
Drosophila Larval Models of Invasive Tumorigenesis for In Vivo Studies on Tumour/Peripheral Host Tissue Interactions during Cancer Cachexia

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
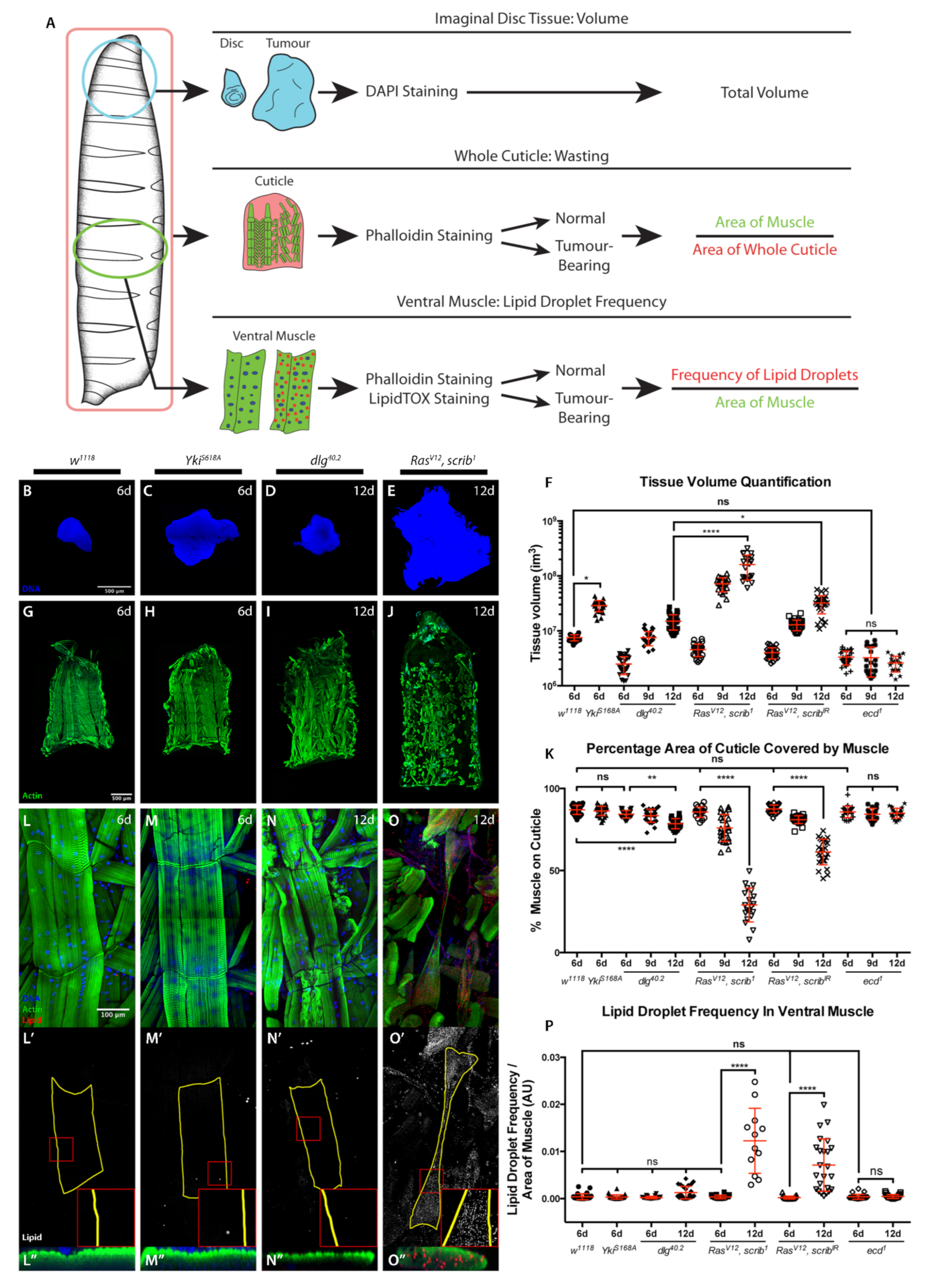
2.1. Cachexia Is Dependent on Tumour Genotype
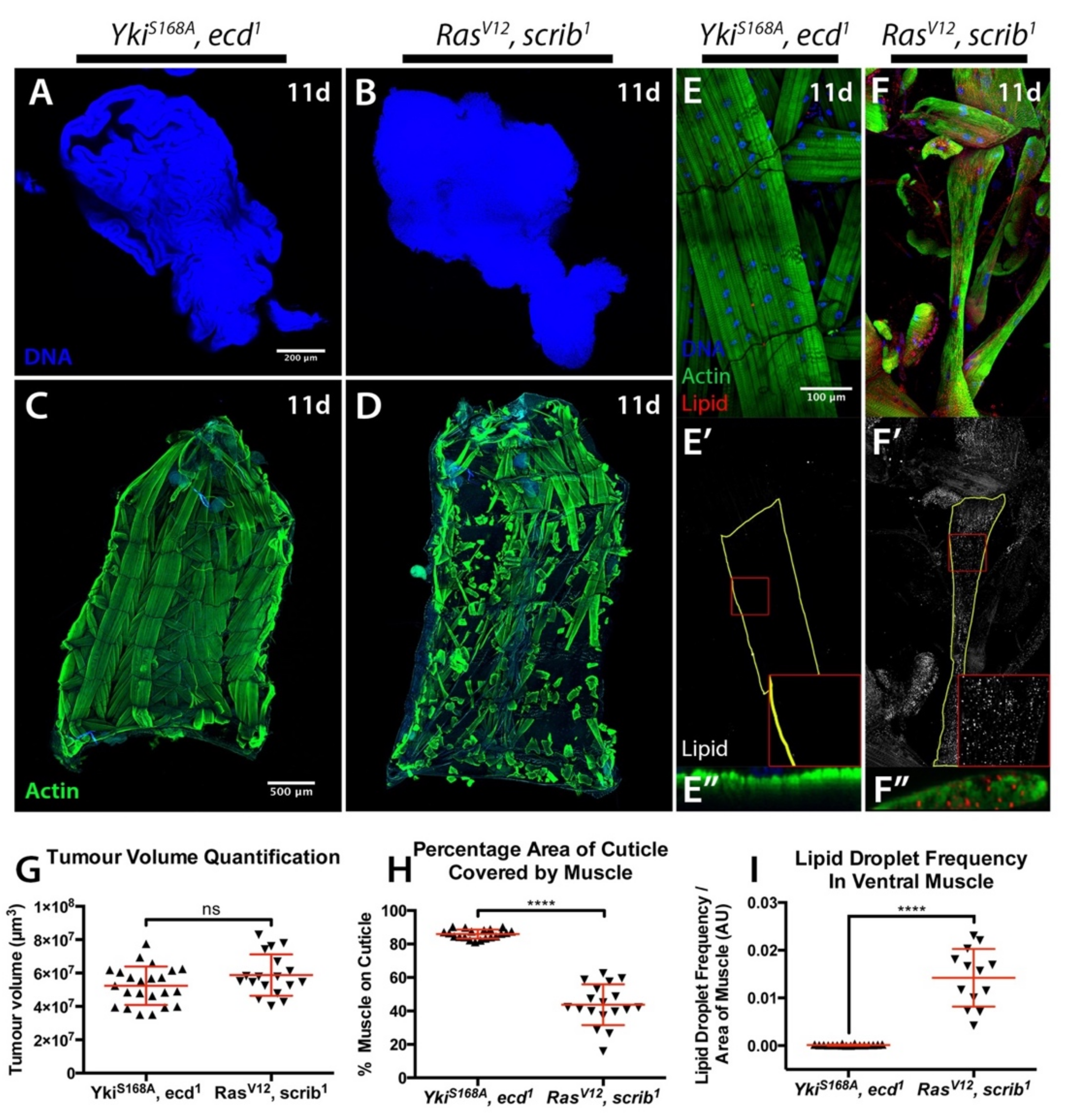
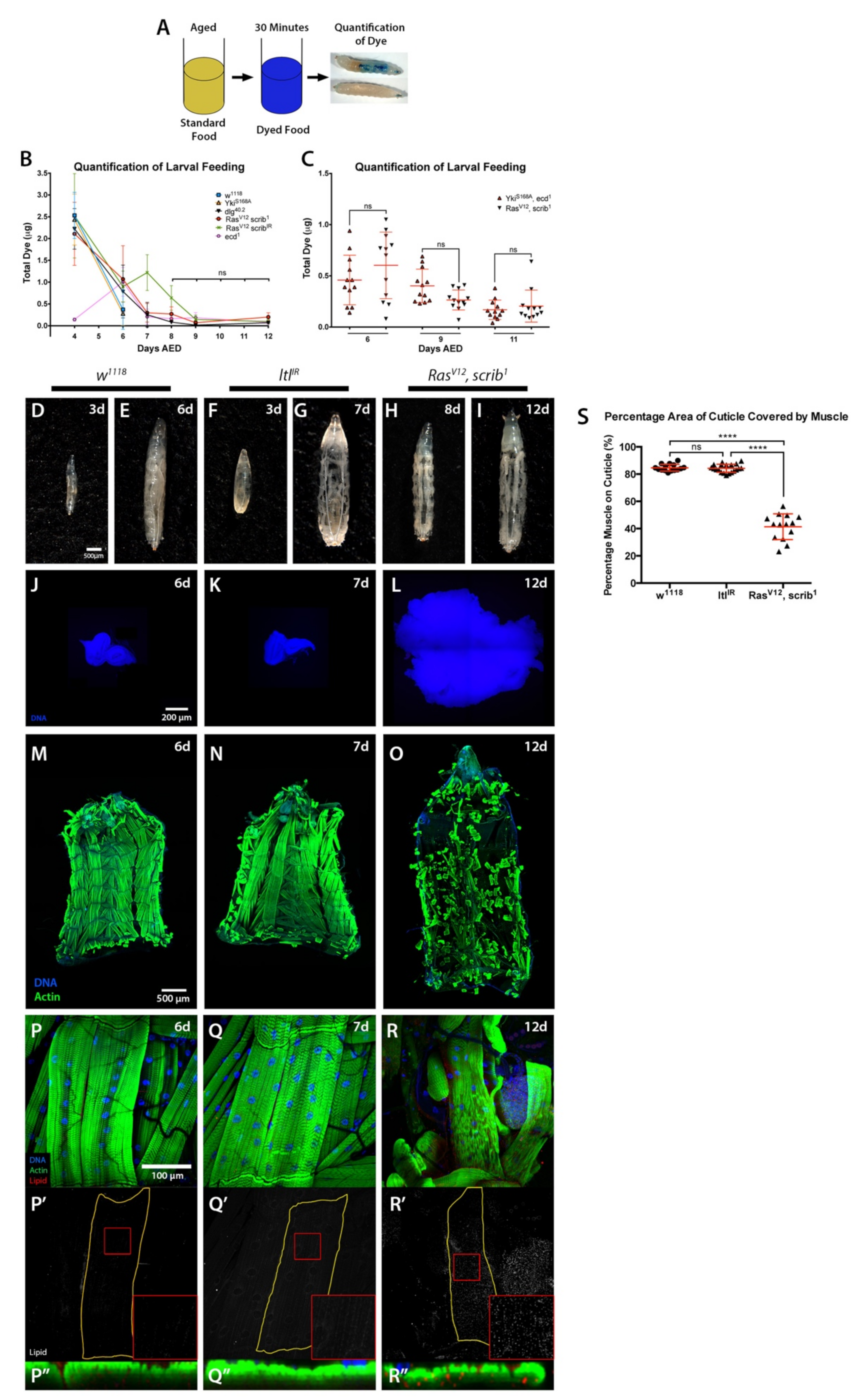
2.2. Starvation and Liquid Retention Are Not Sufficient to Drive Cachexia
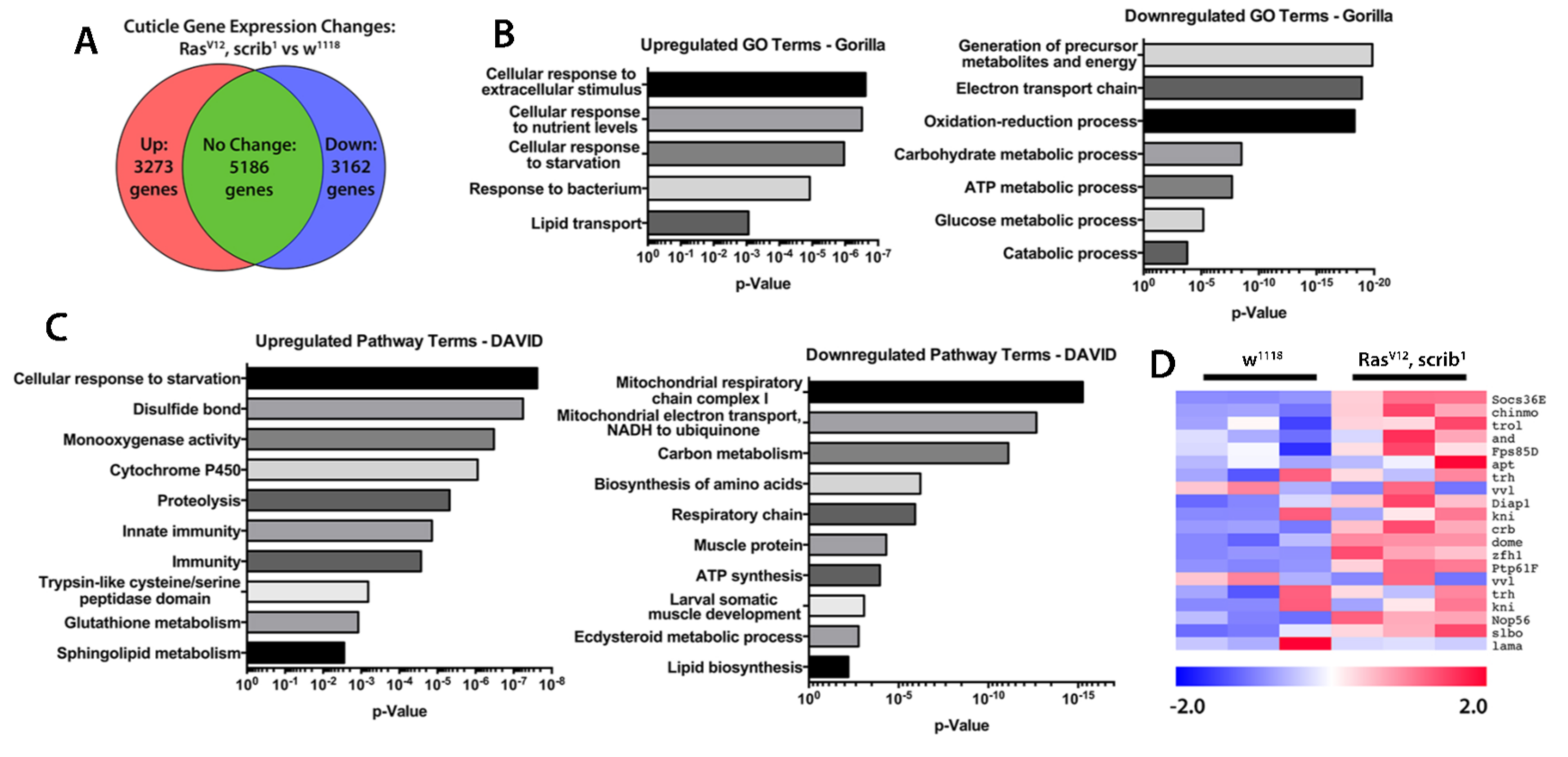
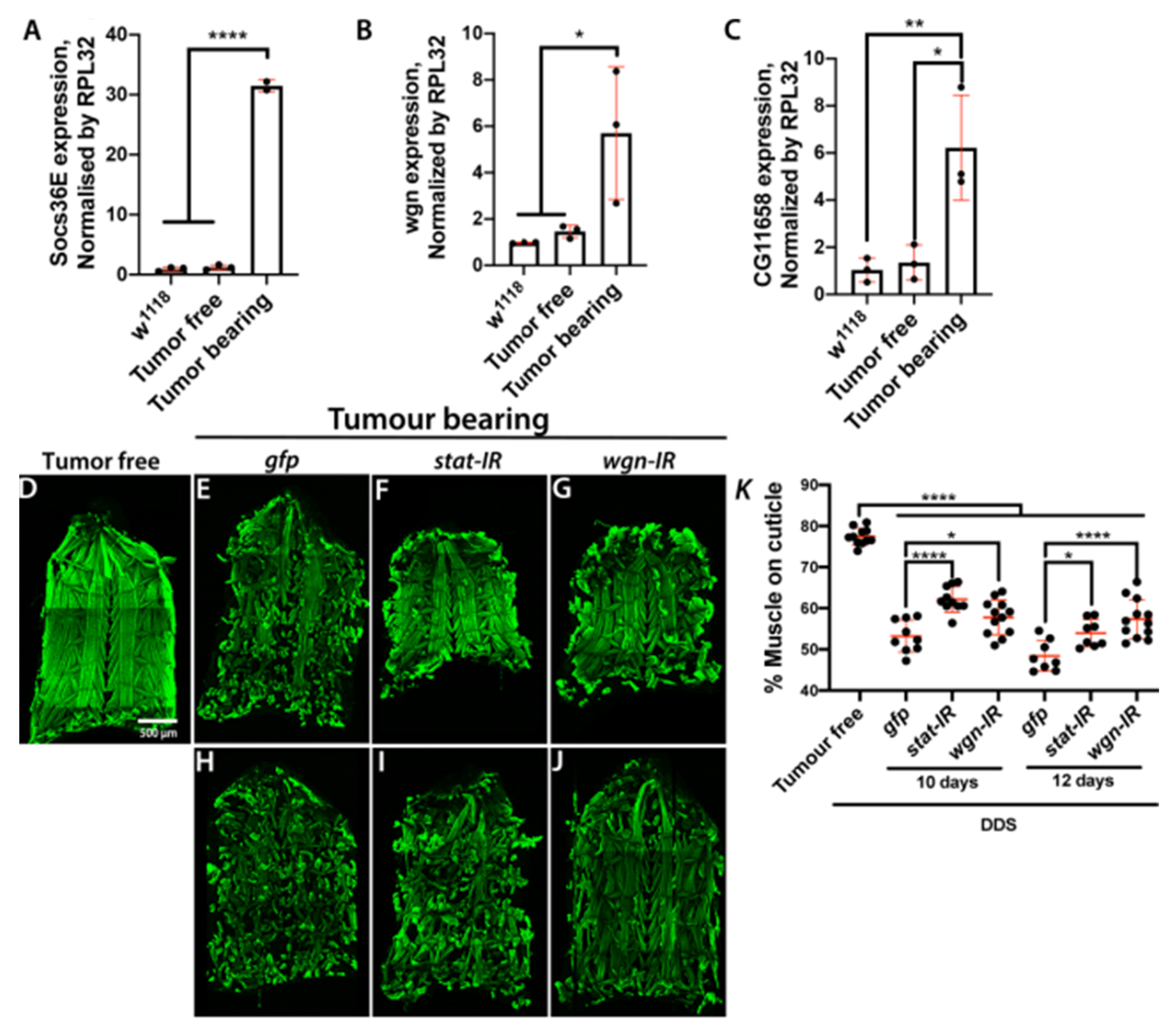
2.3. Inflammation and JAK/STAT Signalling Is Elevated in Cachectic Larval Models
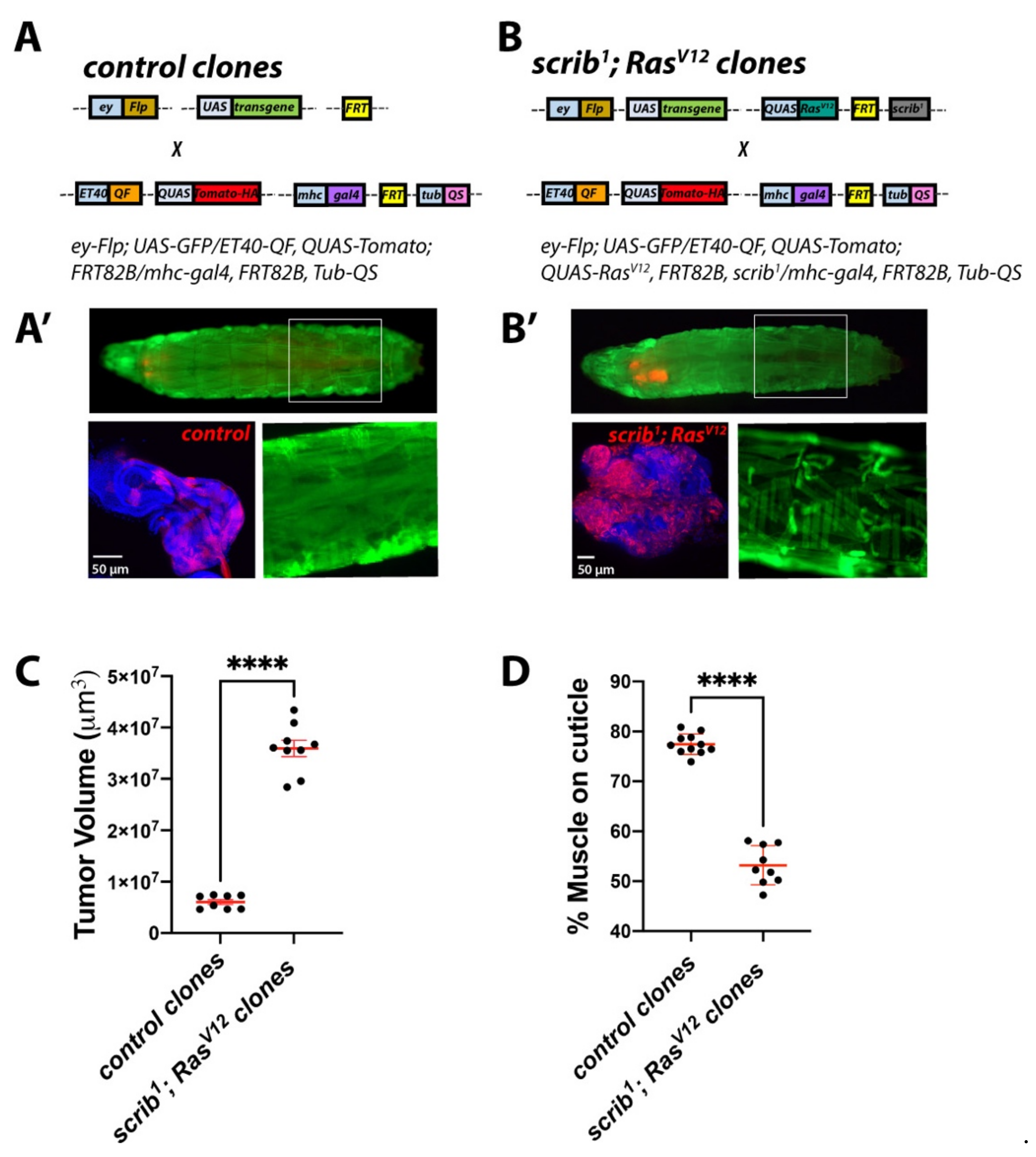
2.4. Establishing a Dual-Driver System to Study Tumour/Host Interactions in Cancer-Induced Cachexia
2.5. Muscle Autonomous JAK/STAT and TNF-α Signalling Drive Tumour-Induced Muscle Tissue Wasting in Drosophila
3. Materials and Methods
3.1. Drosophila Stocks
3.2. Fly Husbandry and Genetics
3.3. Immunofluorescent Staining
3.4. Imaging and Image Processing
3.5. QPCR
3.6. Quantification of the Percentage of Body Wall Muscle Covering the Cuticle
3.7. Quantification of Lipid Droplet Frequency in Ventral Muscle
3.8. Quantification of Tumour Volume
3.9. Larval Feeding Assessment
3.10. RNA Sequencing
3.11. Statistics and Reproducibility
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Couch, M.; Lai, V.; Cannon, T.; Guttridge, D.; Zanation, A.; George, J.; Hayes, D.N.; Zeisel, S.; Shores, C. Cancer cachexia syndrome in head and neck cancer patients: Part I. Diagnosis, impact on quality of life and survival, and treatment. Head Neck 2007, 29, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Von Haehling, S.; Anker, S.D. Cachexia as major underestimated unmet medical need: Facts and numbers. Int. J. Cardiol. 2012, 161, 121–123. [Google Scholar] [CrossRef]
