
Novel High Affinity Sigma-1 Receptor Ligands from Minimal Ensemble Docking-Based Virtual Screening

 , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
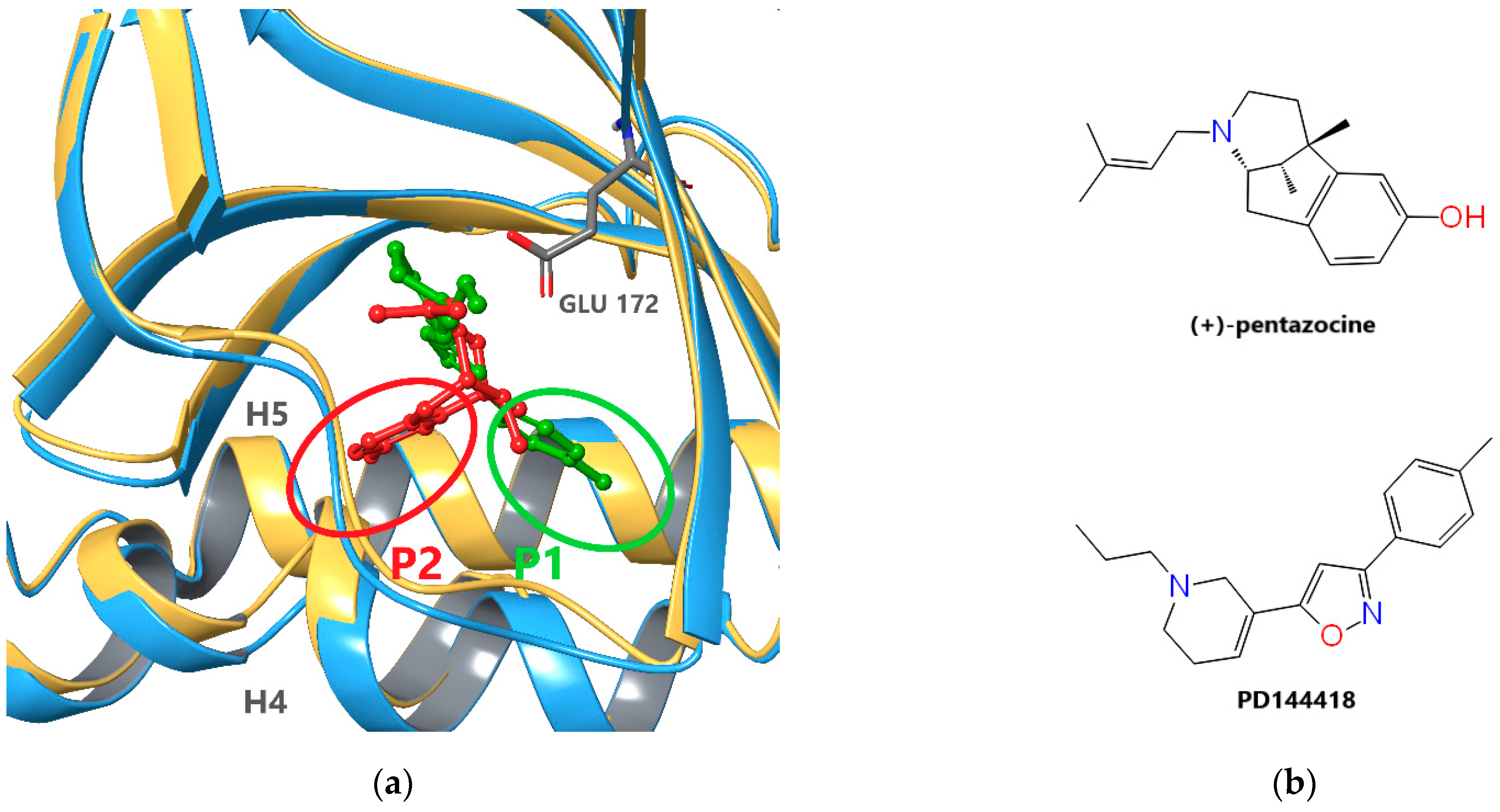
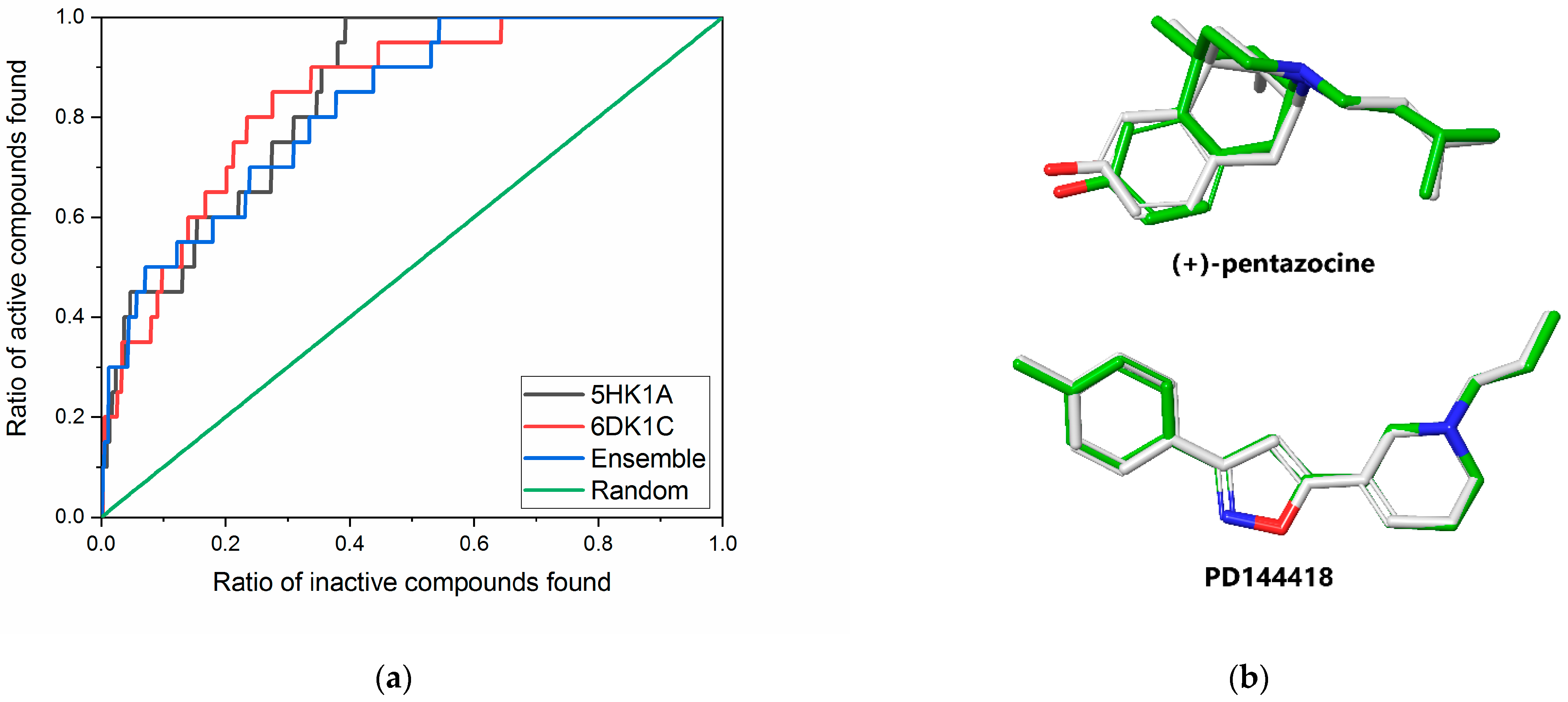
2.1. Receptor Model Selection and Validation
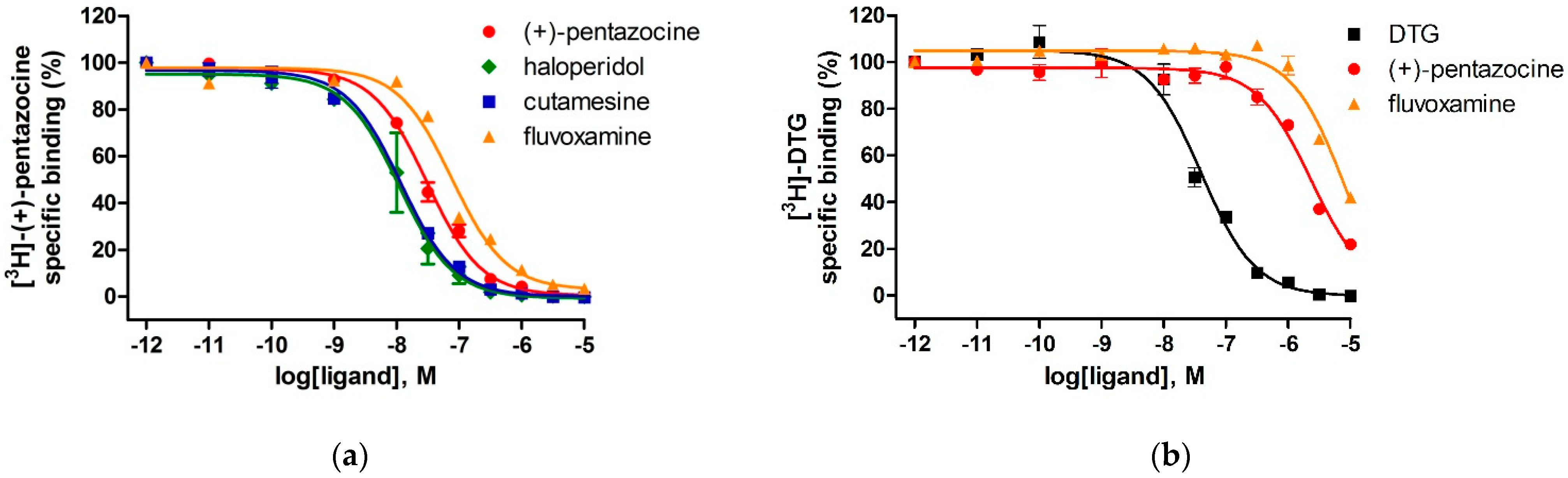
2.2. Setup and Validation of In Vitro Competitive Binding Assays
2.3. Screening of Our in-House Library

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Measured Ki (nM) | Docking Score (kcal/mol) | Chemical Name | Structure of Best Docked Stereoisomer |
|---|---|---|---|---|
| L1 1 | 32.0 | −11.62 | N-Benzyl-6,7-dimethoxy-1,2,3,4-tetrahydro-1-isoquinolineethanamine dihydrochloride |  |
| L2 1 | 91.0 | −12.41 | 3-Amino-N-(2-fluoro-3-(trifluoromethyl)benzyl]-3-phenylpropanamide hydrochloride |  |
| L3 2 | 110.0 | −11.95 | 1-[(4-Methoxyphenoxy)methyl]-2-(1,2,3,4-tetrahydroisoquinolin-2-yl)ethanol |  |
| L4 1 | 420.0 | −12.47 | 3-Amino-N-(3-fluoro-5-(trifluoromethyl)benzyl]-3-phenylpropanamide hydrochloride |  |
| L5 1 | 463.0 | −10.74 | (±)-diendo-3′-amino-1-benzyl-5′,8′-methano-4′a,5′,8′,8′a-tetrahydrospiro[piperidine-4,2′(1′H)-quinazolin]-4′(3′H)-one |  |
| L6 2 | 1036.0 | −11.12 | N-Benzyloxycarbonyl-(9-methyl-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)methanamine |  |
| L7 2 | 1381.0 | −11.56 | (1S,3R,4R,6R)-3-(benzylamino)methyl)-7,7-dimethylbicyclo[4.1.0]heptane-3,4-diol |  |
| L8 2 | 1534.0 | −10.68 | 3-Amino-N-(2-fluoro-3-(trifluoromethyl)benzyl]-2-methylpropanamide trifluoroacetate |  |
| L9 1 | 1716.0 | −10.87 | N-Benzyloxycarbonyl-(2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indol-1-yl)methanamine |  |
| L10 1 | 2154.0 | −11.58 | (4R*,11bR*)-9,10-Diethoxy-4-[4-(dimethylamino)phenyl]-1,3,4,6,7,11b-hexahydro-2H-pyrimido[6,1-a]isoquinoline |  |
| L11 1 | 2381.0 | −10.55 | 1-{[(Benzyloxycarbonyl)amino]methyl}-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline |  |
| L12 2 | 3266.0 | −9.69 | (1R*,9bR*)-1-{[(4-Chlorophenyl)thio]methyl}-7,8-diethoxy-1,4,5,9b-tetrahydro-2H-azeto[2,1a]-isoquinoline |  |
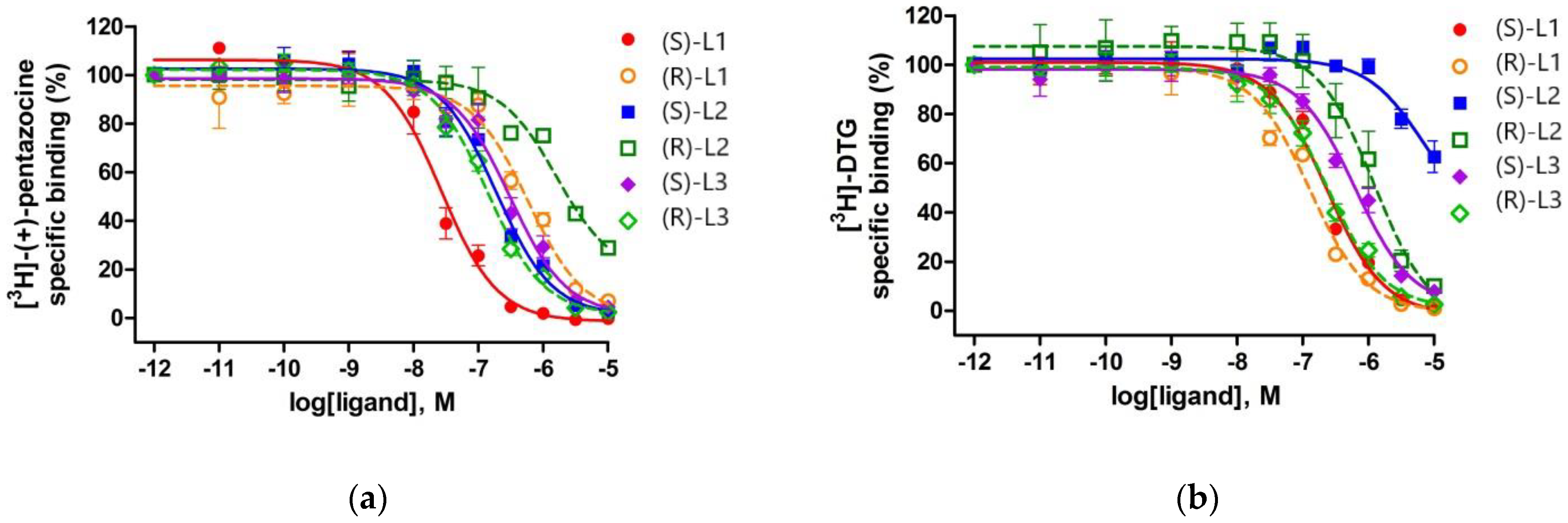
2.4. Binding Affinity of the Enantiomers of the Best Compounds to S1R and S2R
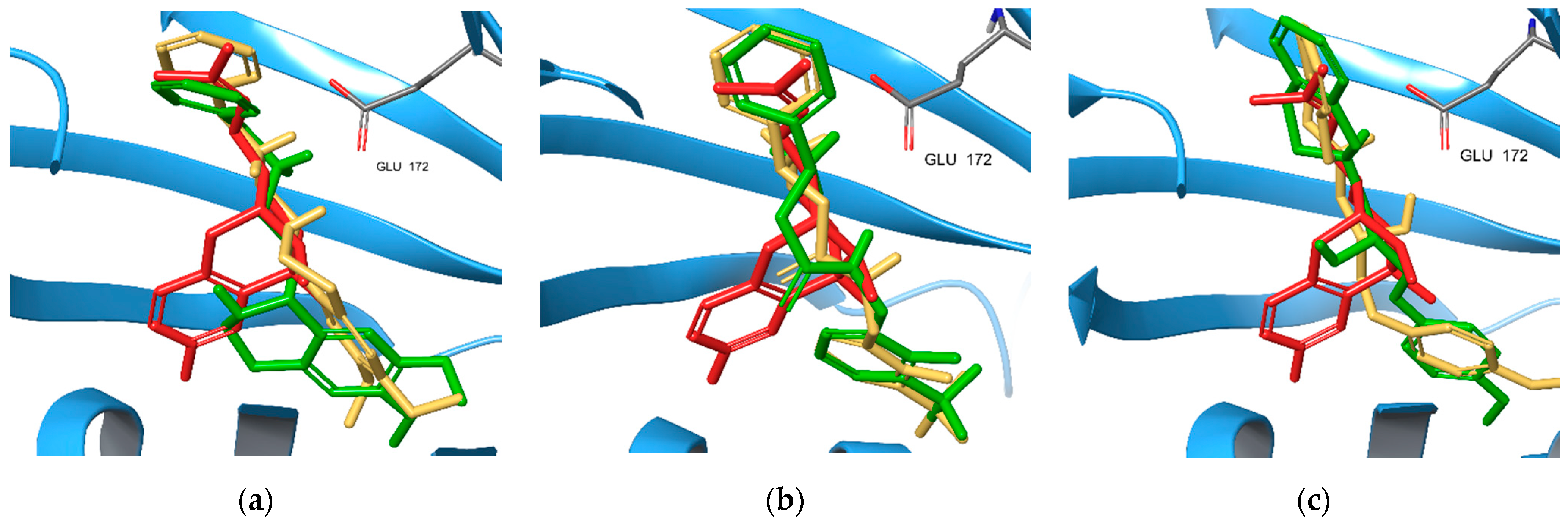
2.5. S1R Binding Poses of Enantiomers of the New Compounds
3. Materials and Methods
3.1. Virtual Screening
3.1.1. Receptor Structures
3.1.2. Model Validation Libraries
3.1.3. Docking Protocol
3.1.4. Model Evaluation
3.2. Binding Assays
3.2.1. Materials
