
In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone as Promising Cyclooxygenase Inhibitors

 , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
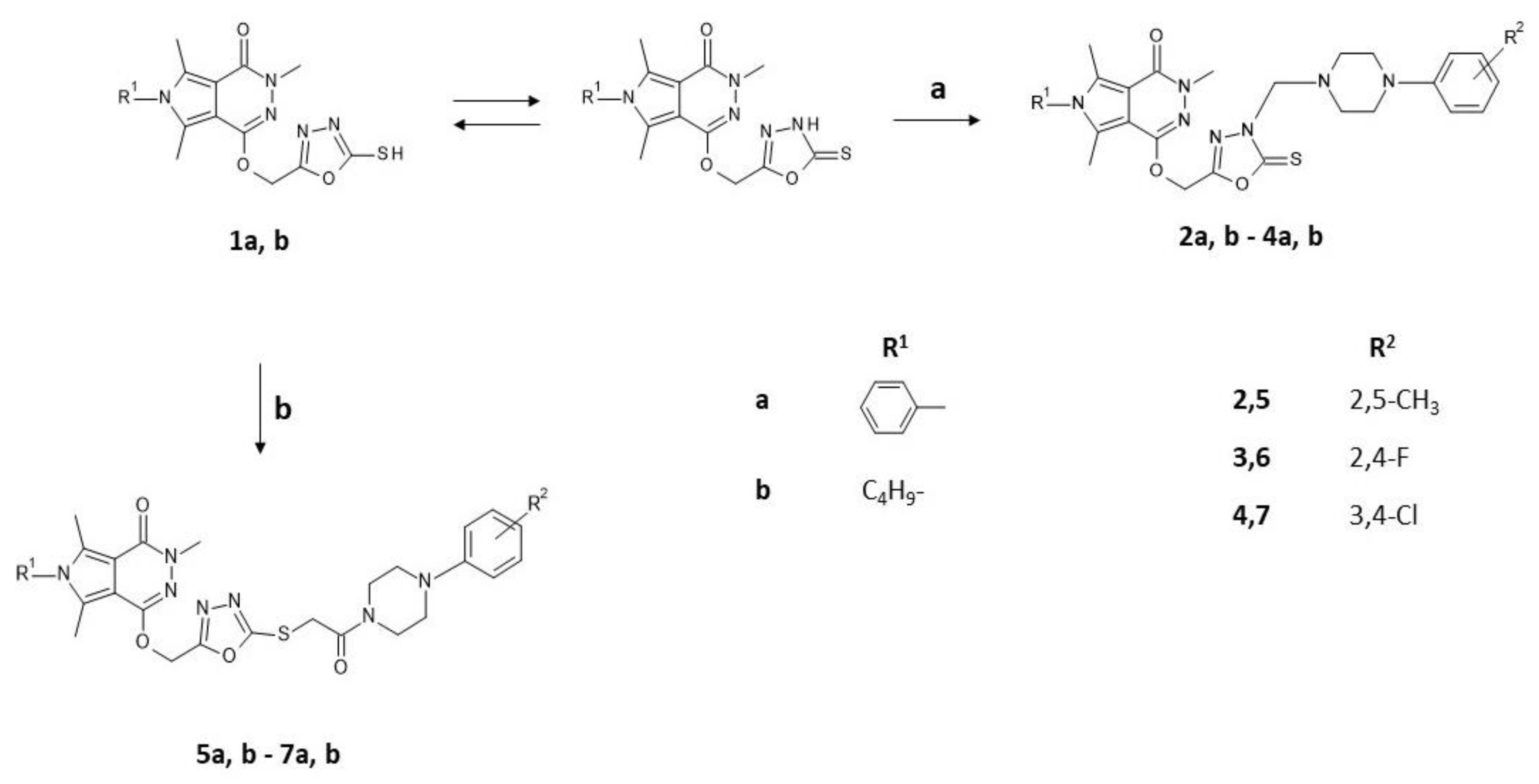
2.1. Chemistry
2.2. Biological Evaluation
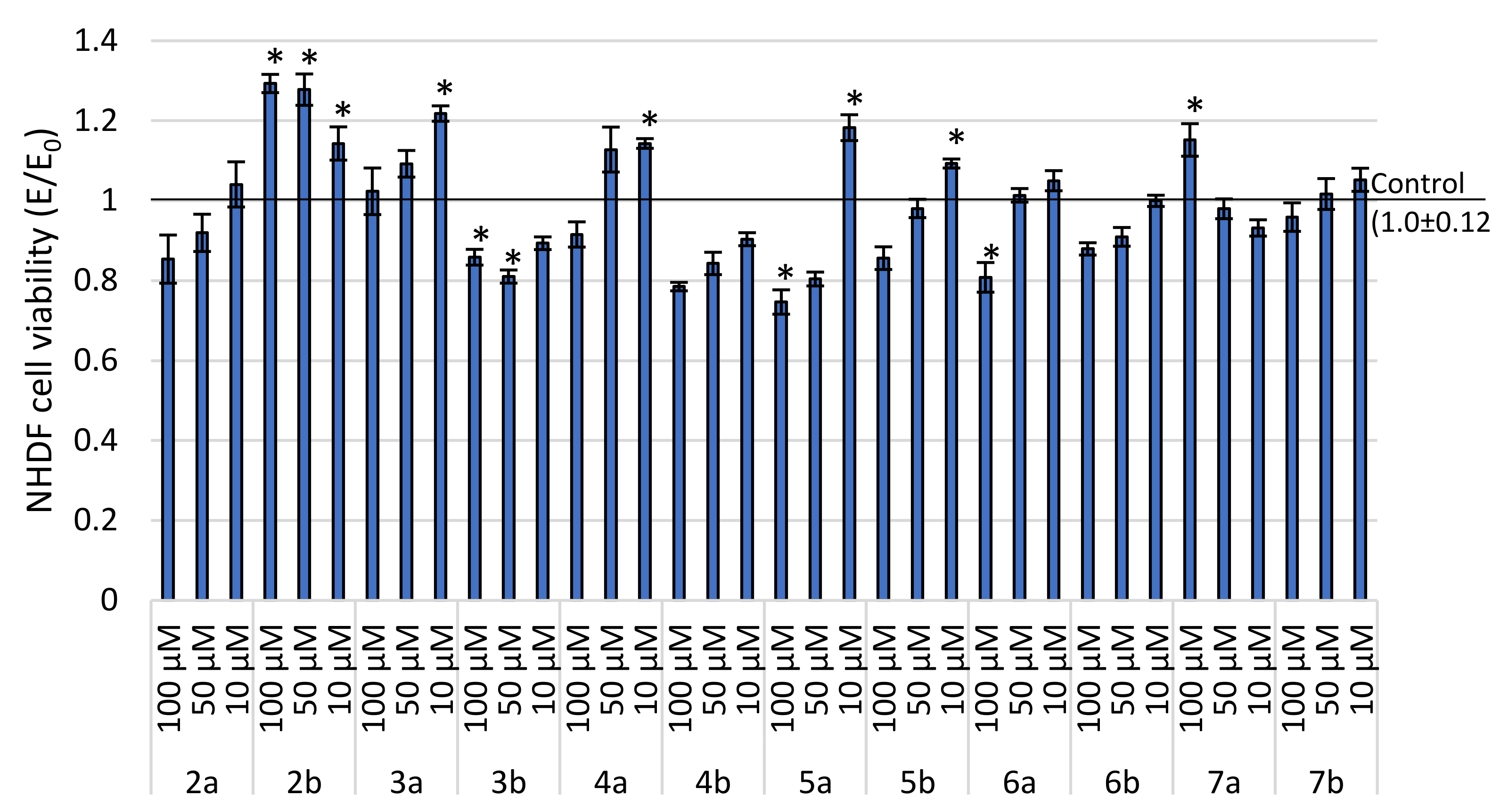
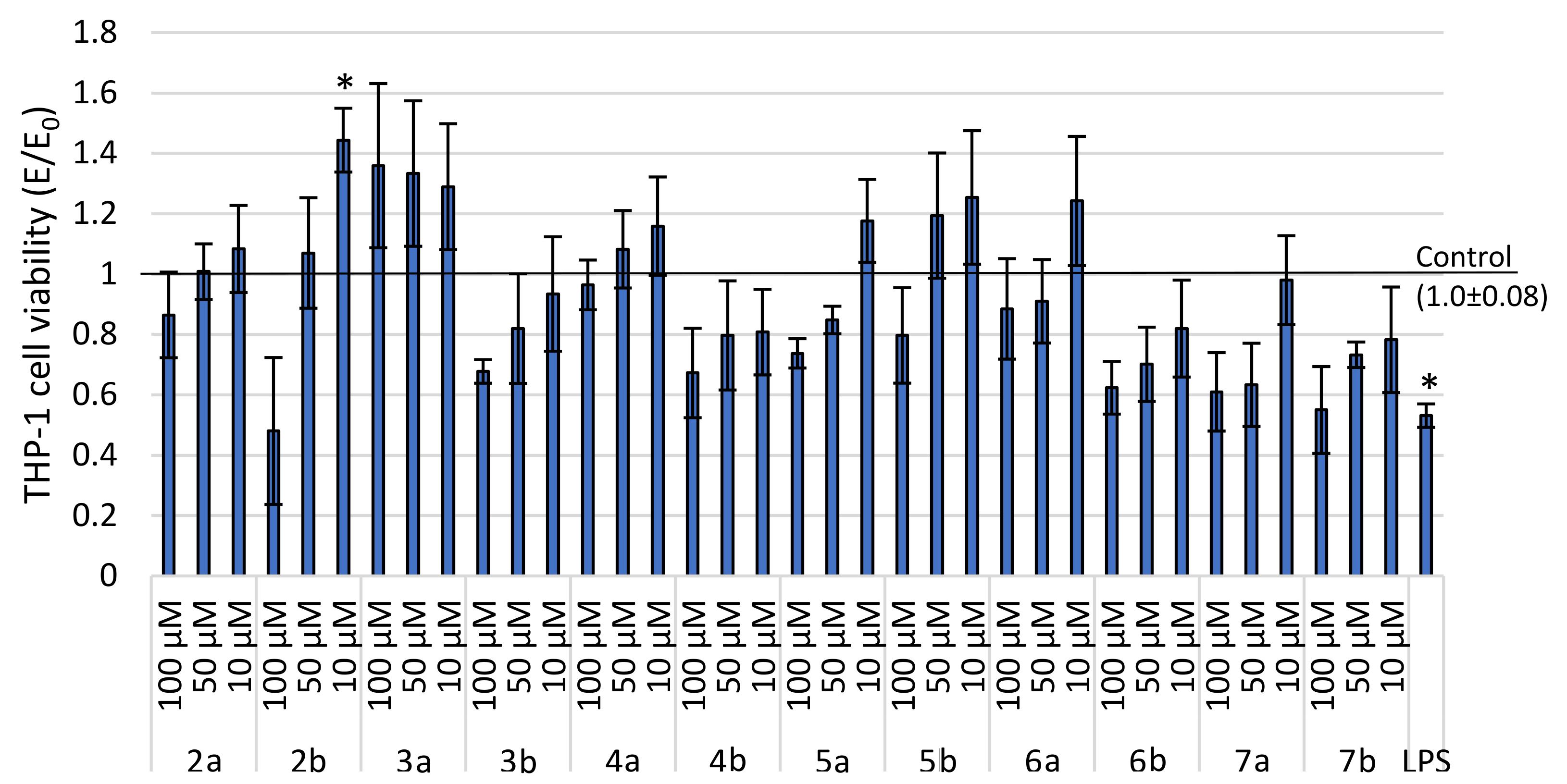
2.2.1. Cytotoxicity Estimation
2.2.2. In Vitro COX Inhibition Assay
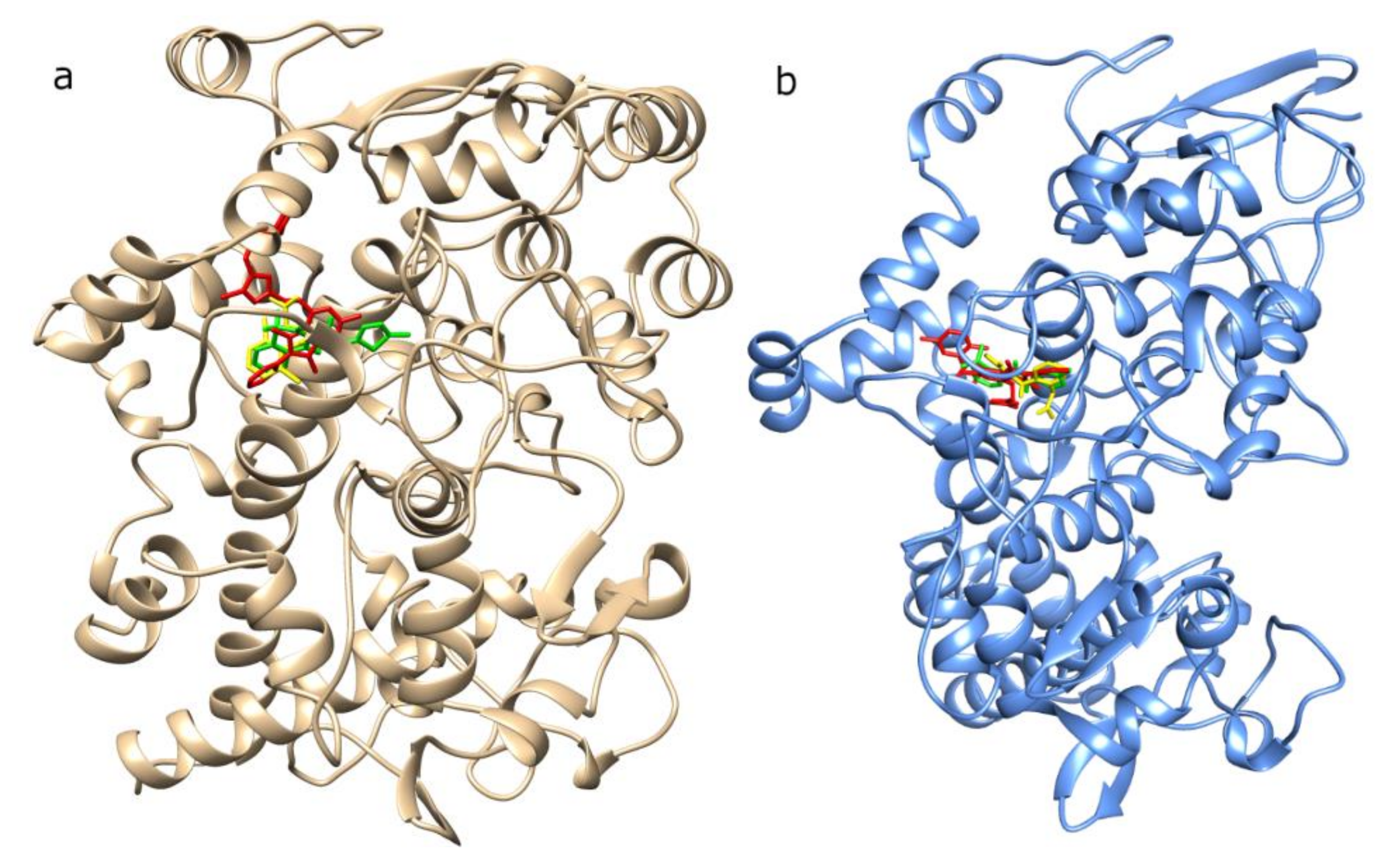
2.2.3. Cyclooxygenase Molecular Docking Study
2.3. Evaluation of Anti-Inflammatory Activity within Cells
2.3.1. Cells Regeneration—MTT Assay
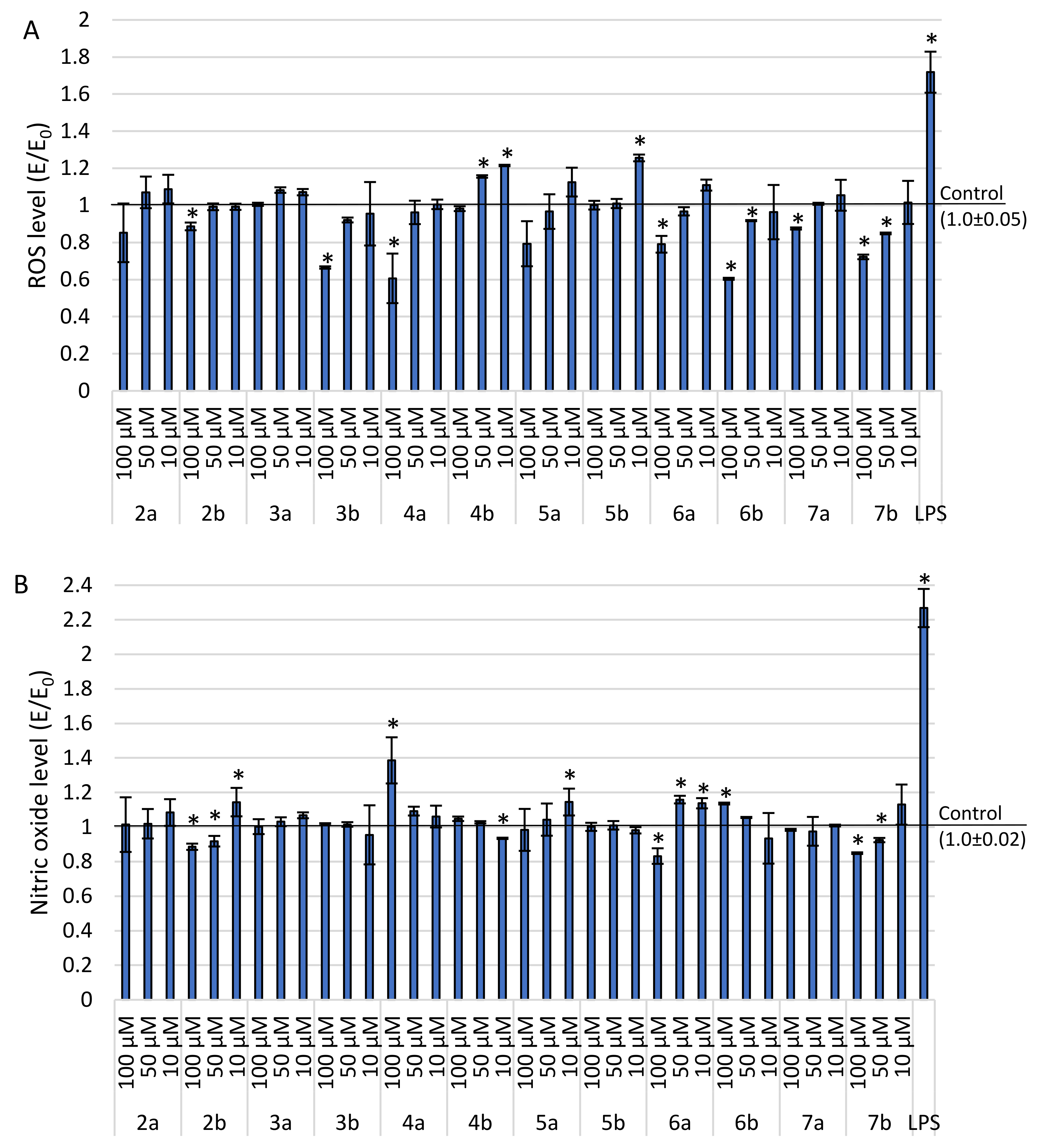
2.3.2. Level of ROS and Nitric Oxide Synthesis
2.4. In Silico Pharmacokinetic Prediction
3. Materials and Methods
3.1. Chemistry
3.1.1. Instruments and Chemicals
3.1.2. Chemical Synthesis
3.2. Cell Line
3.3. Cell Culture Media
3.4. Tested Compounds
3.5. Experimental Design
3.6. MTT Assay
3.7. Level of Reactive Oxygen Species (ROS)—DCF-DA Assay
3.8. Level of Nitrate Ions Synthesis—Griess Assay
3.9. Statistical Analysis
3.10. Molecular Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Holla, B.S.; Gonsalves, R.; Shenoy, S. Synthesis and antibacterial studies of a new series of 1,2-bis(1,3, 4-oxadiazol-2-yl)ethanes and 1,2-bis(4-amino-1,2, 4-triazol-3-yl)ethanes. Eur. J. Med. Chem. 2000, 35, 267–271. [Google Scholar] [CrossRef]
- Macaev, F.; Rusu, G.; Pogrebnoi, S.; Gudima, A.; Stingaci, E.; Vlad, L.; Shvets, N.; Kandemirli, F.; Dimoglo, A.; Reynolds, R. Synthesis of novel 5-aryl-2-thio-1,3,4-oxadiazoles and the study of their structure-anti-mycobacterial activities. Bioorg. Med. Chem. 2005, 13, 4842–4850. [Google Scholar] [CrossRef]
