
Identification of Two Early Folding Stage Prion Non-Local Contacts Suggested to Serve as Key Steps in Directing the Final Fold to Be Either Native or Pathogenic


Abstract
:1. Introduction
2. Results
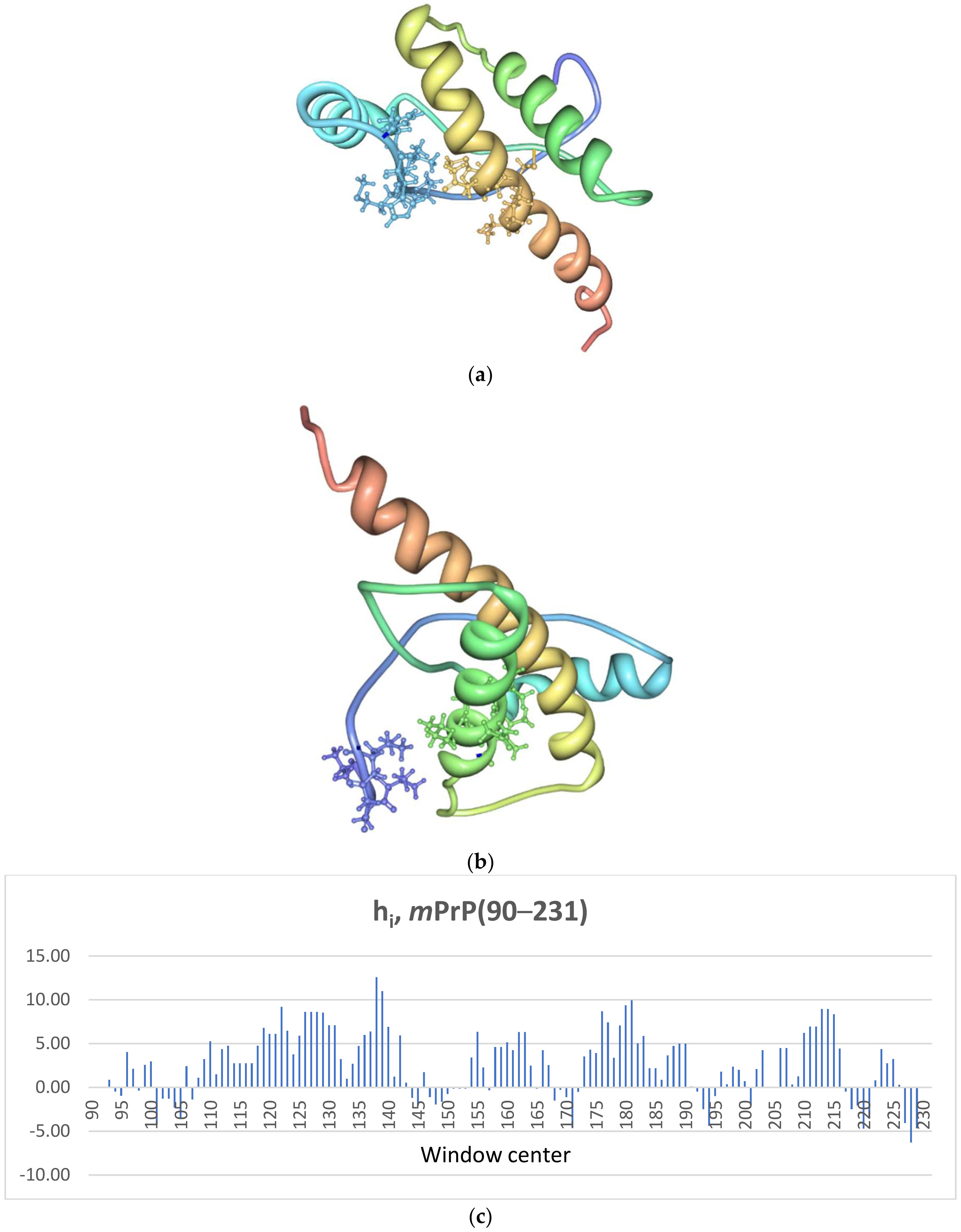
2.1. The Murine Prion Protein mPrP
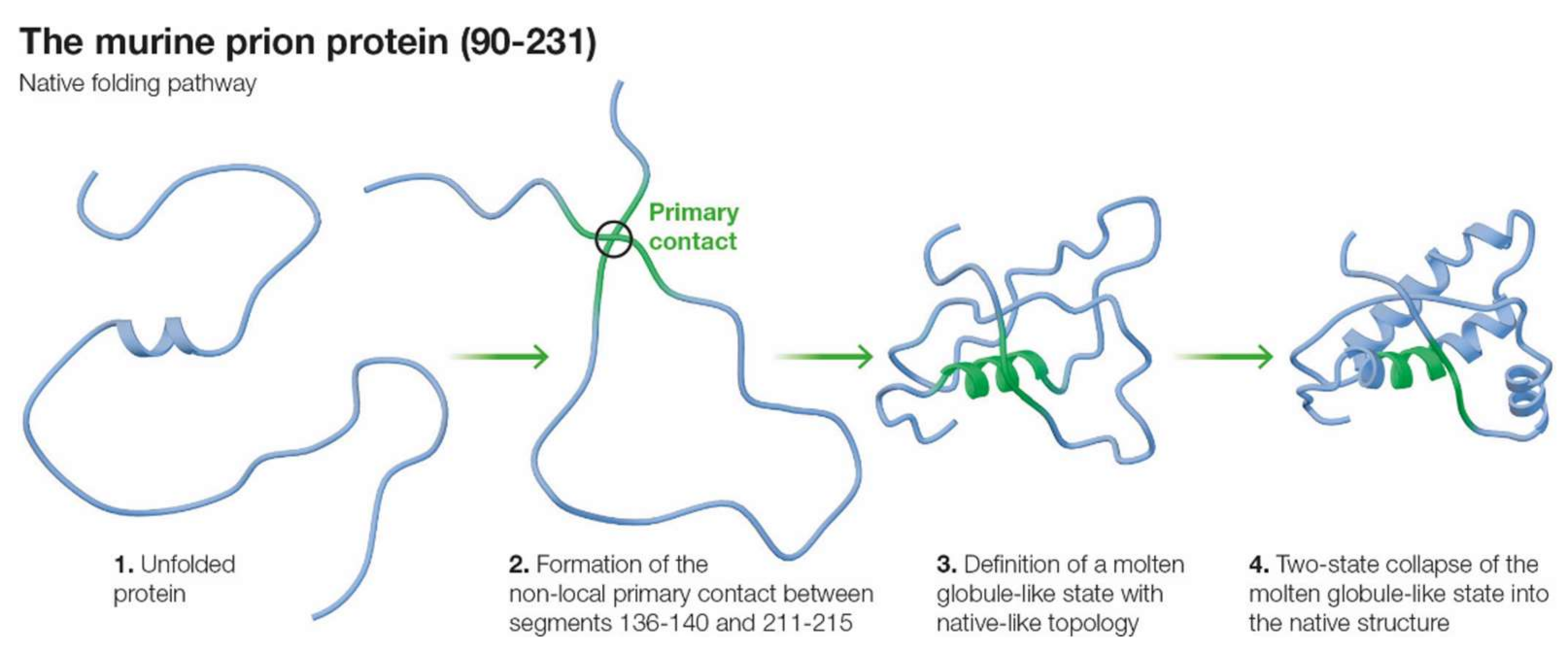
2.2. Primary Contacts for mPrP(90–231)
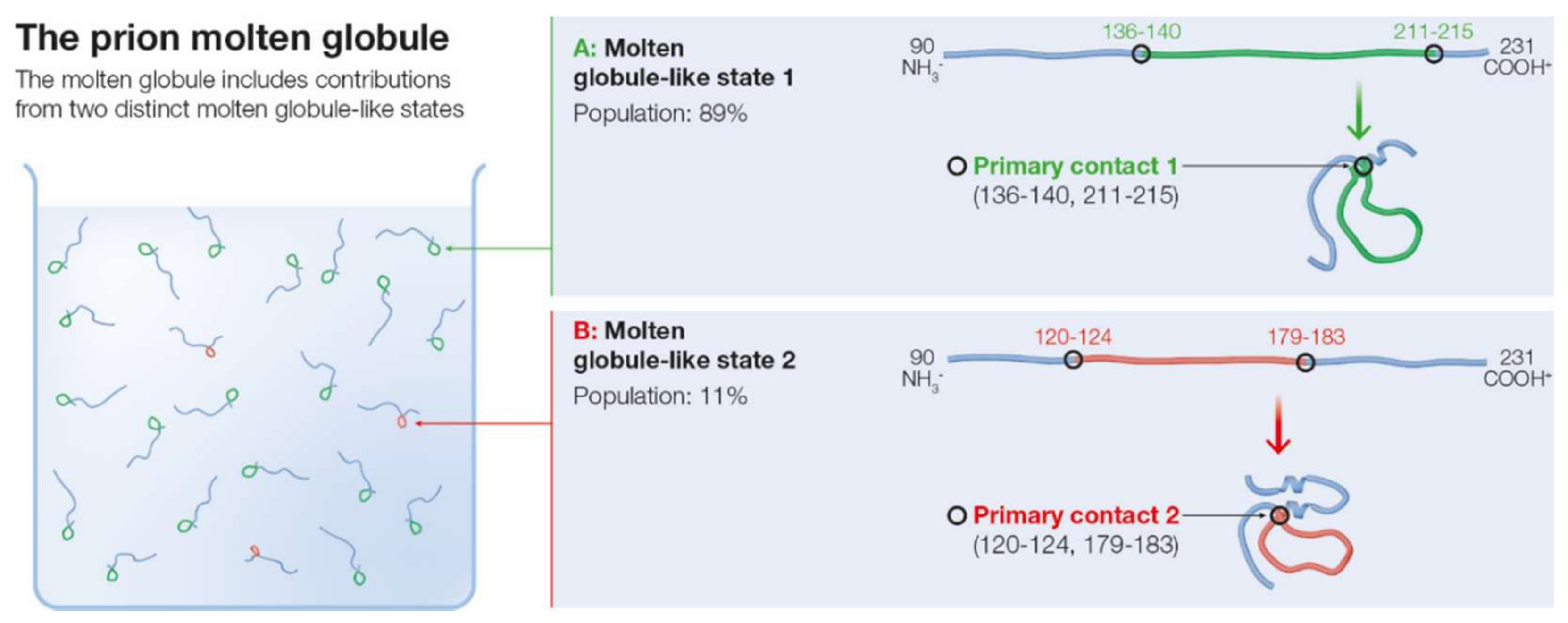
