
The Role of Mitochondrial Function in Peripheral Arterial Disease: Insights from Translational Studies

 , , ,
, , ,
Abstract
:1. Introduction
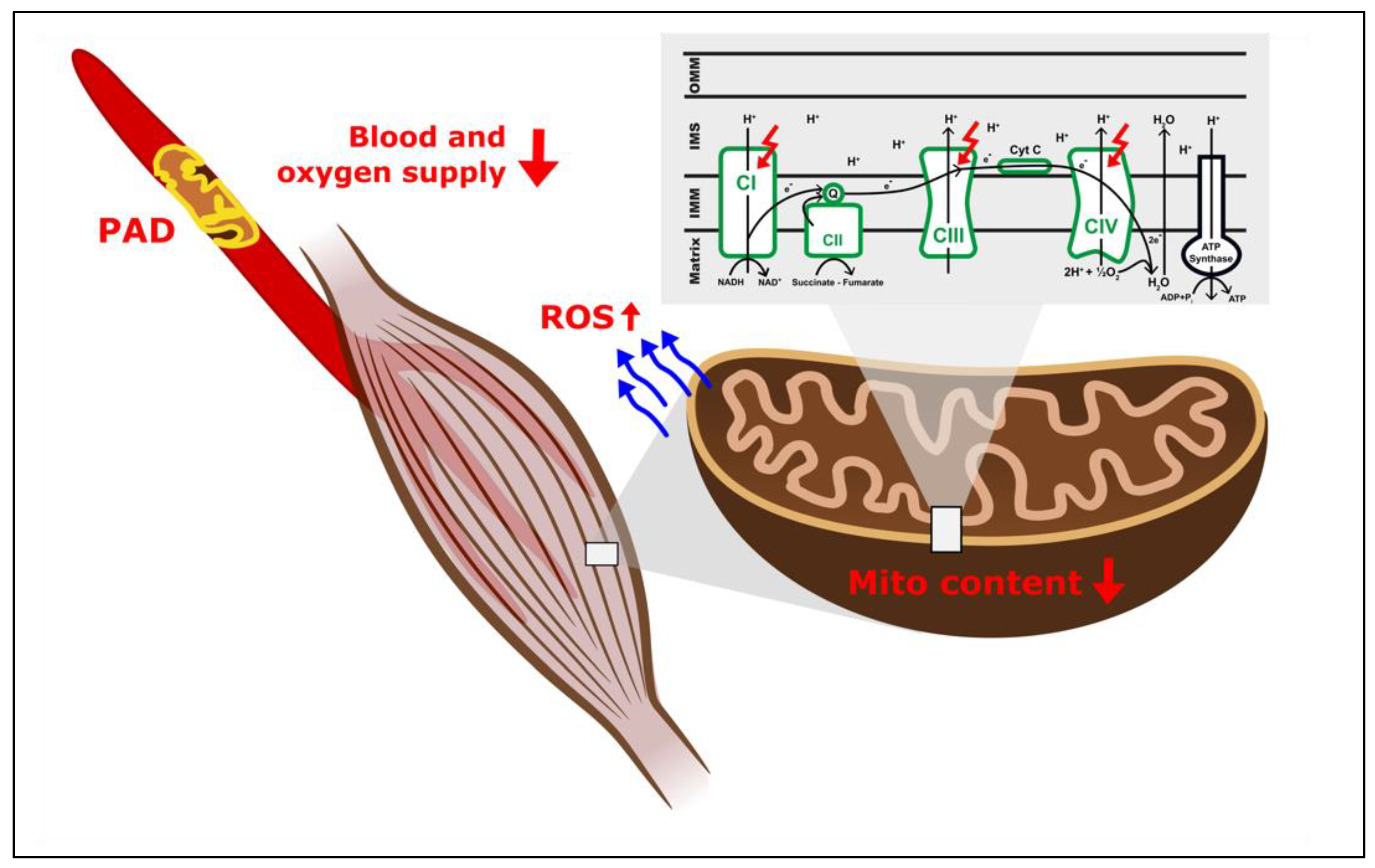
2. Do Mitochondria Play a Role in the Pathogenesis of Ischemic Myopathy in PAD?
3. Effects of Ischemia Reperfusion Injury on Muscle Mitochondria
4. Methods to Assess Mitochondrial Function in PAD
4.1. Near-Infrared Spectroscopy (NIRS)
4.2. 31-Phosphorus Magnetic Resonance Spectroscopy (31P MRS)
4.3. Respirometry
Assessment of Reactive Oxygen Species Production
5. Surrogate Markers to Evaluate Mitochondrial Content in PAD
6. Clinical Relevance of Mitochondrial Recovery in PAD
7. Summary and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Fowkes, F.G.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340. [Google Scholar] [CrossRef]
- Kengne, A.P.; Echouffo-Tcheugui, J.B. Differential burden of peripheral artery disease. Lancet Glob. Health 2019, 7, e980–e981. [Google Scholar] [CrossRef] [Green Version]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.; TASC II Working Group; Bell, K.; Caporusso, J.; Durand-Zaleski, I.; et al. Inter-society consensus for the management of peripheral arterial disease (Tasc Ii). Eur. J. Vasc. Endovasc. Surg. 2007, 33, S1–S75. [Google Scholar] [CrossRef] [Green Version]
- Sampson, U.K.; Norman, P.E.; Fowkes, F.G.; Aboyans, V.; Song, Y.; Harrell, F.E., Jr.; Forouzanfar, M.H.; Naghavi, M.; Denenberg, J.O.; McDermott, M.M.; et al. Estimation of global and regional incidence and prevalence of abdominal aortic aneurysms 1990 to 2010. Glob. Heart 2014, 9, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, R.; Kim, M.; Kieny, R. Surgical treatment of peripheral circulation disorders. Helv. Chir. Acta 1954, 21, 499–533. [Google Scholar] [PubMed]
- Aboyans, V.; Ricco, J.B.; Bartelink, M.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. Editor’s choice—2017 ESC guidelines on the diagnosis and treatment of peripheral arterial diseases, in collaboration with the European society for vascular surgery (ESVS). Eur. J. Vasc. Endovasc. Surg. 2018, 55, 305–368. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.W.; Killewich, L.A. Lack of functional benefits following infrainguinal bypass in peripheral arterial occlusive disease patients. Vasc. Med. 2001, 6, 9–14. [Google Scholar] [CrossRef]
- Regensteiner, J.G.; Hargarten, M.E.; Rutherford, R.B.; Hiatt, W.R. Functional benefits of peripheral vascular bypass surgery for patients with intermittent claudication. Angiology 1993, 44, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Pizzimenti, M.; Riou, M.; Charles, A.L.; Talha, S.; Meyer, A.; Andres, E.; Chakfé, N.; Lejay, A.; Geny, B. The rise of mitochondria in peripheral arterial disease physiopathology: Experimental and clinical data. J. Clin. Med. 2019, 8, 2125. [Google Scholar] [CrossRef] [Green Version]
