
Opioid Addiction and Opioid Receptor Dimerization: Structural Modeling of the OPRD1 and OPRM1 Heterodimer and Its Signaling Pathways



Abstract
:1. Introduction
2. Results and Discussion
2.1. 3D Structure Modeling of OPRM1 and OPRD1 Monomers
2.2. 3D Model of Transmembrane OPRD1–OPRM1 Heterodimer
2.3. 3D Model of OPRD1–OPRM1 Extracellular Domain Complex
2.4. 3D Model of Full-Length OPRD1–OPRM1 Heterodimer within Lipid Bilayer
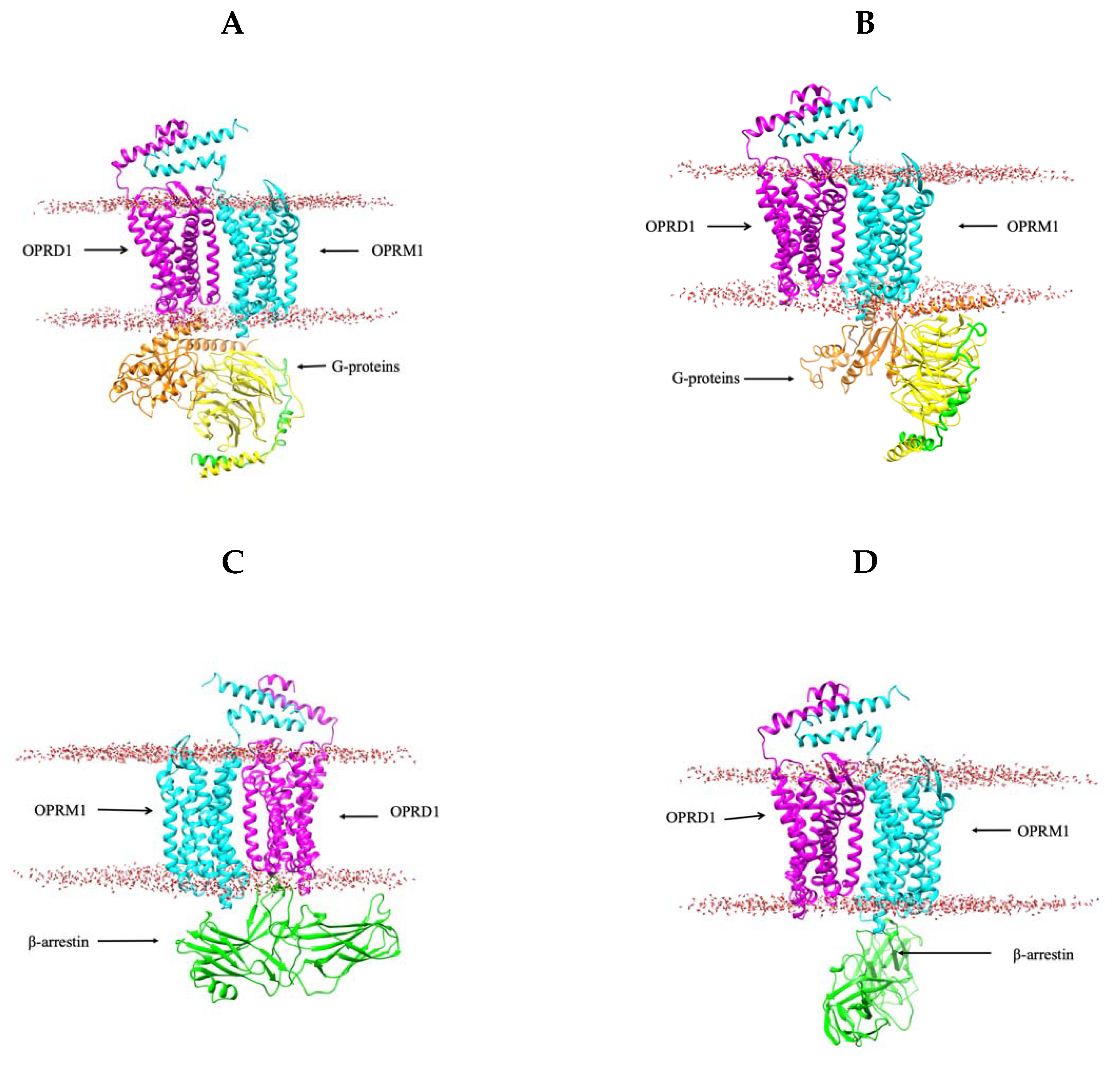
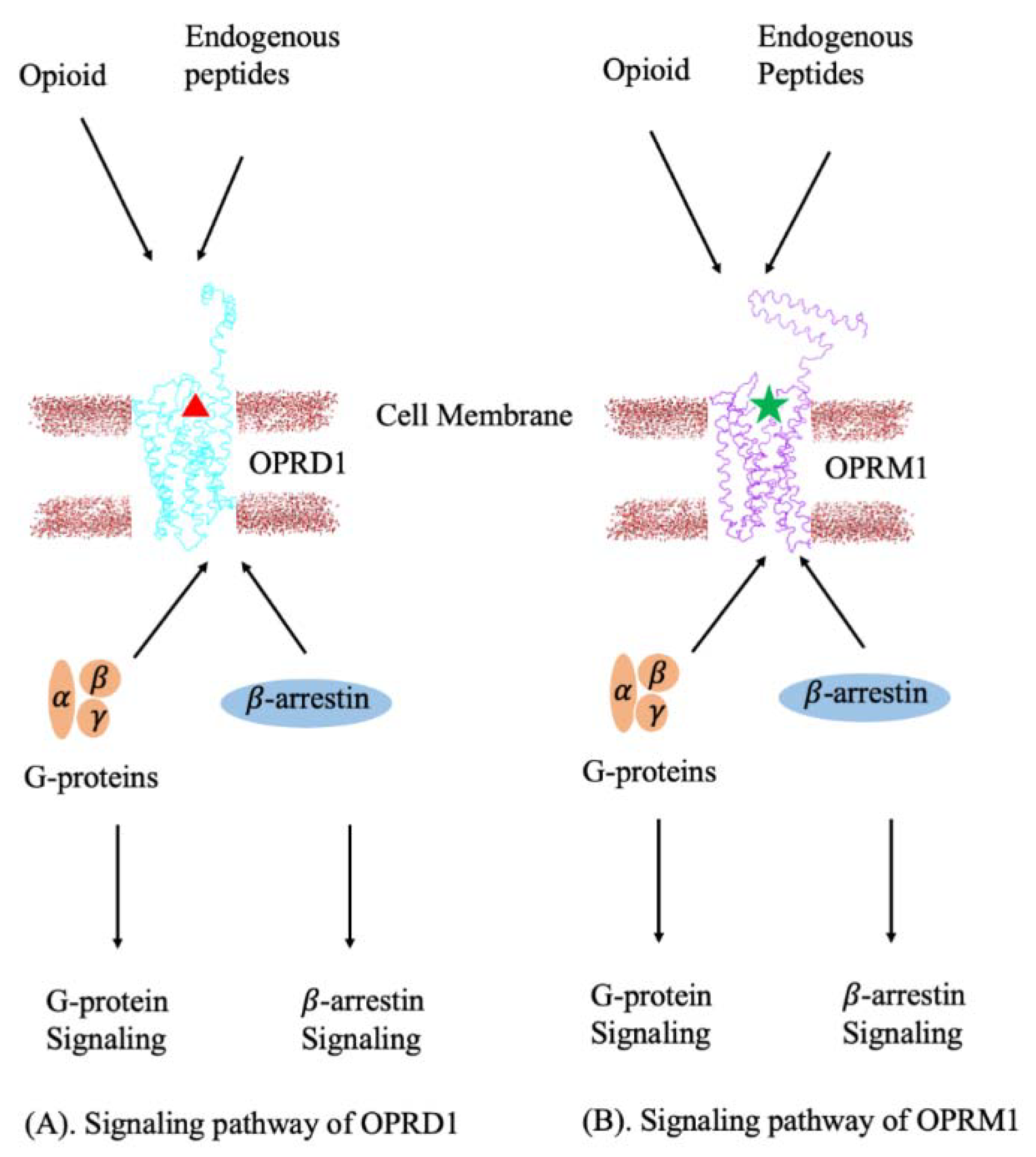
2.5. 3D Modeling of the G-Protein and β-Arrestin Coupled with OPRD1–OPRM1 Heterodimer
2.6. Folding Free Energy Change Due to a Mutation
2.7. Binding Free Energy Change Due to Mutation
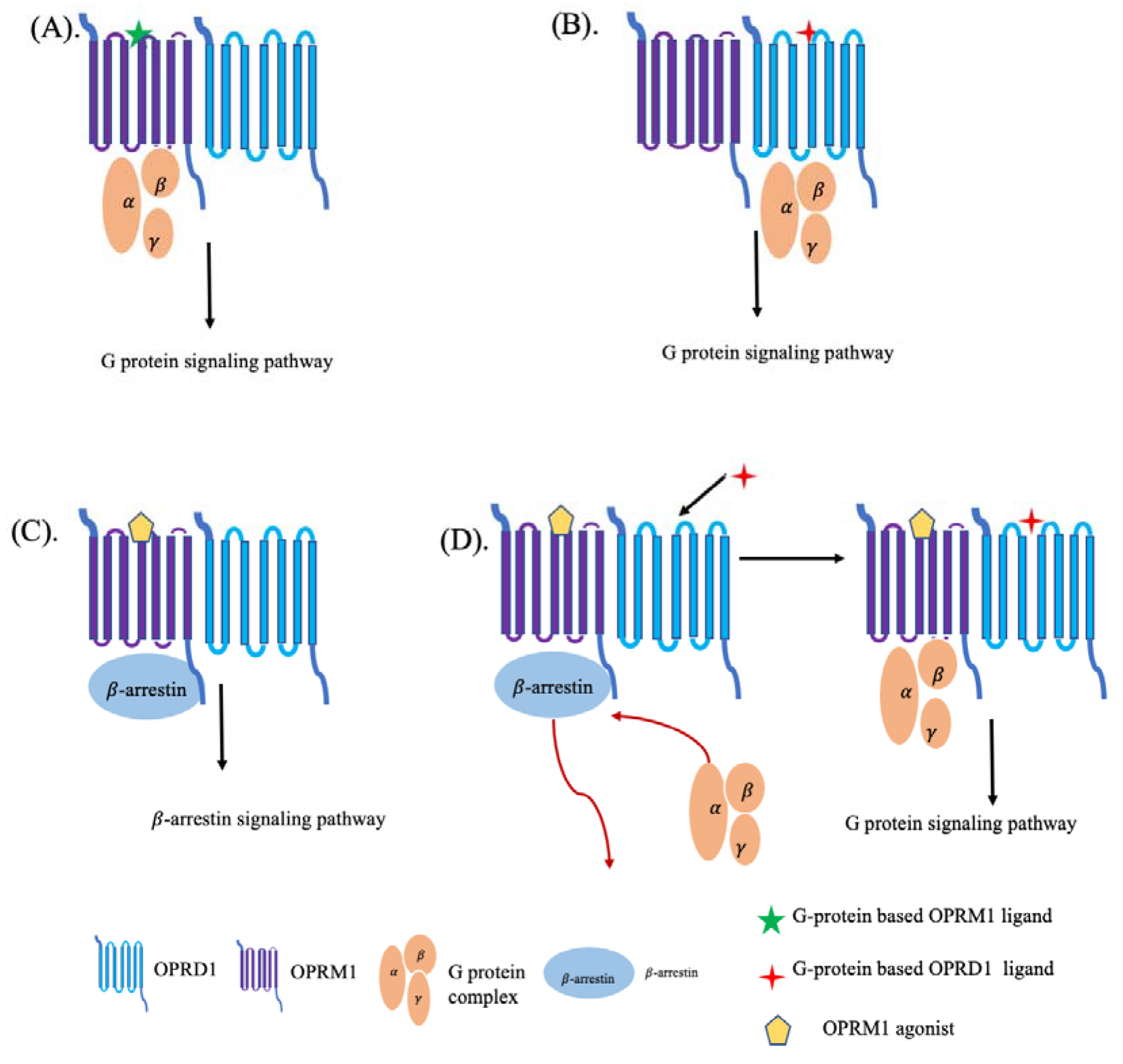
2.8. Structural Insights
3. Methods
3.1. 3D Structure Modeling
3.2. Mapping the Mutations onto the 3D Structure of OPRM1
3.3. Analyzing Folding Free Energy Change Due to Mutation
3.4. Analyzing Binding Free Energy Change Due to Mutation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviation
References
- Coluzzi, F.; Bifulco, F.; Cuomo, A.; Dauri, M.; Leonardi, C.; Melotti, R.M.; Natoli, S.; Romualdi, P.; Savoia, G.; Corcione, A. The challenge of perioperative pain management in opioid-tolerant patients. Ther. Clin. Risk Manag. 2017, 13, 1163–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colvin, L.A.; Bull, F.; Hales, T.G. Perioperative opioid analgesia—when is enough too much? A review of opioid-induced tolerance and hyperalgesia. Lancet 2019, 393, 1558–1568. [Google Scholar] [CrossRef] [Green Version]
- Juurlink, D.N.; Dhalla, I.A. Dependence and Addiction During Chronic Opioid Therapy. J. Med. Toxicol. 2012, 8, 393–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain—Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef]
- Marshall, B.; Bland, M.K.; Hulla, R.; Gatchel, R.J. Considerations in addressing the opioid epidemic and chronic pain within the USA. Pain Manag. 2019, 9, 131–138. [Google Scholar] [CrossRef]
