
Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis


Abstract
:1. Introduction
2. Multiple Sclerosis and Neuroinflammation
2.1. Neuroinflammation during the Course of MS
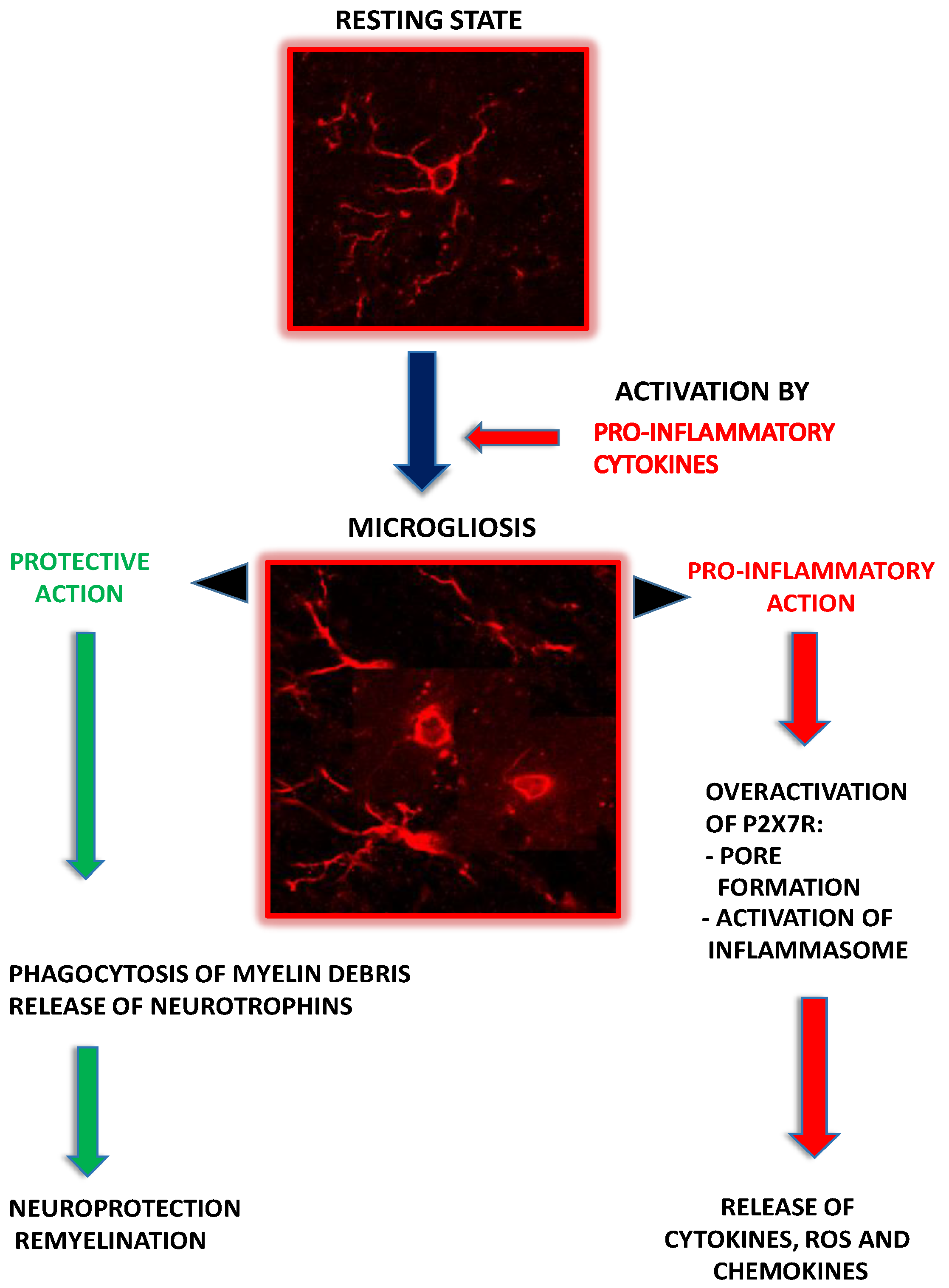
2.2. Microglia as Contributors to MS-Related Neuroinflammation
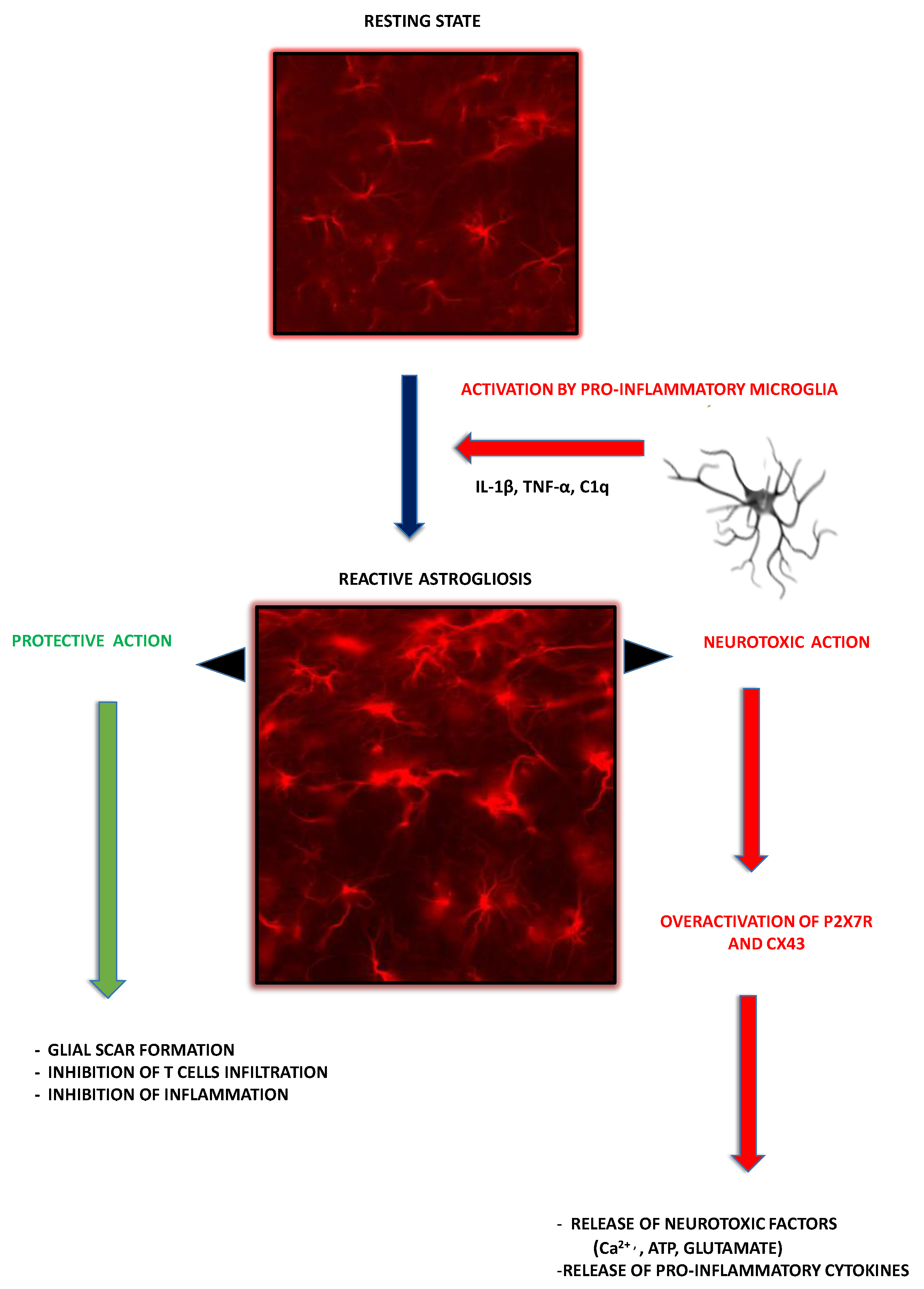
2.3. Astroglia as Contributors to MS-Related Neuroinflammation
3. P2X7R-Mediated Signaling in Glial Cells
3.1. Purinergic P2X7 Receptor—General Characteristic
3.2. P2X7R-Mediated Signaling in Microglia
3.3. P2X7R-Mediated Signaling in Astroglia
4. The Involvement of Glial P2X7R-Dependent Signaling into MS Pathology
4.1. Microglial P2X7R-Mediated Signaling in MS/EAE
4.2. Astroglial P2X7R
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Boissy, A.R.; Cohen, J.A. Multiple sclerosis symptom management. Expert Rev. Neurother. 2007, 7, 1213–1222. [Google Scholar] [CrossRef]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Illes, P. P2X7 receptors amplify cns damage in neurodegenerative diseases. Int. J. Mol. Sci. 2020, 21, 5996. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H. Multiple sclerosis pathology. Cold Spring Harb. Perspect. Med. 2018, 8, a028936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frischer, J.M.; Weigand, S.D.; Guo, Y.; Kale, N.; Parisi, J.E.; Pirko, I.; Mandrekar, J.; Bramow, S.; Metz, I.; Brück, W.; et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 2015, 78, 710–721. [Google Scholar] [CrossRef]
- Hayashi, T.; Morimoto, C.; Burks, J.S.; Kerr, C.; Hauser, S.L. Dual-label immunocytochemistry of the active multiple sclerosis lesion: Major histocompatibility complex and activation antigens. Ann. Neurol. 1988, 24, 523–531. [Google Scholar] [CrossRef]
- Franciotta, D.; Salvetti, M.; Lolli, F.; Serafini, B.; Aloisi, F. B cells and multiple sclerosis. Lancet Neurol. 2008, 7, 852–858. [Google Scholar] [CrossRef]
- Brosnan, C.F.; Bornstein, M.B.; Bloom, B.R. The effects of macrophage depletion on the clinical and pathologic expression of experimental allergic encephalomyelitis. J. Immunol. 1981, 126, 614–620. [Google Scholar]
- Ajami, B.; Bennett, J.L.; Krieger, C.; McNagny, K.M.; Rossi, F.M.V. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat. Neurosci. 2011, 14, 1142–1150. [Google Scholar] [CrossRef]
- Nally, F.K.; De Santi, C.; McCoy, C.E. Nanomodulation of Macrophages in Multiple Sclerosis. Cells 2019, 8, 543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, A.P.D.; Barnett, M.H.; Parratt, J.D.E.; Prineas, J.W. Multiple sclerosis: Distribution of inflammatory cells in newly forming lesions. Ann. Neurol. 2009, 66, 739–753. [Google Scholar] [CrossRef]
- Matthews, P.M. Chronic inflammation in multiple sclerosis—Seeing what was always there. Nat. Rev. Neurol. 2019, 15, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Kleinewietfeld, M.; Hafler, D.A. Regulatory T cells in autoimmune neuroinflammation. Immunol. Rev. 2014, 259, 231–244. [Google Scholar] [CrossRef]
- Barnett, M.H.; Prineas, J.W. Relapsing and Remitting Multiple Sclerosis: Pathology of the Newly Forming Lesion. Ann. Neurol. 2004, 55, 458–468. [Google Scholar] [CrossRef] [PubMed]
