylation. J. Mol. Endocrinol. 2014, 53, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wang, X.; Ren, W.; Ren, J.; Lan, X.; Wang, F.; Zhang, F.; Han, Y.; Song, T.; Holmdahl, R.; et al. High expression of liver histone deacetylase 3 contributes to high-fat-diet-induced metabolic syndrome by suppressing the PPAR-γ and LXR-α-pathways in E3 rats. Mol. Cell. Endocrinol. 2011, 344, 69–80. [Google Scholar] [CrossRef]
- Sathishkumar, C.; Prabu, P.; Balakumar, M.; Lenin, R.; Prabhu, D.; Anjana, R.M.; Mohan, V.; Balasubramanyam, M. Augmentation of histone deacetylase 3 (HDAC3) epigenetic signature at the interface of proinflammation and insulin resistance in patients with type 2 diabetes. Clin. Epigenet. 2016, 8, 125. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.; Behl, T.; Arora, S. Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 2020, 68, 45–50. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, J.; Yuan, Y.; Chen, J.; Cheng, S.; Wang, H.; Xu, Y. Sodium butyrate mitigates type 2 diabetes by inhibiting PERK-CHOP pathway of endoplasmic reticulum stress. Environ. Toxicol. Pharmacol. 2018, 64, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Kaspi, A.; Ziemann, M.; Okabe, J.; Karagiannis, T.C.; El-Osta, A. Systems approach to the pharmacological actions of HDAC inhibitors reveals EP300 activities and convergent mechanisms of regulation in diabetes. Epigenetics 2017, 12, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Fujisaka, S. The role of adipose tissue M1/M2 macrophages in type 2 diabetes mellitus. Diabetol. Int. 2020, 12, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhao, J.; Meng, H.; Zhang, X. Adipose Tissue-Resident Immune Cells in Obesity and Type 2 Diabetes. Front. Immunol. 2019, 10, 1173. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.; de Winther, M.P.; Bossche, J. Epigenetic mechanisms of macrophage activation in type 2 diabetes. Immunobiology 2017, 222, 937–943. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- Bouhlel, M.A.; Brozek, J.; Derudas, B.; Zawadzki, C.; Jude, B.; Staels, B.; Chinetti-Gbaguidi, G. Unlike PPARgamma, PPARalpha or PPARbeta/delta activation does not promote human monocyte differentiation toward alternative macrophages. Biochem. Biophys. Res. Commun. 2009, 386, 459–462. [Google Scholar] [CrossRef]
- Charo, I.F. Macrophage Polarization and Insulin Resistance: PPARγ in Control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Cao, Q.; Yu, L.; Shi, H.; Xue, B.; Shi, H. Epigenetic regulation of macrophage polarization and inflammation by DNA methylation in obesity. JCI Insight 2016, 1, e87748. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, X.; Liu, D.; Yu, L.; Xue, B.; Shi, H. Epigenetic Regulation of Macrophage Polarization by DNA Methyltransferase 3b. Mol. Endocrinol. 2014, 28, 565–574. [Google Scholar] [CrossRef] [Green Version]
- You, D.; Nilsson, E.; Tenen, D.E.; Lyubetskaya, A.; Lo, J.C.; Jiang, R.; Deng, J.; Dawes, B.A.; Vaag, A.; Ling, C.; et al. Dnmt3a is an epigenetic mediator of adipose insulin resistance. Elife 2017, 6, e30766. [Google Scholar] [CrossRef]
- Kim, A.Y.; Park, Y.J.; Pan, X.; Shin, K.C.; Kwak, S.-H.; Bassas, A.F.; Sallam, R.M.; Park, K.S.; Alfadda, A.; Xu, A.; et al. Obesity-induced DNA hypermethylation of the adiponectin gene mediates insulin resistance. Nat. Commun. 2015, 6, 7585. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhang, Y.; Liu, Y.; Zhu, D.; Yu, J.; Li, G.; Sun, Z.; Wang, W.; Jiang, H.; Hong, Z. MiR-27a promotes insulin resistance and mediates glucose metabolism by targeting PPAR-γ-mediated PI3K/AKT signaling. Aging (Albany NY) 2019, 11, 7510–7524. [Google Scholar] [CrossRef]
