
Receptor-Dependent and Independent Regulation of Voltage-Gated Ca2+ Channels and Ca2+-Permeable Channels by Endocannabinoids in the Brain


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Endocannabinoid System in the Brain
2.1. Metabolisms of Endocannabinoids
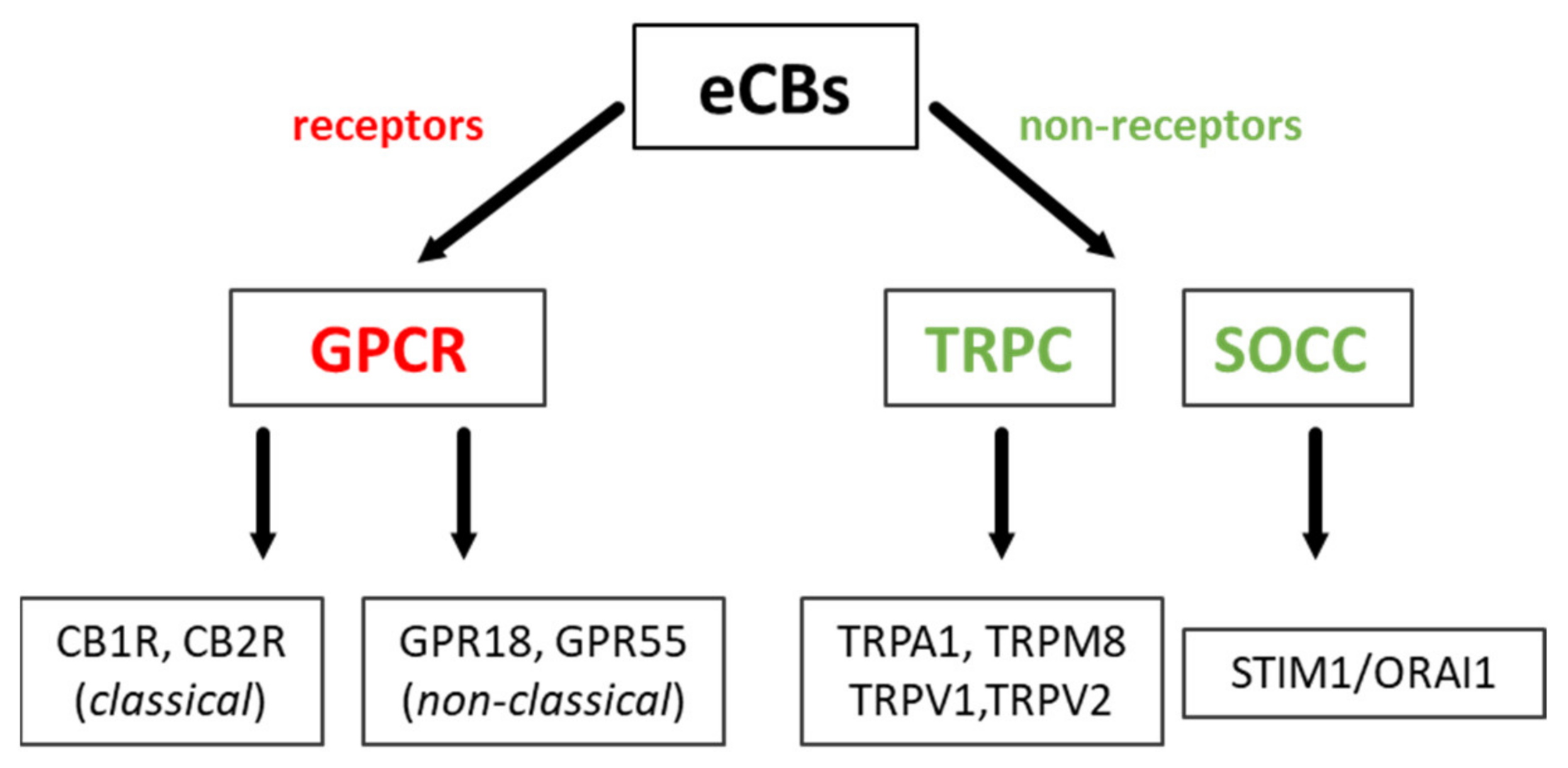
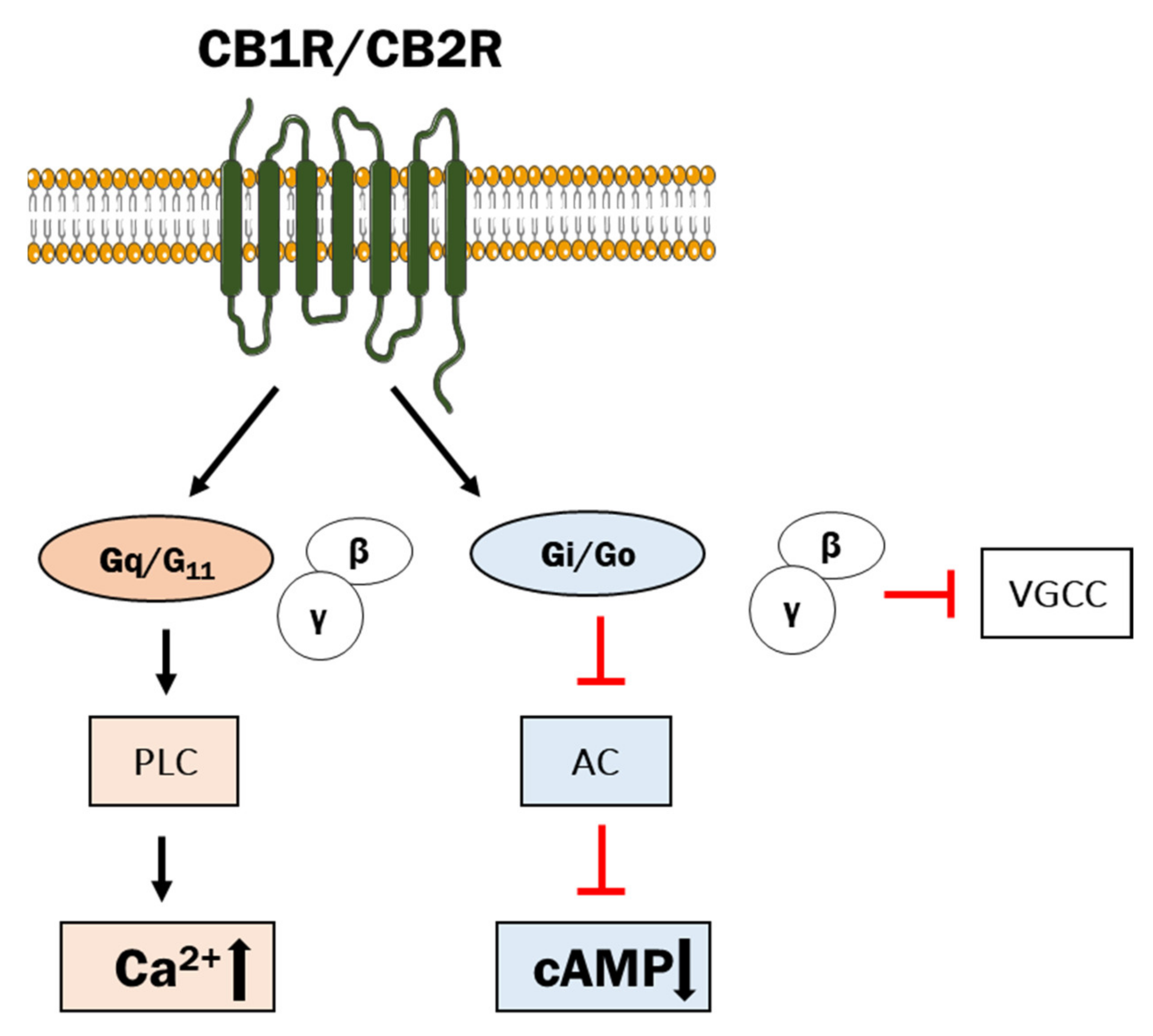
2.2. Classical and Non-Classical GPCRs for eCBs
3. Regulation of Voltage-Gated Ca2+ Channels and Other Ca2+-Permeable Channels by eCBs
3.1. VGCCs
3.2. TRP Channels
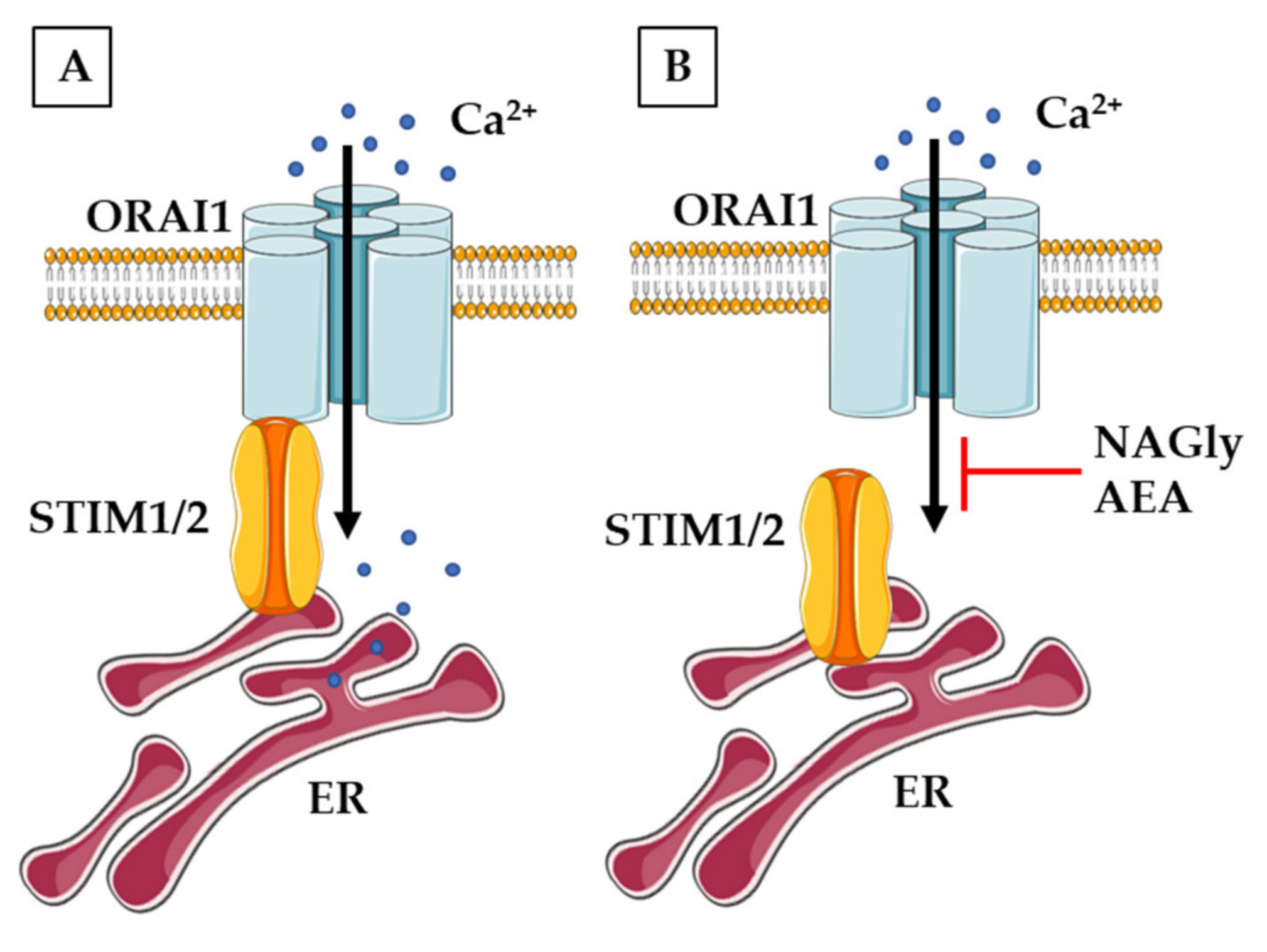
3.3. Calcium-Permeable Channels
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium microdomains: Organization and function. Cell Calcium 2006, 40, 405–412. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar] [CrossRef] [Green Version]
- Kendall, D.A.; Yudowski, G.A. Cannabinoid Receptors in the Central Nervous System: Their Signaling and Roles in Disease. Front. Cell Neurosci. 2016, 10, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno, E.; Cavic, M.; Canela, E.I. Functional Fine-Tuning of Metabolic Pathways by the Endocannabinoid System-Implications for Health and Disease. Int. J. Mol. Sci. 2021, 22, 3661. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Petros, T.J.; Chang, S.Y.; Zavitsanos, P.A.; Zipkin, R.E.; Sivakumar, R.; Coop, A.; Maeda, D.Y.; De Petrocellis, L.; et al. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001, 276, 42639–42644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viader, A.; Blankman, J.L.; Zhong, P.; Liu, X.; Schlosburg, J.E.; Joslyn, C.M.; Liu, Q.S.; Tomarchio, A.J.; Lichtman, A.H.; Selley, D.E.; et al. Metabolic Interplay between Astrocytes and Neurons Regulates Endocannabinoid Action. Cell Rep. 2015, 12, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Schurman, L.D.; Lichtman, A.H. Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Front. Pharmacol. 2017, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where are They Going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef]
- Kruk-Slomka, M.; Dzik, A.; Budzynska, B.; Biala, G. Endocannabinoid System: The Direct and Indirect Involvement in the Memory and Learning Processes—A Short Review. Mol. Neurobiol. 2017, 54, 8332–8347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, D.; Hu, S.S.; Rimmerman, N.; Juknat, A.; Vogel, Z.; Walker, J.M.; Bradshaw, H.B. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010, 11, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katona, I.; Freund, T.F. Multiple functions of endocannabinoid signaling in the brain. Annu. Rev. Neurosci. 2012, 35, 529–558. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K. Cannabinoid receptors: Where they are and what they do. J. Neuroendocrinol. 2008, 20 (Suppl. S1), 10–14. [Google Scholar] [CrossRef]
- Shi, Q.X.; Yang, L.K.; Shi, W.L.; Wang, L.; Zhou, S.M.; Guan, S.Y.; Zhao, M.G.; Yang, Q. The novel cannabinoid receptor GPR55 mediates anxiolytic-like effects in the medial orbital cortex of mice with acute stress. Mol. Brain 2017, 10, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biringer, R.G. Endocannabinoid signaling pathways: Beyond CB1R and CB2R. J. Cell Commun. Signal. 2021, 15, 335–360. [Google Scholar] [CrossRef]
- Dalton, G.D.; Bass, C.E.; Van Horn, C.G.; Howlett, A.C. Signal transduction via cannabinoid receptors. CNS Neurol. Disord. Drug Targets 2009, 8, 422–431. [Google Scholar] [CrossRef]
- Pertwee, R.G.; Howlett, A.C.; Abood, M.E.; Alexander, S.P.; Di Marzo, V.; Elphick, M.R.; Greasley, P.J.; Hansen, H.S.; Kunos, G.; Mackie, K.; et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: Beyond CB₁ and CB₂. Pharmacol. Rev. 2010, 62, 588–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajaraman, G.; Simcocks, A.; Hryciw, D.H.; Hutchinson, D.S.; McAinch, A.J. G protein coupled receptor 18: A potential role for endocannabinoid signaling in metabolic dysfunction. Mol. Nutr. Food Res. 2016, 60, 92–102. [Google Scholar] [CrossRef]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Yang, H.; Zhou, J.; Lehmann, C. GPR55—A putative “type 3” cannabinoid receptor in inflammation. J. Basic Clin. Physiol. Pharmacol. 2016, 27, 297–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.; Abdulrazzaq, G.; Chan, S.L.F.; Penman, J.; Harvey, J.; Alexander, S.P.H. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Adv. Pharmacol. 2017, 80, 223–247. [Google Scholar] [CrossRef] [PubMed]
- Console-Bram, L.; Ciuciu, S.M.; Zhao, P.; Zipkin, R.E.; Brailoiu, E.; Abood, M.E. N-arachidonoyl glycine, another endogenous agonist of GPR55. Biochem. Biophys. Res. Commun. 2017, 490, 1389–1393. [Google Scholar] [CrossRef] [PubMed]
