
The Impact of m1A Methylation Modification Patterns on Tumor Immune Microenvironment and Prognosis in Oral Squamous Cell Carcinoma


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
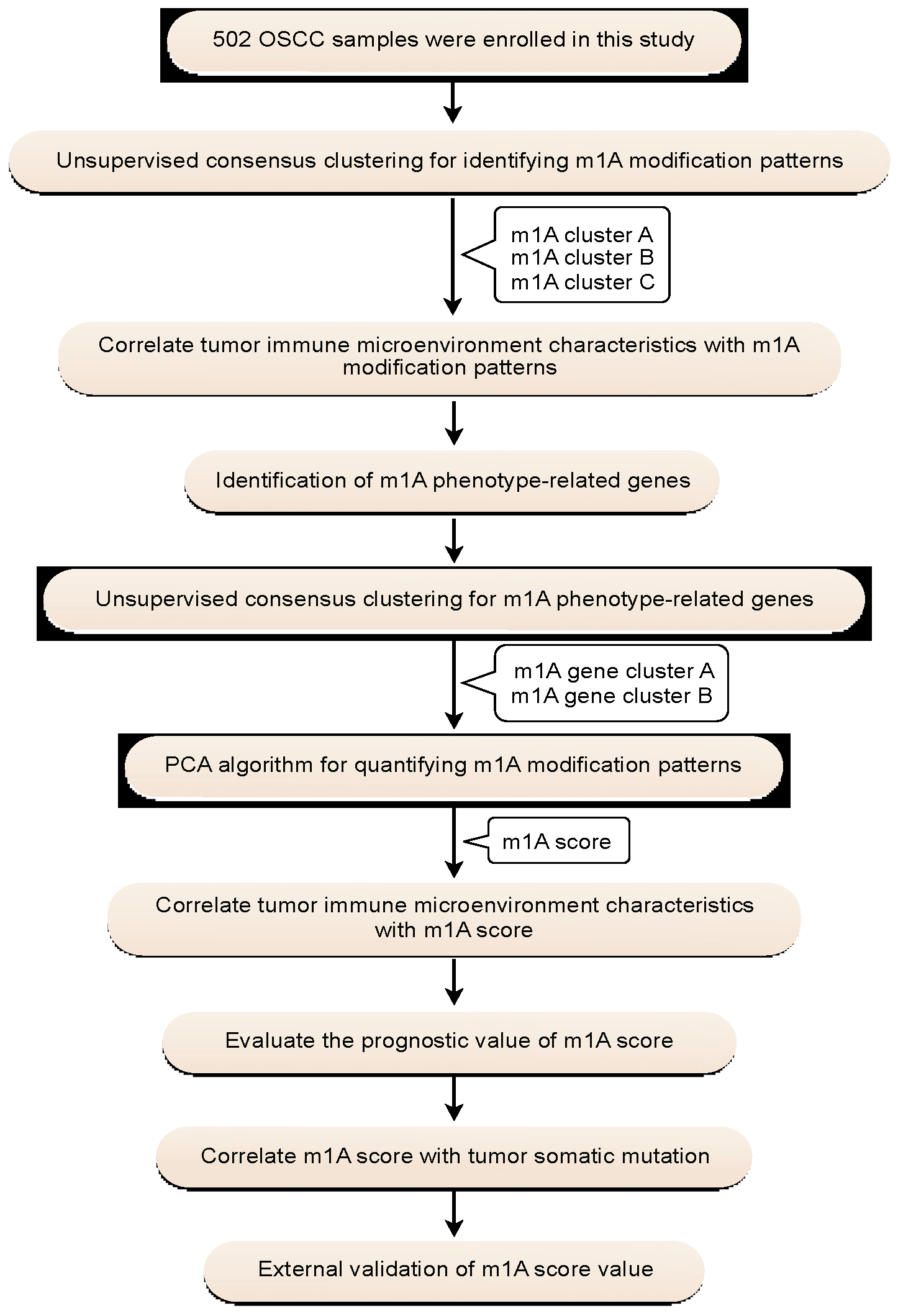
2. Results
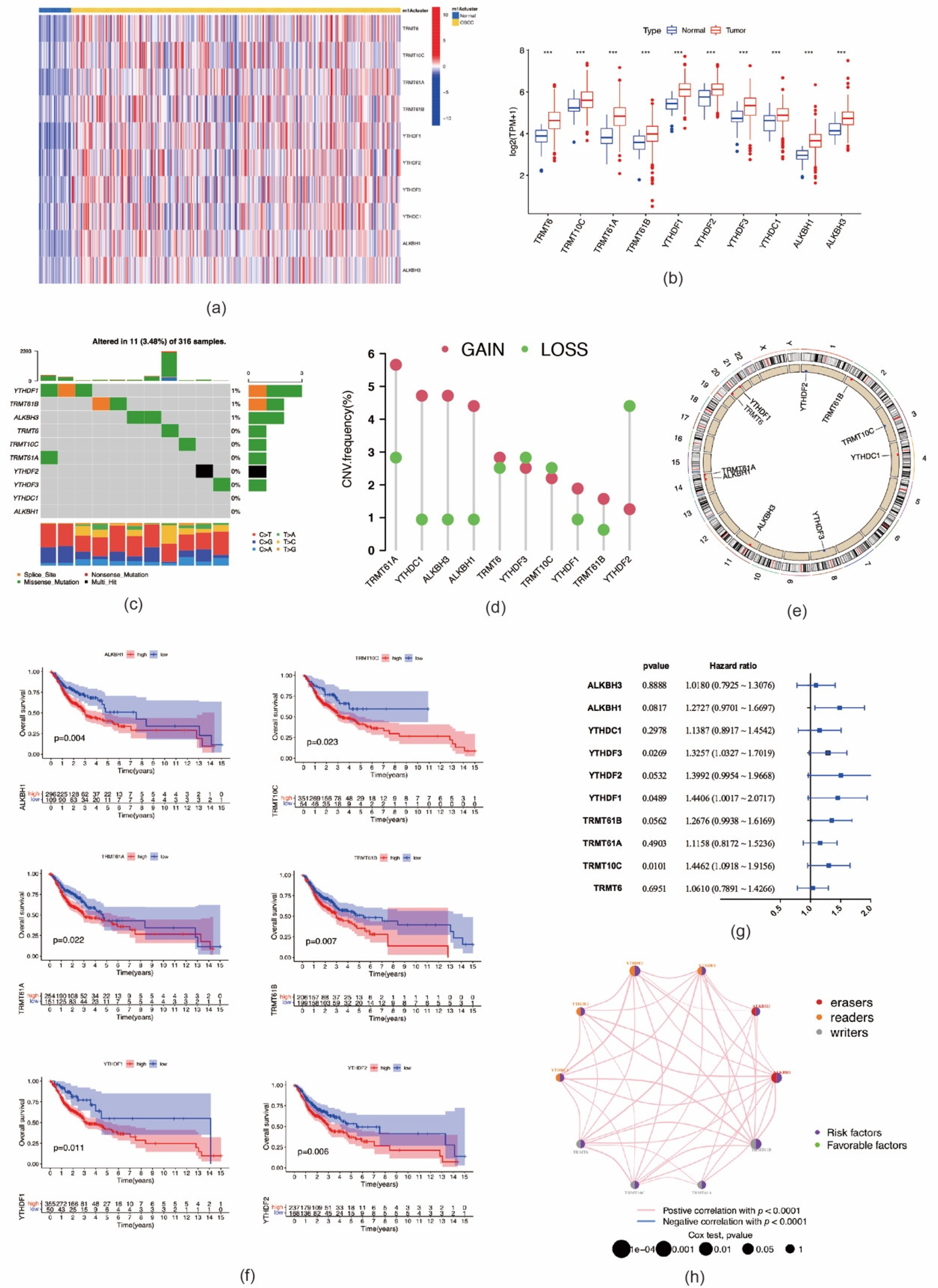
2.1. Genetic Variation and Prognostic Relevance of m1A RNA Methylation Regulators in OSCC
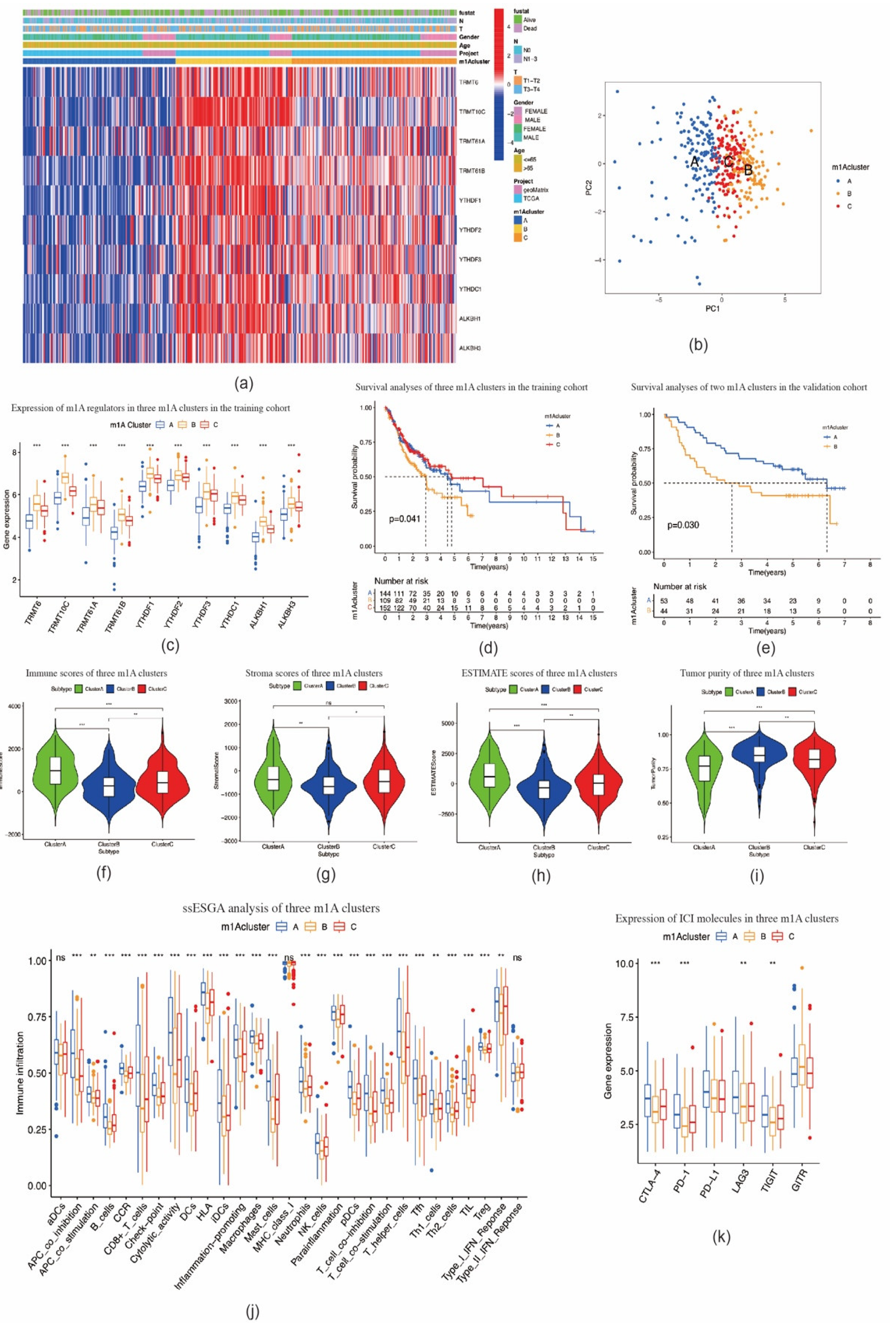
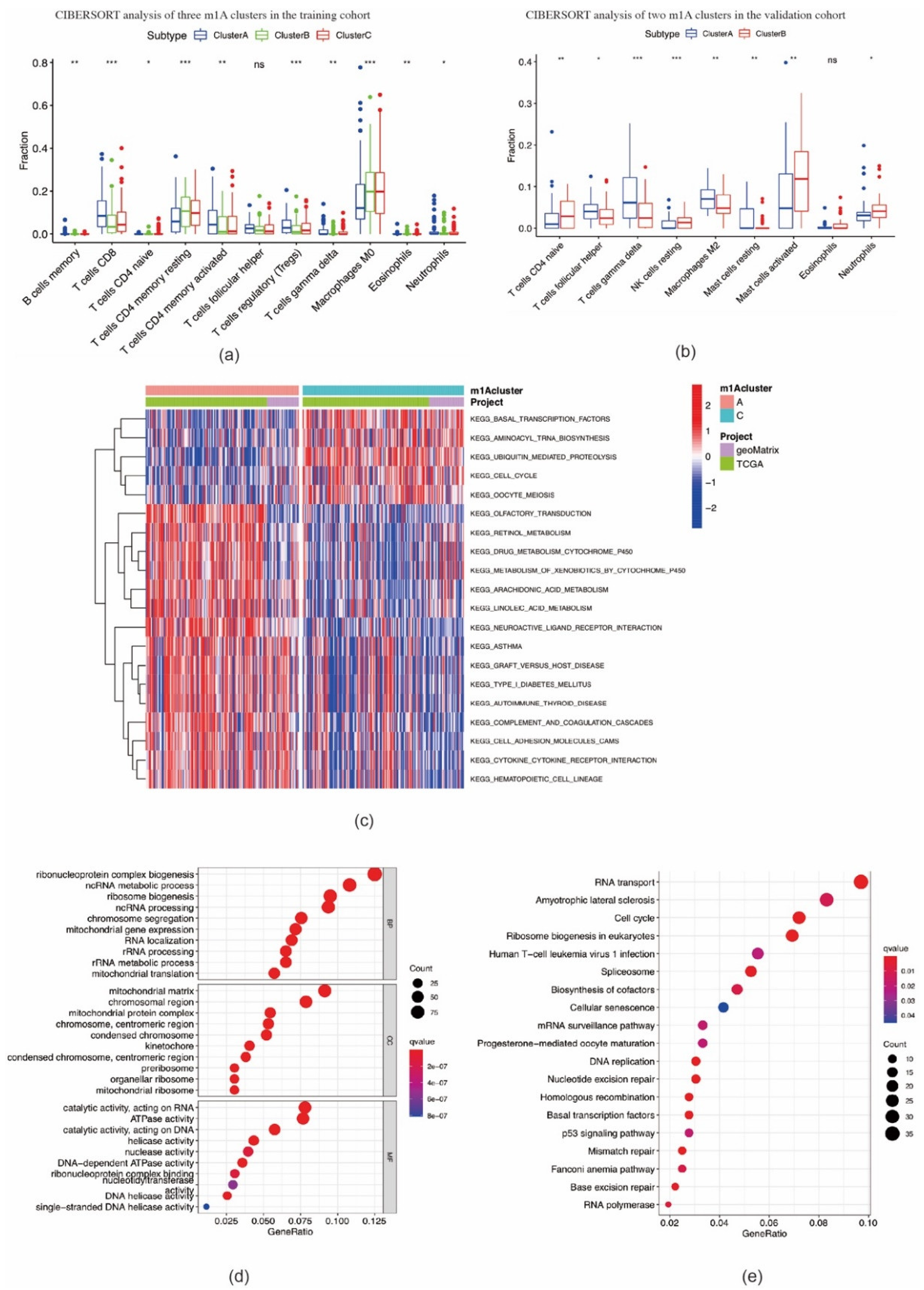
2.2. Consensus Clustering of m1A Regulators in Three Clusters Correlated with OSCC Prognosis and Immune Microenvironment
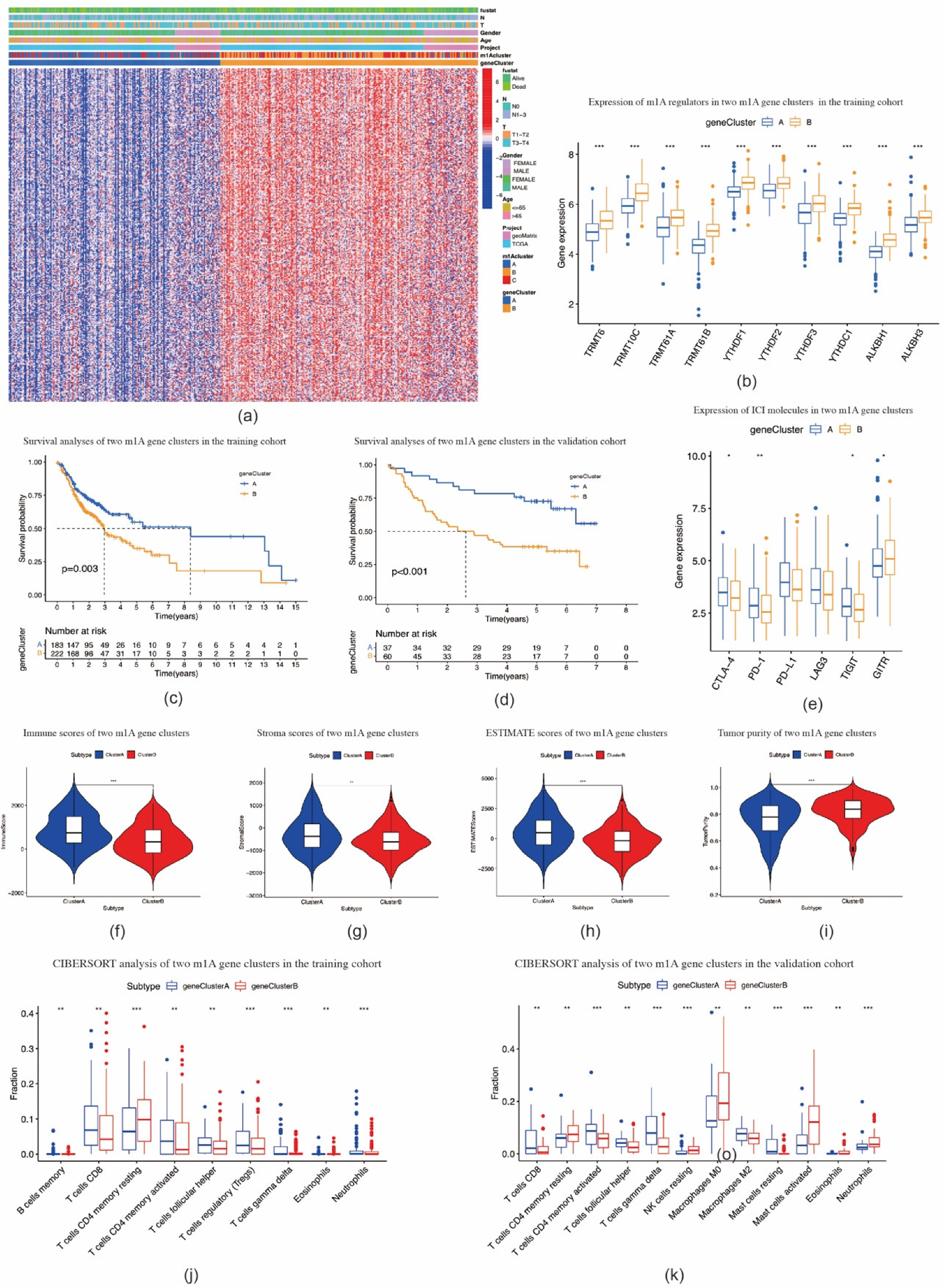
2.3. Identification of m1A Gene Signatures in OSCC
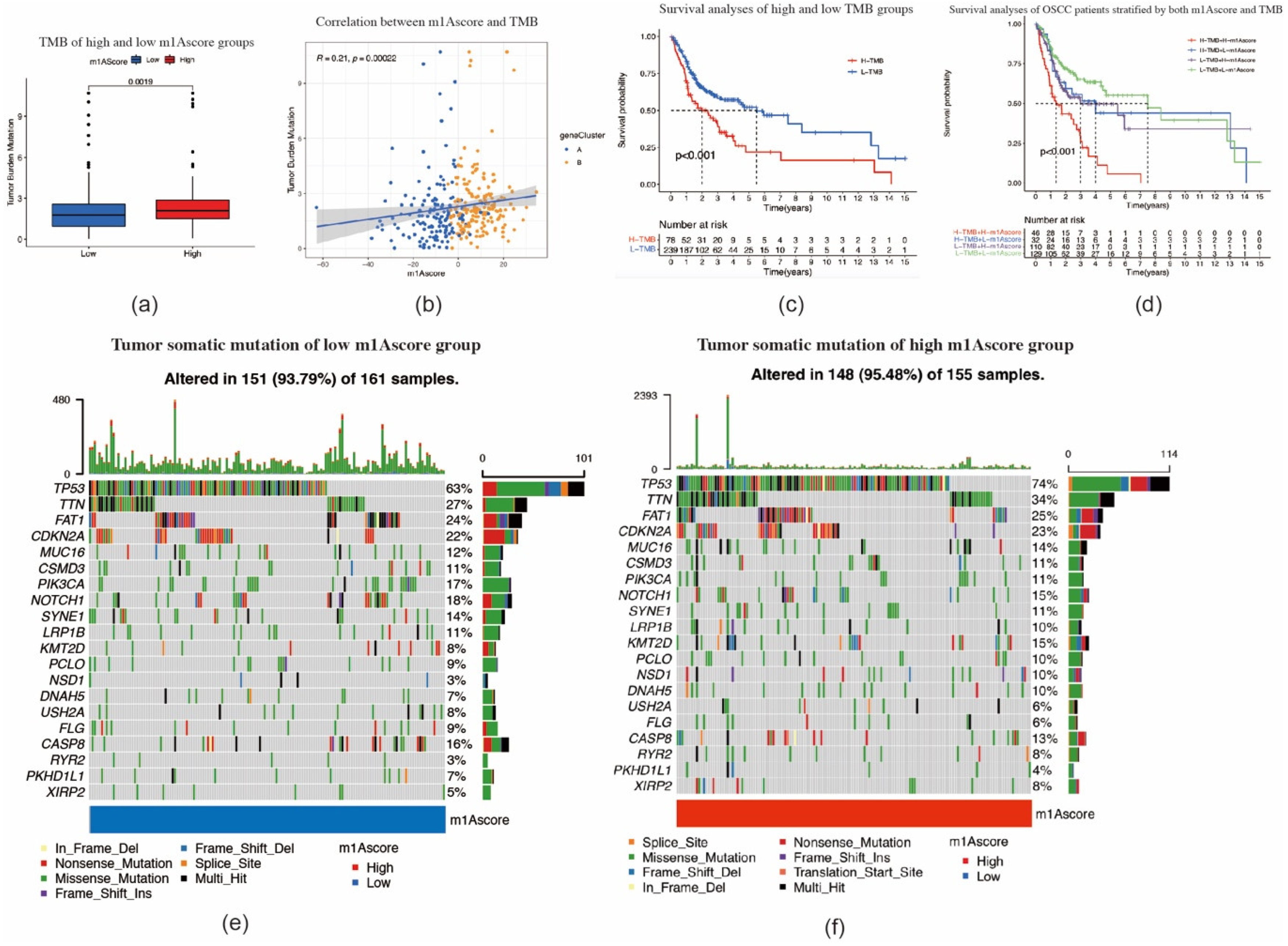
2.4. Construction of the m1A Gene Signature
3. Discussion
4. Materials and Methods
4.1. Dataset Source and Preprocessing
4.2. Unsupervised Consensus Clustering of m1A Methylation Regulators
4.3. TIME Cell Infiltration Evaluation
4.4. Gene Set Variation Analysis
4.5. Identification of m1A-Related Differentially Expressed Genes
4.6. GO and KEGG Pathway Enrichment Analyses
4.7. Construction of m1A Gene Signature
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gődény, M. Prognostic factors in advanced pharyngeal and oral cavity cancer; significance of multimodality imaging in terms of 7th edition of TNM. Cancer Imaging Off. Publ. Int. Cancer Imaging Soc. 2014, 14, 15. [Google Scholar] [CrossRef] [Green Version]
- Warnakulasuriya, S. Global epidemiology of oral and oropharyngeal cancer. Oral Oncol. 2009, 45, 309–316. [Google Scholar] [CrossRef]
- Mashberg, A.; Boffetta, P.; Winkelman, R.; Garfinkel, L. Tobacco smoking, alcohol drinking, and cancer of the oral cavity and oropharynx among U.S. veterans. Cancer 1993, 72, 1369–1375. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Chang, J.T.-C.; Liao, C.-T.; Wang, H.-M.; Yen, T.-C.; Chiu, C.-C.; Lu, Y.-C.; Li, H.-F.; Cheng, A.