
Extracellular Vesicles and Cancer Stem Cells in Tumor Progression: New Therapeutic Perspectives


Abstract
:1. Introduction
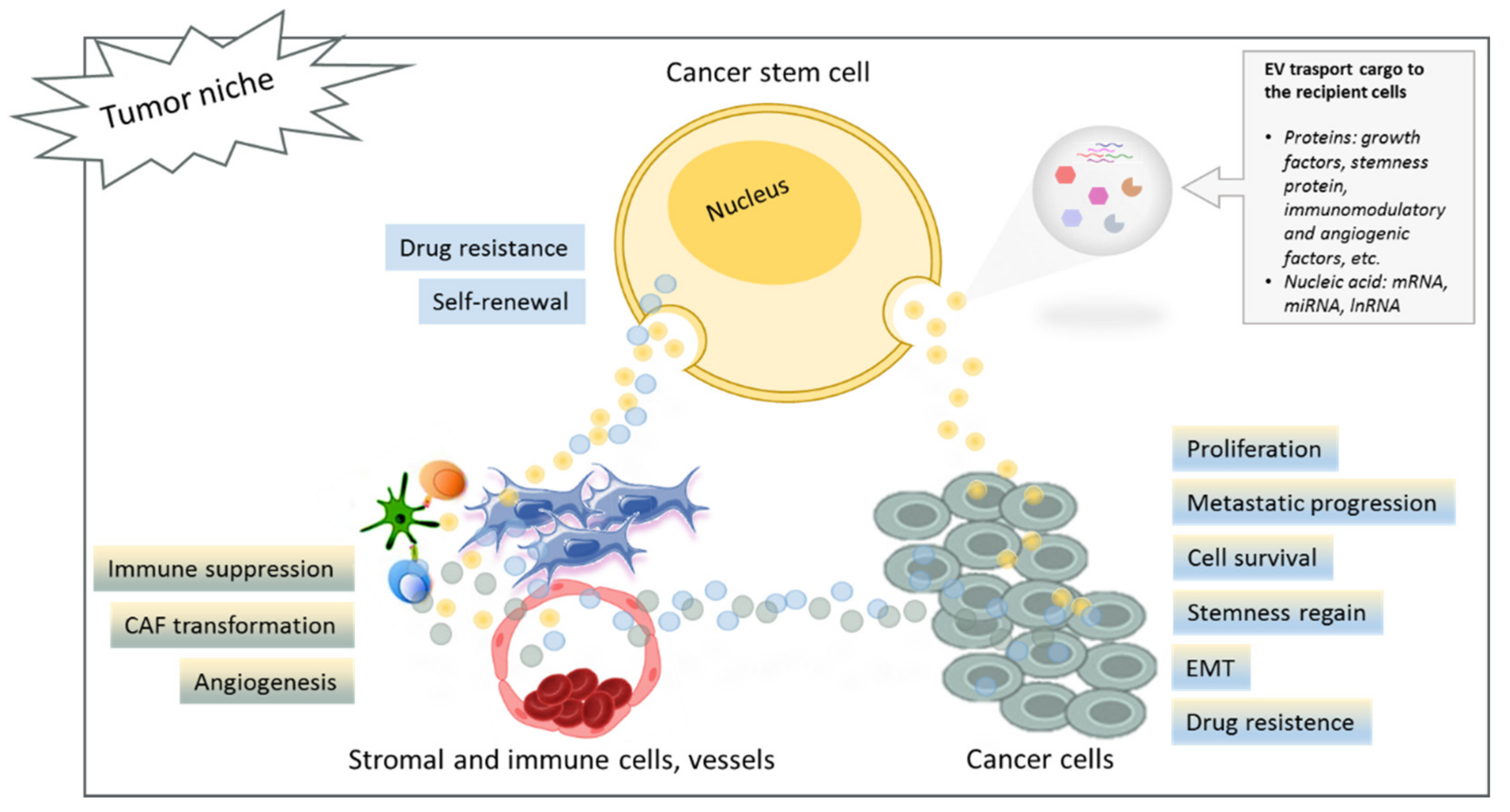
2. Protumoral Effects of CSC-Derived EVs on Non-CSC Tumor Cells
2.1. Stemness and Metastatic Phenotype
2.2. Drug Resistance
3. Cross-Talk between CSCs and the Tumor Microenvironment via EV Transfer
3.1. EV-Mediated Communication between CSCs and Stromal Cells
3.2. The Immunosuppressive Role of CSC-Derived EVs
3.3. The Proangiogenic Properties of CSC-Derived EVs
4. Cross-Talk between Non-CSC Tumor Cells and the Tumor Microenvironment via EV Transfer
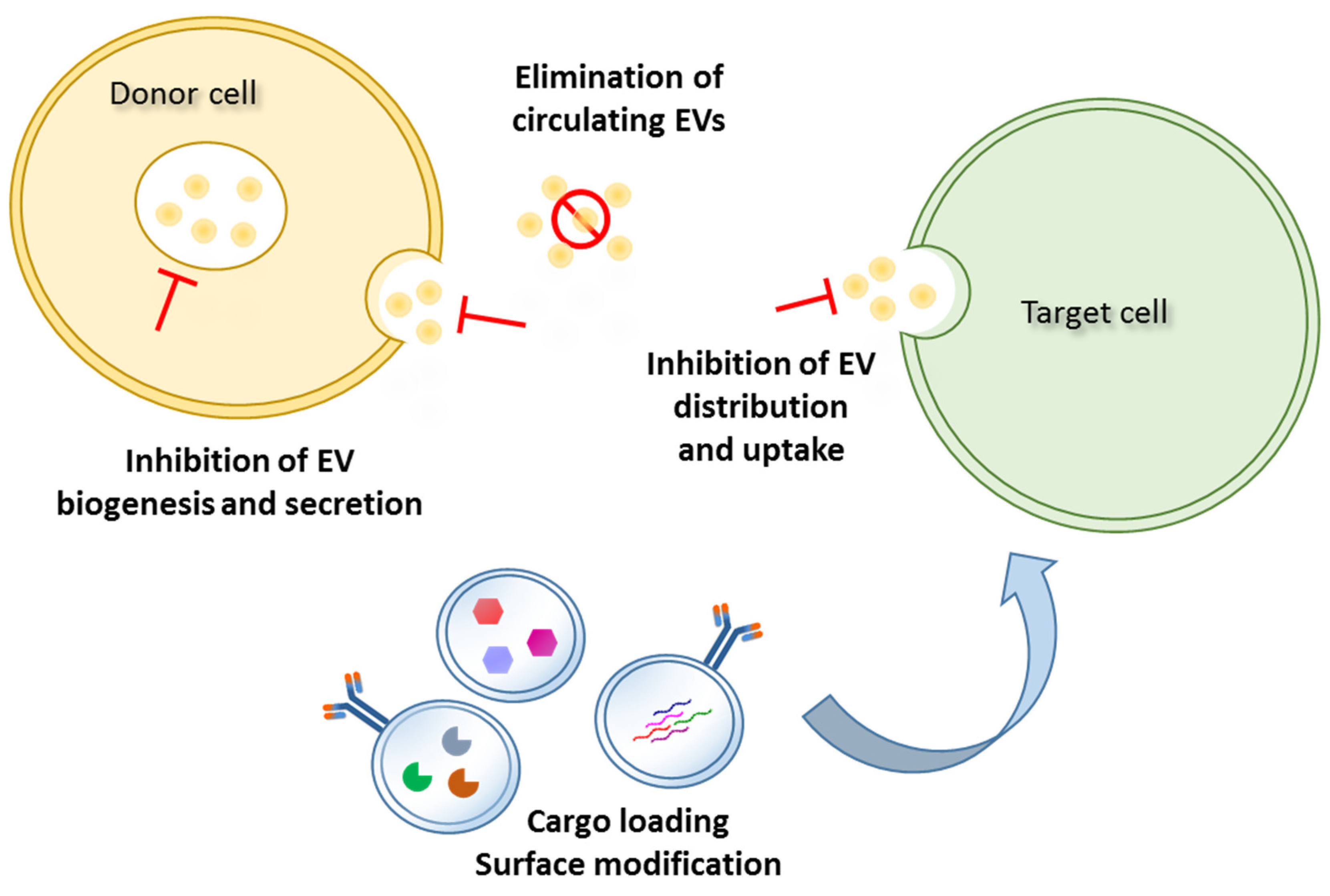
5. EV-Based Strategies for CSC Targeting as Anticancer Therapy
5.1. Inhibition of Biogenesis and Release/Uptake of EVs
5.2. Removal of Circulating Cancer EVs
5.3. Natural Bioengineered EVs for CSC Targeting and Drug Delivery
5.4. Synthetic Nanoparticles for CSC Targeting and Drug Delivery
6. Conclusions and Perspectives
Funding
Conflicts of Interest
References
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar] [CrossRef]
- Prasetyanti, P.R.; Medema, J.P. Intra-tumor heterogeneity from a cancer stem cell perspective. Mol. Cancer 2017, 16, 41. [Google Scholar] [CrossRef] [Green Version]
- Koren, S.; Bentires-Alj, M. Breast Tumor Heterogeneity: Source of Fitness, Hurdle for Therapy. Mol. Cell 2015, 60, 537–546. [Google Scholar] [CrossRef]
- Rich, J.N. Cancer stem cells: Understanding tumor hierarchy and heterogeneity. Medicine 2016, 5 (Suppl. S1), S2–S7. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Lawson, J.C.; Blatch, G.L.; Edkins, A.L. Cancer stem cells in breast cancer and metastasis. Breast Cancer Res. Treat. 2009, 118, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef] [PubMed]
- Capp, J.P. Cancer Stem Cells: From Historical Roots to a New Perspective. J. Oncol. 2019, 11, 5189232. [Google Scholar] [CrossRef] [PubMed]
- Sachs, N.; Clevers, H. Organoid cultures for the analysis of cancer phenotypes. Curr. Opin. Genet. Dev. 2014, 24, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Ryu, C.J. Cancer stem cell surface markers on normal stem cells. BMB Rep. 2017, 50, 285–298. [Google Scholar] [CrossRef] [Green Version]
- Scioli, M.G.; Storti, G.; D’Amico, F.; Gentile, P.; Fabbri, G.; Cervelli, V.; Orlandi, A. The Role of Breast Cancer Stem Cells as a Prognostic Marker and a Target to Improve the Efficacy of Breast Cancer Therapy. Cancers 2019, 11, 1021. [Google Scholar] [CrossRef] [Green Version]
- Asghari, F.; Khademi, R.; Ranjbar, F.E.; Malekshahi, Z.V.; Majidi, R.F. Application of Nanotechnology in Targeting of Cancer Stem Cells: A Review. Int. J. Stem Cells 2019, 12, 227–239. [Google Scholar] [CrossRef]
- Takahashi-Yanaga, F.; Kahn, M. Targeting Wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smit, L.; Berns, K.; Spence, K.; Ryder, W.D.; Zeps, N.; Madiredjo, M.; Beijersbergen, R.; Bernards, R.; Clarke, R.B. An integrated genomic approach identifies that the PI3K/AKT/FOXO pathway is involved in breast cancer tumor initiation. Oncotarget 2016, 7, 2596–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woosley, A.N.; Dalton, A.C.; Hussey, G.S.; Howley, B.V.; Mohanty, B.K.; Grelet, S.; Dincman, T.; Bloos, S.; Olsen, S.K.; Howe, P.H. TGFβ promotes breast cancer stem cell self-renewal through an ILEI/LIFR signaling axis. Oncogene 2019, 38, 3794–3811. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ridgway, L.D.; Wetzel, M.D.; Ngo, J.; Yin, W.; Kumar, D.; Goodman, J.C.; Groves, M.D.; Marchetti, D. The identification and characterization of breast cancer CTCs competent for brain metastasis. Sci. Transl. Med. 2013, 5, 180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baccelli, I.; Schneeweiss, A.; Riethdorf, S.; Stenzinger, A.; Schillert, A.; Vogel, V.; Klein, C.; Saini, M.; Bäuerle, T.; Wallwiener, M.; et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat. Biotechnol. 2013, 31, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast Cancer Stem Cells Transition between Epithelial and Mesenchymal States Reflective of their Normal Counterparts. Stem Cell Rep. 2013, 2, 78–91. [Google Scholar] [CrossRef]
- Yang, F.; Xu, J.; Tang, L.; Guan, X. Breast cancer stem cell: The roles and therapeutic implications. Cell Mol. Life Sci. 2017, 74, 951–966. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Ke, S.Q.; Huang, Z.; Flavahan, W.; Fang, X.; Paul, J.; Wu, L.; Sloan, A.E.; McLendon, R.E.; Li, X.; et al. Periostin secreted by glioblastoma stem cells recruits M2 tumor-associated macrophages and promotes malignant growth. Nat. Cell Biol. 2015, 17, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology 2014, 15, e28862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clara, J.A.; Monge, C.; Yang, Y.; Takebe, N. Targeting signalling pathways and the immune microenvironment of cancer stem cells—A clinical update. Nat. Rev. Clin. Oncol. 2020, 17, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 25. [Google Scholar] [CrossRef]
- Zhao, J. Cancer stem cells and chemoresistance: The smartest survives the raid. Pharmacol. Ther. 2016, 160, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGFβ2 dictates disseminated tumour cell fate in target organs through TGFβ-RIII and p38α/β signaling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef] [Green Version]
- Kurtova, A.V.; Xiao, J.; Mo, Q.; Pazhanisamy, S.; Krasnow, R.; Lerner, S.P.; Chen, F.; Roh, T.T.; Lay, E.; Ho, P.L.; et al. Blocking PGE2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 2015, 517, 209–213. [Google Scholar] [CrossRef]