- Kufe, D.W.; Pollock, R.E.; Weichselbaum, R.R.; Bast, R.C., Jr.; Gansler, T.S.; Holland, J.F.; Frei, E., III (Eds.) Holland-Frei Cancer Medicine, 6th ed.; BC Decker: Hamilton, ON, USA, 2003. [Google Scholar]
- De Matos-Neto, E.M.; Lima, J.D.; de Pereira, W.O.; Figuerêdo, R.G.; Riccardi, D.M.D.R.; Radloff, K.; das Neves, R.X.; Camargo, R.G.; Maximiano, L.F.; Tokeshi, F.; et al. Systemic Inflammation in Cachexia—Is Tumor Cytokine Expression Profile the Culprit? Front. Immunol. 2015, 6, 629. [Google Scholar] [CrossRef]
- Wallengren, O.; Lundholm, K.; Bosaeus, I. Diagnostic criteria of cancer cachexia: Relation to quality of life, exercise capacity and survival in unselected palliative care patients. Support. Care Cancer 2013, 21, 1569–1577. [Google Scholar] [CrossRef] [PubMed]
- Andreyev, H.J.N.; Norman, A.R.; Oates, J.; Cunningham, D. Why do patients with weight loss have a worse outcome when undergoing chemotherapy for gastrointestinal malignancies? Eur. J. Cancer 1998, 34, 503–509. [Google Scholar] [CrossRef]
- O’Gorman, P.; McMillan, D.C.; McArdle, C.S. Impact of weight loss, appetite, and the inflammatory response on quality of life in gastrointestinal cancer patients. Nutr. Cancer 1998, 32, 76–80. [Google Scholar] [CrossRef]
- Lok, C. Cachexia: The last illness. Nature 2015, 528, 182–183. [Google Scholar] [CrossRef] [Green Version]
- Fearon, K.C.H.; Moses, A.G.W. Cancer cachexia. Int. J. Cardiol. 2002, 85, 73–81. [Google Scholar] [CrossRef]
- Dewys, W.D.; Begg, C.; Lavin, P.T.; Band, P.R.; Bennett, J.M.; Bertino, J.R.; Cohen, M.H.; Douglass, H.O., Jr.; Engstrom, P.F.; Ezdinli, E.Z.; et al. Prognostic effect of weight loss prior to chemotherapy in cancer patients. Eastern Cooperative Oncology Group. Am. J. Med. 1980, 69, 491–497. [Google Scholar] [CrossRef]
- Bachmann, J.; Ketterer, K.; Marsch, C.; Fechtner, K.; Krakowski-Roosen, H.; Büchler, M.W.; Friess, H.; Martignoni, M.E. Pancreatic cancerrelated cachexia: Influence on metabolism and correlation to weight loss and pulmonary function. BMC Cancer 2009, 9, 255. [Google Scholar] [CrossRef] [Green Version]