- Pegoretti, V.; Swanson, K.A.; Bethea, J.R.; Probert, L.; Eisel, U.L.M.; Fischer, R. Inflammation and Oxidative Stress in Multiple Sclerosis: Consequences for Therapy Development. Oxid. Med. Cell. Longev. 2020, 2020, 7191080. [Google Scholar] [CrossRef]
- Groom, A.J.; Smith, T.; Turski, L. Multiple sclerosis and glutamate. Ann. N. Y. Acad. Sci. 2003, 993, 229–275. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.; Peruzzotti-Jametti, L.; Bernstock, J.D.; Pluchino, S. The role of immune cells, glia and neurons in white and gray matter pathology in multiple sclerosis. Prog. Neurobiol. 2015, 127, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerrero, B.L.; Sicotte, N.L. Microglia in Multiple Sclerosis: Friend or Foe? Front. Immunol. 2020, 11, 374. [Google Scholar] [CrossRef]
- Monif, M.; Burnstock, G.; Williams, D.A. Microglia: Proliferation and activation driven by the P2X7 receptor. Int. J. Biochem. Cell Biol. 2010, 42, 1753–1756. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease HHS Public Access. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef]
- Lassmann, H.; Van Horssen, J.; Mahad, D. Progressive multiple sclerosis: Pathology and pathogenesis. Nat. Rev. Neurol. 2012, 8, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Voß, E.V.; Škuljec, J.; Gudi, V.; Skripuletz, T.; Pul, R.; Trebst, C.; Stangel, M. Characterisation of microglia during de- and remyelination: Can they create a repair promoting environment? Neurobiol. Dis. 2012, 45, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Jian, C.; Liao, Y.; Huang, Q.; Wu, Y.; Liu, X.; Zou, D.; Wu, Y. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 2017, 13, 1661–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napoli, I.; Neumann, H. Protective effects of microglia in multiple sclerosis. Exp. Neurol. 2010, 225, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Perry, V.H. Microglial physiology: Unique stimuli, specialized responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef]
- Shi, R. Polyethylene glycol repairs membrane damage and enhances functional recovery: A tissue engineering approach to spinal cord injury. Neurosci. Bull. 2013, 29, 460–466. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.-I.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2016, 36, 39–49. [Google Scholar] [CrossRef]
- Öhrfelt, A.; Axelsson, M.; Malmeström, C.; Novakova, L.; Heslegrave, A.; Blennow, K.; Lycke, J.; Zetterberg, H. Soluble TREM-2 in cerebrospinal fluid from patients with multiple sclerosis treated with natalizumab or mitoxantrone. Mult. Scler. J. 2016, 22, 1587–1595. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; Van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 2012, 135, 886–899. [Google Scholar] [CrossRef] [Green Version]
- Haider, L.; Zrzavy, T.; Hametner, S.; Höftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifenbring, S.; Brück, W.; Lassmann, H. The topograpy of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016, 139, 807–815. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, A.F.; Davies, C.L.; Miron, V.E. Microglia: Origins, homeostasis, and roles in myelin repair. Curr. Opin. Neurobiol. 2017, 47, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Grygorowicz, T.; Strużyńska, L. Early P2X7R-dependent activation of microglia during the asymptomatic phase of autoimmune encephalomyelitis. Inflammopharmacology 2019, 27, 129–137. [Google Scholar] [CrossRef] [Green Version]