- Li, Y.; Laterra, J. Cancer stem cells: Distinct entities or dynamically regulated phenotypes? Cancer Res. 2012, 72, 576–580. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Wu, D.; Wu, P.; Chen, Z.; Huang, J. The cancer stem cell niche: Cross talk between cancer stem cells and their microenvironment. Tumour Biol. 2014, 35, 3945–3951. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gercel-Taylor, C. The origin, function, and diagnostic potential of RNA within extracellular vesicles present in human biological fluids. Front. Genet. 2013, 4, 142. [Google Scholar] [CrossRef] [Green Version]
- Mol, E.A.; Goumans, M.J.; Doevendans, P.A.; Sluijter, J.P.G.; Vader, P. Higher functionality of extracellular vesicles isolated using size-exclusion chromatography compared to ultracentrifugation. Nanomedicine 2017, 13, 2061–2065. [Google Scholar] [CrossRef]
- Ludwig, A.K.; De Miroschedji, K.; Doeppner, T.R.; Börger, V.; Ruesing, J.; Rebmann, V.; Durst, S.; Jansen, S.; Bremer, M.; Behrmann, E.; et al. Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. J. Extracell Vesicles 2018, 7, 1528109. [Google Scholar] [CrossRef]
- Février, B.; Raposo, G. Exosomes: Endosomal-derived vesicles shipping extracellular messages. Curr. Opin. Cell Biol. 2004, 16, 415–421. [Google Scholar] [CrossRef]
- De Gassart, A.; Géminard, C.; Hoekstra, D.; Vidal, M. Exosome secretion: The art of reutilizing nonrecycled proteins? Traffic 2004, 5, 896–903. [Google Scholar] [CrossRef]
- Wollert, T.; Hurley, J.H. Molecular mechanism of multivesicular body biogenesis by ESCRT complexes. Nature 2010, 464, 864–869. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Moita, C.; van Niel, G.; Kowal, J.; Vigneron, J.; Benaroch, P.; Manel, N.; Moita, L.F.; Théry, C.; Raposo, G. Analysis of ESCRT functions in exosome biogenesis, composition and secretion highlights the heterogeneity of extracellular vesicles. J. Cell Sci. 2013, 126 Pt 24, 5553–5565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Stoorvogel, W.; Kleijmeer, M.J.; Geuze, H.J.; Raposo, G. The biogenesis and functions of exosomes. Traffic 2002, 3, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neuro-oncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucki, R.; Bachelot-Loza, C.; Zachowski, A.; Giraud, F.; Sulpice, J.C. Calcium induces phospholipid redistribution and microvesicle release in human erythrocyte membranes by independent pathways. Biochemistry 1998, 37, 15383–15391. [Google Scholar] [CrossRef]
- Wang, T.; Gilkes, D.M.; Takano, N.; Xiang, L.; Luo, W.; Bishop, C.J.; Chaturvedi, P.; Green, J.J.; Semenza, G.L. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc. Natl. Acad. Sci. USA 2014, 111, E3234–E3242. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.H.; D’Asti, E.; Magnus, N.; Al-Nedawi, K.; Meehan, B.; Rak, J. Microvesicles as mediators of intercellular communication in cancer--the emerging science of cellular ‘debris’. Semin. Immunopathol. 2011, 33, 455–467. [Google Scholar] [CrossRef] [Green Version]
- Castellana, D.; Zobairi, F.; Martinez, M.C.; Panaro, M.A.; Mitolo, V.; Freyssinet, J.M.; Kunzelmann, C. Membrane microvesicles as actors in the establishment of a favorable prostatic tumoral niche: A role for activated fibroblasts and CX3CL1-CX3CR1 axis. Cancer Res. 2009, 69, 785–793. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Wang, L.; Dong, L.; Wang, X. Emerging role of exosome signalling in maintaining cancer stem cell dynamic equilibrium. J. Cell Mol. Med. 2018, 22, 3719–3728. [Google Scholar] [CrossRef]
- Seo, N.; Akiyoshi, K.; Shiku, H. Exosome-mediated regulation of tumor immunology. Cancer Sci. 2018, 109, 2998–3004. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Li, D.; Shen, L.; Hu, D.; Tang, B.; Guo, W.; Wang, Z.; Zhang, Z.; Wei, G.; He, D. Exosomes derived from Piwil2-induced cancer stem cells transform fibroblasts into cancer-associated fibroblasts. Oncol. Rep. 2020, 43, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular Vesicles in Cancer: Cell-to-Cell Mediators of Metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [Green Version]
- Walcher, L.; Kistenmacher, A.K.; Suo, H.; Kitte, R.; Dluczek, S.; Strauß, A.; Blaudszun, A.R.; Yevsa, T.; Fricke, S.; Kossatz-Boehlert, U. Cancer Stem Cells-Origins and Biomarkers: Perspectives for Targeted Personalized Therapies. Front. Immunol. 2020, 11, 1280. [Google Scholar] [CrossRef]
- Kumar, D.; Gupta, D.; Shankar, S.; Srivastava, R.K. Biomolecular characterization of exosomes released from cancer stem cells: Possible implications for biomarker and treatment of cancer. Oncotarget 2015, 6, 3280–3291. [Google Scholar] [CrossRef] [Green Version]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Pozo Martín, Y.; Park, D.; Ramachandran, A.; Ombrato, L.; Calvo, F.; Chakravarty, P.; Spencer-Dene, B.; Derzsi, S.; Hill, C.S.; Sahai, E.; et al. Mesenchymal Cancer Cell-Stroma Crosstalk Promotes Niche Activation, Epithelial Reversion, and Metastatic Colonization. Cell Rep. 2015, 13, 2456–2469. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Wang, J.; Liu, Y.; Gong, X. Epithelial cell adhesion molecule and epithelial-mesenchymal transition are associated with vasculogenic mimicry, poor prognosis, and metastasis of triple negative breast cancer. Int. J. Clin. Exp. Pathol. 2019, 12, 1678–1689. [Google Scholar] [PubMed]
- Orlandi, A.; Ciucci, A.; Ferlosio, A.; Pellegrino, A.; Chiariello, L.; Spagnoli, L.G. Increased expression and activity of matrix metalloproteinases characterize embolic cardiac myxomas. Am. J. Pathol. 2005, 166, 1619–1628. [Google Scholar] [CrossRef] [Green Version]