- Liu, F.; Luo, X.-Q.; Song, B.-A.; Bhadury, P.S.; Yang, S.; Jin, L.-H.; Xue, W.; Hu, D.-Y. Synthesis and antifungal activity of novel sulfoxide derivatives containing trimethoxyphenyl substituted 1,3,4-thiadiazole and 1,3,4-oxadiazole moiety. Bioorg. Med. Chem. 2008, 16, 3632–3640. [Google Scholar] [CrossRef]
- Liu, K.G.; Smith, J.S.; Ayscue, A.H.; Henke, B.R.; Lambert, M.H.; Leesnitzer, L.M.; Plunket, K.D.; Willson, T.M.; Sternbach, D.D. Identification of a series of oxadiazole-substituted alpha-isopropoxy phenylpropanoic acids with activity on PPARalpha, PPARgamma, and PPARdelta. Bioorg. Med. Chem. Lett. 2001, 11, 2385–2388. [Google Scholar] [CrossRef]
- Zarghi, A.; Tabatabai, S.A.; Faizi, M.; Ahadian, A.; Navabi, P.; Zanganeh, V.; Shafiee, A. Synthesis and anticonvulsant activity of new 2-substituted-5-(2-benzyloxyphenyl)-1,3,4-oxadiazoles. Bioorg. Med. Chem. Lett. 2005, 15, 1863–1865. [Google Scholar] [CrossRef] [PubMed]
- Palaska, E.; Sahin, G.; Kelicen, P.; Durlu, N.T.; Altinok, G. Synthesis and anti-inflammatory activity of 1-acylthiosemicarbazides, 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazole-3-thiones. Farmaco 2002, 57, 101–107. [Google Scholar] [CrossRef]
- Burbuliene, M.M.; Jakubkiene, V.; Mekuskiene, G.; Udrenaite, E.; Smicius, R.; Vainilavicius, P. Synthesis and anti-inflammatory activity of derivatives of 5-[(2-disubstitutedamino-6-methyl-pyrimidin-4-yl)-sulfanylmethyl]-3H-1,3,4-oxadiazole-2-thiones. Farmaco 2004, 59, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Amir, M.; Shikha, K. Synthesis and anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation activities of some new 2-[(2,6-dichloroanilino) phenyl]acetic acid derivatives. Eur. J. Med. Chem. 2004, 39, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Akhter, M.; Akhter, N.; Alam, M.M.; Zaman, M.S.; Saha, R.; Kumar, A.; Akhter, M.; Akhter, N.; Alam, M.M.; Zaman, M.S.; et al. both COX and LOX inhibitory activity Synthesis and