- Melis, M.; Pistis, M. Endocannabinoid signaling in midbrain dopamine neurons: More than physiology? Curr. Neuropharmacol. 2007, 5, 268–277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kushmerick, C.; Price, G.D.; Taschenberger, H.; Puente, N.; Renden, R.; Wadiche, J.I.; Duvoisin, R.M.; Grandes, P.; von Gersdorff, H. Retroinhibition of presynaptic Ca2+ currents by endocannabinoids released via postsynaptic mGluR activation at a calyx synapse. J. Neurosci. 2004, 24, 5955–5965. [Google Scholar] [CrossRef] [Green Version]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid signaling and synaptic function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadas, H.; Gaillet, S.; Beltramo, M.; Venance, L.; Piomelli, D. Biosynthesis of an endogenous cannabinoid precursor in neurons and its control by calcium and cAMP. J. Neurosci. 1996, 16, 3934–3942. [Google Scholar] [CrossRef] [Green Version]
- Komarnytsky, S.; Rathinasabapathy, T.; Wagner, C.; Metzger, B.; Carlisle, C.; Panda, C.; Le Brun-Blashka, S.; Troup, J.P.; Varadharaj, S. Endocannabinoid System and Its Regulation by Polyunsaturated Fatty Acids and Full Spectrum Hemp Oils. Int. J. Mol. Sci. 2021, 22, 5479. [Google Scholar] [CrossRef]
- Watson, J.E.; Kim, J.S.; Das, A. Emerging class of omega-3 fatty acid endocannabinoids & their derivatives. Prostaglandins Other Lipid Mediat. 2019, 143, 106337. [Google Scholar] [CrossRef]
- Bradshaw, H.B.; Rimmerman, N.; Hu, S.S.; Benton, V.M.; Stuart, J.M.; Masuda, K.; Cravatt, B.F.; O’Dell, D.K.; Walker, J.M. The endocannabinoid anandamide is a precursor for the signaling lipid N-arachidonoyl glycine by two distinct pathways. BMC Biochem. 2009, 10, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, N.; Tsuboi, K.; Uyama, T. Metabolism of endocannabinoids and related N-acylethanolamines: Canonical and alternative pathways. FEBS J. 2013, 280, 1874–1894. [Google Scholar] [CrossRef]
- Kano, M.; Ohno-Shosaku, T.; Hashimotodani, Y.; Uchigashima, M.; Watanabe, M. Endocannabinoid-mediated control of synaptic transmission. Physiol. Rev. 2009, 89, 309–380. [Google Scholar] [CrossRef]
- Wang, J.; Ueda, N. Biology of endocannabinoid synthesis system. Prostaglandins Other Lipid Mediat. 2009, 89, 112–119. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef] [Green Version]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Zelasko, S.; Arnold, W.R.; Das, A. Endocannabinoid metabolism by cytochrome P450 monooxygenases. Prostaglandins Other Lipid Mediat. 2015, 116–117, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Béquet, F.; Uzabiaga, F.; Desbazeille, M.; Ludwiczak, P.; Maftouh, M.; Picard, C.; Scatton, B.; Le Fur, G. CB1 receptor-mediated control of the release of endocannabinoids (as assessed by microdialysis coupled with LC/MS) in the rat hypothalamus. Eur. J. Neurosci. 2007, 26, 3458–3464. [Google Scholar] [CrossRef]
- Shohami, E.; Cohen-Yeshurun, A.; Magid, L.; Algali, M.; Mechoulam, R. Endocannabinoids and traumatic brain injury. Br. J. Pharmacol. 2011, 163, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Wang, L.; Harvey-White, J.; Huang, B.X.; Kim, H.Y.; Luquet, S.; Palmiter, R.D.; Krystal, G.; Rai, R.; Mahadevan, A.; et al. Multiple pathways involved in the biosynthesis of anandamide. Neuropharmacology 2008, 54, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, G.M.; Cravatt, B.F. Anandamide biosynthesis catalyzed by the phosphodiesterase GDE1 and detection of glycerophospho-N-acyl ethanolamine precursors in mouse brain. J. Biol. Chem. 2008, 283, 9341–9349. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Stucky, A.; Liu, J.; Shen, C.; Trocme-Thibierge, C.; Morain, P. Dissociating beta-amyloid from alpha 7 nicotinic acetylcholine receptor by a novel therapeutic agent, S 24795, normalizes alpha 7 nicotinic acetylcholine and NMDA receptor function in Alzheimer’s disease brain. J. Neurosci. 2009, 29, 10961–10973. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, T.; Bartsch, J.C.; Beer, A.; Lomazzo, E.; Guggenhuber, S.; Lange, M.D.; Bindila, L.; Pape, H.C.; Lutz, B. Impaired anandamide/palmitoylethanolamide signaling in hippocampal glutamatergic neurons alters synaptic plasticity, learning, and emotional responses. Neuropsychopharmacology 2019, 44, 1377–1388. [Google Scholar] [CrossRef]
- Burstein, S.H.; Rossetti, R.G.; Yagen, B.; Zurier, R.B. Oxidative metabolism of anandamide. Prostaglandins Other Lipid Mediat. 2000, 61, 29–41. [Google Scholar] [CrossRef]
- Tegeder, I. Endocannabinoids as Guardians of Metastasis. Int. J. Mol. Sci. 2016, 17, 230. [Google Scholar] [CrossRef] [Green Version]
- Burstein, S.H.; Huang, S.M.; Petros, T.J.; Rossetti, R.G.; Walker, J.M.; Zurier, R.B. Regulation of anandamide tissue levels by N-arachidonylglycine. Biochem. Pharmacol. 2002, 64, 1147–1150. [Google Scholar] [CrossRef]
- Prusakiewicz, J.J.; Kingsley, P.J.; Kozak, K.R.; Marnett, L.J. Selective oxygenation of N-arachidonylglycine by cyclooxygenase-2. Biochem. Biophys. Res. Commun. 2002, 296, 612–617. [Google Scholar] [CrossRef]