- Al-Sowayan, B.S.; Al-Shareeda, A.T.; Alrfaei, B.M. Cancer Stem Cell-Exosomes, Unexposed Player in Tumorigenicity. Front. Pharmacol. 2020, 11, 384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, D.; Yang, Q.; Dai, Y.; Guo, W.; Du, H.; Song, L.; Peng, X. Oncogenic miR-210-3p promotes prostate cancer cell EMT and bone metastasis via NF-κB signaling pathway. Mol. Cancer 2017, 10, 117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrozza, V.; Pastore, A.L.; Palleschi, G.; Tito, C.; Porta, N.; Ricci, S.; Marigliano, C.; Costantini, M.; Simone, G.; Di Carlo, A.; et al. Secreted miR-210-3p as non-invasive biomarker in clear cell renal cell carcinoma. Oncotarget 2017, 8, 69551–69558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Sai, B.; Wang, F.; Wang, L.; Wang, Y.; Zheng, L.; Li, G.; Tang, J.; Xiang, J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer 2019, 18, 1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; He, J.; Hu, H.; Tu, L.; Sun, Z.; Liu, Y.; Luo, F. Lung CSC-derived exosomal miR-210-3p contributes to a pro-metastatic phenotype in lung cancer by targeting FGFRL1. J. Cell Mol. Med. 2020, 24, 6324–6339. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.C.; Rini, B.I. Treatment of renal cell carcinoma: Current status and future directions. CA Cancer J. Clin. 2017, 67, 507–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marona, P.; Górka, J.; Mazurek, Z.; Wilk, W.; Rys, J.; Majka, M.; Jura, J.; Miekus, K. MCPIP1 Downregulation in Clear Cell Renal Cell Carcinoma Promotes Vascularization and Metastatic Progression. Cancer Res. 2017, 77, 4905–4920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Cai, J.L.; Huang, P.Z.; Kang, L.; Huang, M.J.; Wang, L.; Wang, J.P. miR19b-3p promotes the growth and metastasis of colorectal cancer via directly targeting ITGB8. Am. J. Cancer Res. 2017, 7, 1996–2008. [Google Scholar] [PubMed]
- Wang, L.; Yang, G.; Zhao, D.; Wang, J.; Bai, Y.; Peng, Q.; Wang, H.; Fang, R.; Chen, G.; Wang, Z.; et al. CD103-positive CSC exosome promotes EMT of clear cell renal cell carcinoma: Role of remote MiR-19b-3p. Mol. Cancer 2019, 11, 86. [Google Scholar] [CrossRef] [PubMed]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 7, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Gibb, E.A.; Brown, C.J.; Lam, W.L. The functional role of long non-coding RNA in human carcinomas. Mol. Cancer 2011, 10, 38. [Google Scholar] [CrossRef] [Green Version]
- Tao, L.; Wang, X.; Zhou, Q. Long noncoding RNA SNHG16 promotes the tumorigenicity of cervical cancer cells by recruiting transcriptional factor SPI1 to upregulate PARP9. Cell Biol. Int. 2020, 44, 773–784. [Google Scholar] [CrossRef]
- Wang, X.; Kan, J.; Han, J.; Zhang, W.; Bai, L.; Wu, H. LncRNA SNHG16 Functions as an Oncogene by Sponging MiR-135a and Promotes JAK2/STAT3 Signal Pathway in Gastric Cancer. J. Cancer 2019, 10, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Li, P.; Lv, H.; Li, N.; Ren, S.; Xu, W. Exosomal SNHG16 secreted by CSCs promotes glioma development via TLR7. Stem Cell Res. Ther. 2021, 12, 349. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chefetz, I.; Alvero, A.B.; Holmberg, J.C.; Lebowitz, N.; Craveiro, V.; Yang-Hartwich, Y.; Yin, G.; Squillace, L.; Gurrea Soteras, M.; Aldo, P.; et al. TLR2 enhances ovarian cancer stem cell self-renewal and promotes tumor repair and recurrence. Cell Cycle 2013, 12, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, M.; Taguchi, Y.; Ito-Kureha, T.; Semba, K.; Yamaguchi, N.; Inoue, J. NF-κB non-cell-autonomously regulates cancer stem cell populations in the basal-like breast cancer subtype. Nat. Commun. 2013, 4, 2299. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Wang, L.; Zhou, Y.; Dong, L.; Ma, W.; Lv, L.; Zhang, J.; Wang, X. Glioblastoma Stem Cell-Derived Exosomes Enhance Stemness and Tumorigenicity of Glioma Cells by Transferring Notch1 Protein. Cell Mol. Neurobiol. 2020, 40, 767–784. [Google Scholar] [CrossRef] [PubMed]
- Safa, A.R.; Saadatzadeh, M.R.; Cohen-Gadol, A.A.; Pollok, K.E.; Bijangi-Vishehsaraei, K. Emerging targets for glioblastoma stem cell therapy. J. Biomed. Res. 2016, 30, 19–31. [Google Scholar]
- Nishihara, R.; Wu, K.; Lochhead, P.; Morikawa, T.; Liao, X.; Qian, Z.R.; Inamura, K.; Kim, S.A.; Kuchiba, A.; Yamauchi, M.; et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N. Engl. J. Med. 2013, 369, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.D.; Ren, Y.R.; Gao, Y.X.; Zhang, L.; Ding, Z. Hsa_circ_0005075 predicts a poor prognosis and acts as an oncogene in colorectal cancer via activating Wnt/β-catenin pathway. Eur Rev. Med. Pharmacol. Sci. 2019, 23, 3311–3319. [Google Scholar] [PubMed]
- Zhao, H.; Chen, S.; Fu, Q. Exosomes from CD133 + cells carrying circ-ABCC1 mediate cell stemness and metastasis in colorectal cancer. J. Cell Biochem. 2020, 121, 3286–3297. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Koren, E.; Fuchs, Y. The bad seed: Cancer stem cells in tumor development and resistance. Drug Resist. Updates 2016, 28, 1–12. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Meidhof, S.; Brabletz, S.; Lehmann, W.; Preca, B.T.; Mock, K.; Ruh, M.; Schüler, J.; Berthold, M.; Weber, A.; Burk, U.; et al. ZEB1-associated drug resistance in cancer cells is reversed by the class I HDAC inhibitor mocetinostat. EMBO Mol. Med. 2015, 7, 831–847. [Google Scholar] [CrossRef]
- Uramoto, H.; Iwata, T.; Onitsuka, T.; Shimokawa, H.; Hanagiri, T.; Oyama, T. Epithelial-mesenchymal transition in EGFR-TKI acquired resistant lung adenocarcinoma. Anticancer Res. 2010, 30, 2513–2517. [Google Scholar]
- Seigel, G.M.; Campbell, L.M.; Narayan, M.; Gonzalez-Fernandez, F. Cancer stem cell characteristics in retinoblastoma. Mol. Vis. 2005, 12, 729–737. [Google Scholar]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Nuchtern, J.G.; Jax, T.W.; Gobel, U.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells with high drug efflux capacity in human tumor cells. Proc. Natl. Acad. Sci. USA 2004, 101, 14228–14233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, D.J.; Taussig, D.; Simpson, C.; Allen, K.; Rohatiner, A.Z.; Lister, T.A.; Bonnet, D. Characterization of cells with a high aldehyde dehydrogenase activity from cord blood and acute myeloid leukemia samples. Stem Cells 2005, 23, 752–760. [Google Scholar] [CrossRef]