- Benéitez, M.C.; Gil-Alegre, M.E. Opioid Addiction: Social Problems Associated and Implications of Both Current and Possible Future Treatments, including Polymeric Therapeutics for Giving Up the Habit of Opioid Consumption. BioMed Res. Int. 2017, 2017, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Deroche-Gamonet, V.; Belin, D.; Piazza, P.V. Evidence for Addiction-like Behavior in the Rat. Science 2004, 305, 1014–1017. [Google Scholar] [CrossRef] [Green Version]
- Moran, M.; Blum, K.; Ponce, J.V.; Lott, L.; Gondré–Lewis, M.C.; Badgaiyan, S.; Brewer, R.; Downs, B.W.; Fynman, P.; Weingarten, A.; et al. High Genetic Addiction Risk Score (GARS) in Chronically Prescribed Severe Chronic Opioid Probands Attending Multi-pain Clinics: An Open Clinical Pilot Trial. Mol. Neurobiol. 2021, 58, 1–12. [Google Scholar] [CrossRef]
- Butelman, E.R.; Yuferov, V.; Kreek, M.J. Kappa-opioid receptor/dynorphin system: Genetic and pharmacotherapeutic implications for addiction. Trends Neurosci. 2012, 35, 587–596. [Google Scholar] [CrossRef] [Green Version]
- Crist, R.C.; Reiner, B.C.; Berrettini, W.H. A review of opioid addiction genetics. Curr. Opin. Psychol. 2018, 27, 31–35. [Google Scholar] [CrossRef]
- Berrettini, W. A brief review of the genetics and pharmacogenetics of opioid use disorders. Dialogues Clin. Neurosci. 2017, 19, 229–236. [Google Scholar]
- Le Foll, B.; Gallo, A.; Le Strat, Y.; Lu, L.; Gorwood, P. Genetics of dopamine receptors and drug addiction: A comprehensive review. Behav. Pharmacol. 2009, 20, 1–17. [Google Scholar] [CrossRef]
- Lachman, H.M.; Fann, C.S.; Bartzis, M.; Evgrafov, O.V.; Rosenthal, R.N.; Nunes, E.V.; Miner, C.; Santana, M.; Gaffney, J.; Riddick, A.; et al. Genomewide suggestive linkage of opioid dependence to chromosome 14q. Hum. Mol. Genet. 2007, 16, 1327–1334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bennett, K.G.; Kelley, B.P.; Vick, A.D.; Lee, J.S.; Gunaseelan, V.; Brummett, C.M.; Waljee, J.F. Persistent Opioid Use and High-Risk Prescribing in Body Contouring Patients. Plast. Reconstr. Surg. 2019, 143, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Santosa, K.B.; Hu, H.