biological evaluation of 2, 5-disubstituted 1, 3, 4-oxadiazole derivatives with both COX and LOX inhibitory activity. J. Enzyme Inhib. Med. Chem. 2011, 26, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Atobe, H.; Kushida, H.; Yamamoto, K. In vitro sensitivity of mycoplasmas isolated from various animals and sewage to antibiotics and nitrofurans. J. Antibiot. 1971, 24, 443–451. [Google Scholar] [CrossRef] [Green Version]
- Schlecker, R.; Thieme, P.C. The synthesis of antihypertensive 3-(1,3,4-oxadiazol-2-yl)phenoxypropanolahines. Tetrahedron 1988, 44, 3289–3294. [Google Scholar] [CrossRef]
- Cocohoba, J.; Dong, B.J. Raltegravir: The first HIV integrase inhibitor. Clin. Ther. 2008, 30, 1747–1765. [Google Scholar] [CrossRef] [PubMed]
- Vardan, S.; Smulyan, H.; Mookherjee, S.; Eich, R. Effects of tiodazosin, a new antihypertensive, hemodynamics and clinical variables. Clin. Pharmacol. Ther. 1983, 34, 290–296. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Growcott, J.W. Zibotentan endothelin ETA receptor antagonist oncolytic. Drugs Future 2009, 34, 624–633. [Google Scholar] [CrossRef]
- Tagad, H.D.; Hamada, Y.; Nguyen, J.T.; Hamada, T.; Abdel-Rahman, H.; Yamani, A.; Nagamine, A.; Ikari, H.; Igawa, N.; Hidaka, K.; et al. Design of pentapeptidic BACE1 inhibitors with carboxylic acid bioisosteres at P1′ and P4 positions. Bioorg. Med. Chem. 2010, 18, 3175–3186. [Google Scholar] [CrossRef]
- Kohara, Y.; Kubo, K.; Imamiya, E.; Wada, T.; Inada, Y.; Naka, T. Synthesis and angiotensin II receptor antagonistic activities of benzimidazole derivatives bearing acidic heterocycles as novel tetrazole bioisosteres. J. Med. Chem. 1996, 39, 5228–5235. [Google Scholar] [CrossRef]
- Marnett, L.J. Cyclooxygenase mechanisms. Curr. Opin. Chem. Biol. 2000, 4, 545–552. [Google Scholar] [CrossRef]