- Del Ferraro, C.; Grant, M.; Koczywas, M.; Dorr-Uyemura, L.A. Management of Anorexia-Cachexia in Late-Stage Lung Cancer Patients. J. Hosp. Palliat. Nurs. 2012, 14, 397–402. [Google Scholar] [CrossRef] [Green Version]
- Monitto, C.L.; Berkowitz, D.; Lee, K.M.; Pin, S.; Li, D.; Breslow, M.; O’Malley, B.; Schiller, M. Differential gene expression in a murine model of cancer cachexia. Am. J. Physiol. Metab. 2001, 281, E289–E297. [Google Scholar] [CrossRef] [PubMed]
- Bennani-Baiti, N.; Walsh, D. Animal models of the cancer anorexia–cachexia syndrome. Support. Care Cancer 2010, 19, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Bilder, D.; Li, M.; Perrimon, N. Cooperative Regulation of Cell Polarity and Growth by Drosophila Tumor Suppressors. Science 2000, 289, 113–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Pastor-Pareja, J.C.; Xu, T. Interaction between RasV12 and scribbled clones induces tumour growth and invasion. Nature 2010, 463, 545–548. [Google Scholar] [CrossRef] [Green Version]
- Pagliarini, R.A.; Xu, T. A genetic screen in Drosophila for metastatic behavior. Science 2003, 302, 1227–1231. [Google Scholar] [CrossRef]
- Parisi, F.; Stefanatos, R.; Strathdee, K.; Yu, Y.; Vidal, M. Transformed Epithelia Trigger Non-Tissue-Autonomous Tumor Suppressor Response by Adipocytes via Activation of Toll and Eiger/TNF Signaling. Cell Rep. 2014, 6, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Pastor-Pareja, J.C.; Wu, M.; Xu, T. An innate immune response of blood cells to tumors and tissue damage in Drosophila. Dis. Models Mech. 2008, 1, 144–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero, J.; Macagno, J.P.; Stefanatos, R.; Strathdee, K.E.; Cagan, R.L.; Vidal, M. Oncogenic Ras Diverts a Host TNF Tumor Suppressor Activity into Tumor Promoter. Dev. Cell 2010, 18, 999–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katheder, N.; Khezri, R.; Ofarrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef]