-M.; Brummett, C.M.; Olsen, M.A.; Englesbe, M.J.; Williams, E.A.; Waljee, J.F. New persistent opioid use among older patients following surgery: A Medicare claims analysis. Surgery 2019, 167, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.A.; Dauw, C.A.; Ghani, K.R.; Gunaseelan, V.; Kim, T.; Leavitt, D.A.; Raisky, J.; Yan, P.L.; Hollingsworth, J.M. New Persistent Opioid Use after Outpatient Ureteroscopy for Upper Tract Stone Treatment. Urology 2019, 134, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Welk, B.; McClure, A.; Clarke, C.; Vogt, K.; Campbell, J. An Opioid Prescription for Men Undergoing Minor Urologic Surgery Is Associated with an Increased Risk of New Persistent Opioid Use. Eur. Urol. 2019, 77, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, P. Placebo and Opioid Analgesia—Imaging a Shared Neuronal Network. Science 2002, 295, 1737–1740. [Google Scholar] [CrossRef] [Green Version]
- Fields, H.L. Pain modulation: Expectation, opioid analgesia and virtual pain. Biol. Basis Mind Body Interact. 2000, 122, 245–253. [Google Scholar] [CrossRef]
- Moy, J.K.; Hartung, J.E.; Duque, M.G.; Friedman, R.; Nagarajan, V.; Loeza-Alcocer, E.; Koerber, H.R.; Christoph, T.; Schröder, W.; Gold, M.S. Distribution of functional opioid receptors in human dorsal root ganglion neurons. Pain 2020, 161, 1636–1649. [Google Scholar] [CrossRef]
- Al-Hasani, R.; Bruchas, M.R. Bruchas, Molecular mechanisms of opioid receptor-dependent signaling and behavior. J. Am. Soc. Anesthesiol. 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, R.; Gomes, I.; Devi, L.A. Opioid Receptor Dimerization. In The Opiate Receptors; Humana Press: Totowa, NJ, USA, 2010; pp. 407–437. [Google Scholar] [CrossRef]
- Moller, J.; Isbilir, A.; Sungkaworn, T.; Osberg, B.; Karathanasis, C.; Sunkara, V.; Grushevskyi, E.O.; Bock, A.; Annibale, P.; Heilemann, M.; et al. Single-molecule analysis reveals agonist-specific dimer formation of micro-opioid receptors. Nat. Chem. Biol. 2020, 16, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Filizola, M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol. 2015, 763, 206–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massotte, D. In vivo opioid receptor heteromerization: Where do we stand? Br. J. Pharmacol. 2015, 172, 420–434. [Google Scholar] [CrossRef] [Green Version]