3.2.2. Preparation of Membrane Homogenates for S1R and S2R Binding Assays
3.2.3. Saturation Binding Experiments
3.2.4. Radioligand Binding Assays of Sigma-1 Receptor
3.2.5. Radioligand Binding Assays of Sigma-2 Receptor
3.2.6. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The Effects of Morphine- and Nalorphine- like Drugs in the Nondependent and Morphine-Dependent Chronic Spinal Dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar]
- Su, T.P. Evidence for Sigma Opioid Receptor: Binding of [3H]SKF-10047 to Etorphine-Inaccessible Sites in Guinea-Pig Brain. J. Pharmacol. Exp. Ther. 1982, 223, 284–290. [Google Scholar] [PubMed]
- Tam, S.W.; Cook, L. Sigma Opiates and Certain Antipsychotic Drugs Mutually Inhibit (+)-[3H] SKF 10,047 and [3H]Haloperidol Binding in Guinea Pig Brain Membranes. Proc. Natl. Acad. Sci. USA 1984, 81, 5618–5621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexander, S.P.H.; Benson, H.E.; Faccenda, E.; Pawson, A.J.; Sharman, J.L.; McGrath, J.C.; Catterall, W.A.; Spedding, M.; Peters, J.A.; Harmar, A.J.; et al. The Concise Guide to PHARMACOLOGY 2013/14: Overview: Overview. Br. J. Pharmacol. 2013, 170, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Hellewell, S.B.; Bruce, A.; Feinstein, G.; Orringer, J.; Williams, W.; Bowen, W.D. Rat Liver and Kidney Contain High Densities of Σ1 and Σ2 Receptors: Characterization by Ligand Binding and Photoaffinity Labeling. Eur. J. Pharmacol. Mol. Pharmacol. 1994, 268, 9–18. [Google Scholar] [CrossRef]
- Penke, B.; Fülöp, L.; Szűcs, M.; Frecska, E. The Role of Sigma-1 Receptor, an Intracellular Chaperone in Neurodegenerative Diseases. Curr. Neuropharmacol. 2017, 16, 97–116. [Google Scholar] [CrossRef] [PubMed]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, Molecular Cloning, and Expression of the Mammalian Sigma1-Binding Site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.R.; Zheng, S.; Gurpinar, E.; Koehl, A.; Manglik, A.; Kruse, A.C. Crystal Structure of the Human Σ1 Receptor. Nature 2016, 532, 527–530. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Mach, R.H. The Evolution of the Sigma-2 (σ2) Receptor from Obscure Binding Site to Bona Fide Therapeutic Target. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets; Smith, S.B., Su, T.-P., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 964, pp. 49–61. ISBN 978-3-319-50172-7. [Google Scholar]