- Blobaum, A.L.; Marnett, L.J. Structural and functional basis of cyclooxygenase inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vane, J.R.; Botting, R.M. Mechanism of action of nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998, 104, 2S–8S, discussion 21S–22S. [Google Scholar] [CrossRef]
- Smith, W.L.; Urade, Y.; Jakobsson, P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [Green Version]
- Cashman, J.N. The Mechanisms of Action of NSAIDs in Analgesia. Drugs 1996, 52, 13–23. [Google Scholar] [CrossRef]
- Rodríguez, L.A.G.; Tolosa, L.B.; Traditional, A. Risk of Upper Gastrointestinal Complications Among Users of Traditional NSAIDs and COXIBs in the General Population. Gastroenterology 2007, 498–506. [Google Scholar] [CrossRef]
- Sostres, C.; Gargallo, C.J.; Arroyo, M.T.; Lanas, A. Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs, aspirin and coxibs) on upper gastrointestinal tract. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 121–132. [Google Scholar] [CrossRef]
- Bidaut-Russell, M.; Gabriel, S.E. Adverse gastrointestinal effects of NSAIDs: Consequences and costs. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 739–753. [Google Scholar] [CrossRef]
- Laine, L. Gastrointestinal effects of NSAIDs and coxibs. J. Pain Symptom Manag. 2003, 25, 32–40. [Google Scholar] [CrossRef]
- Wallace, J.L.; Devchand, P.R. Emerging roles for cyclooxygenase-2 in gastrointestinal mucosal defense. Br. J. Pharmacol. 2005, 145, 275–282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soll, A.H.; McCarthy, D. NSAID-related gastrointestinal complications. Clin. Cornerstone 1999, 1, 42–56. [Google Scholar] [CrossRef]
- Takeuchi, K. Pathogenesis of NSAID-induced gastric damage: Importance of cyclooxygenase inhibition and gastric hypermotility. World J. Gastroenterol. 2012, 18, 2147–2160. [Google Scholar] [CrossRef] [PubMed]
