
Regulation of the Golgi Apparatus by p38 and JNK Kinases during Cellular Stress Responses

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
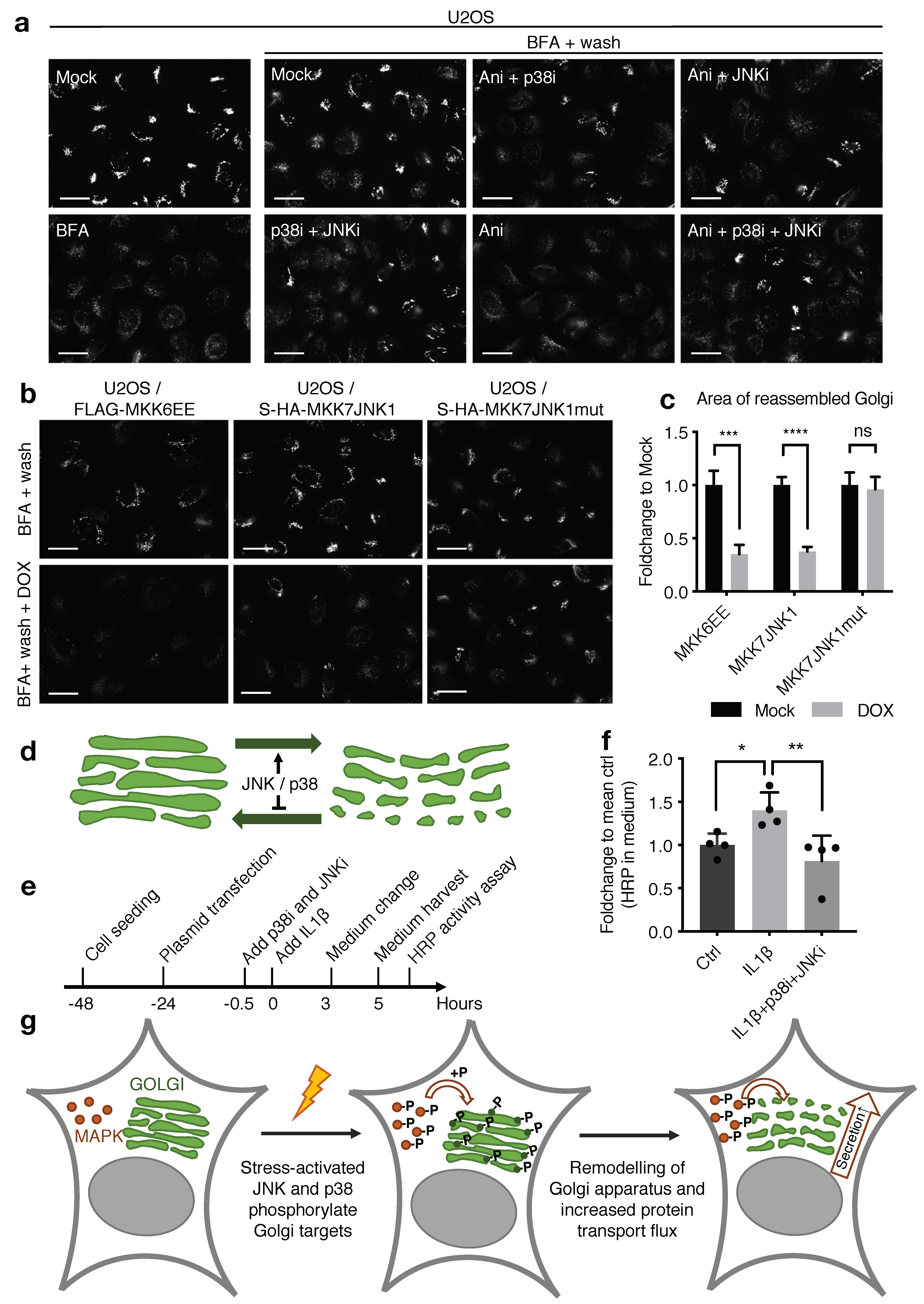{kind=link}
Abstract
:1. Introduction
2. Results
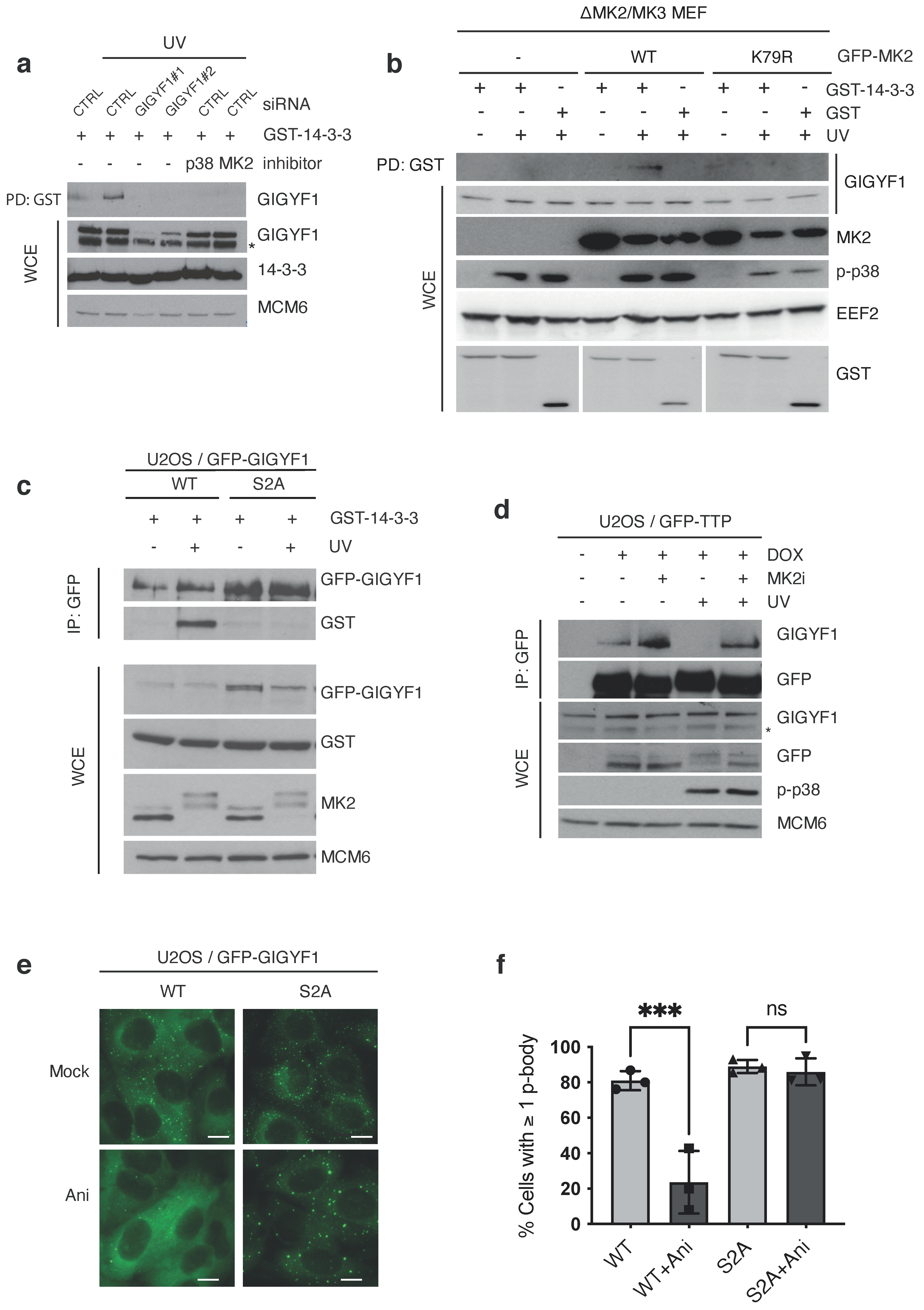
2.1. GIGYF1 Is Phosphorylated and Bound by 14–3–3 after Stress
2.2. MK2 Phosphorylation and 14–3–3 Binding Excludes GIGYF1 from p-Bodies
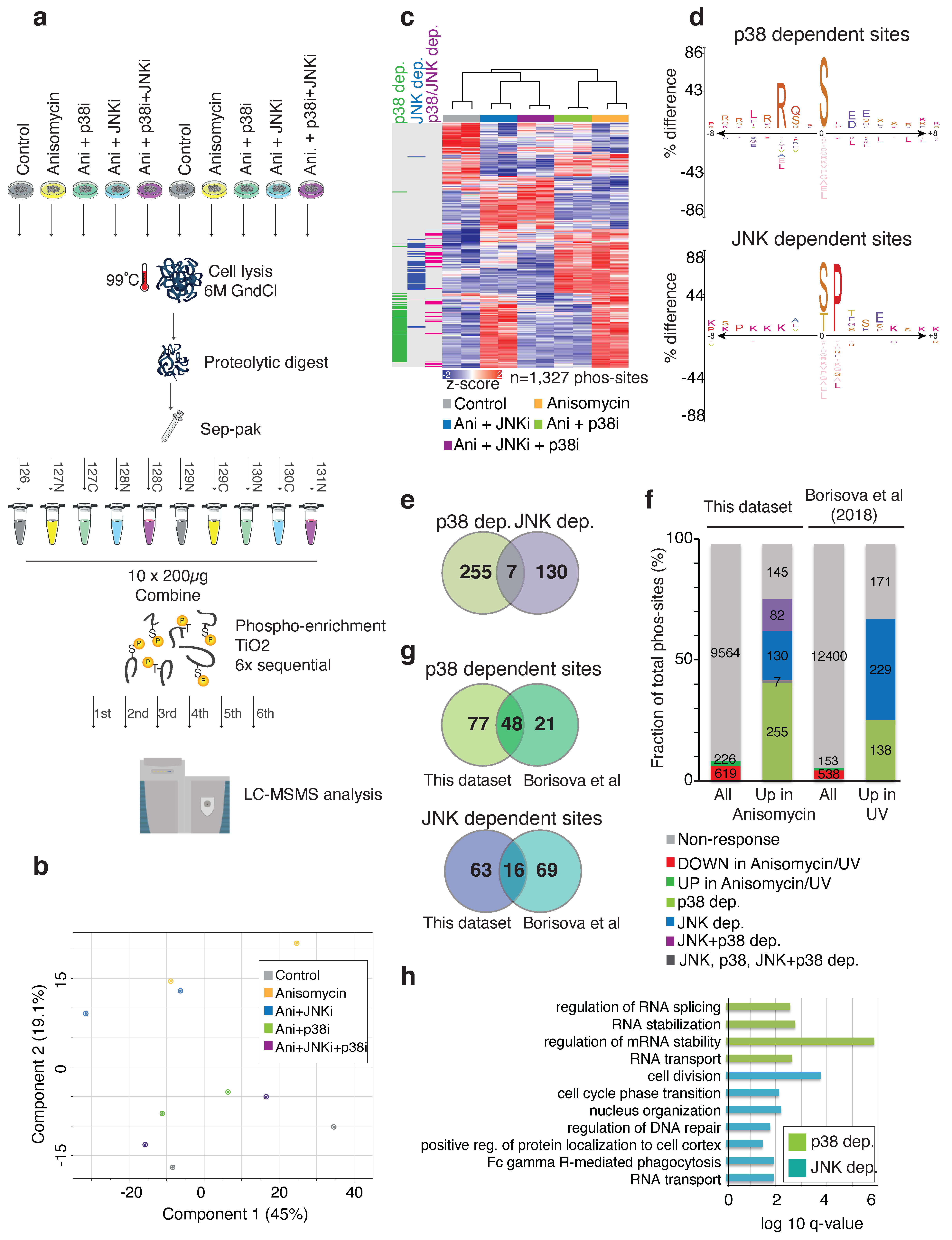
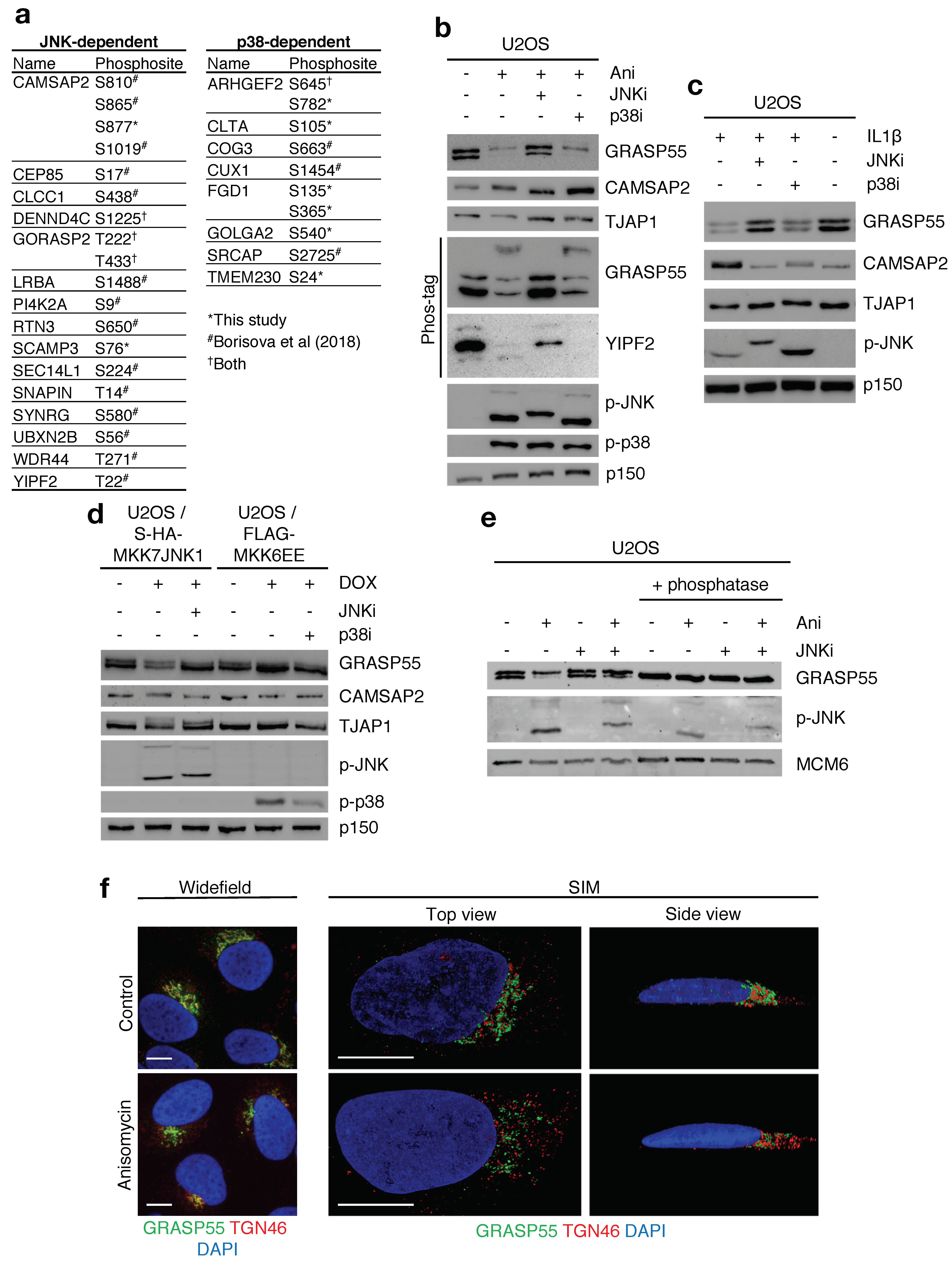
2.3. Multiple Golgi Apparatus-Associated Proteins Are Targets of JNK- and p38-Dependent Phosphorylation
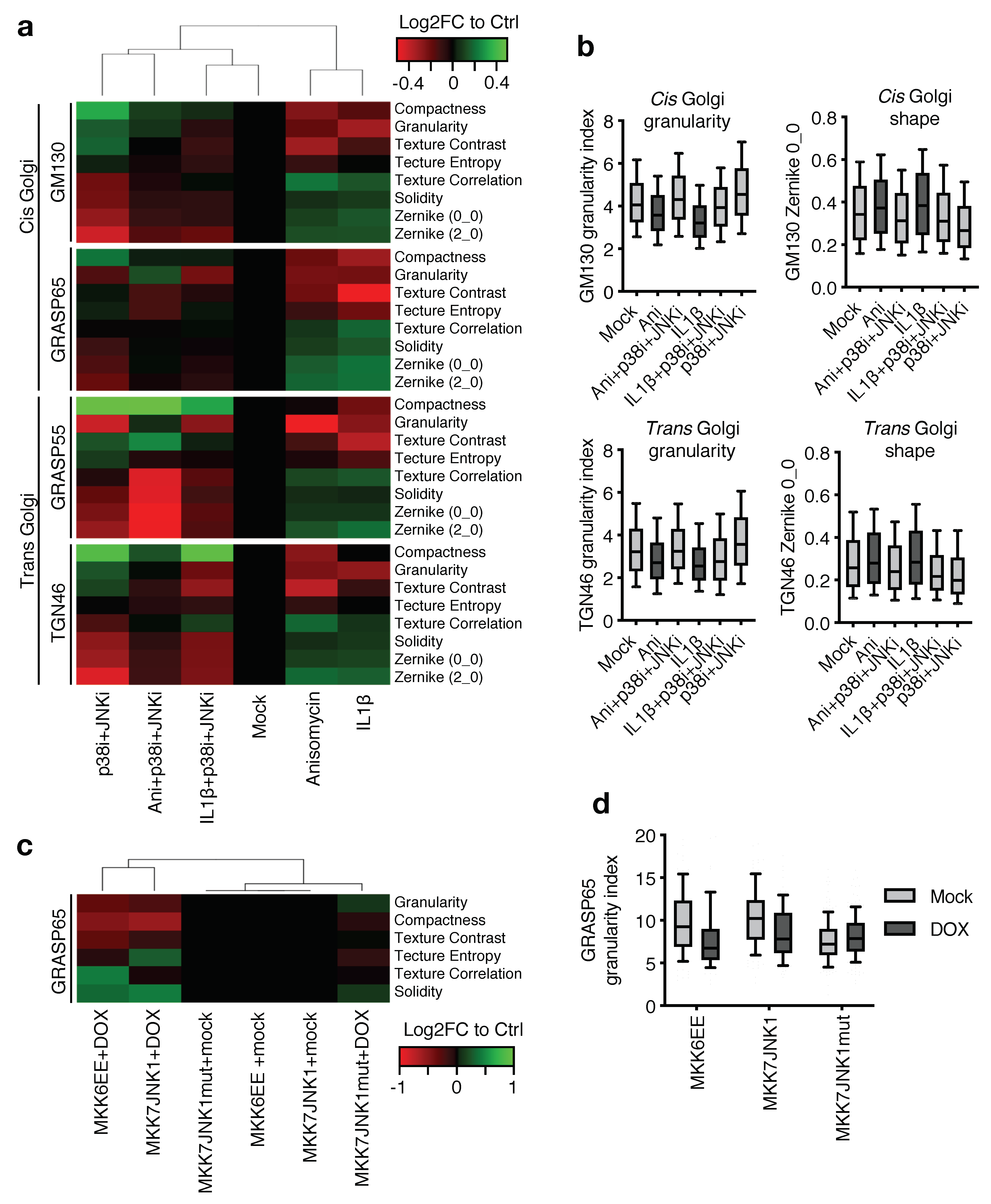
2.4. MAP Kinase Signaling Restructures the Golgi Apparatus
2.5. JNK and p38 Modulate the Dynamic Properties and Secretory Capacity of the Golgi Apparatus
3. Discussion
4. Materials and Methods
4.1. Plasmids and siRNA
4.2. Cell Culture and Treatment
4.3. Proteomics
4.4. Raw Data Processing
4.5. Data Analysis
4.6. Western Blotting
4.7. Phos-Tag Gel
4.8. In Vitro Kinase Assay
4.9. Immunofluorescence Microscopy
4.10. Image Analysis
4.11. Statistical Analysis
4.12. Golgi Reassembly Assay
4.13. Exocytosis Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canovas, B.; Nebreda, A.R. Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 346–366. [Google Scholar] [CrossRef] [PubMed]
- Gaestel, M. MAPKAP kinases—MKs—Two’s company, three’s a crowd. Nat. Rev. Mol. Cell Biol. 2006, 7, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef] [PubMed]
- Manieri, E.; Sabio, G. Stress kinases in the modulation of metabolism and energy balance. J. Mol. Endocrinol. 2015, 55, R11–R22. [Google Scholar] [CrossRef]