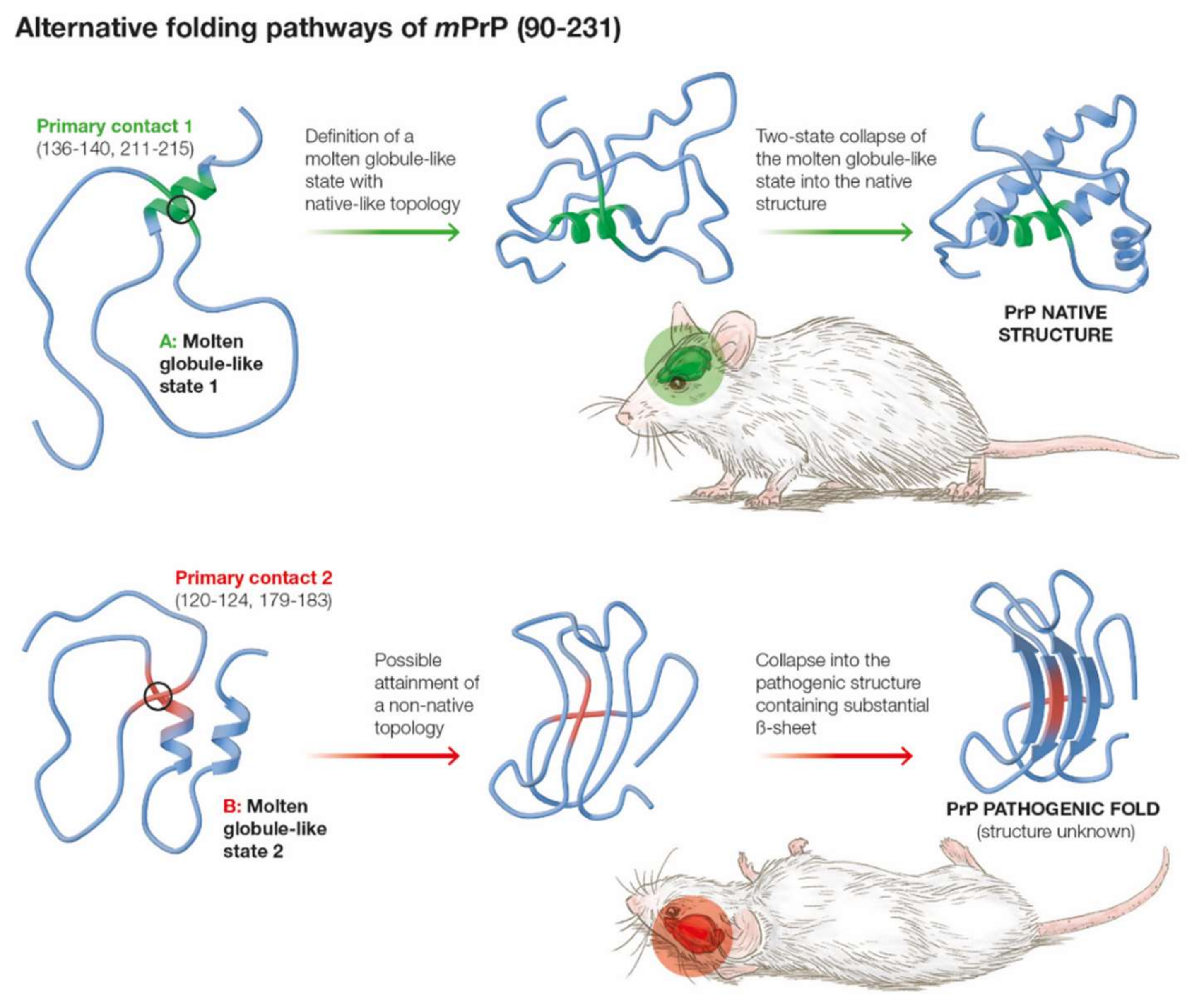
2.3. Molten Globule State for mPrP(90–231)
2.4. Is the Less Populated MGLIS the Branching Point for the Pathogenic Transition from the Molten Globule?
2.5. Kinetics of the Two-State Collapse of the Prion Protein
3. Discussion
4. Methods
4.1. The Multi-State Folding Pathway of Small Proteins in the SCM
4.2. The Molten Globule
- (a)
- The MGLIS has a primary loop in a fluctuating but generally open conformation due to the need to minimize ∆Gloop in the early folding stages.
- (b)
- The MGLIS shows structural fluctuations on a larger scale compared to the native state because of the unfolded primary loop.
- (c)
- The overall dimensions of the MGLIS are larger than those of the native protein because of the open primary loop.