- Alon, A.; Schmidt, H.R.; Wood, M.D.; Sahn, J.J.; Martin, S.F.; Kruse, A.C. Identification of the Gene That Codes for the σ2 Receptor. Proc. Natl. Acad. Sci. USA 2017, 114, 7160–7165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nastasi, G.; Miceli, C.; Pittalà, V.; Modica, M.N.; Prezzavento, O.; Romeo, G.; Rescifina, A.; Marrazzo, A.; Amata, E. S2RSLDB: A Comprehensive Manually Curated, Internet-Accessible Database of the Sigma-2 Receptor Selective Ligands. J. Cheminf. 2017, 9, 3. [Google Scholar] [CrossRef] [Green Version]
- Georgiadis, M.-O.; Karoutzou, O.; Foscolos, A.-S.; Papanastasiou, I. Sigma Receptor (σR) Ligands with Antiproliferative and Anticancer Activity. Molecules 2017, 22, 1408. [Google Scholar] [CrossRef] [Green Version]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.-C. Small Molecules Selectively Targeting Sigma-1 Receptor for the Treatment of Neurological Diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef]
- Langa, F.; Codony, X.; Tovar, V.; Lavado, A.; Gimenez, E.; Cozar, P.; Cantero, M.; Dordal, A.; Hernandez, E.; Perez, R.; et al. Generation and Phenotypic Analysis of Sigma Receptor Type I (Sigma1) Knockout Mice. Eur. J. Neurosci. 2003, 18, 2188–2196. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal Sigma-1 Receptors: Signaling Functions and Protective Roles in Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Kruse, A.C. The Molecular Function of σ Receptors: Past, Present, and Future. Trends Pharmacol. Sci. 2019, 40, 636–654. [Google Scholar] [CrossRef]
- Monassier, L.; Bousquet, P. Sigma Receptors: From Discovery to Highlights of Their Implications in the Cardiovascular System. Fundam. Clin. Pharmacol. 2002, 16, 1–8. [Google Scholar] [CrossRef]
- Hayashi, T.; Su, T.-P. Regulating Ankyrin Dynamics: Roles of Sigma-1 Receptors. Proc. Natl. Acad. Sci. USA 2001, 98, 491–496. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Wilke, R.A.; Aydar, E.; Palmer, C.P.; Chen, Y.; Ruoho, A.E.; Jackson, M.B. Membrane-delimited Coupling between Sigma Receptors and K+ Channels in Rat Neurohypophysial Terminals Requires Neither G-protein nor ATP. J. Physiol. 2000, 526, 527–539. [Google Scholar] [CrossRef]
- Brimson, J.; Brown, C.; Safrany, S. Antagonists Show GTP-sensitive High-affinity Binding to the Sigma-1 Receptor. Br. J. Pharmacol. 2011, 164, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Delprat, B.; Crouzier, L.; Su, T.-P.; Maurice, T. At the Crossing of ER Stress and MAMs: A Key Role of Sigma-1 Receptor. In Calcium Signaling; Islam, M., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2020; Volume 1131, pp. 699–718. ISBN 978-3-030-12456-4. [Google Scholar]