-J. Head and neck cancer in the betel quid chewing area: Recent advances in molecular carcinogenesis. Cancer Sci. 2008, 99, 1507–1514. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, L.; Bennardo, F.; Giudice, A.; Passante, M.; Dastoli, S.; Morrone, P.; Provenzano, E.; Patruno, C.; Nisticò, S.P. Local Chemotherapy as an Adjuvant Treatment in Unresectable Squamous Cell Carcinoma: What Do We Know So Far? Curr. Oncol. 2021, 28, 2317–2325. [Google Scholar] [CrossRef] [PubMed]
- Pentangelo, G.; Nisticò, S.P.; Provenzano, E.; Cisale, G.Y.; Bennardo, L. Topical 5% Imiquimod Sequential to Surgery for HPV-Related Squamous Cell Carcinoma of the Lip. Medicina 2021, 57, 563. [Google Scholar] [CrossRef]
- Zhang, L.; Meng, X.; Zhu, X.-W.; Yang, D.-C.; Chen, R.; Jiang, Y.; Xu, T. Long non-coding RNAs in Oral squamous cell carcinoma: Biologic function, mechanisms and clinical implications. Mol. Cancer 2019, 18, 102. [Google Scholar] [CrossRef]
- Couzin-Frankel, J. Breakthrough of the year 2013. Cancer immunotherapy. Science 2013, 342, 1432–1433. [Google Scholar] [CrossRef] [Green Version]
- Melero, I.; Castanon, E.; Alvarez, M.; Champiat, S.; Marabelle, A. Intratumoural administration and tumour tissue targeting of cancer immunotherapies. Nat. Rev. Clin. Oncol. 2021, 18, 558–576. [Google Scholar] [CrossRef]
- Dong, Y.; Wang, Z.; Mao, F.; Cai, L.; Dan, H.; Jiang, L.; Zeng, X.; Li, T.; Zhou, Y.; Chen, Q. PD-1 blockade prevents the progression of oral carcinogenesis. Carcinogenesis 2021, 42, 891–902. [Google Scholar] [CrossRef]
- Gao, A.; Pan, X.; Yang, X.; Lin, Z. Predictive factors in the treatment of oral squamous cell carcinoma using PD-1/PD-L1 inhibitors. Investig. New Drugs 2021, 39, 1132–1138. [Google Scholar] [CrossRef]