- Kreso, A.; O’Brien, C.A.; van Galen, P.; Gan, O.I.; Notta, F.; Brown, A.M.; Ng, K.; Ma, J.; Wienholds, E.; Dunant, C.; et al. Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 2013, 339, 543–548. [Google Scholar] [CrossRef] [Green Version]
- Prieto-Vila, M.; Takahashi, R.U.; Usuba, W.; Kohama, I.; Ochiya, T. Drug Resistance Driven by Cancer Stem Cells and Their Niche. Int. J. Mol. Sci. 2017, 18, 2574. [Google Scholar] [CrossRef] [Green Version]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Singh, P.K.; Caffery, T.; Ouellette, M.M.; Hollingsworth, M.A. Sonic hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Zhao, N.; Cui, J.; Wu, H.; Xiong, J.; Peng, T. Exosomes derived from cancer stem cells of gemcitabine-resistant pancreatic cancer cells enhance drug resistance by delivering miR-210. Cell Oncol. 2020, 43, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Hui, K.M. Mechanism of cancer drug resistance and the involvement of noncoding RNAs. Curr. Med. Chem. 2014, 21, 3029–3041. [Google Scholar] [CrossRef]
- Ouyang, M.; Li, Y.; Ye, S.; Ma, J.; Lu, L.; Lv, W.; Chang, G.; Li, X.; Li, Q.; Wang, S.; et al. MicroRNA profiling implies new markers of chemoresistance of triple-negative breast cancer. PLoS ONE 2014, 9, e96228. [Google Scholar]
- Kong, W.; He, L.; Coppola, M.; Guo, J.; Esposito, N.N.; Coppola, D.; Cheng, J.Q. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J. Biol. Chem. 2016, 291, 22855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, J.; Berg, T.; Kurzejamska, E.; Pang, M.F.; Tabor, V.; Jansson, M.; Roswall, P.; Pietras, K.; Sund, M.; Religa, P.; et al. MiR-155-mediated loss of C/EBPβ shifts the TGF-β response from growth inhibition to epithelial-mesenchymal transition, invasion and metastasis in breast cancer. Oncogene 2013, 32, 5614–5624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, J.C.; Lima, N.D.S.; Sarian, L.O.; Matheu, A.; Ribeiro, M.L.; Derchain, S.F.M. Exosome-mediated breast cancer chemoresistance via miR-155 transfer. Sci Rep. 2018, 8, 829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langlands, F.E.; Horgan, K.; Dodwell, D.D.; Smith, L. Breast cancer subtypes: Response to radiotherapy and potential radiosensitisation. Br. J. Radiol. 2013, 86, 20120601. [Google Scholar] [CrossRef]
- Qin, F.; Fan, Q.; Yu, P.K.N.; Almahi, W.A.; Kong, P.; Yang, M.; Cao, W.; Nie, L.; Chen, G.; Han, W. Properties and gene expression profiling of acquired radioresistance in mouse breast cancer cells. Ann. Transl. Med. 2021, 9, 628. [Google Scholar] [CrossRef]
- Ko, Y.S.; Jin, H.; Lee, J.S.; Park, S.W.; Chang, K.C.; Kang, K.M.; Jeong, B.K.; Kim, H.J. Radioresistant breast cancer cells exhibit increased resistance to chemotherapy and enhanced invasive properties due to cancer stem cells. Oncol. Rep. 2018, 40, 3752–3762. [Google Scholar] [CrossRef]
- Paramanantham, A.; Jung, E.J.; Go, S.I.; Jeong, B.K.; Jung, J.M.; Hong, S.C.; Kim, G.S.; Lee, W.S. Activated ERK Signaling Is One of the Major Hub Signals Related to the Acquisition of Radiotherapy-Resistant MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 4940. [Google Scholar] [CrossRef]
- Lau, E.Y.; Ho, N.P.; Lee, T.K. Cancer Stem Cells and Their Microenvironment: Biology and Therapeutic Implications. Stem Cells Int. 2017, 2017, 3714190. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Richards, K.E.; Zeleniak, A.E.; Fishel, M.L.; Wu, J.; Littlepage, L.E.; Hill, R. Cancer-associated fibroblast exosomes regulate survival and proliferation of pancreatic cancer cells. Oncogene 2017, 36, 1770–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, C.; Liu, T.; Yin, R. Biomarkers for cancer-associated fibroblasts. Biomark. Res. 2020, 11, 64. [Google Scholar]
- Lopez-de-Andres, J.L.; Jimenez-Garcia, R.; Hernández-Barrera, V.; Jiménez-Trujillo, I.; de Miguel-Yanes, J.M.; Carabantes-Alarcon, D.; de Miguel-Diez, J.; Lopez-Herranz, M. Cancer stem cell secretome in the tumor microenvironment: A key point for an effective personalized cancer treatment. J. Hematol. Oncol. 2020, 15, 136. [Google Scholar]
- Fontana, S.; Saieva, L.; Taverna, S.; Alessandro, R. Contribution of proteomics to understanding the role of tumor-derived exosomes in cancer progression: State of the art and new perspectives. Proteomics 2013, 13, 1581–1594. [Google Scholar]
- Peng, D.; Wang, H.; Li, L.; Ma, X.; Chen, Y.; Zhou, H.; Luo, Y.; Xiao, Y.; Liu, L. miR-34c-5p promotes eradication of acute myeloid leukemia stem cells by inducing senescence through selective RAB27B targeting to inhibit exosome shedding. Leukemia 2018, 32, 1180–1188. [Google Scholar] [CrossRef]
- Sharma, A. Role of stem cell derived exosomes in tumor biology. Int. J. Cancer. 2018, 15, 1086–1092. [Google Scholar] [CrossRef] [Green Version]
- Lindoso, R.S.; Collino, F.; Camussi, G. Extracellular vesicles derived from renal cancer stem cells induce a pro-tumorigenic phenotype in mesenchymal stromal cells. Oncotarget 2015, 6, 7959–7969. [Google Scholar] [CrossRef] [Green Version]
- Bonuccelli, G.; Avnet, S.; Grisendi, G.; Salerno, M.; Granchi, D.; Dominici, M.; Kusuzaki, K.; Baldini, N. Role of mesenchymal stem cells in osteosarcoma and metabolic reprogramming of tumor cells. Oncotarget 2014, 5, 7575–7588. [Google Scholar] [CrossRef]
