
Novel 2-(Adamantan-1-ylamino)Thiazol-4(5H)-One Derivatives and Their Inhibitory Activity towards 11β-HSD1—Synthesis, Molecular Docking and In Vitro Studies

 ,
,  , and
, and
Abstract
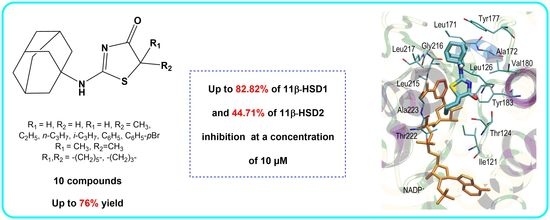
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. In Vitro Studies
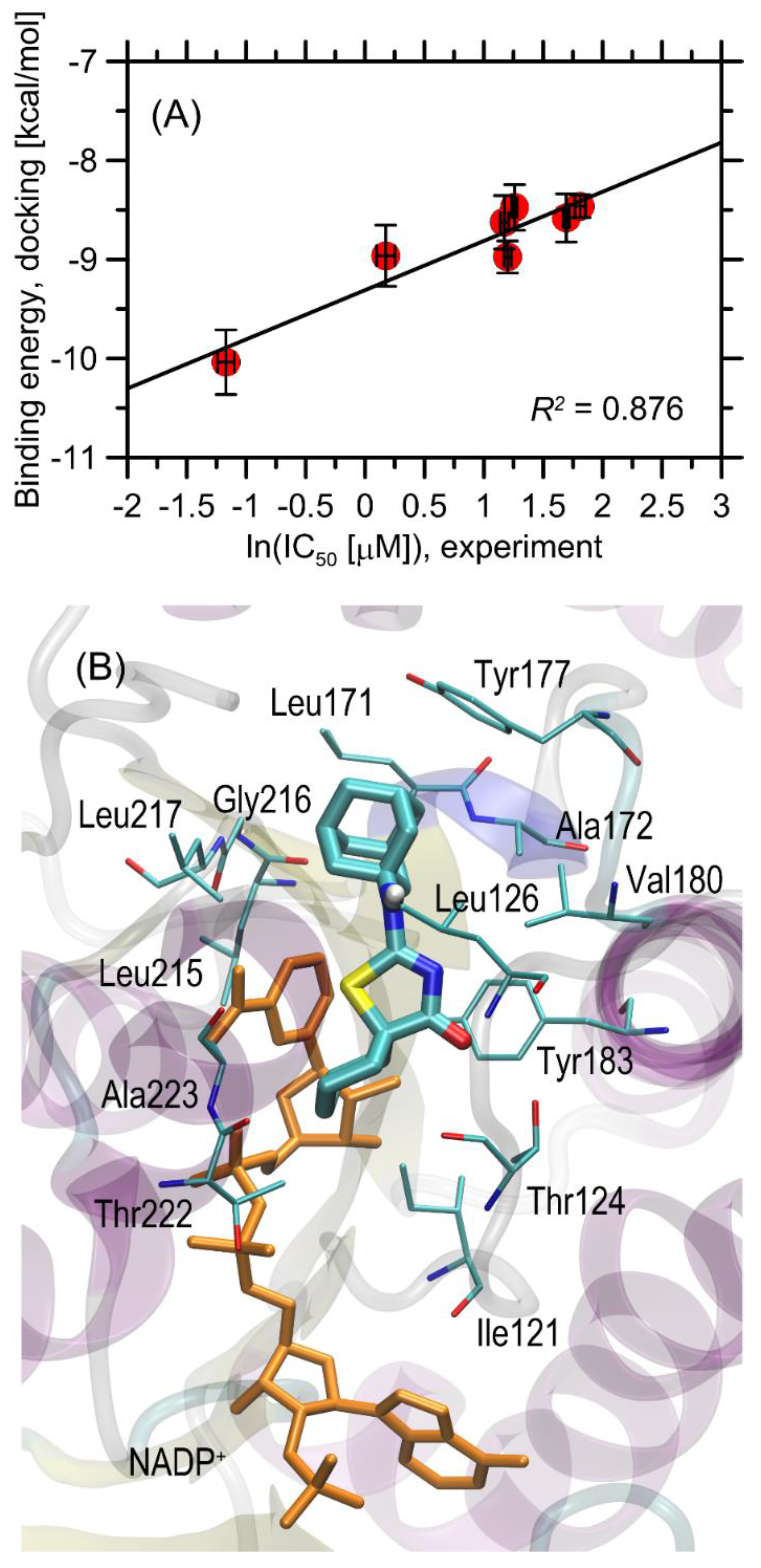
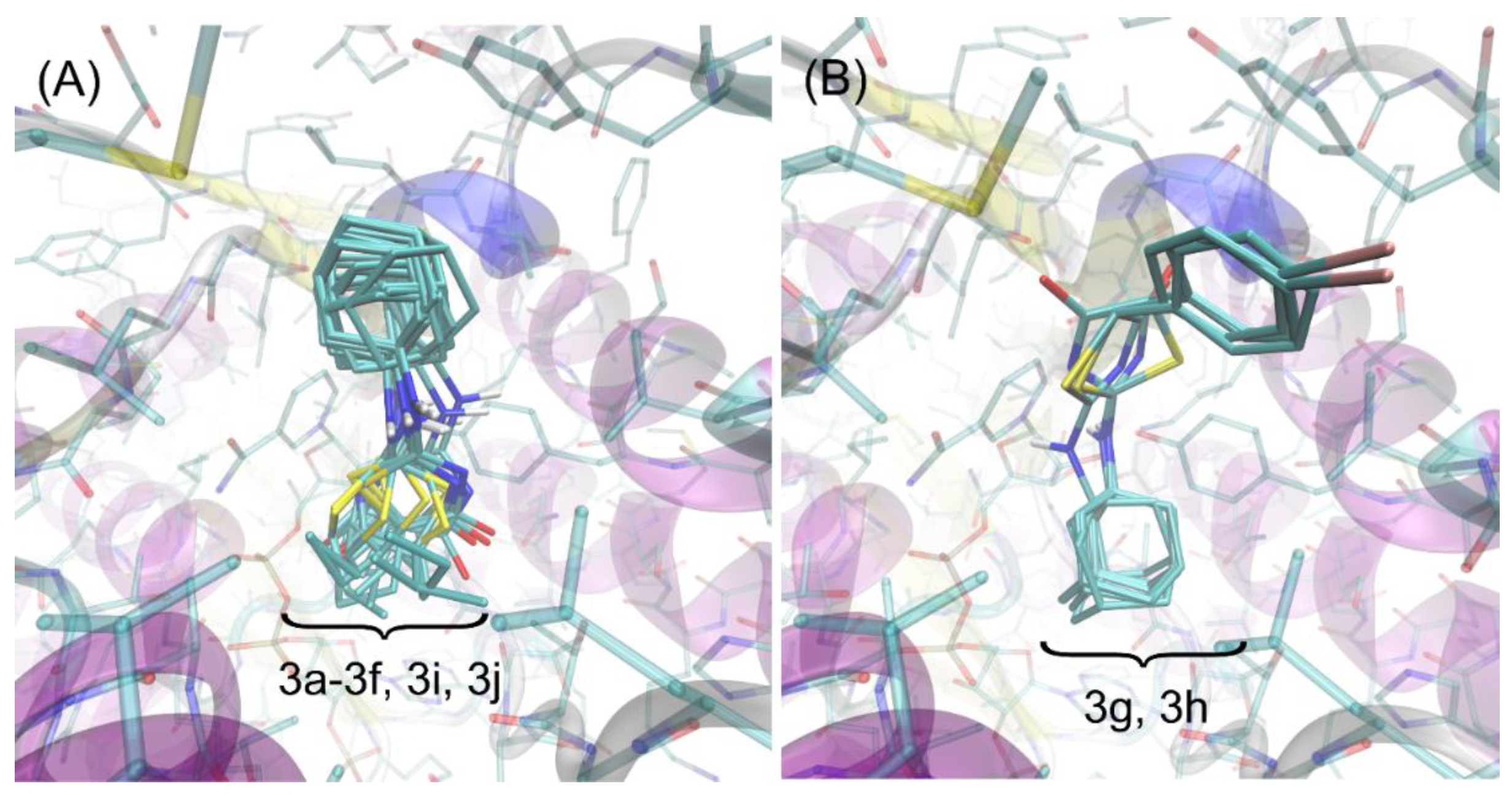
2.3. Molecular Docking
2.4. Bioavailability and Alerts for PAINS—In Silico Simulation
3. Materials and Methods
3.1. General Information
3.2. Reagents and Solvents
3.3. General Procedures of Synthesis
3.3.1. Method A (Synthesis of Compounds 3a–3d and 3g–3h)
3.3.2. Method B (Synthesis of Compounds 3e–3f)
3.3.3. Method C (Synthesis of Compounds 3i–3j)
3.4. Inhibition of 11β-HSD Assays
3.4.1. 11β-HSD1
3.4.2. 11β-HSD2
3.4.3. Determination of IC50
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Obesity and Overweight. Available online: https://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 15 June 2021).