- Heppner, F.L.; Greter, M.; Marino, D.; Falsig, J.; Raivich, G.; Hövelmeyer, N.; Waisman, A.; Rülicke, T.; Prinz, M.; Priller, J.; et al. Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat. Med. 2005, 11, 146–152. [Google Scholar] [CrossRef]
- Bhasin, M.; Wu, M.; Tsirka, S.E. Modulation of microglial/macrophage activation by macrophage inhibitory factor (TKP) or tuftsin (TKPR) attenuates the disease course of experimental autoimmune encephalomyelitis. BMC Immunol. 2007, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- O’Loughlin, E.; Madore, C.; Lassmann, H.; Butovsky, O. Microglial phenotypes and functions in multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018, 8, a028993. [Google Scholar] [CrossRef] [PubMed]
- Patsopoulos, N.A.; Baranzini, S.E.; Santaniello, A.; Shoostari, P.; Cotsapas, C.; Wong, G.; Beecham, A.H.; James, T.; Replogle, J.; Vlachos, I.S.; et al. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 2019, 365, eaav7188. [Google Scholar] [CrossRef] [Green Version]
- Cotrina, M.L.; Lin, J.H.C.; Alves-Rodrigues, A.; Liu, S.; Li, J.; Azmi-Ghadimi, H.; Kang, J.; Naus, C.C.G.; Nedergaard, M. Connexins regulate calcium signaling by controlling ATP release. Proc. Natl. Acad. Sci. USA 1998, 95, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfrieger, F.W.; Barres, B.A. Synaptic efficacy enhanced by glial cells in vitro. Science 1997, 277, 1684–1687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hertz, L.; Gibbs, M.E. What learning in day-old chickens can teach a neurochemist: Focus on astrocyte metabolism. J. Neurochem. 2009, 109, 10–16. [Google Scholar] [CrossRef]
- Perea, J.R.; López, E.; Díez-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular Monomeric Tau Is Internalized by Astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [Green Version]
- Morales, H.P.; Schousboe, A. Volume regulation in astrocytes: A role for taurine as an osmoeffector. J. Neurosci. Res. 1988, 20, 505–509. [Google Scholar] [CrossRef]
- Duan, S.; Anderson, C.M.; Keung, E.C.; Chen, Y.; Chen, Y.; Swanson, R.A. P2X7 receptor-mediated release of excitatory amino acids from astrocytes. J. Neurosci. 2003, 23, 1320–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parpura, V.; Basarsky, T.A.; Liu, F.; Jeftinija, K.; Jeftinija, S.; Haydon, P.G. Glutamate-mediated astrocyte-neuron signalling. Nature 1994, 369, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia HHS Public Access. Ted M Dawson 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Wang, L.Y.; Cai, W.Q.; Chen, P.H.; Deng, Q.Y.; Zhao, C.M. Downregulation of P2X7 receptor expression in rat oligodendrocyte precursor cells after hypoxia ischemia. Glia 2009, 57, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Shih, R.H.; Wang, C.Y.; Yang, C.M. NF-kappaB signaling pathways in neurological inflammation: A mini review. Front. Mol. Neurosci. 2015, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, C.S.; Amici, M.; Bortolotto, Z.A.; Doherty, A.; Csaba, Z.; Fafouri, A.; Dournaud, P.; Gressens, P.; Collingridge, G.L.; Peineau, S. The role of JAK-STAT signaling within the CNS. JAK-STAT 2013, 2, e22925. [Google Scholar] [CrossRef] [Green Version]