- Vilardaga, J.-P.; Agnati, L.F.; Fuxe, K.; Ciruela, F. G-protein-coupled receptor heteromer dynamics. J. Cell Sci. 2010, 123, 4215–4220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietis, N.; Guerrini, R.; Calo, G.; Salvadori, S.; Rowbotham, D.J.; Lambert, D.G. Simultaneous targeting of multiple opioid receptors: A strategy to improve side-effect profile. Br. J. Anaesth. 2009, 103, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Olson, K.M.; Keresztes, A.; Tashiro, J.K.; Daconta, L.V.; Hruby, V.J.; Streicher, J.M. Synthesis and Evaluation of a Novel Bivalent Selective Antagonist for the Mu-Delta Opioid Receptor Heterodimer that Reduces Morphine Withdrawal in Mice. J. Med. Chem. 2018, 61, 6075–6086. [Google Scholar] [CrossRef] [PubMed]
- Pinello, C.; Guerrero, M.; Eberhart, C.; Volmar, C.H.; Saldanha, S.A.; Cayanan, C.; Urbano, M.; Brown, S.J.; Ferguson, J.; Gomes, I.; et al. Characterization of an agonist probe for opioid receptor mu 1 (OPRM1)-opioid receptor delta 1 (OPRD1) heterodimerization. Probe Rep. NIH Mol. Libr. Program. 2013. [Google Scholar]
- Skorpen, F.; von Hofacker, S.; Bjørngaard, M.; Skogholt, A.H.; Dale, O.; Kaasa, S.; Klepstad, P. The rare Arg181Cys mutation in the μ opioid receptor can abolish opioid responses. Acta Anaesthesiol. Scand. 2016, 60, 1084–1091. [Google Scholar] [CrossRef] [Green Version]
- Knapman, A.; Santiago, M.J.; Connor, M. A6V polymorphism of the human μ-opioid receptor decreases signalling of morphine and endogenous opioids in vitro. Br. J. Pharmacol. 2015, 172, 2258–2272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Levran, O.; Londono, D.; O’Hara, K.; Nielsen, D.; Peles, E.; Rotrosen, J.; Casadonte, P.; Linzy, S.; Randesi, M.; Ott, J.; et al. Genetic susceptibility to heroin addiction: A candidate gene association study. Genes Brain Behav. 2008, 7, 720–729. [Google Scholar] [CrossRef] [Green Version]