- Villumsen, B.H.; Danielsen, J.R.; Povlsen, L.; Sylvestersen, K.B.; Merdes, A.; Beli, P.; Yang, Y.G.; Choudhary, C.; Nielsen, M.L.; Mailand, N.; et al. A new cellular stress response that triggers centriolar satellite reorganization and ciliogenesis. EMBO J. 2013, 32, 3029–3040. [Google Scholar] [CrossRef] [PubMed]
- Tollenaere, M.A.X.; Villumsen, B.H.; Blasius, M.; Nielsen, J.C.; Wagner, S.A.; Bartek, J.; Beli, P.; Mailand, N.; Bekker-Jensen, S. p38- and MK2-dependent signalling promotes stress-induced centriolar satellite remodelling via 14-3-3-dependent sequestration of CEP131/AZI1. Nat. Commun. 2015, 6, 10075. [Google Scholar] [CrossRef] [Green Version]
- Swaffer, M.P.; Jones, A.W.; Flynn, H.R.; Snijders, A.P.; Nurse, P. CDK Substrate Phosphorylation and Ordering the Cell Cycle. Cell 2016, 167, 1750–1761.e16. [Google Scholar] [CrossRef] [Green Version]
- Shiloh, Y.; Ziv, Y. The ATM protein kinase: Regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell Biol. 2013, 14, 197–210. [Google Scholar] [CrossRef]
- Borisova, M.E.; Voigt, A.; Tollenaere, M.A.X.; Sahu, S.K.; Juretschke, T.; Kreim, N.; Mailand, N.; Choudhary, C.; Bekker-Jensen, S.; Akutsu, M.; et al. p38-MK2 signaling axis regulates RNA metabolism after UV-light-induced DNA damage. Nat. Commun. 2018, 9, 1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeke, A.; Misheva, M.; Remenyi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [Green Version]
- Day, K.J.; Staehelin, L.A.; Glick, B.S. A three-stage model of Golgi structure and function. Histochem. Cell Biol. 2013, 140, 239–249. [Google Scholar] [CrossRef] [Green Version]
- Glick, B.S.; Luini, A. Models for Golgi traffic: A critical assessment. Cold Spring Harb. Perspect. Biol. 2011, 3, a005215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potelle, S.; Klein, A.; Foulquier, F. Golgi post-translational modifications and associated diseases. J. Inherit. Metab. Dis. 2015, 38, 741–751. [Google Scholar] [CrossRef]