- Kwon, Y.; Song, W.; Droujinine, I.A.; Hu, Y.; Asara, J.M.; Perrimon, N. Systemic Organ Wasting Induced by Localized Expression of the Secreted Insulin/IGF Antagonist ImpL2. Dev. Cell 2015, 33, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Figueroa-Clarevega, A.; Bilder, D. Malignant Drosophila Tumors Interrupt Insulin Signaling to Induce Cachexia-like Wasting. Dev. Cell 2015, 33, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Ng, K.G.-L.; Dombek, K.M.; Eom, D.S.; Kwon, Y.V. Tumors overcome the action of the wasting factor ImpL2 by locally elevating Wnt/Wingless. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Yeom, E.; Shin, H.; Yoo, W.; Jun, E.; Kim, S.; Hong, S.H.; Kwon, D.W.; Ryu, T.H.; Suh, J.M.; Kim, S.C.; et al. Tumour-derived Dilp8/INSL3 induces cancer anorexia by regulating feeding neuropeptides via Lgr3/8 in the brain. Nat. Cell Biol. 2021, 23, 172–183. [Google Scholar] [CrossRef]
- Newton, H.; Wang, Y.-F.; Camplese, L.; Mokochinski, J.B.; Kramer, H.B.; Brown, A.E.X.; Fets, L.; Hirabayashi, S. Systemic muscle wasting and coordinated tumour response drive tumourigenesis. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Brumby, A.M.; Richardson, H.E. Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 2003, 22, 5769–5779. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Irvine, K.D. In vivo analysis of Yorkie phosphorylation sites. Oncogene 2009, 28, 1916–1927. [Google Scholar] [CrossRef] [Green Version]