- Pipinos, I.I.; Judge, A.R.; Selsby, J.T.; Zhu, Z.; Swanson, S.A.; Nella, A.A.; Dodd, S.L. The myopathy of peripheral arterial occlusive disease: Part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc. Endovasc. Surg. 2007, 41, 481–489. [Google Scholar] [CrossRef]
- Brass, E.P.; Hiatt, W.R. Acquired skeletal muscle metabolic myopathy in atherosclerotic peripheral arterial disease. Vasc. Med. 2000, 5, 55–59. [Google Scholar] [CrossRef]
- Vendelin, M.; Beraud, N.; Guerrero, K.; Andrienko, T.; Kuznetsov, A.V.; Olivares, J.; Kay, L.; Saks, V.A. Mitochondrial regular arrangement in muscle cells: A “crystal-like” pattern. Am. J. Physiol. Cell Physiol. 2005, 288, C757–C767. [Google Scholar] [CrossRef] [Green Version]
- Paradis, S.; Charles, A.L.; Meyer, A.; Lejay, A.; Scholey, J.W.; Chakfé, N.; Zoll, J.; Geny, B. Chronology of mitochondrial and cellular events during skeletal muscle ischemia-reperfusion. Am. J. Physiol. Cell Physiol. 2016, 310, C968–C982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farinon, A.M.; Marbini, A.; Gemignani, F.; Govoni, E.; Bragaglia, M.M.; Sianesi, M.; Tedeschi, F. Skeletal muscle and peripheral nerve changes caused by chronic arterial insufficiency—Significance and clinical correlations—Histological, histochemical and ultrastructural study. Clin. Neuropathol. 1984, 3, 240–252. [Google Scholar]
- Hedberg, B.; Angquist, K.A.; Sjöström, M. Peripheral arterial insufficiency and the fine structure of the gastrocnemius muscle. Int. Angiol. 1988, 7, 50–59. [Google Scholar] [PubMed]
- Pesta, D.; Paschke, V.; Hoppel, F.; Kobel, C.; Kremser, C.; Esterhammer, R.; Burtscher, M.; Kemp, G.J.; Schocke, M. Different metabolic responses during incremental exercise assessed by localized 31p mrs in sprint and endurance athletes and untrained individuals. Int. J. Sports Med. 2013, 34, 669–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greiner, A.; Esterhammer, R.; Bammer, D.; Messner, H.; Kremser, C.; Jaschke, W.R.; Fraedrich, G.; Schocke, M.F. High-energy phosphate metabolism in the calf muscle of healthy humans during incremental calf exercise with and without moderate cuff stenosis. Eur. J. Appl. Physiol. 2007, 99, 519–531. [Google Scholar] [CrossRef]
- Greiner, A.; Esterhammer, R.; Messner, H.; Biebl, M.; Mühlthaler, H.; Fraedrich, G.; Jaschke, W.R.; Schocke, M.F. High-energy phosphate metabolism during incremental calf exercise in patients with unilaterally symptomatic peripheral arterial disease measured by phosphor 31 magnetic resonance spectroscopy. J. Vasc. Surg. 2006, 43, 978–986. [Google Scholar] [CrossRef] [Green Version]
- Zatina, M.A.; Berkowitz, H.D.; Gross, G.M.; Maris, J.M.; Chance, B. 31p nuclear magnetic resonance spectroscopy: Noninvasive biochemical analysis of the ischemic extremity. J. Vasc. Surg. 1986, 3, 411–420. [Google Scholar] [CrossRef]