- Gosavi, P.; Gleeson, P.A. The Function of the Golgi Ribbon Structure—An Enduring Mystery Unfolds! Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Kirk, S.J.; Cliff, J.M.; Thomas, J.A.; Ward, T.H. Biogenesis of secretory organelles during B cell differentiation. J. Leukoc. Biol. 2010, 87, 245–255. [Google Scholar] [CrossRef]
- Madeira, F.; Tinti, M.; Murugesan, G.; Berrett, E.; Stafford, M.; Toth, R.; Cole, C.; MacKintosh, C.; Barton, G.J. 14-3-3-Pred: Improved methods to predict 14-3-3-binding phosphopeptides. Bioinformatics 2015, 31, 2276–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaffe, M.B.; Rittinger, K.; Volinia, S.; Caron, P.R.; Aitken, A.; Leffers, H.; Gamblin, S.J.; Smerdon, S.J.; Cantley, L.C. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 1997, 91, 961–971. [Google Scholar] [CrossRef] [Green Version]
- Fu, R.; Olsen, M.T.; Webb, K.; Bennett, E.J.; Lykke-Andersen, J. Recruitment of the 4EHP-GYF2 cap-binding complex to tetraproline motifs of tristetraprolin promotes repression and degradation of mRNAs with AU-rich elements. RNA 2016, 22, 373–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tollenaere, M.A.X.; Tiedje, C.; Rasmussen, S.; Nielsen, J.C.; Vind, A.C.; Blasius, M.; Batth, T.S.; Mailand, N.; Olsen, J.V.; Gaestel, M.; et al. GIGYF1/2-Driven Cooperation between ZNF598 and TTP in Posttranscriptional Regulation of Inflammatory Signaling. Cell Rep. 2019, 26, 3511–3521.e3514. [Google Scholar] [CrossRef] [Green Version]
- Jesch, S.A.; Lewis, T.S.; Ahn, N.G.; Linstedt, A.D. Mitotic phosphorylation of Golgi reassembly stacking protein 55 by mitogen-activated protein kinase ERK2. Mol. Biol. Cell 2001, 12, 1811–1817. [Google Scholar] [CrossRef] [Green Version]