- Pazos, M.R.; Núñez, E.; Benito, C.; Tolón, R.M.; Romero, J. Functional neuroanatomy of the endocannabinoid system. Pharmacol. Biochem. Behav. 2005, 81, 239–247. [Google Scholar] [CrossRef]
- Lovinger, D.M. Presynaptic modulation by endocannabinoids. Handb. Exp. Pharmacol. 2008, 435–477. [Google Scholar] [CrossRef]
- Bosier, B.; Muccioli, G.G.; Hermans, E.; Lambert, D.M. Functionally selective cannabinoid receptor signalling: Therapeutic implications and opportunities. Biochem. Pharmacol. 2010, 80, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ong, W.Y.; Mackie, K. A light and electron microscopic study of the CB1 cannabinoid receptor in primate brain. Neuroscience 1999, 92, 1177–1191. [Google Scholar] [CrossRef]
- Howlett, A.C.; Barth, F.; Bonner, T.I.; Cabral, G.; Casellas, P.; Devane, W.A.; Felder, C.C.; Herkenham, M.; Mackie, K.; Martin, B.R.; et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002, 54, 161–202. [Google Scholar] [CrossRef]
- Hoffman, K.M.; Eisen, M.R.; Chandler, J.K.; Nelson, M.R.; Johnson, E.A.; McNutt, P.M. Retrograde activation of CB1R by muscarinic receptors protects against central organophosphorus toxicity. Neuropharmacology 2019, 155, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Ho, B.C.; Wassink, T.H.; Ziebell, S.; Andreasen, N.C. Cannabinoid receptor 1 gene polymorphisms and marijuana misuse interactions on white matter and cognitive deficits in schizophrenia. Schizophr. Res. 2011, 128, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benyamina, A.; Kebir, O.; Blecha, L.; Reynaud, M.; Krebs, M.O. CNR1 gene polymorphisms in addictive disorders: A systematic review and a meta-analysis. Addict. Biol. 2011, 16, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Wetherill, L.; Dick, D.M.; Xuei, X.; Hinrichs, A.; Hesselbrock, V.; Kramer, J.; Nurnberger, J.I.; Schuckit, M.; Bierut, L.J.; et al. Evidence for association between polymorphisms in the cannabinoid receptor 1 (CNR1) gene and cannabis dependence. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2009, 150, 736–740. [Google Scholar] [CrossRef] [Green Version]
- Tao, R.; Li, C.; Jaffe, A.E.; Shin, J.H.; Deep-Soboslay, A.; Yamin, R.; Weinberger, D.R.; Hyde, T.M.; Kleinman, J.E. Cannabinoid receptor CNR1 expression and DNA methylation in human prefrontal cortex, hippocampus and caudate in brain development and schizophrenia. Transl. Psychiatry 2020, 10, 158. [Google Scholar] [CrossRef]
- Gardner, E.L. Endocannabinoid signaling system and brain reward: Emphasis on dopamine. Pharmacol. Biochem. Behav. 2005, 81, 263–284. [Google Scholar] [CrossRef]
- Eggan, S.M.; Lewis, D.A. Immunocytochemical distribution of the cannabinoid CB1 receptor in the primate neocortex: A regional and laminar analysis. Cereb. Cortex 2007, 17, 175–191. [Google Scholar] [CrossRef]
- Howlett, A.C. Cannabinoid receptor signaling. Handb. Exp. Pharmacol. 2005, 53–79. [Google Scholar] [CrossRef]
- Busquets-Garcia, A.; Bains, J.; Marsicano, G. CB1 Receptor Signaling in the Brain: Extracting Specificity from Ubiquity. Neuropsychopharmacology 2018, 43, 4–20. [Google Scholar] [CrossRef]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids mediate neuron-astrocyte communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Turu, G.; Hunyady, L. Signal transduction of the CB1 cannabinoid receptor. J. Mol. Endocrinol. 2010, 44, 75–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, C.J.; Xi, Z.X. Progress in brain cannabinoid CB. Neurosci. Biobehav. Rev. 2019, 98, 208–220. [Google Scholar] [CrossRef]
- Chen, D.J.; Gao, M.; Gao, F.F.; Su, Q.X.; Wu, J. Brain cannabinoid receptor 2: Expression, function and modulation. Acta Pharmacol. Sin. 2017, 38, 312–316. [Google Scholar] [CrossRef] [PubMed]
- Lutz, B. Neurobiology of cannabinoid receptor signaling. Dialogues Clin. Neurosci. 2020, 22, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.P.; Patel, S.; Perchuk, A.; Meozzi, P.A.; Myers, L.; Mora, Z.; Tagliaferro, P.; Gardner, E.; et al. Discovery of the presence and functional expression of cannabinoid CB2 receptors in brain. Ann. N. Y. Acad. Sci. 2006, 1074, 514–536. [Google Scholar] [CrossRef]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Cabral, G.A.; Griffin-Thomas, L. Emerging role of the cannabinoid receptor CB2 in immune regulation: Therapeutic prospects for neuroinflammation. Expert Rev. Mol. Med. 2009, 11, e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mechoulam, R.; Parker, L.A. The endocannabinoid system and the brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, I.L.; Rodrigues da Silva, N.; Guimarães, F.S.; Gomes, F.V. Are CB2 Receptors a New Target for Schizophrenia Treatment? Front. Psychiatry 2020, 11, 587154. [Google Scholar] [CrossRef]
- Ibsen, M.S.; Connor, M.; Glass, M. Cannabinoid CB. Cannabis Cannabinoid Res. 2017, 2, 48–60. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Pinilla, E.; Varani, K.; Reyes-Resina, I.; Angelats, E.; Vincenzi, F.; Ferreiro-Vera, C.; Oyarzabal, J.; Canela, E.I.; Lanciego, J.L.; Nadal, X.; et al. Binding and Signaling Studies Disclose a Potential Allosteric Site for Cannabidiol in Cannabinoid CB. Front. Pharmacol. 2017, 8, 744. [Google Scholar] [CrossRef]