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V. Obesity and dyslipidemia. Metabolism 2019, 92, 71–81. [Google Scholar] [CrossRef]
- Magliano, D.J.; Shaw, J.E.; Zimmet, P.Z. How to best define the metabolic syndrome. Ann. Med. 2006, 38, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Varughese, A.G.; Nimkevych, O.; Uwaifo, G.I. Hypercortisolism in obesity-associated hypertension. Curr. Hypertens. Rep. 2014, 16, 443. [Google Scholar] [CrossRef]
- Magiakou, M.A.; Smyrnaki, P.; Chrousos, G.P. Hypertension in Cushing’s syndrome. Best Pract. Res. 2006, 20, 467–482. [Google Scholar] [CrossRef] [PubMed]
- Peckett, A.J.; Wright, D.C.; Riddell, M.C. The effects of glucocorticoids on adipose tissue lipid metabolism. Metabolism 2011, 60, 1500–1510. [Google Scholar] [CrossRef] [PubMed]
- Baudrand, R.; Vaidya, A. Cortisol dysregulation in obesity-related metabolic disorders. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Stimson, R.H.; Walker, B.R. The role and regulation of 11β-hydroxysteroid dehydrogenase type 1 in obesity and the metabolic syndrome. Horm. Mol. Biol. Clin. Investig. 2013, 15, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Musiała, N.; Hołyńska-Iwan, I.; Olszewska-Słonina, D. Kortyzol—Nadzór nad ustrojem w fizjologii i stresie. Diagn. Lab. 2018, 54, 29–36. [Google Scholar] [CrossRef]
- Nagalski, A.; Kiersztan, A. Fizjologia i molekularny mechanizm działania glikokortykoidów. Postepy Hig. Med. Dośw. 2010, 64, 133–145. [Google Scholar]
- Draper, N.; Stewart, P.M. 11-Hydroxysteroid dehydrogenase and the pre-receptor regulation of corticosteroid hormone action. J. Endocrinol. 2005, 186, 251–271. [Google Scholar] [CrossRef] [Green Version]
- Wake, D.J.; Walker, B.R. Inhibition of 11beta-hydroxysteroid dehydrogenase type 1 in obesity. Endocrine 2006, 29, 101–108. [Google Scholar] [CrossRef]
- Stomby, A.; Andrew, R.; Walker, B.R.; Olsson, T. Tissue-specific dysregulation of cortisol regeneration by 11βHSD1 in obesity: Has it promised too much? Diabetologia 2014, 57, 1100–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulmyer-Lacroix, O.; Boullu, S.; Oliver, C.; Alessi, M.C.; Grino, M. Expression of the mRNA coding for 11beta-hydroxysteroid dehydrogenase type 1 in adipose tissue from obese patients: An in situ hybridization study. J. Clin. Endocrinol. Metab. 2002, 87, 2701–2705. [Google Scholar] [CrossRef] [Green Version]
- Dube, S.; Norby, B.J.; Pattan, V.; Carter, R.E.; Basu, A.; Basu, R. 11β-hydroxysteroid dehydrogenase types 1 and 2 activity in subcutaneous adipose tissue in humans: Implications in obesity and diabetes. J. Clin. Endocrinol. Metab. 2015, 100, E70–E76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, S.S.; Prashanth, A.; Kumar, C.P.; Reddy, S.J.; Giridharan, N.V.; Vajreswari, A. A novel genetically-obese rat model with elevated 11 beta-hydroxysteroid dehydrogenase type 1 activity in subcutaneous adipose tissue. Lipids Health Dis. 2010, 9, 132. [Google Scholar] [CrossRef] [Green Version]
- Schnackenberg, C.G.; Costell, M.H.; Krosky, D.J.; Cui, J.; Wu, C.W.; Hong, V.S.; Harpel, M.R.; Wilette, R.N.; Yue, T.-L. Chronic inhibition of 11 β -hydroxysteroid dehydrogenase type 1 activity decreases hypertension, insulin resistance, and hypertriglyceridemia in metabolic syndrome. Biomed. Res. Int. 2013, 2013, 427640. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Zhang, X.; Zhang, M. Inhibition of 11β-hydroxysteroid dehydrogenase type 1 ameliorates obesity-related insulin resistance. Biochem. Biophys. Res. Commun. 2016, 478, 474–480. [Google Scholar] [CrossRef]
- Parekh, N.M.; Juddhawala, K.V.; Rawal, B.M. Antimicrobial activity of thiazolyl benzenesulfonamide-condensed 2,4-thiazolidinediones derivatives. Med. Chem. Res. 2013, 22, 2737–2745. [Google Scholar] [CrossRef]