- Stephens, N.A.; Skipworth, R.J.E.; MacDonald, A.J.; Greig, C.A.; Ross, J.A.; Fearon, K.C.H. Intramyocellular lipid droplets increase with progression of cachexia in cancer patients. J. Cachexia Sarcopenia Muscle 2011, 2, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garelli, A.; Gontijo, A.M.; Miguela, V.; Caparros, E.; Dominguez, M. Imaginal Discs Secrete Insulin-Like Peptide 8 to Mediate Plasticity of Growth and Maturation. Science 2012, 336, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Douglas, R.G.; Shaw, J.H.F. Metabolic effects of cancer. Br. J. Surg. 1990, 77, 246–254. [Google Scholar] [CrossRef] [PubMed]
- Morrison, S. Control of food intake in cancer cachexia: A challenge and a tool. Physiol. Behav. 1976, 17, 705–714. [Google Scholar] [CrossRef]
- Tisdale, M.J. Cancer cachexia: Metabolic alterations and clinical manifestations. Nutrition 1997, 13, 1–7. [Google Scholar] [CrossRef]
- Gullett, N.P.; Mazurak, V.; Hebbar, G.; Ziegler, T.R. Nutritional Interventions for Cancer-Induced Cachexia. Curr. Probl. Cancer 2011, 35, 58–90. [Google Scholar] [CrossRef] [Green Version]
- Szuperák, M.; Salah, S.; Meyer, E.J.; Nagarajan, U.; Ikmi, A.; Gibson, M.C. Feedback regulation of Drosophila BMP signaling by the novel extracellular protein Larval Translucida. Development 2011, 138, 715–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baracos, V.; DeVivo, C.; Hoyle, D.H.; Goldberg, A.L. Activation of the ATP-ubiquitin-proteasome pathway in skeletal muscle of cachectic rats bearing a hepatoma. Am. J. Physiol. Content 1995, 268, E996–E1006. [Google Scholar] [CrossRef] [PubMed]
- Cruz, B.; Oliveira, A.; Gomes-Marcondes, M.C.C. L-leucine dietary supplementation modulates muscle protein degradation and increases pro-inflammatory cytokines in tumour-bearing rats. Cytokine 2017, 96, 253–260. [Google Scholar] [CrossRef]
- Fujita, J.; Tsujinaka, T.; Jano, M.; Ebisui, C.; Saito, H.; Katsume, A.; Akamatsu, K.-I.; Ohsugi, Y.; Shiozaki, H.; Monden, M. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int. J. Cancer 1996, 68, 637–643. [Google Scholar] [CrossRef]