- Choi, W.H.; Ji, K.A.; Jeon, S.B.; Yang, M.S.; Kim, H.; Min, K.J.; Shong, M.; Jou, I.; Joe, E.H. Anti-inflammatory roles of retinoic acid in rat brain astrocytes: Suppression of interferon-γ-induced JAK/STAT phosphorylation. Biochem. Biophys. Res. Commun. 2005, 329, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, J.E.; Imura, T.; Song, B.; Qi, J.; Ao, Y.; Nguyen, T.K.; Korsak, R.A.; Takeda, K.; Akira, S.; Sofroniew, M.V. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J. Neurosci. 2008, 28, 7231–7243. [Google Scholar] [CrossRef]
- Haroon, F.; Drögemüller, K.; Händel, U.; Brunn, A.; Reinhold, D.; Nishanth, G.; Mueller, W.; Trautwein, C.; Ernst, M.; Deckert, M.; et al. Gp130-Dependent Astrocytic Survival Is Critical for the Control of Autoimmune Central Nervous System Inflammation. J. Immunol. 2011, 186, 6521–6531. [Google Scholar] [CrossRef]
- Sidoryk-Węgrzynowicz, M.; Struzyńska, L. Astroglial contribution to tau-dependent neurodegeneration. Biochem. J. 2019, 476, 3493–3504. [Google Scholar] [CrossRef] [PubMed]
- Torres, G.E.; Egan, T.M.; Voigt, M.M. Hetero-oligomeric assembly of P2X receptor subunits. Specificities exist with regard to possible partners. J. Biol. Chem. 1999, 274, 6653–6659. [Google Scholar] [CrossRef] [Green Version]
- Gordon, J.L. Extracellular ATP: Effects, sources and fate. Biochem. J. 1986, 233, 309–319. [Google Scholar] [CrossRef]
- Hattori, M.; Gouaux, E. Molecular mechanism of ATP binding and ion channel activation in P2X receptors. Nature 2012, 485, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Karasawa, A.; Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 2016, 5, e22153. [Google Scholar] [CrossRef] [PubMed]
- Coutinho-Silva, R.; Persechini, P.M. P2Z purinoceptor-associated pores induced by extracellular ATP in macrophages and J774 cells. Am. J. Physiol. Cell Physiol. 1997, 273, C1793–C1800. [Google Scholar] [CrossRef]
- Pelegrin, P.; Surprenant, A. Pannexin-1 mediates large pore formation and interleukin-1β release by the ATP-gated P2X7 receptor. EMBO J. 2006, 25, 5071–5082. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.P.; Qin, T.; Seidel, J.L.; Zheng, Y.; Eikermann, M.; Ferrari, M.D.; Van Den Maagdenberg, A.M.J.M.; Moskowitz, M.A.; Ayata, C.; Eikermann-Haerter, K. Inhibition of the P2X7-PANX1 complex suppresses spreading depolarization and neuroinflammation. Brain 2017, 140, 1643–1656. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Sanz, J.M.; Chiozzi, P.; Falzoni, S. The P2Z/P2X7 receptor of microglial cells: A novel immunomodulatory receptor. Prog. Brain Res. 1999, 120, 355–368. [Google Scholar] [CrossRef]
- Deuchars, S.A.; Atkinson, L.; Brooke, R.E.; Musa, H.; Milligan, C.J.; Batten, T.F.C.; Buckley, N.J.; Parson, S.H.; Deuchars, J. Neuronal P2X7 receptors are targeted to presynaptic terminals in the central and peripheral nervous systems. J. Neurosci. 2001, 21, 7143–7152. [Google Scholar] [CrossRef] [Green Version]
- Pannicke, T.; Fischer, W.; Biedermann, B.; Schädlich, H.; Grosche, J.; Faude, F.; Wiedemann, P.; Allgaier, C.; Illes, P.; Burnstock, G.; et al. P2X7 receptors in Muller glial cells from the human retina. J. Neurosci. 2000, 20, 5965–5972. [Google Scholar] [CrossRef] [Green Version]