- Zoratti, C.; Kipmen-Korgun, D.; Osibow, K.; Malli, R.; Graier, W.F. Anandamide initiates Ca(2+) signaling via CB2 receptor linked to phospholipase C in calf pulmonary endothelial cells. Br. J. Pharmacol. 2003, 140, 1351–1362. [Google Scholar] [CrossRef] [Green Version]
- Thompson, K.J.; Tobin, A.B. Crosstalk between the M1 muscarinic acetylcholine receptor and the endocannabinoid system: A relevance for Alzheimer’s disease? Cell Signal. 2020, 70, 109545. [Google Scholar] [CrossRef] [PubMed]
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: A mechanism for receptor cross-talk? Mol. Pharmacol. 2005, 67, 1697–1704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawzdargo, M.; Nguyen, T.; Lee, D.K.; Lynch, K.R.; Cheng, R.; Heng, H.H.; George, S.R.; O’Dowd, B.F. Identification and cloning of three novel human G protein-coupled receptor genes GPR52, PsiGPR53 and GPR55: GPR55 is extensively expressed in human brain. Brain Res. Mol. Brain Res. 1999, 64, 193–198. [Google Scholar] [CrossRef]
- Vassilatis, D.K.; Hohmann, J.G.; Zeng, H.; Li, F.; Ranchalis, J.E.; Mortrud, M.T.; Brown, A.; Rodriguez, S.S.; Weller, J.R.; Wright, A.C.; et al. The G protein-coupled receptor repertoires of human and mouse. Proc. Natl. Acad. Sci. USA 2003, 100, 4903–4908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabiec, U.; Hohmann, T.; Ghadban, C.; Rothgänger, C.; Wong, D.; Antonietti, A.; Groth, T.; Mackie, K.; Dehghani, F. Protective Effect of N-Arachidonoyl Glycine-GPR18 Signaling after Excitotoxical Lesion in Murine Organotypic Hippocampal Slice Cultures. Int. J. Mol. Sci. 2019, 20, 1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes-Resina, I.; Navarro, G.; Aguinaga, D.; Canela, E.I.; Schoeder, C.T.; Załuski, M.; Kieć-Kononowicz, K.; Saura, C.A.; Müller, C.E.; Franco, R. Molecular and functional interaction between GPR18 and cannabinoid CB. Biochem. Pharmacol. 2018, 157, 169–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pertwee, R.G. The pharmacology of cannabinoid receptors and their ligands: An overview. Int. J. Obes. 2006, 30 (Suppl. S1), S13–S18. [Google Scholar] [CrossRef] [Green Version]
- Console-Bram, L.; Brailoiu, E.; Brailoiu, G.C.; Sharir, H.; Abood, M.E. Activation of GPR18 by cannabinoid compounds: A tale of biased agonism. Br. J. Pharmacol. 2014, 171, 3908–3917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, R.A. The enigmatic pharmacology of GPR55. Trends Pharmacol. Sci. 2009, 30, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Hurst, K.; Badgley, C.; Ellsworth, T.; Bell, S.; Friend, L.; Prince, B.; Welch, J.; Cowan, Z.; Williamson, R.; Lyon, C.; et al. A putative lysophosphatidylinositol receptor GPR55 modulates hippocampal synaptic plasticity. Hippocampus 2017, 27, 985–998. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Toshida, T.; Maruyama, K.; Nakajima, K.; Yamashita, A.; Sugiura, T. 2-Arachidonoyl-sn-glycero-3-phosphoinositol: A possible natural ligand for GPR55. J. Biochem. 2009, 145, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Okuno, T.; Yokomizo, T. What is the natural ligand of GPR55? J. Biochem. 2011, 149, 495–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elphick, M.R. The evolution and comparative neurobiology of endocannabinoid signalling. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3201–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heifets, B.D.; Castillo, P.E. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 2009, 71, 283–306. [Google Scholar] [CrossRef] [Green Version]
- Chevaleyre, V.; Takahashi, K.A.; Castillo, P.E. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu. Rev. Neurosci. 2006, 29, 37–76. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, E.M.; Vivar, C.; Camandola, S. Physiology and pathology of calcium signaling in the brain. Front. Pharmacol. 2012, 3, 61. [Google Scholar] [CrossRef] [Green Version]
- Dolphin, A.C. Voltage-gated calcium channels: Their discovery, function and importance as drug targets. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef]
- Zheng, J. Molecular mechanism of TRP channels. Compr. Physiol. 2013, 3, 221–242. [Google Scholar] [CrossRef] [Green Version]
- Samanta, A.; Hughes, T.E.T.; Moiseenkova-Bell, V.Y. Transient Receptor Potential (TRP) Channels. Subcell Biochem. 2018, 87, 141–165. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. The Inositol Trisphosphate/Calcium Signaling Pathway in Health and Disease. Physiol. Rev. 2016, 96, 1261–1296. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, D.; Chung, W.Y.; Yang, D.; Hong, J.H.; Jha, A.; Muallem, S. STIM-TRP Pathways and Microdomain Organization: Ca. Adv. Exp. Med. Biol. 2017, 993, 139–157. [Google Scholar] [CrossRef]