- Sansone, P.; Savini, C.; Kurelac, I.; Chang, Q.; Amato, L.B.; Strillacci, A.; Stepanova, A.; Iommarini, L.; Mastroleo, C.; Daly, L.; et al. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, E9066–E9075. [Google Scholar] [CrossRef] [Green Version]
- Figueroa, J.; Phillips, L.M.; Shahar, T.; Hossain, A.; Gumin, J.; Kim, H.; Bean, A.J.; Calin, G.A.; Fueyo, J.; Walters, E.T.; et al. Exosomes from Glioma-Associated Mesenchymal Stem Cells Increase the Tumorigenicity of Glioma Stem-like Cells via Transfer of miR-1587. Cancer Res. 2017, 77, 5808–5819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prendergast, G.C. Immune escape as a fundamental trait of cancer: Focus on IDO. Oncogene 2008, 27, 3889–3900. [Google Scholar] [CrossRef] [Green Version]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E.; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, J.C.; Gonzalez, G.C.; Zhang, L.; Ibrahim, G.; Kelly, J.J.; Gustafson, M.P.; Lin, Y.; Dietz, A.B.; Forsyth, P.A.; Yong, V.W.; et al. Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro-Oncology 2010, 12, 351–365. [Google Scholar] [CrossRef]
- Wu, A.; Wei, J.; Kong, L.Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro-Oncology 2010, 12, 1113–1125. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 20, 484. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, R.; Codrici, E.; Popescu, I.D.; Mihai, S.; Necula, L.G.; Petrescu, D.; Teodoru, M.; Tanase, C.P. Cytokine patterns in brain tumour progression. Mediat. Inflamm. 2013, 2013, 979748. [Google Scholar] [CrossRef]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C.; et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzaei, R.; Sarkar, S.; Dzikowski, L.; Rawji, K.S.; Khan, L.; Faissner, A.; Bose, P.; Yong, V.W. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology 2018, 7, e1478647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, W.L.; Jiang, J.K.; Yang, S.H.; Huang, T.S.; Lan, H.Y.; Teng, H.W.; Yang, C.Y.; Tsai, Y.P.; Lin, C.H.; Wang, H.W.; et al. MicroRNA-146a directs the symmetric division of Snail-dominant colorectal cancer stem cells. Nat. Cell Biol. 2014, 16, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Luo, Y. Targeting macrophages in cancer immunotherapy. Signal. Transduct. Target. Ther. 2021, 26, 127. [Google Scholar]
- Cheng, H.; Wang, Z.; Fu, L.; Xu, T. Macrophage Polarization in the Development and Progression of Ovarian Cancers: An Overview. Front. Oncol. 2019, 22, 421. [Google Scholar] [CrossRef] [Green Version]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Schaerli, A.R.; Gao, F.; Däster, S.; Trella, E.; Droeser, R.A.; Muraro, M.G.; Zajac, P.; Zanetti, R.; Gillanders, W.E.; et al. Expression of programmed death ligand 1 (PD-L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res. Treat. 2014, 146, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Collino, F.; Vitillo, L.; Damasco, C.; Deregibus, M.C.; Tetta, C.; Bussolati, B.; Camussi, G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer Res. 2011, 71, 5346–5356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vera, N.; Acuña-Gallardo, S.; Grünenwald, F.; Caceres-Verschae, A.; Realini, O.; Acuña, R.; Lladser, A.; Illanes, S.E.; Varas-Godoy, M. Small Extracellular Vesicles Released from Ovarian Cancer Spheroids in Response to Cisplatin Promote the Pro-Tumorigenic Activity of Mesenchymal Stem Cells. Int. J. Mol. Sci. 2019, 20, 4972. [Google Scholar] [CrossRef] [Green Version]
- Conigliaro, A.; Costa, V.; Lo Dico, A.; Saieva, L.; Buccheri, S.; Dieli, F.; Manno, M.; Raccosta, S.; Mancone, C.; Tripodi, M.; et al. CD90+ liver cancer cells modulate endothelial cell phenotype through the release of exosomes containing H19 lncRNA. Mol. Cancer 2015, 14, 155. [Google Scholar] [CrossRef]
- Wang, Z.F.; Liao, F.; Wu, H.; Dai, J. Glioma stem cells-derived exosomal miR-26a promotes angiogenesis of microvessel endothelial cells in glioma. J. Exp. Clin. Cancer Res. 2019, 17, 201. [Google Scholar] [CrossRef] [Green Version]
- Nazarenko, I.; Rana, S.; Baumann, A.; McAlear, J.; Hellwig, A.; Trendelenburg, M.; Lochnit, G.; Preissner, K.T.; Zöller, M. Cell Surface Tetraspanin Tspan8 Contributes to Molecular Pathways of Exosome-Induced Endothelial Cell Activation. Cancer Res. 2010, 70, 1668–1678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Sena-Esteves, M.; Curry, W.T., Jr.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Lin, X.J.; Chong, Y.; Guo, Z.W.; Xie, C.; Yang, X.J.; Zhang, Q.; Li, S.P.; Xiong, Y.; Yuan, Y.; Min, J.; et al. A serum microRNA classifier for early detection of hepatocellular carcinoma: A multicentre, retrospective, longitudinal biomarker identification study with a nested case-control study. Lancet Oncol. 2015, 16, 804–815. [Google Scholar] [CrossRef]
- Lin, X.J.; Fang, J.H.; Yang, X.J.; Zhang, C.; Yuan, Y.; Zheng, L.; Zhuang, S.M. Hepatocellular Carcinoma Cell-Secreted Exosomal MicroRNA-210 Promotes Angiogenesis In Vitro and In Vivo. Mol. Ther. Nucleic Acids 2018, 11, 243–252. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pfannenstiel, L.W.; Bolesta, E.; Montes, C.L.; Zhang, X.; Chapoval, A.I.; Gartenhaus, R.B.; Strome, S.E.; Gastman, B.R. Interleukin-7 Inhibits Tumor-Induced CD27−CD28− Suppressor T Cells: Implications for Cancer Immunotherapy. Clin. Cancer Res. 2011, 17, 4975–4986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer 2017, 5, 65. [Google Scholar] [CrossRef]