- Ibrahim, S.A.; Rizk, H.F. Synthesis and Biological Evaluation of Thiazole Derivatives. In Azoles: Synthesis, Properties, Applications and Perspectives; IntechOpen: London, UK, 2020. [Google Scholar] [CrossRef]
- Petrou, A.; Fesatidou, M.; Athina, A. Thiazole ring—A biologically active scaffold. Molecules 2021, 26, 3166. [Google Scholar] [CrossRef]
- Barf, T.; Vallgårda, J.; Edmont, R.; Häggström, C.; Kurz, G.; Nygren, A.; Larwood, V.; Mosialou, E.; Axelsson, K.; Olsson, R.; et al. Arylsulfonamidothiazoles as a new class of potential antidiabetic drugs. Discovery of potent and selective inhibitors of the 11β-hydroxysteroid dehydrogenase type 1. J. Med. Chem. 2002, 45, 3813–3815. [Google Scholar] [CrossRef] [PubMed]
- Fotsch, C.; Wang, M. Blockade of glucocorticoid excess at the tissue level: Inhibitors of 11β-hydroxysteroid dehydrogenase type 1 as a therapy for type 2 diabetes. J. Med. Chem. 2008, 51, 4852–4857. [Google Scholar] [CrossRef] [PubMed]
- Hale, C.; Véniant, M.; Wang, Z.; Chen, M.; McCormick, J.; Cupples, R.; Hickman, D.; Min, X.; Sudom, A.; Xu, H.; et al. Structural characterization and pharmacodynamic effects of an orally active 11β-hydroxysteroid dehydrogenase type 1 inhibitor. Chem. Biol. Drug Des. 2008, 71, 36–44. [Google Scholar] [CrossRef]
- St Jean, D.J., Jr.; Yuan, C.; Bercot, E.A.; Cupples, R.; Chen, M.; Fretland, J.; Hale, C.; Hungate, R.W.; Komorowski, R.; Veniant, M.; et al. 2-(S)-Phenethylaminothiazolones as potent, orally efficacious inhibitors of 11β-hydroxysteriod dehydrogenase type 1. J. Med. Chem. 2007, 50, 429–432. [Google Scholar] [CrossRef]
- Johansson, L.; Fotsch, C.; Bartberger, D.M.; Castro, V.M.; Chen, M.; Emery, M.; Gustafsson, S.; Hale, C.; Hickman, D.; Homan, E.; et al. 2-Amino-1,3-thiazol-4(5H)-ones as potent and selective 11β-hydroxysteroid dehydrogenase type 1 inhibitors: Enzyme-ligand Co-crystal structure and demonstration of pharmacodynamic effects in C57Bl/6 mice. J. Med. Chem. 2008, 51, 2933–2943. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; St Jean, D.J., Jr.; Liu, Q.; Cai, L.; Li, A.; Han, N.; Moniz, G.; Askew, B.; Hungate, R.W.; Johansson, L.; et al. The discovery of 2-anilinothiazolones as 11β-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 6056–6061. [Google Scholar] [CrossRef]
- Véniant, M.M.; Hale, C.; Hungate, R.W.; Gahm, K.; Emery, M.G.; Jona, J.; Joseph, S.; Adams, J.; Hague, A.; Moniz, G.; et al. Discovery of a potent, orally active 11β-hydroxysteroid dehydrogenase type 1 inhibitor for clinical study: Identification of (S)-2-((1S,2S,4R)-bicyclo[2.2.1]heptan-2-ylamino)-5-isopropyl-5-methylthiazol-4(5H)-one (AMG 221). J. Med. Chem. 2010, 53, 4481–4487. [Google Scholar] [CrossRef]
- Caille, S.; Cui, S.; Hwang, T.L.; Wang, X.; Faul, M.M. Two asymmetric syntheses of AMG 221, an inhibitor of 11β-hydroxysteroid dehydrogenase type 1. J. Org. Chem. 2009, 74, 3833–3842. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Kołodziejska, R.; Kupczyk, D.; Płaziński, W.; Kosmalski, T. A novel derivatives of thiazol 4(5H)one and their activity in the inhibition of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Chem. 2018, 79, 115–121. [Google Scholar] [CrossRef]
- Studzińska, R.; Kołodziejska, R.; Płaziński, W.; Kupczyk, D.; Kosmalski, T.; Jasieniecka, K.; Modzelewska-Banachiewicz, B. Synthesis of the N-methyl derivatives of 2-aminothiazol-4(5H)–one and their interactions with 11βHSD1 - molecular modeling and in vitro studies. Chem. Biodivers. 2019, 16, e1900065. [Google Scholar] [CrossRef] [PubMed]
- Kupczyk, D.; Studzińska, R.; Bilski, R.; Baumgart, S.; Kołodziejska, R.; Woźniak, A. Synthesis of novel 2-(isopropylamino)thiazol-4(5H)-one derivatives and their inhibitory activity of 11β-HSD1 and 11β-HSD2 in aspect of carcinogenesis prevention. Molecules 2020, 25, 4233. [Google Scholar] [CrossRef]