- Simms, B.A.; Zamponi, G.W. Neuronal voltage-gated calcium channels: Structure, function, and dysfunction. Neuron 2014, 82, 24–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Nanou, E.; Catterall, W.A. Calcium Channels, Synaptic Plasticity, and Neuropsychiatric Disease. Neuron 2018, 98, 466–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demuth, D.G.; Molleman, A. Cannabinoid signalling. Life Sci. 2006, 78, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Lovinger, D.M. Presynaptic G Protein-Coupled Receptors: Gatekeepers of Addiction? Front. Cell Neurosci. 2016, 10, 264. [Google Scholar] [CrossRef] [Green Version]
- Naderi, N.; Ahmad-Molaei, L.; Mazar-Atabaki, A.; Ronaghi, A.; Shirazi-Zand, Z.; Motiei-Langroudi, S.M.; Eslahkar, S. L-type calcium channel mediates anticonvulsant effect of cannabinoids in acute and chronic murine models of seizure. Neurochem. Res. 2012, 37, 279–287. [Google Scholar] [CrossRef]
- Zhang, Y.; Xie, H.; Lei, G.; Li, F.; Pan, J.; Liu, C.; Liu, Z.; Liu, L.; Cao, X. Regulatory effects of anandamide on intracellular Ca(2+) concentration increase in trigeminal ganglion neurons. Neural Regen. Res. 2014, 9, 878–887. [Google Scholar] [CrossRef]
- Bruzsik, B.; Biro, L.; Zelena, D.; Sipos, E.; Szebik, H.; Sarosdi, K.R.; Horvath, O.; Farkas, I.; Csillag, V.; Finszter, C.K.; et al. Somatostatin Neurons of the Bed Nucleus of Stria Terminalis Enhance Associative Fear Memory Consolidation in Mice. J. Neurosci. 2021, 41, 1982–1995. [Google Scholar] [CrossRef] [PubMed]
- Gunduz-Cinar, O. The endocannabinoid system in the amygdala and modulation of fear. Prog. Neuropsychopharmacol. Biol. Psychiatry 2021, 105, 110116. [Google Scholar] [CrossRef] [PubMed]
- Gebremedhin, D.; Lange, A.R.; Campbell, W.B.; Hillard, C.J.; Harder, D.R. Cannabinoid CB1 receptor of cat cerebral arterial muscle functions to inhibit L-type Ca2+ channel current. Am. J. Physiol. 1999, 276, H2085–H2093. [Google Scholar] [CrossRef] [Green Version]
- Maccarrone, M.; Rossi, S.; Bari, M.; De Chiara, V.; Fezza, F.; Musella, A.; Gasperi, V.; Prosperetti, C.; Bernardi, G.; Finazzi-Agrò, A.; et al. Anandamide inhibits metabolism and physiological actions of 2-arachidonoylglycerol in the striatum. Nat. Neurosci. 2008, 11, 152–159. [Google Scholar] [CrossRef]
- Puente, N.; Cui, Y.; Lassalle, O.; Lafourcade, M.; Georges, F.; Venance, L.; Grandes, P.; Manzoni, O.J. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat. Neurosci. 2011, 14, 1542–1547. [Google Scholar] [CrossRef]
- Lo, Y.K.; Chiang, H.T.; Wu, S.N. Effect of arvanil (N-arachidonoyl-vanillyl-amine), a nonpungent anandamide-capsaicin hybrid, on ion currents in NG108-15 neuronal cells. Biochem. Pharmacol. 2003, 65, 581–591. [Google Scholar] [CrossRef]
- Caulfield, M.P.; Brown, D.A. Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br. J. Pharmacol. 1992, 106, 231–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackie, K.; Hille, B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. USA 1992, 89, 3825–3829. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K.; Devane, W.A.; Hille, B. Anandamide, an endogenous cannabinoid, inhibits calcium currents as a partial agonist in N18 neuroblastoma cells. Mol. Pharmacol. 1993, 44, 498–503. [Google Scholar]
- Brown, S.P.; Safo, P.K.; Regehr, W.G. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J. Neurosci. 2004, 24, 5623–5631. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Ikeda, S.R.; Lewis, D.L. Rat brain cannabinoid receptor modulates N-type Ca2+ channels in a neuronal expression system. Mol. Pharmacol. 1996, 49, 707–714. [Google Scholar]
- Twitchell, W.; Brown, S.; Mackie, K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997, 78, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.J.; Walsh, F.S.; Doherty, P. The FGF receptor uses the endocannabinoid signaling system to couple to an axonal growth response. J. Cell Biol. 2003, 160, 481–486. [Google Scholar] [CrossRef] [Green Version]
- Lozovaya, N.; Min, R.; Tsintsadze, V.; Burnashev, N. Dual modulation of CNS voltage-gated calcium channels by cannabinoids: Focus on CB1 receptor-independent effects. Cell Calcium 2009, 46, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.C.; Lo, S.W.; Hsu, K.S. Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J. Physiol. 2001, 532, 731–748. [Google Scholar] [CrossRef]
- Shen, M.; Thayer, S.A. The cannabinoid agonist Win55,212-2 inhibits calcium channels by receptor-mediated and direct pathways in cultured rat hippocampal neurons. Brain Res. 1998, 783, 77–84. [Google Scholar] [CrossRef]
- Németh, B.; Ledent, C.; Freund, T.F.; Hájos, N. CB1 receptor-dependent and -independent inhibition of excitatory postsynaptic currents in the hippocampus by WIN 55,212-2. Neuropharmacology 2008, 54, 51–57. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular characterization of T-type calcium channels. Cell Calcium 2006, 40, 89–96. [Google Scholar] [CrossRef]