- Saeed, M.; Gao, J.; Shi, Y.; Lammers, T.; Yu, H. Engineering Nanoparticles to Reprogram the Tumor Immune Microenvironment for Improved Cancer Immunotherapy. Theranostics 2019, 9, 7981–8000. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, B.; Nie, Z.; Duan, L.; Xiong, Q.; Jin, Z.; Yang, C.; Chen, Y. The role of m6A modification in the biological functions and diseases. Signal. Transduct. Target. Ther. 2021, 6, 74. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Chai, P.; Jia, R.; Fan, X. Novel insight into the regulatory roles of diverse RNA modifications: Re-defining the bridge between transcription and translation. Mol. Cancer 2020, 19, 78. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mason, C.E. The pivotal regulatory landscape of RNA modifications. Annu. Rev. Genom. Hum. Genet. 2014, 15, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Jia, G. Reversible RNA Modification N(1)-methyladenosine (m(1)A) in mRNA and tRNA. Genom. Proteom. Bioinform. 2018, 16, 155–161. [Google Scholar] [CrossRef]
- Dominissini, D.; Nachtergaele, S.; Moshitch-Moshkovitz, S.; Peer, E.; Kol, N.; Ben-Haim, M.S.; Dai, Q.; Di Segni, A.; Salmon-Divon, M.; Clark, W.C.; et al. The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 2016, 530, 441–446. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Xiong, X.; Zhang, M.; Wang, K.; Chen, Y.; Zhou, J.; Mao, Y.; Lv, J.; Yi, D.; Chen, X.-W.; et al. Base-Resolution Mapping Reveals Distinct m(1)A Methylome in Nuclear- and Mitochondrial-Encoded Transcripts. Mol. Cell 2017, 68, 993–1005.e9. [Google Scholar] [CrossRef] [Green Version]
- Safra, M.; Sas-Chen, A.; Nir, R.; Winkler, R.; Nachshon, A.; Bar-Yaacov, D.; Erlacher, M.; Rossmanith, W.; Stern-Ginossar, N.; Schwartz, S. The m1A landscape on cytosolic and mitochondrial mRNA at single-base resolution. Nature 2017, 551, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Anreiter, I.; Mir, Q.; Simpson, J.T.; Janga, S.C.; Soller, M. New Twists in Detecting mRNA Modification Dynamics. Trends Biotechnol. 2021, 39, 72–89. [Google Scholar] [CrossRef]