- Kupczyk, D.; Studzińska, R.; Baumgart, S.; Bilski, R.; Kosmalski, T.; Kołodziejska, R.; Woźniak, A. A novel N-tert-butyl derivatives of pseudothiohydantoin as potential target in anti-cancer therapy. Molecules 2021, 26, 2612. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Kupczyk, D.; Płazińska, A.; Kołodziejska, R.; Kosmalski, T.; Modzelewska-Banachiewicz, B. Thiazolo[3,2-a]pyrimidin-5-one derivatives as a novel class of 11β-hydroxysteroid dehydrogenase inhibitors. Bioorg. Chem. 2018, 81, 21–26. [Google Scholar] [CrossRef]
- Liu, J.; Obando, D.; Liao, V.; Lifa, T.; Codd, R. The many faces of the adamantyl group in drug design. Eur. J. Med. Chem. 2011, 46, 1949–1963. [Google Scholar] [CrossRef] [PubMed]
- Olson, S.; Aster, S.D.; Brown, K.; Carbin, L.; Graham, D.W.; Hermanowski-Vosatka, A.; LeGrand, C.B.; Mundt, S.S.; Robbins, M.A.; Schaeffer, J.M.; et al. Adamantyl triazoles as selective inhibitors of 11β-hydroxysteroid dehydrogenase type 1. Bioorg. Med. Chem. Lett. 2005, 15, 4359–4362. [Google Scholar] [CrossRef]
- Rohde, J.J.; Pliushchev, M.A.; Sorensen, B.K.; Wodka, D.; Shuai, Q.; Wang, J.; Fung, S.; Monzon, K.M.; Chiou, W.J.; Pan, L.; et al. Discovery and metabolic stabilization of potent and selective 2-amino-N-(adamant-2-yl) acetamide 11β-hydroxysteroid dehydrogenase type 1 inhibitors. J. Med. Chem. 2007, 50, 149–164. [Google Scholar] [CrossRef]
- Patel, J.R.; Shuai, Q.; Dinges, J.; Winn, M.; Pliushchev, M.; Fung, S.; Monzon, K.; Chiou, W.; Wang, J.; Pan, L.; et al. Discovery of adamantane ethers as inhibitors of 11β-HSD-1: Synthesis and biological evaluation. Bioorg. Med. Chem. Lett. 2007, 17, 750–755. [Google Scholar] [CrossRef]
- Sorensen, B.; Winn, M.; Rohde, J.; Shuai, Q.; Wang, J.; Fung, S.; Monzon, K.; Chiou, W.; Stolarik, D.; Imade, H.; et al. Adamantane sulfone and sulfonamide 11-β-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Hoffman, J.; Le, P.; Nair, S.K.; Cripps, S.; Matthews, J.; Smith, C.; Yang, M.; Stan Kupchinsky, S.; Dress, K.; et al. The development and SAR of pyrrolidine carboxamide 11β-HSD1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Pradaux-Caggiano, F.; Thomas, M.P.; Szeto, M.W.Y.; Halem, H.A.; Culler, M.D.; Vicker, N.; Potter, B.V.L. Discovery of adamantyl ethanone derivatives as potent 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors. ChemMedChem 2010, 5, 1026–1044. [Google Scholar] [CrossRef]
- Xiang-Yang, Y.; Chen, S.Y.; Wu, S.; Yoon, D.S.; Wang, H.; Hong, Z.; O’Connor, S.P.; Li, J.; Li, J.J.; Kennedy, L.J.; et al. Discovery of clinical candidate 2-((2S,6S)-2-phenyl-6-hydroxyadamantan-2-yl)-1-(3′-hydroxyazetidin-1-yl)ethanone [BMS-816336], an orally active novel selective 11β-hydroxysteroid dehydrogenase type 1 inhibitor. J. Med. Chem. 2017, 60, 4932–4948. [Google Scholar] [CrossRef]
- Goldberg, F.W.; Dossetter, A.G.; Scott, J.S.; Robb, G.R.; Boyd, S.; Groombridge, S.D.; Kemmitt, P.D.; Sjögren, T.; Gutierrez, P.M.; de Schoolmeester, J.; et al. Optimization of brain penetrant 11β-hydroxysteroid dehydrogenase type I inhibitors and in vivo testing in diet-induced obese mice. J. Med. Chem. 2014, 57, 970–986. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and premeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Molinspiration. Available online: https://www.molinspiration.com/ (accessed on 1 June 2021).
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. Available online: https://www.cbligand.org/PAINS/search_struct.php (accessed on 25 July 2021). [CrossRef] [Green Version]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. Available online: http://zinc15.docking.org/patterns/home/ (accessed on 25 July 2021). [CrossRef] [PubMed]
- FAF-Drugs4. Available online: https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py?form=FAF-Drugs4#forms::FAF-Drugs4 (accessed on 25 July 2021).