- Türkan, H.; Karahalil, B.; Kadıoğlu, E.; Eren, K.; Gürol, D.T.; Karakaya, A.E. The association between the OPRM1 A118G polymorphism and addiction in a Turkish population. Arch. Ind. Hyg. Toxicol. 2019, 70, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Chatti, I.; Creveaux, I.; Woillard, J.B.; Langlais, S.; Amara, A.; Fatma, L.B.; Saad, A.; Gribaa, M.; Libert, F. Association of the OPRM1 and COMT genes’ polymorphisms with the efficacy of morphine in Tunisian cancer patients: Impact of the high genetic heterogeneity in Tunisia? Therapie 2016, 71, 507–513. [Google Scholar] [CrossRef]
- Asl, S.S.; Roointan, A.; Bergen, H.; Amiri, S.; Mardani, P.; Ashtari, N.; Shabani, R.; Mehdizadeh, M. Opioid Receptors Gene Polymorphism and Heroin Dependence in Iran. Basic Clin. Neurosci. J. 2018, 9, 101–106. [Google Scholar] [CrossRef]
- Halikere, A.; Popova, D.; Scarnati, M.S.; Hamod, A.; Swerdel, M.R.; Moore, J.C.; Tischfield, J.A.; Hart, R.P.; Pang, Z.P. Addiction associated N40D mu-opioid receptor variant modulates synaptic function in human neurons. Mol. Psychiatry 2019, 25, 1406–1419. [Google Scholar] [CrossRef] [PubMed]
- Haerian, B.S.; Haerian, M.S. OPRM1 rs1799971 polymorphism and opioid dependence: Evidence from a meta-analysis. Pharmacogenomics 2013, 14, 813–824. [Google Scholar] [CrossRef]
- Wang, S.-C.; Chen, Y.-C.; Lee, C.-H.; Cheng, C.-M. Opioid Addiction, Genetic Susceptibility, and Medical Treatments: A Review. Int. J. Mol. Sci. 2019, 20, 4294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreek, M.J.; Nielsen, D.A.; LaForge, K.S. Genes Associated With Addiction: Alcoholism, Opiate, and Cocaine Addiction. NeuroMolecular Med. 2004, 5, 085–108. [Google Scholar] [CrossRef]
- Gupta, A.; Décaillot, F.M.; Gomes, I.; Tkalych, O.; Heimann, A.S.; Ferro, E.S.; Devi, L.A. Conformation State-sensitive Antibodies to G-protein-coupled Receptors. J. Biol. Chem. 2007, 282, 5116–5124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deb, I.; Chakraborty, J.; Gangopadhyay, P.K.; Choudhury, S.R.; Das, S. Single-nucleotide polymorphism (A118G) in exon 1 ofOPRM1gene causes alteration in downstream signaling by mu-opioid receptor and may contribute to the genetic risk for addiction. J. Neurochem. 