- Colomar, A.; Amédée, T. ATP stimulation of P2X7 receptors activates three different ionic conductances on cultured mouse Schwann cells. Eur. J. Neurosci. 2001, 14, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Cotrina, M.L.; Lin, J.H.C.; López-García, J.C.; Naus, C.C.G.; Nedergaard, M. ATP-mediated glia signaling. J. Neurosci. 2000, 20, 2835–2844. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Koizumi, S.; Tsuda, M. The role of nucleotides in the neuron-glia communication responsible for the brain functions. J. Neurochem. 2007, 102, 1447–1458. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Vizi, E.S.; Wirkner, K.; Illes, P. P2X7 receptors in the nervous system. Prog. Neurobiol. 2006, 78, 327–346. [Google Scholar] [CrossRef] [PubMed]
- Sperlágh, B.; Illes, P. P2X7 receptor: An emerging target in central nervous system diseases. Trends Pharmacol. Sci. 2014, 35, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Yiangou, Y.; Facer, P.; Durrenberger, P.; Chessell, I.P.; Naylor, A.; Bountra, C.; Banati, R.R.; Anand, P. COX-2, CB2 and P2X7-immunoreactivities are increased in activated microglial cells/macrophages of multiple sclerosis and amyotrophic lateral sclerosis spinal cord. BMC Neurol. 2006, 6, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kobayashi, K.; Takahashi, E.; Miyagawa, Y.; Yamanaka, H.; Noguchi, K. Induction of the P2X7 receptor in spinal microglia in a neuropathic pain model. Neurosci. Lett. 2011, 504, 57–61. [Google Scholar] [CrossRef]
- Monif, M.; Reid, C.A.; Powell, K.L.; Smart, M.L.; Williams, D.A. The P2X7 receptor drives microglial activation and proliferation: A trophic role for P2X7R pore. J. Neurosci. 2009, 29, 3781–3791. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Ido, K.; Kohsaka, S.; Inoue, K.; Nakata, Y. Production and Release of Neuroprotective Tumor Necrosis Factor by P2X 7 Receptor-Activated Microglia. J. Neurosci. 2004, 24, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K. Microglial activation by purines and pyrimidines. Glia 2002, 40, 156–163. [Google Scholar] [CrossRef] [PubMed]
- la Sala, A.; Ferrari, D.; Di Virgilio, F.; Idzko, M.; Norgauer, J.; Girolomoni, G. Alerting and tuning the immune response by extracellular nucleotides. J. Leukoc. Biol. 2003, 73, 339–343. [Google Scholar] [CrossRef]
- Monif, M.; Reid, C.A.; Powell, K.L.; Drummond, K.J.; O’Brien, T.J.; Williams, D.A. Interleukin-1β has trophic effects in microglia and its release is mediated by P2X7R pore. J. Neuroinflamm. 2016, 13, 173. [Google Scholar] [CrossRef] [Green Version]
- Illes, P.; Rubini, P.; Ulrich, H.; Zhao, Y.; Tang, Y. Regulation of Microglial Functions by Purinergic Mechanisms in the Healthy and Diseased CNS. Cells 2020, 9, 1108. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Mingam, R.; De Smedt, V.; Amédée, T.; Bluthé, R.M.; Kelley, K.W.; Dantzer, R.; Layé, S. In vitro and in vivo evidence for a role of the P2X7 receptor in the release of IL-1β in the murine brain. Brain. Behav. Immun. 