- Hitchcock, S.A. Blood–brain barrier permeability considerations forCNS-targeted compound library design. Curr. Opin. Cell Biol. 2008, 12, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Studzińska, R.; Karczmarska-Wódzka, A.; Kozakiewicz, A.; Kołodziejska, R.; Paprocka, R.; Wróblewski, M.; Augustyńska, B.; Modzelewska-Banachiewicz, B. 2-Allylaminothiazole and 2-allylaminodihydrothiazole derivatives: Synthesis, characterization and evaluation of bioactivity. Monatsh. Chem. 2015, 146, 1673–1679. [Google Scholar] [CrossRef]
- Kupczyk, D.; Studzińska, R.; Bilski, R.; Woźniak, A. Application of ELISA technique and human microsomes in the search for 11β-hydroxysteroid dehydrogenase inhibitors. Biomed. Res. Int. 2019, 5747436. [Google Scholar] [CrossRef]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminf. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Rappe, A.K.; Casewit, C.J.; Colwell, K.S.; Goddard, W.A.; Skiff, W.M. UFF, a full periodic table force field for molecular mechanics and molecular dynamics simulations. J. Am. Chem. Soc. 1992, 114, 10024–10035. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]





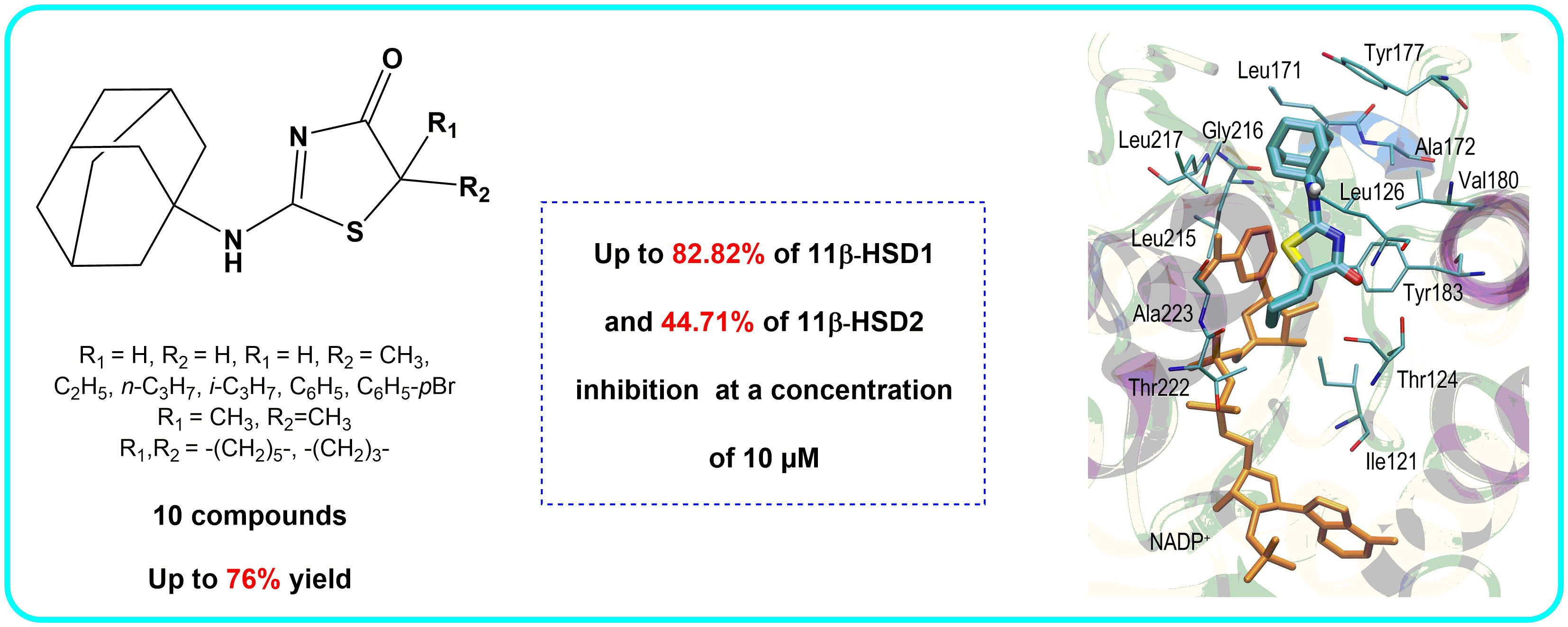{kind=link}
{kind=link}
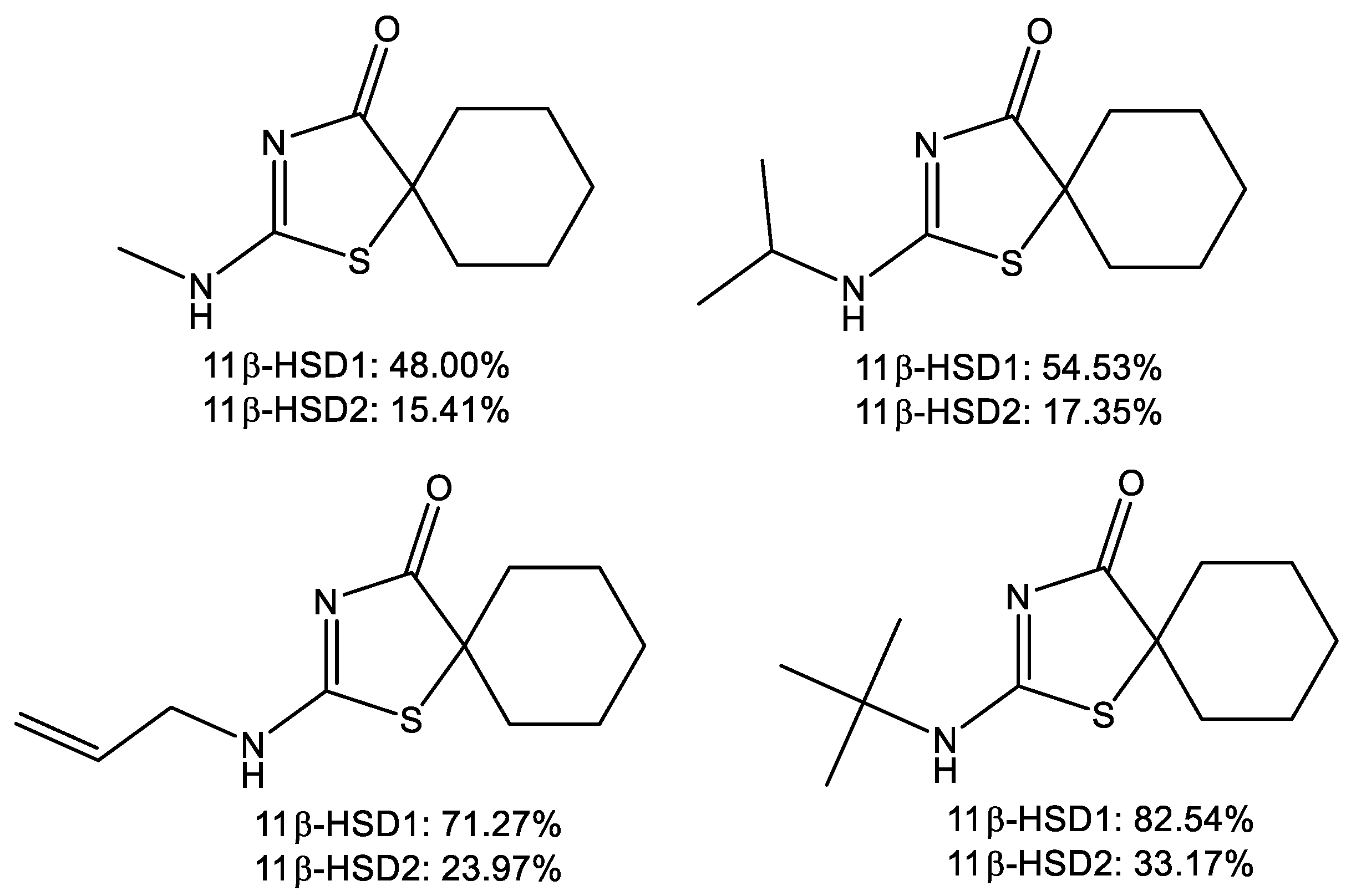{kind=link}
{kind=link}
{kind=link}

| No. | R1 | R2 | Procedure | Isolated Yield [%] | M.p. (°C) |
|---|---|---|---|---|---|
| 3a | H | H | A | 64.1 | 271–273 |
| 3b | H | CH3 | A | 75.8 | 265–267 |
| 3c | H | C2H5 | A | 66.1 | 257–259 |
| 3d | H | C3H7 | A | 60.1 | 247–249 |