- Montes, C.L.; Chapoval, A.I.; Nelson, J.; Orhue, V.; Zhang, X.; Schulze, D.H.; Strome, S.E.; Gastman, B.R. Tumor-induced senescent T cells with suppressor function: A potential form of tumor immune evasion. Cancer Res. 2008, 68, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Filaci., G.; Fenoglio, D.; Fravega, M.; Ansaldo, G.; Borgonovo, G.; Traverso, P.; Villaggio, B.; Ferrera, A.; Kunkl, A.; Rizzi, M.; et al. CD8+ CD28- T regulatory lymphocytes inhibiting T cell proliferative and cytotoxic functions infiltrate human cancers. J. Immunol. 2007, 179, 4323–4334. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.L.; Wang, W.; Lan, X.L.; Zeng, Z.C.; Liang, Y.S.; Yan, Y.R.; Song, F.Y.; Wang, F.F.; Zhu, X.H.; Liao, W.J.; et al. CAFs secreted exosomes promote metastasis and chemotherapy resistance by enhancing cell stemness and epithelial-mesenchymal transition in colorectal cancer. Mol. Cancer 2019, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tan, X.; Tan, Y.; Li, Q.; Ma, J.; Wang, G. Mesenchymal Stem Cell Derived Exosomes in Cancer Progression, Metastasis and Drug Delivery: A Comprehensive Review. J. Cancer 2018, 9, 3129–3137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Stanisavljevic, J.; Loubat-Casanovas, J.; Herrera, M.; Luque, T.; Peña, R.; Lluch, A.; Albanell, J.; Bonilla, F.; Rovira, A.; Peña, C.; et al. Snail1-Expressing Fibroblasts in the Tumor Microenvironment Display Mechanical Properties That Support Metastasis. Cancer Res. 2015, 75, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Quintavalle, C.; Donnarumma, E.; Iaboni, M.; Roscigno, G.; Garofalo, M.; Romano, G.; Fiore, D.; De Marinis, P.; Croce, C.M.; Condorelli, G. Effect of miR-21 and miR-30b/c on TRAIL-induced apoptosis in glioma cells. Oncogene 2013, 32, 4001–4008. [Google Scholar] [CrossRef] [Green Version]
- Fiore, D.; Donnarumma, E.; Roscigno, G.; Iaboni, M.; Russo, V.; Affinito, A.; Adamo, A.; De Martino, F.; Quintavalle, C.; Romano, G.; et al. miR-340 predicts glioblastoma survival and modulates key cancer hallmarks through down-regulation of NRAS. Oncotarget 2016, 7, 19531–19547. [Google Scholar] [CrossRef] [Green Version]
- Donnarumma, E.; Fiore, D.; Nappa, M.; Roscigno, G.; Adamo, A.; Iaboni, M.; Russo, V.; Affinito, A.; Puoti, I.; Quintavalle, C.; et al. Cancer-associated fibroblasts release exosomal microRNAs that dictate an aggressive phenotype in breast cancer. Oncotarget 2017, 8, 19592–19608. [Google Scholar] [CrossRef] [Green Version]
- Josson, S.; Gururajan, M.; Sung, S.Y.; Hu, P.; Shao, C.; Zhau, H.E.; Liu, C.; Lichterman, J.; Duan, P.; Li, Q.; et al. Stromal fibroblast-derived miR-409 promotes epithelial-to-mesenchymal transition and prostate tumorigenesis. Oncogene 2015, 34, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Stadtfeld, M.; Apostolou, E.; Akutsu, H.; Fukuda, A.; Follett, P.; Natesan, S.; Kono, T.; Shioda, T.; Hochedlinger, K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature 2010, 465, 175–181. [Google Scholar] [CrossRef] [Green Version]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell Vesicles. 2019, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Yan, C.; Mu, L.; Huang, K.; Li, X.; Tao, D.; Wu, Y.; Qin, J. Fibroblast-Derived Exosomes Contribute to Chemoresistance through Priming Cancer Stem Cells in Colorectal Cancer. PLoS ONE 2015, 10, e0125625. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.L.; Zhuang, T.; Xing, B.H.; Li, N.; Li, Q. Exosomal DNMT1 mediates cisplatin resistance in ovarian cancer. Cell Biochem. Funct. 2017, 35, 296–303. [Google Scholar] [CrossRef]
- Richards, K.E.; Go, D.B.; Hill, R. Surface Acoustic Wave Lysis and Ion-Exchange Membrane Quantification of Exosomal MicroRNA. Methods Mol. Biol. 2017, 1580, 59–70. [Google Scholar] [PubMed]
- Aung, G.; Niyonsaba, F.; Ushio, H.; Kajiwara, N.; Saito, H.; Ikeda, S.; Ogawa, H.; Okumura, K. Catestatin, a neuroendocrine antimicrobial peptide, induces human mast cell migration, degranulation and production of cytokines and chemokines. Immunology 2011, 132, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.; Aung, T.; Vogel, D.; Chapuy, B.; Wenzel, D.; Becker, S.; Sinzig, U.; Venkataramani, V.; von Mach, T.; Jacob, R.; et al. Nuclear Trapping through Inhibition of Exosomal Export by Indomethacin Increases Cytostatic Efficacy of Doxorubicin and Pixantrone. Clin. Cancer Res. 2016, 22, 395–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, F.M.; Saleh, E.; Alawadhi, H.; Harati, R.; Zimmermann, W.H.; El-Awady, R. Inhibition of exosome release by ketotifen enhances sensitivity of cancer cells to doxorubicin. Cancer Biol. Ther. 2018, 19, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Xing, F.; Liu, Y.; Wu, S.Y.; Wu, K.; Sharma, S.; Mo, Y.Y.; Feng, J.; Sanders, S.; Jin, G.; Singh, R.; et al. Loss of XIST in Breast Cancer Activates MSN-c-Met and Reprograms Microglia via Exosomal miRNA to Promote Brain Metastasis. Cancer Res. 2018, 78, 4316–4330. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.H.; Huang, Y.C.; Lee, Y.H.; Tan, Z.L.; Tsai, C.J.; Chuang, Y.C.; Tu, H.F.; Liu, T.C.; Hsu, F.T. Anticancer Efficacy and Mechanism of Amentoflavone for Sensitizing Oral Squamous Cell Carcinoma to Cisplatin. Anticancer Res. 2020, 40, 6723–6732. [Google Scholar] [CrossRef]
- Ren, W.; Hou, J.; Yang, C.; Wang, H.; Wu, S.; Wu, Y.; Zhao, X.; Lu, C. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J. Exp. Clin. Cancer Res. 2019, 38, 62. [Google Scholar] [CrossRef]
- Zheng, P.; Chen, L.; Yuan, X.; Luo, Q.; Liu, Y.; Xie, G.; Ma, Y.; Shen, L. Exosomal transfer of tumor-associated macrophage-derived miR-21 confers cisplatin resistance in gastric cancer cells. J. Exp. Clin. Cancer Res. 2017, 36, 53. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.H.; Tsao, C.J. Emerging role of microRNA-21 in cancer. Biomed. Rep. 2016, 5, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530. [Google Scholar] [CrossRef]