- Pipinos, I.I.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Boska, M.D. Phosphorus 31 nuclear magnetic resonance spectroscopy suggests a mitochondrial defect in claudicating skeletal muscle. J. Vasc. Surg. 2000, 31, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Pipinos, I.I.; Sharov, V.G.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A.; Todor, A.; Filis, K.A.; Sabbah, H.N. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J. Vasc. Surg. 2003, 38, 827–832. [Google Scholar] [CrossRef] [Green Version]
- Pipinos, I.I.; Judge, A.R.; Zhu, Z.; Selsby, J.T.; Swanson, S.A.; Johanning, J.M.; Baxter, B.T.; Lynch, T.G.; Dodd, S.L. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radic. Biol. Med. 2006, 41, 262–269. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am. Heart J. 2000, 139, S70–S85. [Google Scholar] [CrossRef] [PubMed]
- McLennan, H.R.; Degli Esposti, M. The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J. Bioenerg. Biomembr. 2000, 32, 153–162. [Google Scholar] [CrossRef]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [Green Version]
- Murphy, E.; Steenbergen, C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol. Rev. 2008, 88, 581–609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaeschke, H.; Woolbright, B.L. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transpl. Rev. 2012, 26, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, J.; Konstas, A.A.; Bateman, B.; Ortolano, G.A.; Pile-Spellman, J. Reperfusion injury following cerebral ischemia: Pathophysiology, MR imaging, and potential therapies. Neuroradiology 2007, 49, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burwell, L.S.; Nadtochiy, S.M.; Brookes, P.S. Cardioprotection by metabolic shut-down and gradual wake-up. J. Mol. Cell. Cardiol. 2009, 46, 804–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, M.D.; Reed, A.B.; Leuenberger, U.A.; Sinoway, L.I. Physiology in medicine: Peripheral arterial disease. J. Appl. Physiol. 2013, 115, 1219–1226. [Google Scholar] [CrossRef]
- Cornelis, N.; Chatzinikolaou, P.; Buys, R.; Fourneau, I.; Claes, J.; Cornelissen, V. The use of near infrared spectroscopy to evaluate the effect of exercise on peripheral muscle oxygenation in patients with lower extremity artery disease: A systematic review. Eur. J. Vasc. Endovasc. Surg. 2021, 61, 837–847. [Google Scholar] [CrossRef]
- Grassi, B.; Quaresima, V. Near-infrared spectroscopy and skeletal muscle oxidative function in vivo in health and disease: A review from an exercise physiology perspective. J. Biomed. Opt. 2016, 21, 091313. [Google Scholar] [CrossRef] [Green Version]
- Schocke, M.F.H.; Esterhammer, R.; Greiner, A. High-energy phosphate metabolism in the exercising muscle of patients with peripheral arterial disease. Vasa 2008, 37, 199–210. [Google Scholar] [CrossRef]