- Feinstein, T.N.; Linstedt, A.D. GRASP55 regulates Golgi ribbon formation. Mol. Biol. Cell 2008, 19, 2696–2707. [Google Scholar] [CrossRef] [Green Version]
- Kranjc, T.; Dempsey, E.; Cagney, G.; Nakamura, N.; Shields, D.C.; Simpson, J.C. Functional characterisation of the YIPF protein family in mammalian cells. Histochem. Cell Biol. 2017, 147, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Lei, K.; Nimnual, A.; Zong, W.X.; Kennedy, N.J.; Flavell, R.A.; Thompson, C.B.; Bar-Sagi, D.; Davis, R.J. The Bax subfamily of Bcl2-related proteins is essential for apoptotic signal transduction by c-Jun NH(2)-terminal kinase. Mol. Cell Biol. 2002, 22, 4929–4942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raingeaud, J.; Whitmarsh, A.J.; Barrett, T.; Derijard, B.; Davis, R.J. MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol. Cell Biol. 1996, 16, 1247–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Zhang, X.; Nix, D.B.; Katoh, T.; Aoki, K.; Tiemeyer, M.; Wang, Y. Regulation of protein glycosylation and sorting by the Golgi matrix proteins GRASP55/65. Nat. Commun. 2013, 4, 1659. [Google Scholar] [CrossRef] [Green Version]
- Bekier, M.E., 2nd; Wang, L.; Li, J.; Huang, H.; Tang, D.; Zhang, X.; Wang, Y. Knockout of the Golgi stacking proteins GRASP55 and GRASP65 impairs Golgi structure and function. Mol. Biol. Cell 2017, 28, 2833–2842. [Google Scholar] [CrossRef]
- Bossard, C.; Bresson, D.; Polishchuk, R.S.; Malhotra, V. Dimeric PKD regulates membrane fission to form transport carriers at the TGN. J. Cell Biol. 2007, 179, 1123–1131. [Google Scholar] [CrossRef] [Green Version]