- Palkar, M.B.; Singhai, A.S.; Ronad, P.M.; Vishwanathswamy, A.H.M.; Boreddy, T.S.; Veerapur, V.P.; Shaikh, M.S.; Rane, R.A.; Karpoormath, R. Synthesis, pharmacological screening and in silico studies of new class of Diclofenac analogues as a promising anti-inflammatory agents. Bioorg. Med. Chem. 2014, 22, 2855–2866. [Google Scholar] [CrossRef]
- Manjunatha, K.; Poojary, B.; Lobo, P.L.; Fernandes, J.; Kumari, N.S. Synthesis and biological evaluation of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 2010, 45, 5225–5233. [Google Scholar] [CrossRef]
- Szczukowski, Ł.; Krzyżak, E.; Zborowska, A.; Zając, P.; Potyrak, K.; Peregrym, K.; Wiatrak, B.; Marciniak, A.; Świątek, P. Design, synthesis and comprehensive investigations of pyrrolo[3,4-d]pyridazinone-based 1,3,4-oxadiazole as new class of selective cox-2 inhibitors. Int. J. Mol. Sci. 2020, 21, 9623. [Google Scholar] [CrossRef] [PubMed]
- Szczukowski, Ł.; Redzicka, A.; Wiatrak, B.; Krzyżak, E.; Marciniak, A.; Gębczak, K.; Gębarowski, T.; Świątek, P. Design, synthesis, biological evaluation and in silico studies of novel pyrrolo[3,4-d]pyridazinone derivatives with promising anti-inflammatory and antioxidant activity. Bioorg. Chem. 2020, 102, 104035. [Google Scholar] [CrossRef]
- Dogruer, D.S.; Kupeli, E.; Yesilada, E.; Sahin, M.F. Synthesis of new 2-[1(2H)-phthalazinon-2-yl]acetamide and 3-[1(2H)-phthalazinon-2-yl]propanamide derivatives as antinociceptive and anti-inflammatory agents. Arch. Pharm. 2004, 337, 303–310. [Google Scholar] [CrossRef]
- Dogruer, D.S.; Sahin, M.F.; Unlü, S.; Ito, S. Studies on some 3(2H)-pyridazinone derivatives with antinociceptive activity. Arch. Pharm. 2000, 333, 79–86. [Google Scholar] [CrossRef]