- Liu, Y.; Luo, F.; Wang, B.; Li, H.; Xu, Y.; Liu, X.; Shi, L.; Lu, X.; Xu, W.; Lu, L.; et al. STAT3-regulated exosomal miR-21 promotes angiogenesis and is involved in neoplastic processes of transformed human bronchial epithelial cells. Cancer Lett. 2016, 370, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.J.; Huang, T.H.; Yadav, V.K.; Sumitra, M.R.; Tzeng, D.T.; Wei, P.L.; Shih, J.W.; Wu, A.T. Preclinical investigation of ovatodiolide as a potential inhibitor of colon cancer stem cells via downregulating sphere-derived exosomal β-catenin/STAT3/miR-1246 cargoes. Am. J. Cancer Res. 2020, 10, 2337–2354. [Google Scholar] [PubMed]
- Falzone, L.; Scola, L.; Zanghì, A.; Biondi, A.; Di Cataldo, A.; Libra, M.; Candido, S. Integrated analysis of colorectal cancer microRNA datasets: Identification of microRNAs associated with tumor development. Aging 2018, 10, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.Y.; Su, Y.K.; Liu, H.W.; Chen, C.H.; Chiu, S.C.; Cho, D.Y.; Lin, S.Z.; Chen, Y.S.; Lin, C.M. Preclinical Evidence of STAT3 Inhibitor Pacritinib Overcoming Temozolomide Resistance via Downregulating miR-21-Enriched Exosomes from M2 Glioblastoma-Associated Macrophages. J. Clin. Med. 2019, 8, 959. [Google Scholar] [CrossRef] [Green Version]
- Gernapudi, R.; Wolfson, B.; Zhang, Y.; Yao, Y.; Yang, P.; Asahara, H.; Zhou, Q. MicroRNA 140 Promotes Expression of Long Noncoding RNA NEAT1 in Adipogenesis. Mol. Cell Biol. 2015, 36, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Marleau, A.M.; Chen, C.S.; Joyce, J.A.; Tullis, R.H. Exosome removal as a therapeutic adjuvant in cancer. J. Transl. Med. 2012, 10, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishida-Aoki, N.; Tominaga, N.; Takeshita, F.; Sonoda, H.; Yoshioka, Y.; Ochiya, T. Disruption of Circulating Extracellular Vesicles as a Novel Therapeutic Strategy against Cancer Metastasis. Mol. Ther. 2017, 25, 181–191. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Nie, H.; Zhou, Y.; Lian, S.; Mei, H.; Lu, Y.; Dong, H.; Li, F.; Li, T.; Li, B.; et al. Eliminating blood oncogenic exosomes into the small intestine with aptamer-functionalized nanoparticles. Nat. Commun. 2019, 10, 5476. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Oller, L.; Seras-Franzoso, J.; Andrade, F.; Rafael, D.; Abasolo, I.; Gener, P.; Schwartz, S., Jr. Extracellular Vesicles as Drug Delivery Systems in Cancer. Pharmaceutics 2020, 12, 1146. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Yang, H.W.; Yang, L.C.; Lu, M.Y.; Tsai, L.L.; Yang, S.F.; Huang, Y.F.; Chou, M.Y.; Yu, C.C.; Hu, F.W. DHFR and MDR1 upregulation is associated with chemoresistance in osteosarcoma stem-like cells. Oncol. Lett. 2017, 14, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Naseri, Z.; Oskuee, R.K.; Forouzandeh-Moghadam, M.; Jaafari, M.R. Delivery of LNA-antimiR-142-3p by Mesenchymal Stem Cells-Derived Exosomes to Breast Cancer Stem Cells Reduces Tumorigenicity. Stem Cell Rev. Rep. 2020, 16, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Isobe, T.; Hisamori, S.; Hogan, D.J.; Zabala, M.; Hendrickson, D.G.; Dalerba, P.; Cai, S.; Scheeren, F.; Kuo, A.H.; Sikandar, S.S.; et al. miR-142 regulates the tumorigenicity of human breast cancer stem cells through the canonical WNT signaling pathway. Elife 2014, 18, e01977. [Google Scholar]
- Scioli, M.G.; Artuso, S.; D’Angelo, C.; Porru, M.; D’Amico, F.; Bielli, A.; Gentile, P.; Cervelli, V.; Leonetti, C.; Orlandi, A. Adipose-derived stem cell-mediated paclitaxel delivery inhibits breast cancer growth. PLoS ONE 2018, 13, e0203426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielli, A.; Scioli, M.G.; Gentile, P.; Agostinelli, S.; Tarquini, C.; Cervelli, V.; Orlandi, A. Adult adipose-derived stem cells and breast cancer: A controversial relationship. Springerplus 2014, 8, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielli, A.; Scioli, M.G.; Gentile, P.; Cervelli, V.; Orlandi, A. Adipose Tissue-Derived Stem Cell Therapy for Post-Surgical Breast Reconstruction—More Light than Shadows. Adv. Clin. Exp. Med. 2015, 24, 545–548. [Google Scholar] [CrossRef]
- Li, B.; Li, Q.; Mo, J.; Dai, H. Drug-Loaded Polymeric Nanoparticles for Cancer Stem Cell Targeting. Front. Pharmacol. 2017, 14, 5151. [Google Scholar] [CrossRef] [Green Version]
- Kunath, K.; Merdan, T.; Hegener, O.; Häberlein, H.; Kissel, T. Integrin targeting using RGD-PEI conjugates for in vitro gene transfer. J. Gene Med. 2003, 5, 588–599. [Google Scholar] [CrossRef]
- Yong, T.; Zhang, X.; Bie, N.; Zhang, H.; Zhang, X.; Li, F.; Hakeem, A.; Hu, J.; Gan, L.; Santos, H.A.; et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019, 10, 3838. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.M.; Wang, Y.C.; Wang, F.; Du, J.Z.; Mao, C.Q.; Sun, C.Y.; Tang, R.Z.; Liu, Y.; Zhu, J.; Zhu, Y.H.; et al. Cancer stem cell therapy using doxorubicin conjugated to gold nanoparticles via hydrazone bonds. Biomaterials 2014, 35, 836–845. [Google Scholar] [CrossRef]
- Ni, M.; Xiong, M.; Zhang, X.; Cai, G.; Chen, H.; Zeng, Q.; Yu, Z. Poly(lactic-co-glycolic acid) nanoparticles conjugated with CD133 aptamers for targeted salinomycin delivery to CD133+ osteosarcoma cancer stem cells. Int. J. Nanomed. 2015, 10, 2537–2554. [Google Scholar]
- Muntimadugu, E.; Kumar, R.; Saladi, S.; Rafeeqi, T.A.; Khan, W. CD44 targeted chemotherapy for co-eradication of breast cancer stem cells and cancer cells using polymeric nanoparticles of salinomycin and paclitaxel. Colloids Surf. B Biointerfaces 2016, 143, 532–546. [Google Scholar] [CrossRef]
- Swaminathan, S.K.; Roger, E.; Toti, U.; Niu, L.; Ohlfest, J.R.; Panyam, J. CD133-targeted paclitaxel delivery inhibits local tumor recurrence in a mouse model of breast cancer. J. Control. Release 2013, 171, 280–287. [Google Scholar] [CrossRef] [Green Version]