- Zamponi, G.W.; Striessnig, J.; Koschak, A.; Dolphin, A.C. The Physiology, Pathology, and Pharmacology of Voltage-Gated Calcium Channels and Their Future Therapeutic Potential. Pharmacol. Rev. 2015, 67, 821–870. [Google Scholar] [CrossRef] [Green Version]
- Chemin, J.; Nargeot, J.; Lory, P. Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J. Biol. Chem. 2007, 282, 2314–2323. [Google Scholar] [CrossRef] [Green Version]
- Chemin, J.; Cazade, M.; Lory, P. Modulation of T-type calcium channels by bioactive lipids. Pflug. Arch. 2014, 466, 689–700. [Google Scholar] [CrossRef]
- Gadotti, V.M.; You, H.; Petrov, R.R.; Berger, N.D.; Diaz, P.; Zamponi, G.W. Analgesic effect of a mixed T-type channel inhibitor/CB2 receptor agonist. Mol. Pain 2013, 9, 32. [Google Scholar] [CrossRef] [Green Version]
- Chemin, J.; Monteil, A.; Perez-Reyes, E.; Nargeot, J.; Lory, P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anandamide. EMBO J. 2001, 20, 7033–7040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, H.R.; Gilmore, A.J.; Connor, M. Inhibition of human recombinant T-type calcium channels by the endocannabinoid N-arachidonoyl dopamine. Br. J. Pharmacol. 2009, 156, 740–750. [Google Scholar] [CrossRef] [Green Version]
- Barbara, G.; Alloui, A.; Nargeot, J.; Lory, P.; Eschalier, A.; Bourinet, E.; Chemin, J. T-type calcium channel inhibition underlies the analgesic effects of the endogenous lipoamino acids. J. Neurosci. 2009, 29, 13106–13114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akopian, A.N.; Ruparel, N.B.; Jeske, N.A.; Patwardhan, A.; Hargreaves, K.M. Role of ionotropic cannabinoid receptors in peripheral antinociception and antihyperalgesia. Trends Pharmacol. Sci. 2009, 30, 79–84. [Google Scholar] [CrossRef] [Green Version]
- Caterina, M.J. TRP channel cannabinoid receptors in skin sensation, homeostasis, and inflammation. ACS Chem. Neurosci. 2014, 5, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Bisogno, T.; Davis, J.B.; Pertwee, R.G.; Di Marzo, V. Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: Inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett. 2000, 483, 52–56. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Bisogno, T.; Maccarrone, M.; Davis, J.B.; Finazzi-Agro, A.; Di Marzo, V. The activity of anandamide at vanilloid VR1 receptors requires facilitated transport across the cell membrane and is limited by intracellular metabolism. J. Biol. Chem. 2001, 276, 12856–12863. [Google Scholar] [CrossRef] [Green Version]
- Di Marzo, V.; De Petrocellis, L.; Fezza, F.; Ligresti, A.; Bisogno, T. Anandamide receptors. Prostaglandins Leukot. Essent. Fatty Acids 2002, 66, 377–391. [Google Scholar] [CrossRef] [PubMed]
- De Petrocellis, L.; Ligresti, A.; Moriello, A.S.; Allarà, M.; Bisogno, T.; Petrosino, S.; Stott, C.G.; Di Marzo, V. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br. J. Pharmacol. 2011, 163, 1479–1494. [Google Scholar] [CrossRef] [Green Version]
- Smart, D.; Gunthorpe, M.J.; Jerman, J.C.; Nasir, S.; Gray, J.; Muir, A.I.; Chambers, J.K.; Randall, A.D.; Davis, J.B. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br. J. Pharmacol. 2000, 129, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Declèves, X.; Cisternino, S. Transient Receptor Potential Vanilloid in the Brain Gliovascular Unit: Prospective Targets in Therapy. Pharmaceutics 2021, 13, 334. [Google Scholar] [CrossRef]
- Staruschenko, A.; Jeske, N.A.; Akopian, A.N. Contribution of TRPV1-TRPA1 interaction to the single channel properties of the TRPA1 channel. J. Biol. Chem. 2010, 285, 15167–15177. [Google Scholar] [CrossRef] [Green Version]
- Akopian, A.N. Regulation of nociceptive transmission at the periphery via TRPA1-TRPV1 interactions. Curr. Pharm. Biotechnol. 2011, 12, 89–94. [Google Scholar] [CrossRef]
- Tóth, A.; Blumberg, P.M.; Boczán, J. Anandamide and the vanilloid receptor (TRPV1). Vitam. Horm. 2009, 81, 389–419. [Google Scholar] [CrossRef] [PubMed]
- Kauer, J.A.; Gibson, H.E. Hot flash: TRPV channels in the brain. Trends Neurosci. 2009, 32, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Maione, S.; Cristino, L.; Migliozzi, A.L.; Georgiou, A.L.; Starowicz, K.; Salt, T.E.; Di Marzo, V. TRPV1 channels control synaptic plasticity in the developing superior colliculus. J. Physiol. 2009, 587, 2521–2535. [Google Scholar] [CrossRef]
- Cui, Y.; Perez, S.; Venance, L. Endocannabinoid-LTP Mediated by CB1 and TRPV1 Receptors Encodes for Limited Occurrences of Coincident Activity in Neocortex. Front. Cell Neurosci. 2018, 12, 182. [Google Scholar] [CrossRef] [PubMed]