- Ozanick, S.; Krecic, A.; Andersland, J.; Anderson, J.T. The bipartite structure of the tRNA m1A58 methyltransferase from S. cerevisiae is conserved in humans. RNA 2005, 11, 1281–1290. [Google Scholar] [CrossRef] [Green Version]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase--extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012, 40, 11583–11593. [Google Scholar] [CrossRef] [PubMed]
- Chujo, T.; Suzuki, T. Trmt61B is a methyltransferase responsible for 1-methyladenosine at position 58 of human mitochondrial tRNAs. RNA 2012, 18, 2269–2276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Qi, M.; Shen, B.; Luo, G.; Wu, Y.; Li, J.; Lu, Z.; Zheng, Z.; Dai, Q.; Wang, H. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019, 47, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Wang, T.; Gonzalez, G.; Wang, Y. Identification of YTH Domain-Containing Proteins as the Readers for N1-Methyladenosine in RNA. Anal. Chem. 2018, 90, 6380–6384. [Google Scholar] [CrossRef]
- Esteve-Puig, R.; Climent, F.; Piñeyro, D.; Domingo-Domènech, E.; Davalos, V.; Encuentra, M.; Rea, A.; Espejo-Herrera, N.; Soler, M.; Lopez, M.; et al. Epigenetic loss of m1A RNA demethylase ALKBH3 in Hodgkin lymphoma targets collagen, conferring poor clinical outcome. Blood 2021, 137, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xue, C.; Yuan, X.; He, Y.; Yu, Z. Gene signatures and prognostic values of m1A-related regulatory genes in hepatocellular carcinoma. Sci. Rep. 2020, 10, 15083. [Google Scholar] [CrossRef]
- Ali, A.T.; Idaghdour, Y.; Hodgkinson, A. Analysis of mitochondrial m1A/G RNA modification reveals links to nuclear genetic variants and associated disease processes. Commun. Biol. 2020, 3, 147. [Google Scholar] [CrossRef]
- Ueda, Y.; Ooshio, I.; Fusamae, Y.; Kitae, K.; Kawaguchi, M.; Jingushi, K.; Hase, H.; Harada, K.; Hirata, K.; Tsujikawa, K. AlkB homolog 3-mediated tRNA demethylation promotes protein synthesis in cancer cells. Sci. Rep. 2017, 7, 42271. [Google Scholar] [CrossRef]