- Smith, K.; Tisdale, M. Mechanism of muscle protein degradation in cancer cachexia. Br. J. Cancer 1993, 68, 314–318. [Google Scholar] [CrossRef] [Green Version]
- Vanderveen, B.N.; Fix, D.K.; Carson, J.A. Disrupted Skeletal Muscle Mitochondrial Dynamics, Mitophagy, and Biogenesis during Cancer Cachexia: A Role for Inflammation. Oxid. Med. Cell. Longev. 2017, 2017, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sukari, A.; Muqbil, I.; Mohammad, M.R.; Philip, P.A.; Azmi, A.S. F-BOX proteins in cancer cachexia and muscle wasting: Emerging regulators and therapeutic opportunities. Semin Cancer Biol. 2016, 36, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Han, J.; Meng, Q.; Xi, Q.; Zhuang, Q.; Jiang, Y.; Han, Y.; Zhang, B.; Fang, J.; Wu, G. Muscle-specific E3 ubiquitin ligases are involved in muscle atrophy of cancer cachexia: An in vitro and in vivo study. Oncol. Rep. 2015, 33, 2261–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonetto, A.; Aydogdu, T.; Jin, X.; Zhang, Z.; Zhan, R.; Puzis, L.; Koniaris, L.G.; Zimmers, T.A. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Metab. 2012, 303, E410–E421. [Google Scholar] [CrossRef] [Green Version]
- Baltgalvis, K.A.; Berger, F.G.; Peña, M.M.O.; Davis, J.M.; White, J.P.; Carson, J.A. Muscle wasting and interleukin-6-induced atrogin-I expression in the cachectic Apc Min/+ mouse. Pflug. Arch. Eur. J. Physiol. 2008, 457, 989–1001. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.; McLeod, L.; Alhayyani, S.; Szczepny, A.; Watkins, D.N.; Chen, W.; Enriori, P.; Ferlin, W.; Ruwanpura, S.; Jenkins, B. Blockade of the IL-6 trans-signalling/STAT3 axis suppresses cachexia in Kras-induced lung adenocarcinoma. Oncogene 2016, 36, 3059–3066. [Google Scholar] [CrossRef]