- Yao, F.; Yu, Y.; Feng, L.; Li, J.; Zhang, M.; Lan, X.; Yan, X.; Liu, Y.; Guan, F.; Zhang, M.; et al. Adipogenic miR-27a in adipose tissue upregulates macrophage activation via inhibiting PPARγ of insulin resistance induced by high-fat diet-associated obesity. Exp. Cell Res. 2017, 355, 105–112. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Marcos, P.J.; Auwerx, J. Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am. J. Clin. Nutr. 2011, 93, 884S–890S. [Google Scholar] [CrossRef] [Green Version]
- Jeninga, E.H.; Schoonjans, K.; Auwerx, J. Reversible acetylation of PGC-1: Connecting energy sensors and effectors to guarantee metabolic flexibility. Oncogene 2010, 29, 4617–4624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodgers, J.; Lerin, C.; Gerhart-Hines, Z.; Puigserver, P. Metabolic adaptations through the PGC-1α and SIRT1 pathways. FEBS Lett. 2007, 582, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhart-Hines, Z.; Rodgers, J.; Bare, O.; Lerin, C.; Kim, S.-H.; Mostoslavsky, R.; Alt, F.W.; Wu, Z.; Puigserver, P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α. EMBO J. 2007, 26, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Sharabi, K.; Lin, H.; Tavares, C.D.; Dominy, J.E.; Camporez, J.P.; Perry, R.J.; Schilling, R.; Rines, A.K.; Lee, J.; Hickey, M.; et al. Selective Chemical Inhibition of PGC-1α Gluconeogenic Activity Ameliorates Type 2 Diabetes. Cell 2017, 169, 148–160.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Scheme: PPAR epigenome implications in diseases. C, cytosine; Me, methyl group, ncRNAs, non-coding RNAs; K, histone lysine; S, histone serine; Ac, acetylation mark; P, phosphorylation mark.
Figure 1.
Scheme: PPAR epigenome implications in diseases. C, cytosine; Me, methyl group, ncRNAs, non-coding RNAs; K, histone lysine; S, histone serine; Ac, acetylation mark; P, phosphorylation mark.

Table 3.
PPAR epigenetics in non-alcoholic steatohepatitis.
| Condition | PPAR Isoform | Epigenetic Player | Effect | References |
|---|---|---|---|---|
| NASH | PPARα | miR-21 | Diminished PPARα expression and activation of HSCs in obesogenic models | [119] |
| TET1 and TET2 | Downregulated enzymes under high fat diet conditions, promoting Ppara hypermethylation | [121] | ||
| Ascorbic acid | Cofactor of TET enzymes. Its lack promotes PPARα target genes hypermethylation | [122] | ||
| JMJD3 | Phosphorylated upon fasting-induced FGF21 signaling. Direct interaction with PPARα for the upregulation of autophagy-related genes | [123] | ||
| PRMT5 | Downregulation of Ppara expression | [124] | ||
| PPARγ | miR-132 | miR-132 downregulation induces the expression of MeCP2 in HSCs | [125] | |
| miR-29a | Expressed upon Rosiglitazone-mediated PPARγ activation. Repression of profibrotic genes | [126] | ||
| MeCP2 | H3K9 and H3K27 methylation and HP1α repressor recruitment in Pparg locus of HSCs. MeCP2 also induces the expression of EZH2 and ASH1 in HSCs. | [125] | ||
| Pparg promoter CpG methylation | Downregulation of PPARγ. Potential non-invasive fibrosis marker in cell-free DNA in plasma. | [127] | ||
| PRMT6 | Repression of PPARγ activity | [128] | ||
| JMJD1A and JMJD2B | Upregulation of Pparg and increased lipid uptake | [129,130] | ||
| LncRNA-H19 | Control of hepatic lipogenesis through mi-130A/PPARγ axis | [126] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Porcuna, J.; Mínguez-Martínez, J.; Ricote, M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. Int. J. Mol. Sci. 2021, 22, 10573. https://doi.org/10.3390/ijms221910573
AMA Style
Porcuna J, Mínguez-Martínez J, Ricote M. The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders. International Journal of Molecular Sciences. 2021; 22(19):10573. https://doi.org/10.3390/ijms221910573
Chicago/Turabian StylePorcuna, Jesús, Jorge Mínguez-Martínez, and Mercedes Ricote. 2021. "The PPARα and PPARγ Epigenetic Landscape in Cancer and Immune and Metabolic Disorders" International Journal of Molecular Sciences 22, no. 19: 10573. https://doi.org/10.3390/ijms221910573
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.