- Pilžys, T.; Marcinkowski, M.; Kukwa, W.; Garbicz, D.; Dylewska, M.; Ferenc, K.; Mieczkowski, A.; Kukwa, A.; Migacz, E.; Wołosz, D.; et al. ALKBH overexpression in head and neck cancer: Potential target for novel anticancer therapy. Sci. Rep. 2019, 9, 13249. [Google Scholar] [CrossRef] [Green Version]
- Yeon, S.Y.; Jo, Y.S.; Choi, E.J.; Kim, M.S.; Yoo, N.J.; Lee, S.H. Frameshift Mutations in Repeat Sequences of ANK3, HACD4, TCP10L, TP53BP1, MFN1, LCMT2, RNMT, TRMT6, METTL8 and METTL16 Genes in Colon Cancers. Pathol. Oncol. Res. 2018, 24, 617–622. [Google Scholar] [CrossRef]
- Chen, X.; Sun, Y.-Z.; Liu, H.; Zhang, L.; Li, J.-Q.; Meng, J. RNA methylation and diseases: Experimental results, databases, Web servers and computational models. Brief. Bioinform. 2019, 20, 896–917. [Google Scholar] [CrossRef]
- Boxberg, M.; Leising, L.; Steiger, K.; Jesinghaus, M.; Alkhamas, A.; Mielke, M.; Pfarr, N.; Götz, C.; Wolff, K.D.; Weichert, W.; et al. Composition and Clinical Impact of the Immunologic Tumor Microenvironment in Oral Squamous Cell Carcinoma. J. Immunol. 2019, 202, 278–291. [Google Scholar] [CrossRef] [Green Version]
- Quan, H.; Fang, L.; Pan, H.; Deng, Z.; Gao, S.; Liu, O.; Wang, Y.; Hu, Y.; Fang, X.; Yao, Z.; et al. An adaptive immune response driven by mature, antigen-experienced T and B cells within the microenvironment of oral squamous cell carcinoma. Int. J. Cancer 2016, 138, 2952–2962. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, J.; Li, H.; Yang, Z.; Zhang, X.; Li, X.; Wang, J.; Zhang, Y.; Chen, S.; Song, M. Single-cell transcriptomics reveal the intratumoral landscape of infiltrated T-cell subpopulations in oral squamous cell carcinoma. Mol. Oncol. 2021, 15, 866–886. [Google Scholar] [CrossRef] [PubMed]
- Nakano, O.; Sato, M.; Naito, Y.; Suzuki, K.; Orikasa, S.; Aizawa, M.; Suzuki, Y.; Shintaku, I.; Nagura, H.; Ohtani, H. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: Clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001, 61, 5132–5136. [Google Scholar] [PubMed]