- Świątek, P.; Strzelecka, M.; Urniaz, R.; Gębczak, K.; Gębarowski, T.; Gąsiorowski, K.; Malinka, W. Synthesis, COX-1/2 inhibition activities and molecular docking study of isothiazolopyridine derivatives. Bioorg. Med. Chem. 2017, 25, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Gautam, R.; Jachak, S.M.; Kumar, V.; Mohan, C.G. Synthesis, biological evaluation and molecular docking studies of stellatin derivatives as cyclooxygenase (COX-1, COX-2) inhibitors and anti-inflammatory agents. Bioorg. Med. Chem. Lett. 2011, 21, 1612–1616. [Google Scholar] [CrossRef]
- Malkowski, M.G.; Ginell, S.L.; Smith, W.L.; Garavito, R.M. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Vecchio, A.J.; Simmons, D.M.; Malkowski, M.G. Structural basis of fatty acid substrate binding to cyclooxygenase-2. J. Biol. Chem. 2010, 285, 22152–22163. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, J.R.; Pawlitz, J.L.; Moreland, K.T.; Stegeman, R.A.; Hood, W.F.; Gierse, J.K.; Stevens, A.M.; Goodwin, D.C.; Rowlinson, S.W.; Marnett, L.J.; et al. Structural insights into the stereochemistry of the cyclooxygenase reaction. Nature 2000, 405, 97–101. [Google Scholar] [CrossRef]
- Loll, P.J.; Picot, D.; Garavito, R.M. The structural basis of aspirin activity inferred from the crystal structure of inactivated prostaglandin H2 synthase. Nat. Struct. Biol. 1995, 2, 637–643. [Google Scholar] [CrossRef]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with Ser-530 and Tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Hermanson, D.J.; Banerjee, S.; Ghebreselasie, K.; Clayton, G.M.; Garavito, R.M.; Marnett, L.J. Oxicams bind in a novel mode to the cyclooxygenase active site via a two-water-mediated H-bonding Network. J. Biol. Chem. 2014, 289, 6799–6808. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Citation; Gaussian. Inc.: Wallingford, CT, USA; p. 2016.