- Hermann, H.; De Petrocellis, L.; Bisogno, T.; Schiano Moriello, A.; Lutz, B.; Di Marzo, V. Dual effect of cannabinoid CB1 receptor stimulation on a vanilloid VR1 receptor-mediated response. Cell Mol. Life Sci. 2003, 60, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Chávez, A.E.; Chiu, C.Q.; Castillo, P.E. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat. Neurosci. 2010, 13, 1511–1518. [Google Scholar] [CrossRef] [Green Version]
- Grueter, B.A.; Brasnjo, G.; Malenka, R.C. Postsynaptic TRPV1 triggers cell type-specific long-term depression in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1519–1525. [Google Scholar] [CrossRef] [PubMed]
- Zygmunt, P.M.; Ermund, A.; Movahed, P.; Andersson, D.A.; Simonsen, C.; Jönsson, B.A.; Blomgren, A.; Birnir, B.; Bevan, S.; Eschalier, A.; et al. Monoacylglycerols activate TRPV1—A link between phospholipase C and TRPV1. PLoS ONE 2013, 8, e81618. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Schiano Moriello, A.; Cerrato, S.; Fusco, M.; Puigdemont, A.; De Petrocellis, L.; Di Marzo, V. The anti-inflammatory mediator palmitoylethanolamide enhances the levels of 2-arachidonoyl-glycerol and potentiates its actions at TRPV1 cation channels. Br. J. Pharmacol. 2016, 173, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Trieu, B.H.; Palmer, L.C.; Jia, Y.; Pham, D.T.; Jung, K.M.; Karsten, C.A.; Merrill, C.B.; Mackie, K.; Gall, C.M.; et al. A Primary Cortical Input to Hippocampus Expresses a Pathway-Specific and Endocannabinoid-Dependent Form of Long-Term Potentiation. eNeuro 2016, 3. [Google Scholar] [CrossRef] [Green Version]
- Maglio, L.E.; Noriega-Prieto, J.A.; Maraver, M.J.; Fernández de Sevilla, D. Endocannabinoid-Dependent Long-Term Potentiation of Synaptic Transmission at Rat Barrel Cortex. Cereb. Cortex 2018, 28, 1568–1581. [Google Scholar] [CrossRef]
- Cui, Y.; Prokin, I.; Xu, H.; Delord, B.; Genet, S.; Venance, L.; Berry, H. Endocannabinoid dynamics gate spike-timing dependent depression and potentiation. eLife 2016, 5, e13185. [Google Scholar] [CrossRef]
- Piette, C.; Cui, Y.; Gervasi, N.; Venance, L. Lights on Endocannabinoid-Mediated Synaptic Potentiation. Front. Mol. Neurosci. 2020, 13, 132. [Google Scholar] [CrossRef]
- Cui, Y.; Paillé, V.; Xu, H.; Genet, S.; Delord, B.; Fino, E.; Berry, H.; Venance, L. Endocannabinoids mediate bidirectional striatal spike-timing-dependent plasticity. J. Physiol. 2015, 593, 2833–2849. [Google Scholar] [CrossRef] [Green Version]
- Raboune, S.; Stuart, J.M.; Leishman, E.; Takacs, S.M.; Rhodes, B.; Basnet, A.; Jameyfield, E.; McHugh, D.; Widlanski, T.; Bradshaw, H.B. Novel endogenous N-acyl amides activate TRPV1-4 receptors, BV-2 microglia, and are regulated in brain in an acute model of inflammation. Front. Cell Neurosci. 2014, 8, 195. [Google Scholar] [CrossRef] [Green Version]
- Muller, C.; Morales, P.; Reggio, P.H. Cannabinoid Ligands Targeting TRP Channels. Front. Mol. Neurosci. 2018, 11, 487. [Google Scholar] [CrossRef]
- De Petrocellis, L.; Starowicz, K.; Moriello, A.S.; Vivese, M.; Orlando, P.; Di Marzo, V. Regulation of transient receptor potential channels of melastatin type 8 (TRPM8): Effect of cAMP, cannabinoid CB(1) receptors and endovanilloids. Exp. Cell Res. 2007, 313, 1911–1920. [Google Scholar] [CrossRef]
- Ambudkar, I.S.; Bandyopadhyay, B.C.; Liu, X.; Lockwich, T.P.; Paria, B.; Ong, H.L. Functional organization of TRPC-Ca2+ channels and regulation of calcium microdomains. Cell Calcium 2006, 40, 495–504. [Google Scholar] [CrossRef]
- Bollimuntha, S.; Pani, B.; Singh, B.B. Neurological and Motor Disorders: Neuronal Store-Operated Ca. Adv. Exp. Med. Biol. 2017, 993, 535–556. [Google Scholar] [CrossRef] [Green Version]
- Parekh, A.B.; Putney, J.W. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef] [Green Version]
- Putney, J.W.; Steinckwich-Besançon, N.; Numaga-Tomita, T.; Davis, F.M.; Desai, P.N.; D’Agostin, D.M.; Wu, S.; Bird, G.S. The functions of store-operated calcium channels. Biochim. Biophys. Acta Mol. Cell Res. 2017, 1864, 900–906. [Google Scholar] [CrossRef]
- Lewis, R.S. Store-Operated Calcium Channels: From Function to Structure and Back Again. Cold Spring Harb. Perspect. Biol. 2020, 12, a035055. [Google Scholar] [CrossRef]
- Yen, M.; Lewis, R.S. Numbers count: How STIM and Orai stoichiometry affect store-operated calcium entry. Cell Calcium 2019, 79, 35–43. [Google Scholar] [CrossRef]
- Kraft, R. STIM and ORAI proteins in the nervous system. Channels 2015, 9, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deak, A.T.; Groschner, L.N.; Alam, M.R.; Seles, E.; Bondarenko, A.I.; Graier, W.F.; Malli, R. The endocannabinoid N-arachidonoyl glycine (NAGly) inhibits store-operated Ca2+ entry by preventing STIM1-Orai1 interaction. J. Cell Sci. 2013, 126, 879–888. [Google Scholar] [CrossRef] [Green Version]
- Bouron, A. Phyto and endocannabinoids exert complex actions on calcium and zinc signaling in mouse cortical neurons. Biochem. Pharmacol. 2018, 152, 244–251. [Google Scholar] [CrossRef]