- Sato, E.; Olson, S.H.; Ahn, J.; Bundy, B.; Nishikawa, H.; Qian, F.; Jungbluth, A.A.; Frosina, D.; Gnjatic, S.; Ambrosone, C.; et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 18538–18543. [Google Scholar] [CrossRef] [Green Version]
- Jewett, A.; Head, C.; Cacalano, N.A. Emerging mechanisms of immunosuppression in oral cancers. J. Dent. Res. 2006, 85, 1061–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, N.K.; Das, S.; Maitra, A.; Sarin, R.; Majumder, P.P. Somatic mutations in arachidonic acid metabolism pathway genes enhance oral cancer post-treatment disease-free survival. Nat. Commun. 2014, 5, 5835. [Google Scholar] [CrossRef] [Green Version]
- Tormoen, G.W.; Crittenden, M.R.; Gough, M.J. Role of the immunosuppressive microenvironment in immunotherapy. Adv. Radiat. Oncol. 2018, 3, 520–526. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, Y.; Katou, F.; Ohtani, H.; Nakayama, T.; Yoshie, O.; Hashimoto, K. Tumor-infiltrating lymphocytes, particularly the balance between CD8(+) T cells and CCR4(+) regulatory T cells, affect the survival of patients with oral squamous cell carcinoma. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2010, 109, 744–752. [Google Scholar] [CrossRef] [PubMed]
- Quan, H.; Shan, Z.; Liu, Z.; Liu, S.; Yang, L.; Fang, X.; Li, K.; Wang, B.; Deng, Z.; Hu, Y.; et al. The repertoire of tumor-infiltrating lymphocytes within the microenvironment of oral squamous cell carcinoma reveals immune dysfunction. Cancer Immunol. Immunother. 2020, 69, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.E.W.; Bell, R.B.; Bifulco, C.B.; Burtness, B.; Gillison, M.L.; Harrington, K.J.; Le, Q.-T.; Lee, N.Y.; Leidner, R.; Lewis, R.L.; et al. The Society for Immunotherapy of Cancer consensus statement on immunotherapy for the treatment of squamous cell carcinoma of the head and neck (HNSCC). J. Immunother. Cancer 2019, 7, 184. [Google Scholar] [CrossRef] [Green Version]