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velázquez-Libera, J.L.; Durán-Verdugo, F.; Valdés-Jiménez, A.; Núñez-Vivanco, G.; Caballero, J. LigRMSD: A web server for automatic structure matching and RMSD calculations among identical and similar compounds in protein-ligand docking. Bioinformatics 2020, 36, 2912–2914. [Google Scholar] [CrossRef] [PubMed]
- Redzicka, A.; Szczukowski, Ł.; Kochel, A.; Wiatrak, B.; Gębczak, K.; Czyżnikowska, Ż. COX-1/COX-2 inhibition activities and molecular docking study of newly designed and synthesized pyrrolo[3,4-c]pyrrole Mannich bases. Bioorg. Med. Chem. 2019, 27, 3918–3928. [Google Scholar] [CrossRef]
- Szczęśniak-Sięga, B.M.; Wiatrak, B.; Czyżnikowska, Ż.; Janczak, J.; Wiglusz, R.J.; Maniewska, J. Synthesis and biological evaluation as well as in silico studies of arylpiperazine-1,2-benzothiazine derivatives as novel anti-inflammatory agents. Bioorg. Chem. 2021, 106, 104476. [Google Scholar] [CrossRef]
- Glomb, T.; Wiatrak, B.; Gębczak, K.; Gębarowski, T.; Bodetko, D.; Czyżnikowska, Ż.; Świątek, P. New 1,3,4-Oxadiazole Derivatives of Pyridothiazine-1,1-Dioxide with Anti-Inflammatory Activity. Int. J. Mol. Sci. 2020, 21, 9122. [Google Scholar] [CrossRef]
- Chen, D.; Menche, G.; Power, T.D.; Sower, L.; Peterson, J.W.; Schein, C.H. Accounting for ligand-bound metal ions in docking small molecules on adenylyl cyclase toxins. Proteins 2007, 67, 593–605. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 [µM] (SD) | COX-2/COX-1 Selectivity Ratio | |
|---|---|---|---|
| COX-1 | COX-2 | ||
| 1a 1b 2a | 130.5 (18.5) 138.1 (12.7) 50.3 (0.01) | 43.6 (1.3) 63.4 (7.2) 49.7 (0.03) | 0.33 0.46 0.99 |
| 2b | 50.0 (0.01) | 49.4 (0.02) | 0.99 |
| 3a | 50.3 (0.01) | 49.7 (0.02) | 0.99 |
| 3b | 54.3 (0.02) | 56.0 (0.01) | 1.03 |
| 4a | 50.2 (0.01) | 49.2 (0.01) | 0.98 |
| 4b | 56.8 (0.01) | 57.3 (0.01) | 1.01 |
| 5a | 53.2 (0.02) | 50.1 (0.08) | 0.94 |
| 5b | 51.8 (0.03) | 53.8 (0.01) | 1.04 |
| 6a | 52.3 (0.01) | 50.2 (0.02) | 0.96 |
| 6b | 51.1 (0.01) | 50.1 (0.01) | 0.98 |
| 7a | 51.6 (0.02) | 50.3 (0.02) | 0.97 |
| 7b | 50.9 (0.04) | 49.5 (0.02) | 0.97 |
| Meloxicam | 83.7 (0.03) | 59.2 (0.06) | 0.71 |
| Celecoxib | 56 (0.1) | 0.30 (0.08) | 0.005 |
| Diclofenac | 3.5 (0.04) | 16.6 (0.03) | 4.74 |
| a | ΔGbind | ΔEint | ΔE1 | ΔE2 |
| 1a | −7.1 | −9.2 | −9.2 | 0.0 |
| 1b | −6.9 | −9.0 | −8.9 | −0.1 |
| 2a | −8.4 | −10.5 | −10.4 | −0.1 |
| 2b | −7.5 | −9.6 | −9.4 | −0.2 |
| 3a | −10.2 | −12.9 | −12.8 | −0.1 |
| 3b | −9.6 | −11.2 | −11.2 | 0.0 |
| 4a | −8.6 | −10.0 | −10.1 | 0.1 |
| 4b | −8.4 | −10.8 | −11.1 | 0.3 |
| 5a | −7.1 | −9.1 | −9.1 | 0.0 |
| 5b | −9.4 | −12.4 | −12.4 | 0.0 |
| 6a | −11.2 | −13.3 | −13.2 | −0.1 |
| 6b | −10.7 | −13.1 | −13.1 | 0.0 |
| 7a | −9.7 | −11.1 | −11.3 | 0.2 |
| 7b | −9.3 | −11.3 | −11.3 | 0.0 |
| Meloxicam | −8.6 | −10.0 | −9.9 | −0.1 |
| Diclofenac | −9.7 | −10.9 | −10.9 | 0.0 |
| b | ΔGbind | ΔEint | ΔE1 | ΔE2 |
| 1a | −12.5 | −13.7 | −13.7 | 0.0 |
| 1b | −12.7 | −13.8 | −13.8 | 0.0 |