- Peter, D.; Weber, R.; Sandmeir, F.; Wohlbold, L.; Helms, S.; Bawankar, P.; Valkov, E.; Igreja, C.; Izaurralde, E. GIGYF1/2 proteins use auxiliary sequences to selectively bind to 4EHP and repress target mRNA expression. Genes Dev. 2017, 31, 1147–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, R.; Chung, M.Y.; Keskeny, C.; Zinnall, U.; Landthaler, M.; Valkov, E.; Izaurralde, E.; Igreja, C. 4EHP and GIGYF1/2 Mediate Translation-Coupled Messenger RNA Decay. Cell Rep. 2020, 33, 108262. [Google Scholar] [CrossRef] [PubMed]
- Sinha, N.K.; Ordureau, A.; Best, K.; Saba, J.A.; Zinshteyn, B.; Sundaramoorthy, E.; Fulzele, A.; Garshott, D.M.; Denk, T.; Thoms, M.; et al. EDF1 coordinates cellular responses to ribosome collisions. Elife 2020, 9, e58828. [Google Scholar] [CrossRef] [PubMed]
- Hickey, K.L.; Dickson, K.; Cogan, J.Z.; Replogle, J.M.; Schoof, M.; D’Orazio, K.N.; Sinha, N.K.; Hussmann, J.A.; Jost, M.; Frost, A.; et al. GIGYF2 and 4EHP Inhibit Translation Initiation of Defective Messenger RNAs to Assist Ribosome-Associated Quality Control. Mol. Cell 2020, 79, 950–962.e6. [Google Scholar] [CrossRef]
- Juszkiewicz, S.; Slodkowicz, G.; Lin, Z.; Freire-Pritchett, P.; Peak-Chew, S.Y.; Hegde, R.S. Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. Elife 2020, 9, e60038. [Google Scholar] [CrossRef] [PubMed]
- Ruscica, V.; Bawankar, P.; Peter, D.; Helms, S.; Igreja, C.; Izaurralde, E. Direct role for the Drosophila GIGYF protein in 4EHP-mediated mRNA repression. Nucleic Acids Res. 2019, 47, 7035–7048. [Google Scholar] [CrossRef]