- Cohen, E.E.W.; Soulières, D.; Le Tourneau, C.; Dinis, J.; Licitra, L.; Ahn, M.-J.; Soria, A.; Machiels, J.-P.; Mach, N.; Mehra, R.; et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): A randomised, open-label, phase 3 study. Lancet 2019, 393, 156–167. [Google Scholar] [CrossRef]
- Burtness, B.; Harrington, K.J.; Greil, R.; Soulières, D.; Tahara, M.; de Castro, G.J.; Psyrri, A.; Basté, N.; Neupane, P.; Bratland, Å.; et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): A randomised, open-label, phase 3 study. Lancet 2019, 394, 1915–1928. [Google Scholar] [CrossRef]
- Ferris, R.L.; Blumenschein, G.J.; Fayette, J.; Guigay, J.; Colevas, A.D.; Licitra, L.; Harrington, K.; Kasper, S.; Vokes, E.E.; Even, C.; et al. Nivolumab for Recurrent Squamous-Cell Carcinoma of the Head and Neck. N. Engl. J. Med. 2016, 375, 1856–1867. [Google Scholar] [CrossRef]
- Park, R.; Winnicki, M.; Liu, E.; Chu, W.-M. Immune checkpoints and cancer in the immunogenomics era. Brief. Funct. Genom. 2019, 18, 133–139. [Google Scholar] [CrossRef]
- Chen, S.-H.; Hsiao, S.-Y.; Chang, K.-Y.; Chang, J.-Y. New Insights into Oral Squamous Cell Carcinoma: From Clinical Aspects to Molecular Tumorigenesis. Int. J. Mol. Sci. 2021, 22, 2252. [Google Scholar] [CrossRef] [PubMed]
- Kujan, O.; van Schaijik, B.; Farah, C.S. Immune Checkpoint Inhibitors in Oral Cavity Squamous Cell Carcinoma and Oral Potentially Malignant Disorders: A Systematic Review. Cancers 2020, 12, 1937. [Google Scholar] [CrossRef]
- Liu, X.; Li, Q.; Zhou, Y.; He, X.; Fang, J.; Lu, H.; Wang, X.; Wang, D.; Ma, D.; Cheng, B.; et al. Dysfunctional role of elevated TIGIT expression on T cells in oral squamous cell carcinoma patients. Oral Dis. 2020. [Google Scholar] [CrossRef]