- Christ, M.G.; Clement, A.M.; Behl, C. The Sigma-1 Receptor at the Crossroad of Proteostasis, Neurodegeneration, and Autophagy. Trends Neurosci. 2020, 43, 79–81. [Google Scholar] [CrossRef]
- Hayashi, T. The Sigma-1 Receptor in Cellular Stress Signaling. Front. Neurosci. 2019, 13, 733. [Google Scholar] [CrossRef] [Green Version]
- Su, T.-P.; Su, T.-C.; Nakamura, Y.; Tsai, S.-Y. The Sigma-1 Receptor as a Pluripotent Modulator in Living Systems. Trends Pharmacol. Sci. 2016, 37, 262–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, U.B.; Ruoho, A.E. Biochemical Pharmacology of the Sigma-1 Receptor. Mol. Pharmacol. 2016, 89, 142–153. [Google Scholar] [CrossRef] [Green Version]
- Yano, H.; Bonifazi, A.; Xu, M.; Guthrie, D.A.; Schneck, S.N.; Abramyan, A.M.; Fant, A.D.; Hong, W.C.; Newman, A.H.; Shi, L. Pharmacological Profiling of Sigma 1 Receptor Ligands by Novel Receptor Homomer Assays. Neuropharmacology 2018, 133, 264–275. [Google Scholar] [CrossRef]
- Miki, Y.; Mori, F.; Kon, T.; Tanji, K.; Toyoshima, Y.; Yoshida, M.; Sasaki, H.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Accumulation of the Sigma-1 Receptor Is Common to Neuronal Nuclear Inclusions in Various Neurodegenerative Diseases: SIGMAR1 in Neuronal Nuclear Inclusions. Neuropathology 2014, 34, 148–158. [Google Scholar] [CrossRef]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.A.; Cavendish, J.Z.; Robson, M.J.; Scandinaro, A.L.; Matsumoto, R.R. Role of Sigma-1 Receptors in Neurodegenerative Diseases. J. Pharmacol. Sci. 2015, 127, 17–29. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Lucke-Wold, B.P.; Mookerjee, S.; Kaushal, N.; Matsumoto, R.R. Sigma-1 Receptors and Neurodegenerative Diseases: Towards a Hypothesis of Sigma-1 Receptors as Amplifiers of Neurodegeneration and Neuroprotection. In Sigma Receptors: Their Role in Disease and as Therapeutic Targets; Smith, S.B., Su, T.-P., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; Volume 964, pp. 133–152. ISBN 978-3-319-50172-7. [Google Scholar]
- Maurice, T.; Goguadze, N. Sigma-1 (σ1) Receptor in Memory and Neurodegenerative Diseases. In Sigma Proteins: Evolution of the Concept of Sigma Receptors; Kim, F.J., Pasternak, G.W., Eds.; Handbook of Experimental Pharmacology; Springer International Publishing: Cham, Switzerland, 2017; Volume 244, pp. 81–108. ISBN 978-3-319-65851-3. [Google Scholar]
- Ruscher, K.; Wieloch, T. The Involvement of the Sigma-1 Receptor in Neurodegeneration and Neurorestoration. J. Pharmacol. Sci. 2015, 127, 30–35. [Google Scholar] [CrossRef]