- (d)
- The extra volume of the MGLIS with respect to the native state is mostly water due to the open conformation of the loop.
- (e)
- Side chains in the primary loop of the MGLIS retain much of their torsional freedom, because the loop is not a fully folded structure.
4.3. Determining the Primary Contact
4.4. Calculating the Effect of Mutations of the Primary Contact on the Stability of the MGLIS
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| mPrP(90–231) | Murine prion protein (90–231) |
| SCM | Sequential collapse model |
| MGLIS | Molten globule-like state |
References
- Prusiner, S.B. (Ed.) Prion Biology and Diseases; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2014. [Google Scholar]
- Ramirez-Alvarado, M.; Kelly, J.W.; Dobson, C.M. Protein Misfolding Diseases, Current and Emerging Principles and Therapies; John Wiley & Sons: Hoboken, NJ, USA, 2010. [Google Scholar]
- Ji, H.-F.; Zhang, H.-Y. ß-Sheet Constitution of Prion Proteins. Trends Biochem. Sci. 2010, 35, 129–134. [Google Scholar] [CrossRef]
- Yamaguchi, K.-I.; Kuwata, K. Formation and Properties of Amyloid Fibrils of Prion Protein. Biophys. Rev. 2018, 10, 517–525. [Google Scholar] [CrossRef] [Green Version]
- Cescatti, M.; Saverioni, D.; Capellani, S.; Tagliavini, F.; Kitamoto, T.; Ironside, J.; Giese, A.; Parchi, P. Abalysis of Conformational Stability of Abnormal Protein Aggregates Across the Spectrum of Creutzfeldt-Jakob Disease Prions. J. Virol. 2016, 90, 6244–6254. [Google Scholar] [CrossRef] [Green Version]
- Kondo, J.; Ihara, Y.; Sairoh, T. Molecular Cloning of cDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11287. [Google Scholar]
- Uversky, V.N. α-Synuclein Misfolding and Neurodegenerative Diseases. Curr. Protein Pept. Sci. 2009, 9, 507–540. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy Bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Navalkar, A.; Ghosh, S.; Pandey, S.; Paul, A.; Datta, D.; Maji, S.K. Prion-like p53 Amyloids in Cancer. Biochemistry 2020, 59, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; Rangel, L.P.; Costa, D.C.F.; Cordeiro, Y.; De Moura Gallo, C.V. Expanding the prion concept to cancer biology: Dominant-negative effect of aggregates of mutant p53 tumor suppressor. Biosci. Rep. 2013, 33, e00054. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.W. The Environmental Dependency of Protein Folding Best Explains Prion and Amyloid Diseases. Proc. Natl. Acad. Sci. USA 1998, 95, 930–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, D.O.V.; DeArmond, S.J.; Cohen, F.E.; Daggett, V. Mapping the Early Steps in the pH-Induced Conformational Conversion of the Prion Protein. Proc. Natl. Acad. Sci. USA 2001, 98, 2985–2989. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of Environmental Factors on the Kinetics of Insulin Fibril Formation: Elucidation of the Molecular Mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar] [CrossRef]
- Hamada, D.; Dobson, C.M. A Kinetic Study of β-Lactoglobulin Amyloid Fibril Formation Promoted by Urea. Protein Sci. 2002, 11, 2417–2426. [Google Scholar] [CrossRef]
- Safar, J.; Roller, P.P.; Gajdusek, D.C.; Gibbs, C.J. Scrapie Amyloid (Prion) Protein has the Conformational Characteristics of an Aggregated Molten Globule Folding Intermediate. Biochemistry 1994, 33, 8375–8383. [Google Scholar] [CrossRef]
- Apetri, A.C.; Maki, K.; Roder, H.; Surewicz, W.K. Early Intermediate in Human Prion Protein as Evidenced by Ultrarapid Mixing Experiments. J. Am. Chem. Soc. 2006, 128, 11673–11678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apetri, A.C.; Surewicz, K.; Surewicz, W.K. The Effect of Disease-Associated Mutations on the Folding Pathway of Human Prion Protein. J. Biol. Chem. 2004, 279, 18008–18014. [Google Scholar] [CrossRef] [Green Version]
- Jenkins, D.C.; Sylvester, I.D.; Pinheiro, T.J.T. The Elusive Intermediate on the Folding Pathway of the Prion Protein. FEBS J. 2008, 275, 1323–1335. [Google Scholar] [CrossRef] [PubMed]
- Honda, R.P.; Xu, M.; Yamaguchi, K.; Roder, H.; Kuwata, K. A Native-like Intermediate Serves as a Branching Point between the Folding and Aggregation Pathways of the Mouse Prion Protein. Structure 2015, 23, 1735–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahn, T.R.; Parker, M.J.; Homans, S.W.; Radford, S.E. Amyloid Formation Under Physiological Conditions Proceeds Via a Native-like Folding Intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Mukaiyama, A.; Nakamura, T.; Makabe, K.; Maki, K.; Goto, Y.; Kuwajima, K. The Molten Globule of β(2)-Microglobulin Accumulated at pH 4 and its Role in Protein Folding. J. Mol. Biol. 2013, 425, 273–291. [Google Scholar] [CrossRef]
- Gerber, R.; Tahiri-Alaoui, A.; Hore, P.J.; James, W. Oligomerization of the Human Prion Protein Proceeds via a Molten Globule Intermediate. J. Biol. Chem. 2007, 282, 6300–6307. [Google Scholar] [CrossRef] [Green Version]