| 3e | H | CH(CH3)2 | B | 18.7 | 239–241 |
| 3f | CH3 | CH3 | B | 15.8 | 208–210 |
| 3g | H | C6H5 | A | 66.2 | 252–254 |
| 3h | H | C6H5p-Br | A | 25.4 | 320 (dec.) |
| 3i | -(CH2)5- | C | 14.6 | 268–270 | |
| 3j | -(CH2)3- | C | 28.3 | 265–266 | |
| No. | R1 | R2 | % of 11β-HSD1 Inhibition 10 μM | IC50 11β-HSD1 [µM] | Binding Energy [kcal/mol] | % of 11β-HSD2 Inhibition 10 μM |
|---|---|---|---|---|---|---|
| 3a | H | H | 22.27 ± 5.31 | nd | −7.91 ± 0.20 | 29.81 ± 4.21 |
| 3b | H | CH3 | 62.15 ± 3.59 | 3.52 ± 0.19 | −8.48 ± 0.23 c | 44.71 ± 4.19 |
| 3c | H | C2H5 | 69.22 ± 5.12 | 5.44 ± 0.32 | −8.58 ± 0.24 c | 14.42 ± 1.16 |
| 3d | H | C3H7 | 72.37 ± 2.63 | 3.23 ± 0.26 | −8.63 ± 0.27 c | 28.85 ± 4.11 |
| 3e | H | CH(CH3)2 | 76.40 ± 2.55 | 1.19 ± 0.21 | −8.96 ± 0.31 c | 16.35 ± 2.06 |
| 3f | CH3 | CH3 | 65.99 ± 1.12 | 6.11 ± 0.62 | −8.46 ± 0.11 | 24.52 ± 3.24 |
| 3g | H | C6H5 | 46.32 ± 5.53 | nd | −9.99 ± 0.45 c | 18.27 ± 3.18 |
| 3h | H | C6H5p-Br | 15.30 ± 0.49 | nd | −10.14 ± 0.46 c | 41.83 ± 6.12 |
| 3i | -(CH2)5- | 82.82 ± 2.05 | 0.31 ± 0.05 | −10.04 ± 0.33 | 44.71 ± 3.37 | |
| 3j | -(CH2)3- | 74.13 ± 2.85 | 3.32 ± 0.24 | −8.98 ± 0.16 | 41.35 ± 4.22 | |
| control | - | 84.78 ± 6.23 a | 0.16 ± 0.15 a | - | 46.15 ± 3.16 b/ 55.77 ± 4.28 a | |
| Compound | miLogP | tPSA [A2] | Molecular Weight [g/mol] | nON | nOHNH | Number Rotatable Bonds | PAINS * |
|---|---|---|---|---|---|---|---|
| 3a | 2.64 | 41.46 | 250.37 | 3 | 1 | 2 | 0 |
| 3b | 3.00 | 41.46 | 264.39 | 3 | 1 | 2 | 0 |