- Pettersen, K.; Andersen, S.; Degen, S.; Tadini, V.; Grosjean, J.; Hatakeyama, S.; Tesfahun, A.N.; Moestue, S.A.; Kim, J.; Nonstad, U.; et al. Cancer cachexia associates with a systemic autophagy-inducing activity mimicked by cancer cell-derived IL-6 trans-signaling. Sci. Rep. 2017, 7, 2046. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.E.; Kunze, P.; Hildebrandt, W.; Künzli, B.; Berberat, P.; Giese, T.; Klöters, O.; Hammer, J.; Büchler, M.W.; Giese, N.A.; et al. Role of Mononuclear Cells and Inflammatory Cytokines in Pancreatic Cancer-Related Cachexia. Clin. Cancer Res. 2005, 11, 5802–5808. [Google Scholar] [CrossRef] [Green Version]
- Ross, J.A.; Moses, A.G.; Maingay, J.; Sangster, K.; Fearon, K.C. Pro-inflammatory cytokine release by peripheral blood mononuclear cells from patients with advanced pancreatic cancer: Relationship to acute phase response and survival. Oncol. Rep. 2009, 21, 1091–1095. [Google Scholar] [CrossRef]
- Narsale, A.A.; Carson, J.A. Role of IL-6 In Cachexia—Therapeutic Implications. Curr. Opin. Support. Palliat. Care 2014, 8, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Pop, V.-V.; Seicean, A.; Lupan, I.; Samasca, G.; Burz, C.-C. IL-6 roles—Molecular pathway and clinical implication in pancreatic cancer—A systemic review. Immunol. Lett. 2017, 181, 45–50. [Google Scholar] [CrossRef]
- Goldberg, R.M.; Loprinzi, C.L.; Mailliard, J.A.; O’Fallon, J.R.; Krook, J.E.; Ghosh, C.; Hestorff, R.D.; Chong, S.F.; Reuter, N.F.; Shanahan, T.G. Pentoxifylline for treatment of cancer anorexia and cachexia? A randomized, double-blind, placebo-controlled trial. J. Clin. Oncol. 1995, 13, 2856–2859. [Google Scholar] [CrossRef]
- Yakovenko, A.; Cameron, M.; Trevino, J.G. Molecular therapeutic strategies targeting pancreatic cancer induced cachexia. World J. Gastrointest. Surg. 2018, 10, 95–106. [Google Scholar] [CrossRef]