2009, 112, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, D.; Johnson, A.D.; Papp, A.C.; Sadée, W. Allelic expression imbalance of human mu opioid receptor (OPRM1) caused by variant A118G. J. Biol. Chem. 2005, 280, 32618–32624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burley, S.K.; Bhikadiya, C.; Bi, C.; Bittrich, S.; Chen, L.; Crichlow, G.V.; Christie, C.H.; Dalenberg, K.; Di Costanzo, L.; Duarte, J.M.; et al. RCSB Protein Data Bank: Powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2020, 49, D437–D451. [Google Scholar] [CrossRef] [PubMed]
- Bairoch, A. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2004, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Zhang, C.; Bell, E.; Zhang, Y. I-TASSER gateway: A protein structure and function prediction server powered by XSEDE. Futur. Gener. Comput. Syst. 2019, 99, 73–85. [Google Scholar] [CrossRef]
- Pierce, B.; Hourai, Y.; Weng, Z. Accelerating Protein Docking in ZDOCK Using an Advanced 3D Convolution Library. PLoS ONE 2011, 6, e24657. [Google Scholar] [CrossRef]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2011, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Kai, M.; Jin, L.; Wang, R. Computational study of the heterodimerization between mu and delta receptors. J. Comput. Aided Mol. Des. 2009, 23, 321–332. [Google Scholar] [CrossRef]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Pomorska, D.K.; Gach, K.; Janecka, A. Immunomodulatory effects of endogenous and synthetic peptides activating opioid receptors. Mini Rev. Med. Chem. 2014, 14, 1148–1155. [Google Scholar] [CrossRef] [PubMed]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacology 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groer, C.E.; Schmid, C.L.; Jaeger, A.M.; Bohn, L.M. Agonist-directed interactions with specific β-arrestins determine μ-opioid receptor trafficking, ubiquitination, and dephosphorylation. J. Biol. Chem. 2011, 286, 31731–31741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, R.; Devi, L.A. Receptor heteromerization and drug discovery. Trends Pharmacol. Sci. 2010, 31, 124–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rozenfeld, R.; Abul-Husn, N.; Gomez, I.; Devi, L.A. An