- Crouzier, L.; Couly, S.; Roques, C.; Peter, C.; Belkhiter, R.; Arguel Jacquemin, M.; Bonetto, A.; Delprat, B.; Maurice, T. Sigma-1 (σ1) Receptor Activity Is Necessary for Physiological Brain Plasticity in Mice. Eur. Neuropsychopharmacol. 2020, 39, 29–45. [Google Scholar] [CrossRef]
- Atzmon, A.; Herrero, M.; Sharet-Eshed, R.; Gilad, Y.; Senderowitz, H.; Elroy-Stein, O. Drug Screening Identifies Sigma-1-Receptor as a Target for the Therapy of VWM Leukodystrophy. Front. Mol. Neurosci. 2018, 11, 336. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhu, L.; Liu, D.; Chi, T.; Ji, X.; Liu, P.; Yang, X.; Tian, X.; Zou, L. Sigma-1 Receptor Protects against Endoplasmic Reticulum Stress-Mediated Apoptosis in Mice with Cerebral Ischemia/Reperfusion Injury. Apoptosis 2019, 24, 157–167. [Google Scholar] [CrossRef]
- Mitsuda, T.; Omi, T.; Tanimukai, H.; Sakagami, Y.; Tagami, S.; Okochi, M.; Kudo, T.; Takeda, M. Sigma-1Rs Are Upregulated via PERK/EIF2α/ATF4 Pathway and Execute Protective Function in ER Stress. Biochem. Biophys. Res. Commun. 2011, 415, 519–525. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Iacomino, A.; Steardo, L.; Scuderi, C. Targeting Neuroinflammation in Alzheimer’s Disease. J. Inflamm. Res. 2016, 9, 199–208. [Google Scholar] [CrossRef] [Green Version]
- Jia, J.; Cheng, J.; Wang, C.; Zhen, X. Sigma-1 Receptor-Modulated Neuroinflammation in Neurological Diseases. Front. Cell. Neurosci. 2018, 12, 314. [Google Scholar] [CrossRef] [Green Version]
- Stracina, T.; Novakova, M. Cardiac Sigma Receptors—An Update. Physiol. Res. 2018, S561–S576. [Google Scholar] [CrossRef] [PubMed]
- Oyer, H.M.; Sanders, C.M.; Kim, F.J. Small-Molecule Modulators of Sigma1 and Sigma2/TMEM97 in the Context of Cancer: Foundational Concepts and Emerging Themes. Front. Pharmacol. 2019, 10, 1141. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Su, T.-P. Sigma-1 Receptor Ligands: Potential in the Treatment of Neuropsychiatric Disorders. CNS Drugs 2004, 18, 269–284. [Google Scholar] [CrossRef]
- Glennon, R.A.; Ablordeppey, S.Y.; Ismaiel, A.M.; El-Ashmawy, M.B.; Fischer, J.B.; Howie, K.B. Structural Features Important for Sigma.1 Receptor Binding. J. Med. Chem. 1994, 37, 1214–1219. [Google Scholar] [CrossRef]
- Glennon, R. Pharmacophore Identification for Sigma-1 (σ1) Receptor Binding: Application of the “Deconstruction—Reconstruction—Elaboration” Approach. Mini-Rev. Med. Chem. 2005, 5, 927–940. [Google Scholar] [CrossRef]