2008, 22, 234–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korcok, J.; Raimundo, L.N.; Ke, H.Z.; Sims, S.M.; Dixon, S.J. Extracellular nucleotides act through P2X7 receptors to activate NF-κB in osteoclasts. J. Bone Miner. Res. 2004, 19, 642–651. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.D.; Soltoff, S.P. P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem. J. 2002, 366, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Shiratori, M.; Tozaki-Saitoh, H.; Yoshitake, M.; Tsuda, M.; Inoue, K. P2X7 receptor activation induces CXCL2 production in microglia through NFAT and PKC/MAPK pathways. J. Neurochem. 2010, 114, 810–819. [Google Scholar] [CrossRef]
- Luo, Y.; Fischer, F.R.; Hancock, W.W.; Dorf, M.E. Macrophage Inflammatory Protein-2 and KC Induce Chemokine Production by Mouse Astrocytes. J. Immunol. 2000, 165, 4015–4023. [Google Scholar] [CrossRef] [Green Version]
- Butt, A.M. ATP: A ubiquitous gliotransmitter integrating neuron—Glial networks. Semin. Cell Dev. Biol. 2011, 22, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Fields, R.D.; Burnstock, G. Purinergic signalling in neuron—Glia interactions. Nat. Rev. Neurosci. 2006, 7, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2018, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Abbracchio, M.P.; Burnstock, G.; Verkhratsky, A.; Zimmermann, H. Purinergic signalling in the nervous system: An overview. Trends Neurosci. 2009, 32, 19–29. [Google Scholar] [CrossRef]
- Fellin, T.; Pozzan, T.; Carmignoto, G. Purinergic receptors mediate two distinct glutamate release pathways in hippocampal astrocytes. J. Biol. Chem. 2006, 281, 4274–4284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, L.; Dinh, T.; Stella, N. ATP induces a rapid and pronounced increase in 2-arachidonoylglycerol production by astrocytes, a response limited by monoacylglycerol lipase. J. Neurosci. 2004, 24, 8068–8074. [Google Scholar] [CrossRef] [Green Version]
- Murakami, K.; Nakamura, Y.; Yoneda, Y. Potentiation by ATP of lipopolysaccharide-stimulated nitric oxide production in cultured astrocytes. Neuroscience 2003, 117, 37–42. [Google Scholar] [CrossRef]
- Gendron, F.P.; Neary, J.T.; Theiss, P.M.; Sun, G.Y.; Gonzalez, F.A.; Weisman, G.A. Mechanisms of P2X7 receptor-mediated ERK1/2 phosphorylation in human astrocytoma cells. Am. J. Physiol. Cell Physiol. 2003, 284, C571–C581. [Google Scholar] [CrossRef]
- Jacques-Silva, M.C.; Rodnight, R.; Lenz, G.; Liao, Z.; Kong, Q.; Tran, M.; Kang, Y.; Gonzalez, F.A.; Weisman, G.A.; Neary, J.T. P2X 7 receptors stimulate AKT phosphorylation in astrocytes. Br. J. Pharmacol. 2004, 141, 1106–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucher, B.M.; Neary, J.T. Bi-functional effects of ATP/P2 receptor activation on tumor necrosis factor-alpha release in lipopolysaccharide-stimulated astrocytes. J. Neurochem. 2005, 92, 525–535. [Google Scholar] [CrossRef]
- Potolicchio, I.; Chitta, S.; Xu, X.; Fonseca, D.; Crisi, G.; Horejsi, V.; Strominger, J.L.; Stern, L.J.; Raposo, G.; Santambrogio, L. Conformational Variation of Surface Class II MHC Proteins during Myeloid Dendritic Cell Differentiation Accompanies Structural Changes in Lysosomal MIIC. J. Immunol. 2005, 175, 4935–4947. [Google Scholar] [CrossRef]