- Amaya Ramirez, C.C.; Hubbe, P.; Mandel, N.; Bethune, J. 4EHP-independent repression of endogenous mRNAs by the RNA-binding protein GIGYF2. Nucleic Acids Res. 2018, 46, 5792–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vind, A.C.; Genzor, A.V.; Bekker-Jensen, S. Ribosomal stress-surveillance: Three pathways is a magic number. Nucleic Acids Res. 2020, 48, 10648–10661. [Google Scholar] [CrossRef] [PubMed]
- Joazeiro, C.A.P. Mechanisms and functions of ribosome-associated protein quality control. Nat. Rev. Mol. Cell Biol. 2019, 20, 368–383. [Google Scholar] [CrossRef]
- Martin, P.B.; Kigoshi-Tansho, Y.; Sher, R.B.; Ravenscroft, G.; Stauffer, J.E.; Kumar, R.; Yonashiro, R.; Muller, T.; Griffith, C.; Allen, W.; et al. NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat. Commun. 2020, 11, 4625. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Hong, N.A.; Masuda, C.A.; Jenkins, B.V.; Nelms, K.A.; Goodnow, C.C.; Glynne, R.J.; Wu, H.; Masliah, E.; Joazeiro, C.A.; et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl. Acad. Sci. USA 2009, 106, 2097–2103. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Sun, Q.Y.; Yu, R.H.; Guo, J.F.; Tang, B.S.; Yan, X.X. The contribution of GIGYF2 to Parkinson’s disease: A meta-analysis. Neurol. Sci. 2015, 36, 2073–2079. [Google Scholar] [CrossRef]
- Ruiz-Martinez, J.; Krebs, C.E.; Makarov, V.; Gorostidi, A.; Marti-Masso, J.F.; Paisan-Ruiz, C. GIGYF2 mutation in late-onset Parkinson’s disease with cognitive impairment. J. Hum. Genet. 2015, 60, 637–640. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.C.; Peterson, A.; Zinshteyn, B.; Regot, S.; Green, R. Ribosome Collisions Trigger General Stress Responses to Regulate Cell Fate. Cell 2020, 182, 404–416.e14. [Google Scholar] [CrossRef] [PubMed]