- Kemp, G.J.; Taylor, D.J.; Thompson, C.H.; Hands, L.J.; Rajagopalan, B.; Styles, P.; Radda, G.K. Quantitative analysis by 31p magnetic resonance spectroscopy of abnormal mitochondrial oxidation in skeletal muscle during recovery from exercise. NMR Biomed. 1993, 6, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Oliveira, P.J. Evaluation of respiration with clark type electrode in isolated mitochondria and permeabilized animal cells. Methods Mol. Biol. 2012, 810, 7–24. [Google Scholar] [PubMed]
- Clark, L.C., Jr.; Lyons, C. Electrode systems for continuous monitoring in cardiovascular surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Gnaiger, E. High-resolution respirometry: Oxphos protocols for human cells and permeabilized fibers from small biopsies of human muscle. Methods Mol. Biol. 2012, 810, 25–58. [Google Scholar]
- Stephens, J.W.; Khanolkar, M.P.; Bain, S.C. The biological relevance and measurement of plasma markers of oxidative stress in diabetes and cardiovascular disease. Atherosclerosis 2009, 202, 321–329. [Google Scholar] [CrossRef]
- Dikalov, S.I.; Harrison, D.G. Methods for detection of mitochondrial and cellular reactive oxygen species. Antioxid. Redox Signal. 2014, 20, 372–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makrecka-Kuka, M.; Krumschnabel, G.; Gnaiger, E. High-resolution respirometry for simultaneous measurement of oxygen and hydrogen peroxide fluxes in permeabilized cells, tissue homogenate and isolated mitochondria. Biomolecules 2015, 5, 1319–1338. [Google Scholar] [CrossRef] [Green Version]
- Starkov, A.A. Measurement of mitochondrial ROS production. In Protein Misfolding and Cellular Stress in Disease and Aging: Concepts and Protocols; Bross, P., Gregersen, N., Eds.; Humana Press: Totowa, NJ, USA, 2010; pp. 245–255. [Google Scholar]
- Eisner, V.; Picard, M.; Hajnóczky, G. Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat. Cell Biol. 2018, 20, 755–765. [Google Scholar] [CrossRef]
- Weibel, E.R. Stereological methods in cell biology: Where are we—Where are we going? J. Histochem. Cytochem. 1981, 29, 1043–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.; Nielsen, J.; Hansen, C.N.; Nielsen, L.B.; Wibrand, F.; Stride, N.; Schroder, H.D.; Boushel, R.; Helge, J.W.; Dela, F.; et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J. Physiol. 2012, 590, 3349–3360. [Google Scholar] [CrossRef]
- Groennebaek, T.; Billeskov, T.B.; Schytz, C.T.; Jespersen, N.R.; Bøtker, H.E.; Olsen, R.K.J.; Eldrup, N.; Nielsen, J.; Farup, J.; De Paoli, F.V.; et al. Mitochondrial structure and function in the metabolic myopathy accompanying patients with critical limb ischemia. Cells 2020, 9, 570. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.E.; Yamaguchi, D.J.; Schmidt, C.A.; Zeczycki, T.N.; Shaikh, S.R.; Brophy, P.; Green, T.D.; Tarpey, M.D.; Karnekar, R.; Goldberg, E.J.; et al. Extensive skeletal muscle cell mitochondriopathy distinguishes critical limb ischemia patients from claudicants. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Angquist, K.A.; Sjöström, M. Intermittent claudication and muscle fiber fine structure: Morphometric data on mitochondrial volumes. Ultrastruct. Pathol. 1980, 1, 461–470. [Google Scholar] [CrossRef]