- Lee, M.H.; Lee, S.J.; Choi, H.J.; Jung, Y.W.; Frøkiær, J.; Nielsen, S.; Kwon, T.H. Regulation of AQP4 protein expression in rat brain astrocytes: Role of P2X7 receptor activation. Brain Res. 2008, 1195, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kamatsuka, Y.; Ohishi, A.; Nishida, K.; Nagasawa, K. P2X7 receptors regulate engulfing activity of non-stimulated resting astrocytes. Biochem. Biophys. Res. Commun. 2013, 439, 90–95. [Google Scholar] [CrossRef]
- Gu, B.J.; Saunders, B.M.; Jursik, C.; Wiley, J.S. The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood 2010, 115, 1621–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasawa, K.; Escartin, C.; Swanson, R.A. Astrocyte cultures exhibit P2X7 receptor channel opening in the absence of exogenous ligands. Glia 2009, 57, 622–633. [Google Scholar] [CrossRef]
- Verkhrasky, A.; Krishtal, O.A.; Burnstock, G. Purinoceptors on neuroglia. Mol. Neurobiol. 2009, 39, 190–208. [Google Scholar] [CrossRef] [PubMed]
- Peña-Altamira, L.E.; Polazzi, E.; Giuliani, P.; Beraudi, A.; Massenzio, F.; Mengoni, I.; Poli, A.; Zuccarini, M.; Ciccarelli, R.; Di Iorio, P.; et al. Release of soluble and vesicular purine nucleoside phosphorylase from rat astrocytes and microglia induced by pro-inflammatory stimulation with extracellular ATP via P2X7 receptors. Neurochem. Int. 2018, 115, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Matute, C.; Torre, I.; Pérez-Cerdá, F.; Pérez-Samartín, A.; Alberdi, E.; Etxebarria, E.; Arranz, A.M.; Ravid, R.; Rodríguez-Antigüedad, A.; Sánchez-Gómez, M.V.; et al. P2X7 receptor blockade prevents ATP excitotoxicity in oligodendrocytes and ameliorates experimental autoimmune encephalomyelitis. J. Neurosci. 2007, 27, 9525–9533. [Google Scholar] [CrossRef] [PubMed]
- Amadio, S.; Parisi, C.; Piras, E.; Fabbrizio, P.; Apolloni, S.; Montilli, C.; Luchetti, S.; Ruggieri, S.; Gasperini, C.; Laghi-Pasini, F.; et al. Modulation of P2X7 receptor during inflammation in multiple sclerosis. Front. Immunol. 2017, 8, 1529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, A.J.; Polak, P.E.; Simonini, V.; Lin, S.X.; Richardson, J.C.; Bongarzone, E.R.; Feinstein, D.L. P2x7 deficiency suppresses development of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2008, 5, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, P.A.; Merkler, D.; Funkner, P.; Shaabani, N.; Meryk, A.; Krings, C.; Barthuber, C.; Recher, M.; Brück, W.; Häussinger, D.; et al. Oxidized ATP inhibits T-cell-mediated autoimmunity. Eur. J. Immunol. 2010, 40, 2401–2408. [Google Scholar] [CrossRef]
- Chen, L.; Brosnan, C.F. Exacerbation of Experimental Autoimmune Encephalomyelitis in P2X 7 R −/− Mice: Evidence for Loss of Apoptotic Activity in Lymphocytes. J. Immunol. 2006, 176, 3115–3126. [Google Scholar] [CrossRef] [Green Version]
- Bernal-Chico, A.; Manterola, A.; Cipriani, R.; Katona, I.; Matute, C.; Mato, S. P2x7 receptors control demyelination and inflammation in the cuprizone model. Brain Behav. Immun. Health 2020, 4, 100062. [Google Scholar] [CrossRef]
- Grygorowicz, T.; Struzyńska, L.; Sulkowski, G.; Chalimoniuk, M.; Sulejczak, D. Temporal expression of P2X7 purinergic receptor during the course of experimental autoimmune encephalomyelitis. Neurochem. Int. 2010, 57, 823–829. [Google Scholar] [CrossRef]