- Moulick, R.; Das, R.; Udgaonkar, J.B. Partially Unfolded Forms of the Prion Protein Populated Under Misfolding-promoting Conditions: Characterization by Hydrogen Exchange Mass Spectroscopy and NMR. J. Biol. Chem. 2015, 290, 25227–25240. [Google Scholar] [CrossRef] [Green Version]
- Hosszu, L.L.P.; Wells, M.A.; Jackson, G.S.; Jonse, S.; Batchelor, M.; Clarke, A.R.; Craven, C.J.; Waltho, J.P.; Collinge, J. Definable Equilibrium States in the Folding of Human Prion Protein. Biochemistry 2005, 44, 16649–16657. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Ronneberg, T.A.; Rabitz, H.A. Sequential Collapse Model for Protein Folding Pathways. J. Phys. Chem. B 1999, 103, 9749–9758. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Haas, E.; Rabitz, H.A. Nature’s Shortcut to Protein Folding. J. Phys. Chem. B 2019, 123, 4463–4476. [Google Scholar] [CrossRef] [PubMed]
- Stepney, S. The neglected pillar of material computation. Phys. D 2008, 237, 1157–1164. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Low Entropic Barrier to the Hydrophobic Collapse of the Prion Protein: Effects of Intermediate States and Conformational Flexibility. J. Phys. Chem. A 2010, 114, 6978–6982. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. A Simple Quantitative Model of Macromolecular Crowding Effects on Protein Folding: Application to the Murine Prion Protein (121–231). Chem. Phys. Lett. 2013, 574, 112–115. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Flexibility Damps Macromolecular Crowding Effects on Protein Folding Dynamics: Application to the Murine Prion Protein (121–231). Chem. Phys. Lett. 2014, 591, 207–211. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Macromolecular Crowding Facilitates the Conformational Transition of Molten Globule States of the Prion Protein. J. Phys. Chem. B 2016, 120, 11093–11101. [Google Scholar] [CrossRef] [PubMed]
- Bergasa-Caceres, F.; Rabitz, H. Predicting the location of the non-local contacts in alpha-synuclein Biochimica et Biophysica Acta Proteins and Proteomics. Biochim. Biophys. Acta Proteins Proteom. 2018, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, L.; Bruni, A.C. Mutations in Prion Protein Gene: Pathogenic Mechanisms in C-Terminal vs. N-Terminal Domain, a Review. Int. J. Mol. Sci. 2019, 20, 3606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, J.; Udgaonkar, J.B. Molecular Mechanism of the Misfolding and Oligomerization of the Prion Protein: Current Understanding and Its Implication. J. Phys. Chem. B 2015, 54, 4431–4442. [Google Scholar] [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wüthrich, K. NMR Structure of the Mouse Prion Protein Domain PrP(121-231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef]
- Gossert, A.D.; Bonjour, S.; Lysek, D.A.; Fiorito, F.; Wüthrich, K. Prion Protein NMR Structures of Elk and of Mouse/Elk Hybrids. Proc. Natl. Acad. Sci. USA 2005, 102, 646–650. [Google Scholar] [CrossRef] [Green Version]
- Wildegger, G.; Glockshuber, R. Extremely Rapid Folding of the C-Terminal Domain of the Prion Protein Without Kinetic Intermediates. Nat. Struct. Biol. 1999, 6, 550–553. [Google Scholar]
- Kachel, N.; Kremer, W.; Zahn, R.; Kalbitzer, H.R. Observation of intermediate states of the human prion protein by high pressure NMR spectroscopy. BMC Struct. Biol. 2006, 6, 16. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.B.D.; Jones, C.E.; Abdelraheim, S.R.; Thompsett, A.R.; Brazier, M.W.; Toms, H.; Brown, D.R.; Viles, J.H. NMR characterization of the pH 4 β-intermediate of the prion protein: The N-terminal half of the protein remains unstructured and retains a high degree of flexibility. Biochem. J. 2007, 401, 533–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingant, E.; Fontes, P.; Alvarez-Martinez, M.T.; Arnaud, J.-D.; Liautard, J.-P.; Pujo-Menjouet, L. A Micellar On-Pathway Intermediate Step Explains the Kinetics of Prion Amyloid Formation. PLoS Comput. Biol. 2014, 10, e1003735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monad, B.; Reddy, G. A Transient Intermediate Populated in Prion Folding Leads to Domain Swapping. Biochemistry 2020, 59, 114–124. [Google Scholar]
- Dunker, A.K.; Garner, E.; Guilliot, S.; Romero, P.; Albrecht, K.; Hart, J.; Obradovic, Z.; Kissinger, C.; Villafranca, J.E. Protein disorder and the evolution of molecular recognition: Theory, predictions and observations. Pac. Symp. Biocomput. 1998, 3, 473–484. [Google Scholar]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Moreland, J.L.; Gramada, A.; Buzko, O.V.; Zhang, Q.; Bourne, P.E. The Molecular Biology Toolkit (MBT): A Modular Platform for Developing Molecular Visualization Applications. BMC Bioinform. 2005, 6, 21. [Google Scholar] [CrossRef] [Green Version]