Emerging Role for the Delta Opioid Receptor in the Regulation of Mu Opioid Receptor Function. Sci. World J. 2007, 7, 64–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pires, D.E.; Ascher, D.; Blundell, T.L. DUET: A server for predicting effects of mutations on protein stability using an integrated computational approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of protein stability upon point mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Myers, R.; Zhang, W.; Alexov, E. Computational Investigation of the Missense Mutations in DHCR7 Gene Associated with Smith-Lemli-Opitz Syndrome. Int. J. Mol. Sci. 2018, 19, 141. [Google Scholar] [CrossRef] [Green Version]
- Darcq, E.; Kieffer, B.L. Opioid receptors: Drivers to addiction? Nat. Rev. Neurosci. 2018, 19, 499–514. [Google Scholar] [CrossRef]
- Crist, R.C.; Berrettini, W.H. The Role of the δ Opioid Receptor Gene, OPRD1, in Addiction. In Neuropathology of Drug Addictions and Substance Misuse; Elsevier: New York, NY, USA, 2016; pp. 899–908. [Google Scholar]
- Décaillot, F.M.; Rozenfeld, R.; Gupta, A.; Devi, L.A. Cell surface targeting of μ-δ opioid receptor heterodimers by RTP. Proc. Nat. Acad. Sci. USA 2008, 105, 16045–16050. [Google Scholar] [CrossRef] [Green Version]
- Šali, A.; Blundell, T.L. Comparative Protein Modelling by Satisfaction of Spatial Restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Li, G.; Panday, S.K.; Alexov, E. SAAFEC-SEQ: A Sequence-Based Method for Predicting the Effect of Single Point Mutations on Protein Thermodynamic Stability. Int. J. Mol. Sci. 2021, 22, 606. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.; Blundell, T.L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 2013, 30, 335–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef] [Green Version]
- Capriotti, E.; Altman, R.B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform. 2011, 12, S3. [Google Scholar] [CrossRef] [Green Version]
- Pahari, S.; Li, G.; Murthy, A.K.; Liang, S.; Fragoza, R.; Yu, H.; Alexov, E. SAAMBE-3D: Predicting Effect of Mutations on Protein–Protein Interactions. Int. J. Mol. Sci. 2020, 21, 2563. [Google Scholar] [CrossRef] [Green Version]