- Pascual, R.; Almansa, C.; Plata-Salamán, C.; Vela, J.M. A New Pharmacophore Model for the Design of Sigma-1 Ligands Validated on a Large Experimental Dataset. Front. Pharmacol. 2019, 10, 519. [Google Scholar] [CrossRef]
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the Worldwide Protein Data Bank. Nat. Struct. Mol. Biol. 2003, 10, 980. [Google Scholar] [CrossRef]
- Schmidt, H.R.; Betz, R.M.; Dror, R.O.; Kruse, A.C. Structural Basis for Σ1 Receptor Ligand Recognition. Nat. Struct. Mol. Biol. 2018, 25, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Rossino, G.; Rui, M.; Pozzetti, L.; Schepmann, D.; Wünsch, B.; Zampieri, D.; Pellavio, G.; Laforenza, U.; Rinaldi, S.; Colombo, G.; et al. Setup and Validation of a Reliable Docking Protocol for the Development of Neuroprotective Agents by Targeting the Sigma-1 Receptor (S1R). Int. J. Mol. Sci. 2020, 21, 7708. [Google Scholar] [CrossRef]
- Greenfield, D.A.; Schmidt, H.R.; Skiba, M.A.; Mandler, M.D.; Anderson, J.R.; Sliz, P.; Kruse, A.C. Virtual Screening for Ligand Discovery at the σ 1 Receptor. ACS Med. Chem. Lett. 2020, 11, 1555–1561. [Google Scholar] [CrossRef] [PubMed]
- Carlson, H.A.; Masukawa, K.M.; McCammon, J.A. Method for Including the Dynamic Fluctuations of a Protein in Computer-Aided Drug Design. J. Phys. Chem. A 1999, 103, 10213–10219. [Google Scholar] [CrossRef] [Green Version]
- Amaro, R.E.; Baudry, J.; Chodera, J.; Demir, Ö.; McCammon, J.A.; Miao, Y.; Smith, J.C. Ensemble Docking in Drug Discovery. Biophys. J. 2018, 114, 2271–2278. [Google Scholar] [CrossRef] [Green Version]
- Tarcsay, Á.; Paragi, G.; Vass, M.; Jójárt, B.; Bogár, F.; Keserű, G.M. The Impact of Molecular Dynamics Sampling on the Performance of Virtual Screening against GPCRs. J. Chem. Inf. Model. 2013, 53, 2990–2999. [Google Scholar] [CrossRef]
- Swift, R.V.; Jusoh, S.A.; Offutt, T.L.; Li, E.S.; Amaro, R.E. Knowledge-Based Methods To Train and Optimize Virtual Screening Ensembles. J. Chem. Inf. Model. 2016, 56, 830–842. [Google Scholar] [CrossRef]
- Liu, T.; Lin, Y.; Wen, X.; Jorissen, R.N.; Gilson, M.K. BindingDB: A Web-Accessible Database of Experimentally Determined Protein-Ligand Binding Affinities. Nucleic Acids Res. 2007, 35, D198–D201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Schrödinger Release 2019-4; Schrödinger, LLC: New York, NY, USA, 2019.