- Vind, A.C.; Snieckute, G.; Blasius, M.; Tiedje, C.; Krogh, N.; Bekker-Jensen, D.B.; Andersen, K.L.; Nordgaard, C.; Tollenaere, M.A.X.; Lund, A.H.; et al. ZAKalpha Recognizes Stalled Ribosomes through Partially Redundant Sensor Domains. Mol. Cell 2020, 78, 700–713.e7. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: Activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell Biol. 1997, 17, 3373–3381. [Google Scholar] [CrossRef] [Green Version]
- Meydan, S.; Guydosh, N.R. Disome and Trisome Profiling Reveal Genome-wide Targets of Ribosome Quality Control. Mol. Cell 2020, 79, 588–602.e6. [Google Scholar] [CrossRef]
- Valente, C.; Colanzi, A. Mechanisms and Regulation of the Mitotic Inheritance of the Golgi Complex. Front. Cell Dev. Biol. 2015, 3, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cervigni, R.I.; Bonavita, R.; Barretta, M.L.; Spano, D.; Ayala, I.; Nakamura, N.; Corda, D.; Colanzi, A. JNK2 controls fragmentation of the Golgi complex and the G2/M transition through phosphorylation of GRASP65. J. Cell Sci. 2015, 128, 2249–2260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasius, M.; Wagner, S.A.; Choudhary, C.; Bartek, J.; Jackson, S.P. A quantitative 14-3-3 interaction screen connects the nuclear exosome targeting complex to the DNA damage response. Genes Dev. 2014, 28, 1977–1982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronkina, N.; Kotlyarov, A.; Dittrich-Breiholz, O.; Kracht, M.; Hitti, E.; Milarski, K.; Askew, R.; Marusic, S.; Lin, L.L.; Gaestel, M.; et al. The mitogen-activated protein kinase (MAPK)-activated protein kinases MK2 and MK3 cooperate in stimulation of tumor necrosis factor biosynthesis and stabilization of p38 MAPK. Mol. Cell Biol. 2007, 27, 170–181. [Google Scholar] [CrossRef] [Green Version]