| 2a | −10.8 | −13.0 | −13.0 | 0.0 |
| 2b | −11.7 | −13.8 | −13.7 | −0.1 |
| 3a | −11.0 | −13.1 | −13.1 | 0.0 |
| 3b | −12.9 | −14.7 | −14.7 | 0.0 |
| 4a | −10.3 | −13.0 | −13.0 | 0.0 |
| 4b | −11.3 | −10.6 | −10.7 | 0.1 |
| 5a | −11.6 | −14.2 | −14.2 | 0.0 |
| 5b | −11.4 | −13.3 | −13.3 | 0.0 |
| 6a | −11.6 | −14.2 | −14.2 | 0.0 |
| 6b | −10.9 | −13.7 | −13.7 | 0.0 |
| 7a | −11.5 | −14.1 | −14.1 | 0.0 |
| 7b | −11.8 | −13.1 | −13.1 | 0.0 |
| Meloxicam | −9.9 | −10.0 | −9.8 | −0.2 |
| Diclofenac | −10.9 | −11.0 | −11.0 | 0.0 |
| Compound | Pharmacokinetics | ||||
|---|---|---|---|---|---|
| MW | Log Po/w | GI Absorption | BBB Permeant | P-gp Substrate | |
| 2a | 585.72 g/mol | 4.63 | High | No | Yes |
| 2b | 565.73 g/mol | 4.36 | High | No | Yes |
| 3a | 593.65 g/mol | 4.40 | High | No | Yes |
| 3b | 573.66 g/mol | 4.39 | High | No | Yes |
| 4a | 626.56 g/mol | 4.86 | High | No | Yes |
| 4b | 606.57 g/mol | 4.81 | High | No | Yes |
| 5a | 613.73 g/mol | 4.19 | Low | No | Yes |
| 5b | 593.74 g/mol | 4.02 | Low | No | Yes |
| 6a | 621.66 g/mol | 4.09 | Low | No | Yes |
| 6b | 601.67 g/mol | 4.05 | Low | No | Yes |
| 7a | 654.57 g/mol | 4.51 | Low | No | Yes |
| 7b | 634.58 g/mol | 4.41 | Low | No | Yes |
| Compound | Druglikeness | |||
|---|---|---|---|---|
| Lipinski | Veber | Bioavailability Score | TPSA | |
| 2a | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 2b | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 3a | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 3b | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 4a | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 4b | Yes; 1 violation | Yes | 0.55 | 118.58 Å2 |
| 5a | No; 2 violations | Yes | 0.17 | 136.82 Å2 |
| 5b | No; 2 violations | No; 1 violation | 0.17 | 136.82 Å2 |
| 6a | No; 2 violations | Yes | 0.17 | 136.82 Å2 |
| 6b | No; 2 violations | No; 1 violation | 0.17 | 136.82 Å2 |
| 7a | No; 2 violations | Yes | 0.17 | 136.82 Å2 |
| 7b | No; 2 violations | No; 1 violation | 0.17 | 136.82 Å2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peregrym, K.; Szczukowski, Ł.; Wiatrak, B.; Potyrak, K.; Czyżnikowska, Ż.; Świątek, P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone as Promising Cyclooxygenase Inhibitors. Int. J. Mol. Sci. 2021, 22, 9130. https://doi.org/10.3390/ijms22179130
Peregrym K, Szczukowski Ł, Wiatrak B, Potyrak K, Czyżnikowska Ż, Świątek P. In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone as Promising Cyclooxygenase Inhibitors. International Journal of Molecular Sciences. 2021; 22(17):9130. https://doi.org/10.3390/ijms22179130
Chicago/Turabian StylePeregrym, Krzysztof, Łukasz Szczukowski, Benita Wiatrak, Katarzyna Potyrak, Żaneta Czyżnikowska, and Piotr Świątek. 2021. "In Vitro and In Silico Evaluation of New 1,3,4-Oxadiazole Derivatives of Pyrrolo[3,4-d]pyridazinone as Promising Cyclooxygenase Inhibitors" International Journal of Molecular Sciences 22, no. 17: 9130. https://doi.org/10.3390/ijms22179130