- Fontanilla, D.; Hajipour, A.R.; Pal, A.; Chu, U.B.; Arbabian, M.; Ruoho, A.E. Probing the Steroid Binding Domain-like I (SBDLI) of the Sigma-1 Receptor Binding Site Using N-Substituted Photoaffinity Labels. Biochemistry 2008, 47, 7205–7217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanilla, D.; Johannessen, M.; Hajipour, A.R.; Cozzi, N.V.; Jackson, M.B.; Ruoho, A.E. The Hallucinogen N,N-Dimethyltryptamine (DMT) Is an Endogenous Sigma-1 Receptor Regulator. Science 2009, 323, 934–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, A.; Hajipour, A.R.; Fontanilla, D.; Ramachandran, S.; Chu, U.B.; Mavlyutov, T.; Ruoho, A.E. Identification of Regions of the Sigma-1 Receptor Ligand Binding Site Using a Novel Photoprobe. Mol. Pharmacol. 2007, 72, 921–933. [Google Scholar] [CrossRef] [PubMed]
- De Costa, B.R.; Bowen, W.D.; Hellewell, S.B.; Walker, J.M.; Thurkauf, A.; Jacobson, A.E.; Rice, K.C. Synthesis and Evaluation of Optically Pure [3H]-(+)-Pentazocine, a Highly Potent and Selective Radioligand for Sigma Receptors. FEBS Lett. 1989, 251, 53–58. [Google Scholar] [CrossRef] [Green Version]
- Ramachandran, S.; Lu, H.; Prabhu, U.; Ruoho, A.E. Purification and Characterization of the Guinea Pig Sigma-1 Receptor Functionally Expressed in Escherichia Coli. Protein Expr. Purif. 2007, 51, 283–292. [Google Scholar] [CrossRef] [Green Version]
- Chu, U.B.; Ruoho, A.E. Sigma Receptor Binding Assays. Curr. Protoc. Pharmacol. 2015, 71, 1.34.1–1.34.21. [Google Scholar] [CrossRef] [Green Version]
- Fishback, J.A.; Rosen, A.; Bhat, R.; McCurdy, C.R.; Matsumoto, R.R. A 96-Well Filtration Method for Radioligand Binding Analysis of σ Receptor Ligands. J. Pharm. Biomed. Anal. 2012, 71, 157–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebreselassie, D.; Bowen, W.D. Sigma-2 Receptors Are Specifically Localized to Lipid Rafts in Rat Liver Membranes. Eur. J. Pharmacol. 2004, 493, 19–28. [Google Scholar] [CrossRef] [Green Version]
- Quirion, R.; Bowen, W.D.; Itzhak, Y.; Junien, J.L.; Musacchio, J.M.; Rothman, R.B.; Su, T.P.; Tam, S.W.; Taylor, D.P. A Proposal for the Classification of Sigma Binding Sites. Trends Pharmacol. Sci. 1992, 13, 85–86. [Google Scholar] [CrossRef]
- Cobos, E.; Entrena, J.; Nieto, F.; Cendan, C.; Pozo, E. Pharmacology and Therapeutic Potential of Sigma1 Receptor Ligands. Curr. Neuropharmacol. 2008, 6, 344–366. [Google Scholar] [CrossRef]
- Narita, N.; Hashimoto, K.; Tomitaka, S.; Minabe, Y. Interactions of Selective Serotonin Reuptake Inhibitors with Subtypes of σ Receptors in Rat Brain. Eur. J. Pharmacol. 1996, 307, 117–119. [Google Scholar] [CrossRef]
- Lever, J.R.; Gustafson, J.L.; Xu, R.; Allmon, R.L.; Lever, S.Z. Σ1 and Σ2 Receptor Binding Affinity and Selectivity of SA4503 and Fluoroethyl SA4503. Synapse 2006, 59, 350–358. [Google Scholar] [CrossRef]