- Kansy, K.; Thiele, O.; Freier, K. The role of human papillomavirus in oral squamous cell carcinoma: Myth and reality. Oral Maxillofac. Surg. 2014, 18, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Charoentong, P.; Finotello, F.; Angelova, M.; Mayer, C.; Efremova, M.; Rieder, D.; Hackl, H.; Trajanoski, Z. Pan-cancer Immunogenomic Analyses Reveal Genotype-Immunophenotype Relationships and Predictors of Response to Checkpoint Blockade. Cell Rep. 2017, 18, 248–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Jiang, Z.; Chen, C.; Wang, X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J. Exp. Clin. Cancer Res. 2018, 37, 327. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. m(6)A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef] [PubMed]
- Sotiriou, C.; Wirapati, P.; Loi, S.; Harris, A.; Fox, S.; Smeds, J.; Nordgren, H.; Farmer, P.; Praz, V.; Haibe-Kains, B.; et al. Gene expression profiling in breast cancer: Understanding the molecular basis of histologic grade to improve prognosis. J. Natl. Cancer Inst. 2006, 98, 262–272. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, L.; Chen, R.; Sugimoto, M.; Mizuta, M.; Kishimoto, Y.; Omori, K. The Impact of m1A Methylation Modification Patterns on Tumor Immune Microenvironment and Prognosis in Oral Squamous Cell Carcinoma. Int. J. Mol. Sci. 2021, 22, 10302. https://doi.org/10.3390/ijms221910302
Gao L, Chen R, Sugimoto M, Mizuta M, Kishimoto Y, Omori K. The Impact of m1A Methylation Modification Patterns on Tumor Immune Microenvironment and Prognosis in Oral Squamous Cell Carcinoma. International Journal of Molecular Sciences. 2021; 22(19):10302. https://doi.org/10.3390/ijms221910302
Chicago/Turabian StyleGao, Li, Ru Chen, Masahiro Sugimoto, Masanobu Mizuta, Yo Kishimoto, and Koichi Omori. 2021. "The Impact of m1A Methylation Modification Patterns on Tumor Immune Microenvironment and Prognosis in Oral Squamous Cell Carcinoma" International Journal of Molecular Sciences 22, no. 19: 10302. https://doi.org/10.3390/ijms221910302