- Dehouck, Y.; Kwasigroch, J.M.; Rooman, M.; Gilis, D. BeAtMuSiC: Prediction of changes in protein–protein binding affinity on mutations. Nucleic Acids Res. 2013, 41, W333–W339. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.H.M.; Myung, Y.; Pires, D.E.; Ascher, D.B. mCSM-PPI2: Predicting the effects of mutations on protein-protein interactions. Nucleic Acids Res. 2019, 47, W338–W344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, N.; Chen, Y.; Lu, H.; Zhao, F.; Alvarez, R.V.; Goncearenco, A.; Panchenko, A.R.; Li, M. MutaBind2: Predicting the Impacts of Single and Multiple Mutations on Protein-Protein Interactions. iScience 2020, 23, 100939. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| SAAFEC-SEQ | mCSM | SDM | DUET | CUPSAT | I-Mutant 2.0 | Avg (kcal/mol) | SD | |
|---|---|---|---|---|---|---|---|---|
| A6V | −0.86 | −0.216 | −0.24 | −0.045 | 0.26 | −0.16 | −0.211 | 0.367 |
| N40D | −0.73 | −1.58 | −0.03 | −1.227 | −0.86 | −0.8 | −0.608 | 0.522 |
| SAAMBE-3D | mCSM | BeAtMusic | Mutabind 2 | Avg(kcal/mol) | SD | |
|---|---|---|---|---|---|---|
| A6V | −0.01 | −0.389 | −0.34 | −0.38 | −0.27975 | 0.181 |
| N40D | −0.23 | −0.163 | −0.28 | −0.44 | −0.27825 | 0.118 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, B.; Hand, W.; Alexov, E. Opioid Addiction and Opioid Receptor Dimerization: Structural Modeling of the OPRD1 and OPRM1 Heterodimer and Its Signaling Pathways. Int. J. Mol. Sci. 2021, 22, 10290. https://doi.org/10.3390/ijms221910290
Wu B, Hand W, Alexov E. Opioid Addiction and Opioid Receptor Dimerization: Structural Modeling of the OPRD1 and OPRM1 Heterodimer and Its Signaling Pathways. International Journal of Molecular Sciences. 2021; 22(19):10290. https://doi.org/10.3390/ijms221910290
Chicago/Turabian StyleWu, Bohua, William Hand, and Emil Alexov. 2021. "Opioid Addiction and Opioid Receptor Dimerization: Structural Modeling of the OPRD1 and OPRM1 Heterodimer and Its Signaling Pathways" International Journal of Molecular Sciences 22, no. 19: 10290. https://doi.org/10.3390/ijms221910290