- Honda, R.P.; Yamaguchi, K.; Kuwata, K. Acid-Induced Molten Globule of a Prion Protein: Crucial Role of Strand 1-Helix 1-Strand 2 Segment. J. Biol. Chem. 2014, 289, 30355–30363. [Google Scholar] [CrossRef] [Green Version]
- Hart, T.; Hosszu, L.L.P.; Trevitt, C.R.; Jackson, G.S.; Waltho, J.P.; Collinge, J.; Clarke, A.R. Folding kinetics of the human prion protein probed by temperature jump. Proc. Natl. Acad. Sci. USA 2009, 106, 5651–5656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liemann, S.; Glockshuber, R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999, 38, 3258–3267. [Google Scholar] [CrossRef] [PubMed]
- Chakroun, N.; Fornili, A.; Prigent, S.; Kleinjung, J.; Dreiss, C.A.; Rezaei, H.; Fraternali, F. Decrypting Prion Protein Conversion into a β-Rich Conformer by Molecular Dynamics. J. Chem. Theor. Comp. 2012, 9, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Dima, R.I.; Thirumalai, D. Probing the Instabilities in the Dynamics of Helical Fragments from Mouse PrPC. Proc. Natl. Acad. Sci. USA 2004, 101, 15335–15340. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Thirumalai, D. Helices 2 and 3 Are the Initiation Sites in the PrP(C) → PrP(SC) Transition. Biochemistry 2013, 52, 310–331. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.-H.; Legname, G.; Serban, A.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Prion Proteins with Pathogenic and Protective Mutations Show Similar Structure and Dynamics. Biochemistry 2009, 48, 8120–8128. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, N. Hot Spot of Structural Ambivalence in Prion Protein Revealed by Secondary Structure Principal Component Analysis. J. Phys. Chem. B 2014, 118, 9826–9833. [Google Scholar] [CrossRef]
- Liu, A.; Riek, P.; Zahn, R.; Hornemann, S.; Glockshuber, R.; Wuthrich, K. Peptides and Proteins in Neurodegenerative Disease: Helix Propensity of a Polypeptide Containing Helix 1 of the Mouse Prion Protein Studied by NMR and CD Spectroscopy. Biopolymers 1999, 51, 145–152. [Google Scholar] [CrossRef]
- Ziegler, J.; Sticht, H.; Marx, U.; Muller, W.; Rosch, P.; Schwarzinger, S. CD and NMR Studies of Prion Protein (PrP) Helix 1. J. Biol. Chem. 2003, 278, 50175–50181. [Google Scholar] [CrossRef] [Green Version]
- Honda, R.; Kuwata, K. Evidence for a central role of PrP helix 2 in the nucleation of amyloid fibrils. FASEB J. 2018, 32, 3641–3652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kei-Ichi, Y.; Matsumoto, T.; Kuwata, K. Critical Region for Amyloid Fibril Formation of Mouse Prion Protein: Unusual Amyloidogenic Properties of the Helix 2 Peptide. Biochemistry 2008, 47, 13242–13251. [Google Scholar]
- Bagyinszky, E.; Van Giau, V.; Youn, Y.C.; An, S.S.A.; Kim, S. Characterization of mutations in PRNP (prion) gene and their possible roles in neurodegenerative diseases. Neuropsychiatr. Dis. Treat. 2018, 14, 2067–2085. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Udgaonkar, J.B. The Pathogenic Mutation T182A Converts the Prion Protein into a Molten Globule-like Conformation Whose Misfolding to Oligomers but Not to Fibrils is Drastically Accelerated. J. Phys. Chem. B 2015, 55, 459–469. [Google Scholar] [CrossRef]
- Hernández, M.S.; Barritt, J.D.; Sobek, J.; Hornermann, S.; Aguzzi, A.; De Simone, A. Mechanism of Misfolding of the Human Prion Protein Revealed by a Pathological Mutation. Proc. Natl. Acad. Sci. USA 2021, 118, e2019631118. [Google Scholar] [CrossRef]
- Grasbon-Frodl, E.; Lorenz, H.; Mann, U.; Nitsch, R.M.; Windl, O.; Kretzschmar, H.A. Loss of glycosylation associated with the T183A mutation in human prion disease. Acta Neuropathol. 2004, 476–484. [Google Scholar] [CrossRef]
- Muñoz-Nieto, M.; Ramonet, N.; López-Gastón, J.I.; Cuadrado-Corrales, N.; Calero, O.; Díaz-Hurtado, M.; Ipiens, J.R.; Ramón y Cajal, S.; De Pedro-Cuesta, J.; Calero, M. A Novel Mutation I215V in the PRNP Gene Associated with Creutzfeldt-Jakob and Alzheimer’s Diseases in Three Patients with Divergent Clinical Phenotypes. J. Neurol. 2013, 260, 77–84. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, P.; Della Longa, S.; Arcovito, A.; Mancini, G.; Zitolo, A.; Chillemi, G.; Giachin, G.; Legname, G.; Benetti, F. Effects of the Pathological Q212P Mutation on Human Prion Protein Non-Octarepeat Copper-Binding Site. Biochemistry 2012, 51, 6068–6079. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H. Relating contact order to the rate of cooperative collapse in the sequential collapse model for protein folding pathways. Chem. Phys. Lett. 2003, 376, 612–617. [Google Scholar] [CrossRef]
- Plaxco, K.W.; University of California, Santa Barbara, CA, USA; Lawrence, C.; University of California, Santa Barbara, CA, USA. Personal Communication of Internal Computational Result for RCO in 1AG2, 2010.