| 3c | 3.51 | 41.46 | 278.42 | 3 | 1 | 3 | 0 |
| 3d | 4.07 | 41.46 | 292.45 | 3 | 1 | 4 | 0 |
| 3e | 3.75 | 41.46 | 292.45 | 3 | 1 | 3 | 0 |
| 3f | 3.45 | 41.46 | 278.42 | 3 | 1 | 2 | 0 |
| 3g | 4.22 | 41.46 | 326.46 | 3 | 1 | 3 | 0 |
| 3h | 5.03 | 41.46 | 405.36 | 3 | 1 | 3 | 0 |
| 3i | 4.62 | 41.46 | 318.49 | 3 | 1 | 2 | 0 |
| 3j | 3.37 | 41.46 | 290.43 | 3 | 1 | 2 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Studzińska, R.; Kupczyk, D.; Płaziński, W.; Baumgart, S.; Bilski, R.; Paprocka, R.; Kołodziejska, R. Novel 2-(Adamantan-1-ylamino)Thiazol-4(5H)-One Derivatives and Their Inhibitory Activity towards 11β-HSD1—Synthesis, Molecular Docking and In Vitro Studies. Int. J. Mol. Sci. 2021, 22, 8609. https://doi.org/10.3390/ijms22168609
Studzińska R, Kupczyk D, Płaziński W, Baumgart S, Bilski R, Paprocka R, Kołodziejska R. Novel 2-(Adamantan-1-ylamino)Thiazol-4(5H)-One Derivatives and Their Inhibitory Activity towards 11β-HSD1—Synthesis, Molecular Docking and In Vitro Studies. International Journal of Molecular Sciences. 2021; 22(16):8609. https://doi.org/10.3390/ijms22168609
Chicago/Turabian StyleStudzińska, Renata, Daria Kupczyk, Wojciech Płaziński, Szymon Baumgart, Rafał Bilski, Renata Paprocka, and Renata Kołodziejska. 2021. "Novel 2-(Adamantan-1-ylamino)Thiazol-4(5H)-One Derivatives and Their Inhibitory Activity towards 11β-HSD1—Synthesis, Molecular Docking and In Vitro Studies" International Journal of Molecular Sciences 22, no. 16: 8609. https://doi.org/10.3390/ijms22168609