- Thanigaimani, S.; Phie, J.; Sharma, C.; Wong, S.; Ibrahim, M.; Huynh, P.; Moxon, J.; Jones, R.; Golledge, J. Network meta-analysis comparing the outcomes of treatments for intermittent claudication tested in randomized controlled trials. J. Am. Heart Assoc. 2021, 10, e019672. [Google Scholar] [CrossRef] [PubMed]
- Schunk, K.; Romaneehsen, B.; Rieker, O.; Duber, C.; Kersjes, W.; Schadmand-Fischer, S.; Schmiedt, W.; Thelen, M. Dynamic phosphorus-31 magnetic resonance spectroscopy in arterial occlusive disease: Effects of vascular therapy on spectroscopic results. Investig. Radiol. 1998, 33, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Pipinos, I.I.; Boska, M.D.; Shepard, A.D.; Anagnostopoulos, P.V.; Katsamouris, A. Pentoxifylline reverses oxidative mitochondrial defect in claudicating skeletal muscle. J. Surg. Res. 2002, 102, 126–132. [Google Scholar] [CrossRef]
- Murphy, T.P.; Cutlip, D.E.; Regensteiner, J.G.; Mohler, E.R.; Cohen, D.J.; Reynolds, M.R.; Massaro, J.M.; Lewis, B.A.; Cerezo, J.; Oldenburg, N.C.; et al. Supervised exercise versus primary stenting for claudication resulting from aortoiliac peripheral artery disease: Six-month outcomes from the claudication: Exercise versus endoluminal revascularization (clever) study. Circulation 2012, 125, 130–139. [Google Scholar] [CrossRef] [Green Version]
- Holloszy, J.O.; Coyle, E.F. Adaptations of skeletal muscle to endurance exercise and their metabolic consequences. J. Appl. Physiol. Respir. Environ. Exerc. Physiol. 1984, 56, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.; Hoppel, F.; Macek, C.; Messner, H.; Faulhaber, M.; Kobel, C.; Parson, W.; Burtscher, M.; Schocke, M.; Gnaiger, E. Similar qualitative and quantitative changes of mitochondrial respiration following strength and endurance training in normoxia and hypoxia in sedentary humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 301, R1078–R1087. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.W.; Poehlman, E.T. Exercise rehabilitation programs for the treatment of claudication pain. A meta-analysis. JAMA 1995, 274, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Stewart, K.J.; Hiatt, W.R.; Regensteiner, J.G.; Hirsch, A.T. Exercise training for claudication. N. Engl. J. Med. 2002, 347, 1941–1951. [Google Scholar] [CrossRef]
- van Schaardenburgh, M.; Wohlwend, M.; Rognmo, Ø.; Mattsson, E.J.R. Exercise in claudicants increase or decrease walking ability and the response relates to mitochondrial function. J. Transl. Med. 2017, 15, 130. [Google Scholar] [CrossRef]