- Banzato, A.; Bobisse, S.; Rondina, M.; Renier, D.; Bettella, F.; Esposito, G.; Quintieri, L.; Meléndez-Alafort, L.; Mazzi, U.; Zanovello, P.; et al. A paclitaxel-hyaluronan bioconjugate targeting ovarian cancer affords a potent in vivo therapeutic activity. Clin. Cancer Res. 2008, 14, 3598–3606. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.Y.; Saravanakumar, G.; Park, J.H.; Park, K. Hyaluronic acid-based nanocarriers for intracellular targeting: Interfacial interactions with proteins in cancer. Colloids Surf. B Biointerfaces 2012, 99, 82–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, S.; Du, X.J.; Liu, J.; Sun, R.; Zhu, Y.H.; Wang, J. Delivery of bortezomib with nanoparticles for basal-like triple-negative breast cancer therapy. J. Control. Release 2015, 208, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, R.T.; Qian, H.Q.; Wei, J.; Xie, L.; Shen, J.; Yang, M.; Qian, X.P.; Yu, L.X.; Jiang, X.Q.; et al. Targeted delivery of miR-200c/DOC to inhibit cancer stem cells and cancer cells by the gelatinases-stimuli nanoparticles. Biomaterials 2013, 34, 7191–7203. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Jiang, Y.; Zhang, H.; Sun, B.; Hou, C.; Zheng, J.; Liu, Y.; Zuo, P. Active targeting docetaxel-PLA nanoparticles eradicate circulating lung cancer stem-like cells and inhibit liver metastasis. Mol. Pharm. 2015, 12, 232–239. [Google Scholar] [CrossRef]
- Lopez-Bertoni, H.; Kozielski, K.L.; Rui, Y.; Lal, B.; Vaughan, H.; Wilson, D.R.; Mihelson, N.; Eberhart, C.G.; Laterra, J.; Green, J.J. Bioreducible Polymeric Nanoparticles Containing Multiplexed Cancer Stem Cell Regulating miRNAs Inhibit Glioblastoma Growth and Prolong Survival. Nano Lett. 2018, 18, 4086–4094. [Google Scholar] [CrossRef]
- Zuo, Z.Q.; Chen, K.G.; Yu, X.Y.; Zhao, G.; Shen, S.; Cao, Z.T.; Luo, Y.L.; Wang, Y.C.; Wang, J. Promoting tumor penetration of nanoparticles for cancer stem cell therapy by TGF-β signaling pathway inhibition. Biomaterials 2016, 82, 48–59. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Strategy | Agent/Method | Tumor Type | Inhibition | References |
|---|---|---|---|---|
| EV biogenesis and release/uptake | U0126 | Human pancreatic adenocarcinoma cells Suit-2 | Gemcitabine resistance | [149] |
| GW4869 | Colorectal, pancreatic, and ovarian cancer cells | Chemoresistance | [151,152,153] | |
| Indomethacin | Lymphoma cells | Chemoresistance | [155] | |
| Ketotifen | HeLa, MCF-7, and BT549 breast cancer cells | Doxorubicin resistance | [156] | |
| Fludarabine | Metastatic breast cancer | M2 polarization | [157] | |
| Ovatodiolide | Oral squamous cell carcinoma | Cisplatin resistance | [158] | |
| Colon cancer HCT116 and HT29 cells | 5-FU resistance, CAF transformation, and M2 polarization | [164] | ||
| Pacritinib | Glioblastoma multiforme | Temozolomide resistance, M2 polarization | [166] | |
| Removal of circulating EVs | Shikonin | Breast cancer MCF10DCIS cells | Differentiation, stemness, and migration of cancer cells | [167] |
| Anti-EV marker antibodies | Mouse model of breast cancer | Metastasis | [170] | |
| Hemofiltration in conjunction with pembrolizumab | Advanced head and neck cancer patients (NCT04453046) | Metastasis | [168] | |
| Use of natural bioengineered EVs | EVs from osteogenic differentiated hASCs | MG63 osteosarcoma cells | Osteogenic differentiation of CSCs | [172] |
| EVs from mMSCs loaded with LNA-antimiR-142-3p | MCF7-derived cancer-stem-like cells | Mammosphere formation in vitro and tumorigenicity in vivo | [173] | |
| CM from PTX-primed hASCs | CG5 breast cancer cells | Proliferation in vitro and tumor growth in vivo | [175] | |
| Use of synthetic NPs | DOX@E-PSiNPs | Mouse hepatocellular carcinoma cell line H22 | Tumor growth | [180] |
| DOXO-tethered gold nanoparticles | Mouse breast cancer cells | Sphere formation | [181] | |
| CD133-tethered PLGA nanoparticles—salinomycin loaded | CD133+ osteosarcoma and breast cancer | Proliferation, drug resistance, and relapse of breast CSCs | [182] | |
| PTX-hyaluronan bioconjugate (ONCOFIDTM-P) | Human ovarian cancer IGROV-1 and OVCAR-3 overexpressing CD44 | Tumor growth in vivo | [185] | |
| PEG-b-PLA nanoparticles loaded with bortezomib | Breast cancer cells | Proliferation and survival | [187] | |
| PEG-peptide-poly(e-caprolactone) nanoparticles loaded with DOXO and miR-200c | Gastric carcinoma cell line BGC-823 | Tumor growth in vivo | [188] | |
| PLA nanoparticles—peptide CVKTPAQSC—docetaxel loaded | Lung-cancer-stem-like cells | Liver metastasis in a mouse model | [189] | |
| Poly (beta-amino ester) nanoparticles loaded with miR-148a/miR-296-5p | Glioblastoma cells | Stemness in vitro, tumor growth, and sensitizing to γ-radiation in vivo | [190] | |
| Nanoparticles with siRNAs targeting Plk1 in combination with LY364947 | Breast CSCs | Proliferation in vitro and tumor growth in vivo | [191] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scioli, M.G.; Terriaca, S.; Fiorelli, E.; Storti, G.; Fabbri, G.; Cervelli, V.; Orlandi, A. Extracellular Vesicles and Cancer Stem Cells in Tumor Progression: New Therapeutic Perspectives. Int. J. Mol. Sci. 2021, 22, 10572. https://doi.org/10.3390/ijms221910572
Scioli MG, Terriaca S, Fiorelli E, Storti G, Fabbri G, Cervelli V, Orlandi A. Extracellular Vesicles and Cancer Stem Cells in Tumor Progression: New Therapeutic Perspectives. International Journal of Molecular Sciences. 2021; 22(19):10572. https://doi.org/10.3390/ijms221910572
Chicago/Turabian StyleScioli, Maria Giovanna, Sonia Terriaca, Elena Fiorelli, Gabriele Storti, Giulia Fabbri, Valerio Cervelli, and Augusto Orlandi. 2021. "Extracellular Vesicles and Cancer Stem Cells in Tumor Progression: New Therapeutic Perspectives" International Journal of Molecular Sciences 22, no. 19: 10572. https://doi.org/10.3390/ijms221910572