- Kanda, H.; Igaki, T.; Kanuka, H.; Yagi, T.; Miura, M. Wengen, a Member of the Drosophila Tumor Necrosis Factor Receptor Superfamily, Is Required for Eiger Signaling. J. Biol. Chem. 2002, 277, 28372–28375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kauppila, S.; Maaty, W.S.A.; Chen, P.; Tomar, R.S.; Eby, M.T.; Chapo, J.; Chew, S.; Rathore, N.; Zachariah, S.; Sinha, S.K.; et al. Eiger and its receptor, Wengen, comprise a TNF-like system in Drosophila. Oncogene 2003, 22, 4860–4867. [Google Scholar] [CrossRef] [Green Version]
- Belizário, J.E.; Fontes-Oliveira, C.C.; Borges, J.P.; Kashiabara, J.A.; Vannier, E. Skeletal muscle wasting and renewal: A pivotal role of myokine IL-6. SpringerPlus 2016, 5, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Onesti, J.K.; Guttridge, D.C. Inflammation Based Regulation of Cancer Cachexia. BioMed Res. Int. 2014, 2014, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Potter, C.; Tasic, B.; Russler, E.V.; Liang, L.; Luo, L. The Q System: A Repressible Binary System for Transgene Expression, Lineage Tracing, and Mosaic Analysis. Cell 2010, 141, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hodgson, J.A.; Parvy, J.-P.; Yu, Y.; Vidal, M.; Cordero, J.B. Drosophila Larval Models of Invasive Tumorigenesis for In Vivo Studies on Tumour/Peripheral Host Tissue Interactions during Cancer Cachexia. Int. J. Mol. Sci. 2021, 22, 8317. https://doi.org/10.3390/ijms22158317
Hodgson JA, Parvy J-P, Yu Y, Vidal M, Cordero JB. Drosophila Larval Models of Invasive Tumorigenesis for In Vivo Studies on Tumour/Peripheral Host Tissue Interactions during Cancer Cachexia. International Journal of Molecular Sciences. 2021; 22(15):8317. https://doi.org/10.3390/ijms22158317
Chicago/Turabian StyleHodgson, Joseph A., Jean-Philippe Parvy, Yachuan Yu, Marcos Vidal, and Julia B. Cordero. 2021. "Drosophila Larval Models of Invasive Tumorigenesis for In Vivo Studies on Tumour/Peripheral Host Tissue Interactions during Cancer Cachexia" International Journal of Molecular Sciences 22, no. 15: 8317. https://doi.org/10.3390/ijms22158317