- Gratl, A.; Frese, J.; Speichinger, F.; Pesta, D.; Frech, A.; Omran, S.; Greiner, A. Regeneration of mitochondrial function in gastrocnemius muscle in peripheral arterial disease after successful revascularisation. Eur. J. Vasc. Endovasc. Surg. 2020, 59, 109–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gratl, A.; Pesta, D.; Gruber, L.; Speichinger, F.; Raude, B.; Omran, S.; Greiner, A.; Frese, J.P. The effect of revascularization on recovery of mitochondrial respiration in peripheral artery disease: A case control study. J. Transl. Med. 2021, 19, 244. [Google Scholar] [CrossRef]


{kind=link}
| Reference | Methods | PAD Severity | Outcome/Conclusion |
|---|---|---|---|
| Zatina et al. 1986 [19] | 31P MRS before and after revascularization; | Not specified; | 31P MRS successfully measures impaired bioenergetics of ischemic limbs during exercise and recovery; |
| Pipinos et al. 2000 [20] | 31P MRS before and after isometric exercise; | Stage IIB PAD; | ↑phosphocreatine and ADP recovery time constants in claudicating calf muscle; |
| Pipinos et al. 2002 [51] | 31P MRS before and after 12 weeks of interval exercise training; | Stage IIB PAD; | Pentoxifilline improves mitochondriopathy of claudicating muscle; |
| Pipinos et al. 2002 [21] | In-vitro respirometry; | Advanced PAD; | ↓mitochondrial respiration in patients suffering from advanced PAD; |
| Greiner et al. 2006 [18] | Serial 31P MRS during incremental exercise; | Symptomatic unilateral PAD; | ↑PCr recovery time after unilateral exercise in claudicating calf muscle; |
| Pipinos et al. 2006 [22] | Enzymatic activity measurement, in-vitro respirometry; | Severe PAD; | ↓mitochondrial respiration and enzymatic activities of complexes I, III, and IV in PAD; ↑oxidative stress biomarkers, ↓antioxidative enzymes; |
| Van Schaardenburgh et al. [57] | In-vitro respirometry; | Stage IIB PAD; | Changes in walking performances relate to changes in mitochondrial function after exercise; |
| Gratl et al. 2020 [58] | In-vitro respirometry; | Stage IIB/III PAD | Mitochondrial recovery after successful revascularization; |
| Gratl et al. 2021 [59] | In-vitro respirometry; | Stage IIB/III PAD | Restoration of blood supply is more important to mitochondrial recovery than increased physical activity |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gratl, A.; Wipper, S.; Frese, J.P.; Raude, B.; Greiner, A.; Pesta, D. The Role of Mitochondrial Function in Peripheral Arterial Disease: Insights from Translational Studies. Int. J. Mol. Sci. 2021, 22, 8478. https://doi.org/10.3390/ijms22168478
Gratl A, Wipper S, Frese JP, Raude B, Greiner A, Pesta D. The Role of Mitochondrial Function in Peripheral Arterial Disease: Insights from Translational Studies. International Journal of Molecular Sciences. 2021; 22(16):8478. https://doi.org/10.3390/ijms22168478
Chicago/Turabian StyleGratl, Alexandra, Sabine Wipper, Jan Paul Frese, Ben Raude, Andreas Greiner, and Dominik Pesta. 2021. "The Role of Mitochondrial Function in Peripheral Arterial Disease: Insights from Translational Studies" International Journal of Molecular Sciences 22, no. 16: 8478. https://doi.org/10.3390/ijms22168478