- Moulick, R.; Udgaonkar, J.B. Thermodynamic Characterization of the Unfolding of the Prion Protein. Biophys J. 2014, 106, 410–420. [Google Scholar] [CrossRef] [Green Version]
- Sabareesan, A.T.; Udgaonkar, J.B. The G126V Mutation of the Mouse Prion Protein Hinders Nucleation-Dependent Fibril Formation by Slowing Initial Fibril Growth and by Increasing the Critical Concentration. Biochemistry 2017, 56, 5931–5942. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.N.A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E. Conversion of α-helices into β-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, R.; Sawaya, M.R.; Balbirnie, M.; Madsen, A.O.; Riekel, C.; Grothe, R.; Eisenberg, D. Structure of the cross-β spine of amyloid-like fibrils. Nature 2005, 435, 773–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, D.E.; Keller, C.A.; Plaxo, K.W.; Metiu, H. How the folding rate constant of simple, single-domain proteins depends on the number of native contacts. Proc. Natl. Acad. Sci. USA 2002, 99, 3535–3539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandner, S.; Jaunmukiane, Z. Prion Disease: Experimental Models and Reality. Acta Neuropathol. 2017, 133, 197–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergasa-Caceres, F.; Rabitz, H.A. Interdiction of Protein Folding for Therapeutic Drug Development in SARS-CoV-2. J. Phys. Chem. B 2020, 124, 8201–8208. [Google Scholar] [CrossRef]
- Mustazza, C.; Sbriccoli, M.; Minosi, P.; Raggi, C. Small Molecules with Anti-Prion Activity. Curr. Med. Chem. 2020, 27, 5446–5479. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.K.; Wieland, B.; Swayampakula, M.; Polymenidou, M.; Rahman, M.H.; Kv, N.N.V.; Aguzzi, A.; James, M.N.G. Structural studies of the folded domain of the prion protein bound to the Fab fragment of the antibody POM1. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 1501–1512. [Google Scholar] [CrossRef]
- Spagnolli, G.; Massignan, T.; Astolfi, A.; Biggi, S.; Rigoli, M.; Brunelli, P.; Libergoli, M.; Ianeselli, A.; Orioli, S.; Boldrini, A. Pharmacological Inactivation of the Prion Protein by Targeting a Folding Intermediate. Nat. Comm. Biol. 2021, 4, 62. [Google Scholar] [CrossRef] [PubMed]
- Bertoncini, C.W.; Jung, Y.S.; Fernandez, C.O.; Hoyer, W.; Griesinger, C.; Jovin, T.M.; Zwecktetter, M. Release of Long-Range Interactions Potentiates Aggregation of Natively Unstructured Alpha-Synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 1430–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Lai, B.T.; Kozak, J.J.; Gray, H.B.; Winkler, J.R. Alpha-Synuclein Tertiary Contact Dynamics. J. Phys. Chem. B 2007, 111, 2107–2112. [Google Scholar] [CrossRef] [Green Version]
- Dedmon, M.M.; Lindorff-Larsen, K.; Christodolou, J.; Vendruscolo, M.; Dobson, C.M. Mapping Non-local Interactions in α-Synuclein using Spin-Label NMR and Ensemble Molecular Dynamics Simulations. J. Am. Chem. Soc. 2005, 127, 476–477. [Google Scholar] [CrossRef]
- Ranjan, P.; Kumar, A. Perturbation in Long-Range Contacts Modulates the Kinetics of amyloid Formation in α-Synuclein Familial Mutants. ACS Chem. Neurol. 2017, 8, 2235–2246. [Google Scholar] [CrossRef] [PubMed]
- Angot, E.; Steiner, J.A.; Hansen, C.; Li, J.-Y.; Brundin, P. Are synucleinopathies prion-like diseases? Lancet Neurol. 2010, 9, 1128–1138. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H. Sequential Collapse Folding Pathway of Staphylococcal Nuclease: Entropic Activation Barriers to Hydrophobic Collapse of the Protein Core. J. Phys. Chem. B 2004, 108, 8023–8030. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H. Role of Topology in the Cooperative Collapse of the Protein Core in the Sequential Collapse Model. Folding Pathway of r-Lactalbumin and Hen Lysozyme. J. Phys.Chem.B 2001, 105, 2874–2880. [Google Scholar] [CrossRef]
- Bergasa-Caceres, F.; Rabitz, H.A. Sequential Collapse Folding Pathway of β-Lactoglobulin: Parallel Pathways and Non-Native Secondary Structure. J. Phys. Chem. B 2003, 107, 3606–3612. [Google Scholar] [CrossRef]
- Karplus, M.; Weaver, D.L. Diffusion–collision model for protein folding. Biopolymers 1979, 18, 1421–1437. [Google Scholar] [CrossRef]