- Hogrebe, A.; von Stechow, L.; Bekker-Jensen, D.B.; Weinert, B.T.; Kelstrup, C.D.; Olsen, J.V. Benchmarking common quantification strategies for large-scale phosphoproteomics. Nat. Commun. 2018, 9, 1045. [Google Scholar] [CrossRef] [Green Version]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Wieczorek, S.; Combes, F.; Lazar, C.; Giai Gianetto, Q.; Gatto, L.; Dorffer, A.; Hesse, A.M.; Coute, Y.; Ferro, M.; Bruley, C.; et al. DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 2017, 33, 135–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colaert, N.; Helsens, K.; Martens, L.; Vandekerckhove, J.; Gevaert, K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods 2009, 6, 786–787. [Google Scholar] [CrossRef]
- Nielsen, J.C.; Nordgaard, C.; Tollenaere, M.A.X.; Bekker-Jensen, S. Osmotic Stress Blocks Mobility and Dynamic Regulation of Centriolar Satellites. Cells 2018, 7, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nordgaard, C.; Tollenaere, M.A.X.; Val, A.M.D.; Bekker-Jensen, D.B.; Blasius, M.; Olsen, J.V.; Bekker-Jensen, S. Regulation of the Golgi Apparatus by p38 and JNK Kinases during Cellular Stress Responses. Int. J. Mol. Sci. 2021, 22, 9595. https://doi.org/10.3390/ijms22179595
Nordgaard C, Tollenaere MAX, Val AMD, Bekker-Jensen DB, Blasius M, Olsen JV, Bekker-Jensen S. Regulation of the Golgi Apparatus by p38 and JNK Kinases during Cellular Stress Responses. International Journal of Molecular Sciences. 2021; 22(17):9595. https://doi.org/10.3390/ijms22179595
Chicago/Turabian StyleNordgaard, Cathrine, Maxim A. X. Tollenaere, Ana Martinez Del Val, Dorte B. Bekker-Jensen, Melanie Blasius, Jesper V. Olsen, and Simon Bekker-Jensen. 2021. "Regulation of the Golgi Apparatus by p38 and JNK Kinases during Cellular Stress Responses" International Journal of Molecular Sciences 22, no. 17: 9595. https://doi.org/10.3390/ijms22179595