- Deveci, A.; Hasna, J.; Bouron, A. Inhibition of store-operated calcium channels by N-arachidonoyl glycine (NAGly): No evidence for the involvement of lipid-sensing G protein coupled receptors. Sci. Rep. 2020, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.J.; Vandenberg, R.J.; Vaughan, C.W. N-arachidonyl-glycine modulates synaptic transmission in superficial dorsal horn. Br. J. Pharmacol. 2010, 161, 925–935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 2012, 4, a005710. [Google Scholar] [CrossRef] [Green Version]
- Calton, J.L.; Kang, M.H.; Wilson, W.A.; Moore, S.D. NMDA-Receptor-dependent synaptic activation of voltage-dependent calcium channels in basolateral amygdala. J. Neurophysiol. 2000, 83, 685–692. [Google Scholar] [CrossRef]
- Paoletti, P. Molecular basis of NMDA receptor functional diversity. Eur. J. Neurosci. 2011, 33, 1351–1365. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, N.G.; Siegler Retchless, B.; Johnson, J.W. Molecular bases of NMDA receptor subtype-dependent properties. J. Physiol. 2015, 593, 83–95. [Google Scholar] [CrossRef]
- Regan, M.C.; Romero-Hernandez, A.; Furukawa, H. A structural biology perspective on NMDA receptor pharmacology and function. Curr. Opin. Struct. Biol. 2015, 33, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Hampson, R.E.; Miller, F.; Palchik, G.; Deadwyler, S.A. Cannabinoid receptor activation modifies NMDA receptor mediated release of intracellular calcium: Implications for endocannabinoid control of hippocampal neural plasticity. Neuropharmacology 2011, 60, 944–952. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Muñoz, M.; Sánchez-Blázquez, P.; Merlos, M.; Garzón-Niño, J. Endocannabinoid control of glutamate NMDA receptors: The therapeutic potential and consequences of dysfunction. Oncotarget 2016, 7, 55840–55862. [Google Scholar] [CrossRef] [Green Version]
- Hashimotodani, Y.; Ohno-Shosaku, T.; Watanabe, M.; Kano, M. Roles of phospholipase Cbeta and NMDA receptor in activity-dependent endocannabinoid release. J. Physiol. 2007, 584, 373–380. [Google Scholar] [CrossRef]
- Liu, Q.; Bhat, M.; Bowen, W.D.; Cheng, J. Signaling pathways from cannabinoid receptor-1 activation to inhibition of N-methyl-D-aspartic acid mediated calcium influx and neurotoxicity in dorsal root ganglion neurons. J. Pharmacol. Exp. Ther. 2009, 331, 1062–1070. [Google Scholar] [CrossRef] [Green Version]
- Van der Stelt, M.; Di Marzo, V. Cannabinoid receptors and their role in neuroprotection. Neuromol. Med. 2005, 7, 37–50. [Google Scholar] [CrossRef]
- Khaspekov, L.G.; Brenz Verca, M.S.; Frumkina, L.E.; Hermann, H.; Marsicano, G.; Lutz, B. Involvement of brain-derived neurotrophic factor in cannabinoid receptor-dependent protection against excitotoxicity. Eur. J. Neurosci. 2004, 19, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Won, S.J.; Mao, X.O.; Jin, K.; Greenberg, D.A. Molecular mechanisms of cannabinoid protection from neuronal excitotoxicity. Mol. Pharmacol. 2006, 69, 691–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohno-Shosaku, T.; Hashimotodani, Y.; Ano, M.; Takeda, S.; Tsubokawa, H.; Kano, M. Endocannabinoid signalling triggered by NMDA receptor-mediated calcium entry into rat hippocampal neurons. J. Physiol. 2007, 584, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Sánchez, A.; Sánchez-Blázquez, P.; Rodríguez-Muñoz, M.; Garzón, J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol. Brain 2013, 6, 42. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; See, Y.M.; Lee, J. A Systematic Review of the Effectiveness of Medical Cannabis for Psychiatric, Movement and Neurodegenerative Disorders. Clin. Psychopharmacol. Neurosci. 2017, 15, 301–312. [Google Scholar] [CrossRef] [Green Version]
- Kluger, B.; Triolo, P.; Jones, W.; Jankovic, J. The therapeutic potential of cannabinoids for movement disorders. Mov. Disord. 2015, 30, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Babayeva, M.; Assefa, H.; Basu, P.; Chumki, S.; Loewy, Z. Marijuana Compounds: A Nonconventional Approach to Parkinson’s Disease Therapy. Parkinson’s Dis. 2016, 2016, 1279042. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boczek, T.; Zylinska, L. Receptor-Dependent and Independent Regulation of Voltage-Gated Ca2+ Channels and Ca2+-Permeable Channels by Endocannabinoids in the Brain. Int. J. Mol. Sci. 2021, 22, 8168. https://doi.org/10.3390/ijms22158168
Boczek T, Zylinska L. Receptor-Dependent and Independent Regulation of Voltage-Gated Ca2+ Channels and Ca2+-Permeable Channels by Endocannabinoids in the Brain. International Journal of Molecular Sciences. 2021; 22(15):8168. https://doi.org/10.3390/ijms22158168
Chicago/Turabian StyleBoczek, Tomasz, and Ludmila Zylinska. 2021. "Receptor-Dependent and Independent Regulation of Voltage-Gated Ca2+ Channels and Ca2+-Permeable Channels by Endocannabinoids in the Brain" International Journal of Molecular Sciences 22, no. 15: 8168. https://doi.org/10.3390/ijms22158168