- Ittah, V.; Haas, E. Nonlocal interactions stabilize long range loops in the initial folding intermediates of reduced bovine pancreatic trypsin inhibitor. Biochemistry 1995, 34, 4493–4506. [Google Scholar] [CrossRef]
- Sali, A.; Shakhnovich, E.; Karplus, M. How does a protein fold? Nature 1994, 369, 248–251. [Google Scholar] [PubMed]
- Vendruscolo, M.; Paci, E.; Dobson, C.M.; Karplus, M. Three key residues form a critical contact network in a protein folding transition state. Nature 2001, 409, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N.; Trifonov, E.N. Loop fold structure of proteins: Resolution of Levinthal’s paradox. J. Biomol. Struct. Dyn. 2002, 20, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Berezovsky, I.N.; Guarnera, E.; Zheng, Z. Basic units of protein structure, folding, and function. Prog. Byophys. Mol. Biol. 2016, 128, 85–99. [Google Scholar] [CrossRef] [PubMed]
- Alm, E.; Baker, D. Prediction of Protein-Folding Mechanisms from Free-energy Landscapes Derived from Native Structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11305–11310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, V.; Eaton, W.A. A Simple Model for Calculating the Kinetics of Protein Folding from Three-dimensional Structures. Proc. Natl. Acad. Sci. USA 1999, 96, 11311–11316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makarov, D.E.; Plaxco, K.W. The Topomer Search Model: A Simple, Quantitative Theory of Two-State Protein Folding Kinetics. Protein Sci 2003, 12, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Kuwajima, K. The molten globule state as a clue for understanding the folding and cooperativity of globular-protein structure. Proteins Struct. Funct. Genet. 1989, 6, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Dill, K.A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. [Google Scholar] [CrossRef]
- Baldwin, R.L.; Rose, G.D. How the Hydrophobic Effect Drives Protein Folding. Proc. Natl. Acad. Sci. USA 2016, 113, 12462–12466. [Google Scholar] [CrossRef] [Green Version]
- Shiraki, K.; Nishikawa, K.; Goto, Y. Trifluoroethanol-induced Stabilization of the α-Helical Structure of β-Lactoglobulin: Implication for Non-hierarchical Protein Folding. J. Mol. Biol. 1995, 245, 180–194. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, S.B.; Wu, G.A.; Chodera, J.D.; Dill, K.A. Protein Folding by Zipping and Assembly. Proc. Natl. Acad. Sci. USA 2007, 104, 11987–11992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauchere, J.L.; Pliska, V. Hydrophobic Parameters II of Amino-Acid Side Chains from the Partitioning of N-Acetyl-Amino-Acid Amides. Eur. J. Med. Chem. 1983, 18, 369–375. [Google Scholar]
- Jacobson, H.; Stockmeyer, W.H. Intramolecular Reactions in Polycondensations. I. The Theory of Linear Systems. J. Chem. Phys. 1950, 18, 1600–1607. [Google Scholar] [CrossRef]
- O’Neil, K.T.; DeGrado, W.F. A Thermodynamic Scale for the Helix-Forming Tendencies of the Commonly Occurring Amino. Acids Sci. 1990, 250, 646–651. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Contact | Stability | Native-like |
|---|---|---|
| PC1 (138 on 213) | −4.2 | Yes |
| PC2 (122 on 181) | −2.1 | Yes |
| Mutant | MGLIS | ΔΔGMGLIS,hyd | ΔΔGMGLIS,α | ΔΔGMGLIS |
|---|---|---|---|---|
| T182A | MGLIS2 | −0.1 | −1.1 | −1.2 |
| I214V | MGLIS1 | 0.6 | 0.2 | 0.8 |
| Q211P | MGLIS1 | −0.9 | 4.5 | 3.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bergasa-Caceres, F.; Rabitz, H.A. Identification of Two Early Folding Stage Prion Non-Local Contacts Suggested to Serve as Key Steps in Directing the Final Fold to Be Either Native or Pathogenic. Int. J. Mol. Sci. 2021, 22, 8619. https://doi.org/10.3390/ijms22168619
Bergasa-Caceres F, Rabitz HA. Identification of Two Early Folding Stage Prion Non-Local Contacts Suggested to Serve as Key Steps in Directing the Final Fold to Be Either Native or Pathogenic. International Journal of Molecular Sciences. 2021; 22(16):8619. https://doi.org/10.3390/ijms22168619
Chicago/Turabian StyleBergasa-Caceres, Fernando, and Herschel A. Rabitz. 2021. "Identification of Two Early Folding Stage Prion Non-Local Contacts Suggested to Serve as Key Steps in Directing the Final Fold to Be Either Native or Pathogenic" International Journal of Molecular Sciences 22, no. 16: 8619. https://doi.org/10.3390/ijms22168619