- Kerstetter, A.E.; Padovani-Claudio, D.A.; Bai, L.; Miller, R.H. Inhibition of CXCR2 signaling promotes recovery in models of multiple sclerosis. Exp. Neurol. 2009, 220, 44–56. [Google Scholar] [CrossRef] [Green Version]
- Simpson, J.E.; Newcombe, J.; Cuzner, M.L.; Woodroofe, M.N. Expression of monocyte chemoattractant protein-1 and other β- chemokines by resident glia and inflammatory cells in multiple sclerosis lesions. J. Neuroimmunol. 1998, 84, 238–249. [Google Scholar] [CrossRef]
- Grygorowicz, T.; Wełniak-Kamińska, M.; Struzyńska, L. Early P2X7R-related astrogliosis in autoimmune encephalomyelitis. Mol. Cell. Neurosci. 2016, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Brambilla, R.; D’Ambrosi, N.; Volonté, C.; Matteoli, M.; Verderio, C.; Abbracchio, M.P. Nucleotide-mediated calcium signaling in rat cortical astrocytes: Role of P2X and P2Y receptors. Glia 2003, 43, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, N.; Vayro, S.; Kirchhoff, F.; Verkhratsky, A.; Robbins, J.; Gorecki, D.C.; Butt, A.M. Mechanisms of ATP- and glutamate-mediated calcium signaling in white matter astrocytes. Glia 2008, 56, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Bazargani, N.; Attwell, D. Astrocyte calcium signaling: The third wave. Nat. Neurosci. 2016, 19, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Bijelić, D.D.; Milićević, K.D.; Lazarević, M.N.; Miljković, D.M.; Pristov, J.J.B.; Savić, D.Z.; Petković, B.B.; Andjus, P.R.; Momčilović, M.B.; Nikolić, L.M. Central nervous system-infiltrated immune cells induce calcium increase in astrocytes via astroglial purinergic signaling. J. Neurosci. Res. 2020, 98, 2317–2332. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N. Inflammatory mediators and modulation of blood—Brain barrier permeability. Cell. Mol. Neurobiol. 2000, 20, 131–147. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| P2X7 Targeting Approach | Model | Effect | Reference |
|---|---|---|---|
| Brilliant blue G (antagonist) | Acute EAE rats | Reduced onset of the disease; reduced astrogliosis and microglia proliferation | [35] [109] |
| Brilliant blue G (antagonist) | Chronic EAE mice | Reduced demyelination; ameliorated neurological abnormalities | [103] |
| Oxidized ATP (oxATP antagonist) | Chronic EAE mice | Inhibition of clinical symptoms and demyelination; reduce antigen T cell | [106] |
| Cuprizone (demyelination inducer) | P2X7 null mice | Inhibition of astrogliosis and microglia activation | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidoryk-Węgrzynowicz, M.; Strużyńska, L. Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis. Int. J. Mol. Sci. 2021, 22, 8404. https://doi.org/10.3390/ijms22168404
Sidoryk-Węgrzynowicz M, Strużyńska L. Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis. International Journal of Molecular Sciences. 2021; 22(16):8404. https://doi.org/10.3390/ijms22168404
Chicago/Turabian StyleSidoryk-Węgrzynowicz, Marta, and Lidia Strużyńska. 2021. "Astroglial and Microglial Purinergic P2X7 Receptor as a Major Contributor to Neuroinflammation during the Course of Multiple Sclerosis" International Journal of Molecular Sciences 22, no. 16: 8404. https://doi.org/10.3390/ijms22168404