- Ishima, T.; Fujita, Y.; Hashimoto, K. Interaction of New Antidepressants with Sigma-1 Receptor Chaperones and Their Potentiation of Neurite Outgrowth in PC12 Cells. Eur. J. Pharmacol. 2014, 727, 167–173. [Google Scholar] [CrossRef]
- Roos, K.; Wu, C.; Damm, W.; Reboul, M.; Stevenson, J.M.; Lu, C.; Dahlgren, M.K.; Mondal, S.; Chen, W.; Wang, L.; et al. OPLS3e: Extending Force Field Coverage for Drug-Like Small Molecules. J. Chem. Theory Comput. 2019, 15, 1863–1874. [Google Scholar] [CrossRef] [PubMed]
- Truchon, J.-F.; Bayly, C.I. Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef]
- Dvorácskó, S.; Keresztes, A.; Mollica, A.; Stefanucci, A.; Macedonio, G.; Pieretti, S.; Zádor, F.; Walter, F.R.; Deli, M.A.; Kékesi, G.; et al. Preparation of Bivalent Agonists for Targeting the Mu Opioid and Cannabinoid Receptors. Eur. J. Med. Chem. 2019, 178, 571–588. [Google Scholar] [CrossRef] [Green Version]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]





| Model | RIE | BEDROC (α = 160.9) | BEDROC (α = 80.5) | EF1% | EF2% | ROC |
|---|---|---|---|---|---|---|
| 6DK1A | 5.00 | 0.290 | 0.245 | 15.3 | 10.2 | 0.82 |
| 6DK1B | 4.74 | 0.261 | 0.219 | 10.2 | 7.6 | 0.81 |
| 6DK1C | 5.77 | 0.318 | 0.291 | 20.4 | 10.2 | 0.84 |
| 5HK1A | 6.08 | 0.315 | 0.284 | 10.2 | 10.2 | 0.84 |
| 5HK1B | 5.92 | 0.174 | 0.201 | 5.1 | 7.6 | 0.86 |
| 5HK1C | 5.30 | 0.212 | 0.203 | 10.2 | 7.6 | 0.85 |
| 6DK1C-5HK1A | 6.37 | 0.328 | 0.316 | 15.3 | 15.3 | 0.82 |
| 6DK1C-5HK1B | 5.89 | 0.179 | 0.221 | 10.2 | 10.2 | 0.81 |
| 6DK1C-5HK1C | 6.11 | 0.208 | 0.238 | 5.1 | 20.2 | 0.85 |
| Ligand | S1R Ki ± S.E.M. (nM) | S2R Ki ± S.E.M. (nM) | Selectivity (S2R/S1R) |
|---|---|---|---|
| (+)-pentazocine | 4.8 ± 0.4 | 1698 ± 103 | 354 |
| DTG | n.d. | 29 ± 4 | n.d. |
| fluvoxamine | 31 ± 3 | 6187 ± 296 | 200 |
| haloperidol | 5.2 ± 1.3 | n.d. | n.d. |
| cutamesine | 5.5 ± 1.1 | n.d. | n.d. |
| Ligand | S1R Ki ± S.E.M. (nM) | S2R Ki ± S.E.M. (nM) | Selectivity (S2R/S1R) |
|---|---|---|---|
| (S)-L1 | 11 ± 3 | 169 ± 15 | 15.4 |
| (R)-L1 | 252 ± 12 | 94 ± 5.5 | 0.4 |
| (S)-L2 | 81 ± 3 | 5108 ± 960 | 63 |
| (R)-L2 | 699 ± 57 | 920 ± 25 | 1.3 |
| (S)-L3 | 132 ± 23 | 463 ± 33 | 3.5 |
| (R)-L3 | 58 ± 3 | 176 ± 21 | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dvorácskó, S.; Lázár, L.; Fülöp, F.; Palkó, M.; Zalán, Z.; Penke, B.; Fülöp, L.; Tömböly, C.; Bogár, F. Novel High Affinity Sigma-1 Receptor Ligands from Minimal Ensemble Docking-Based Virtual Screening. Int. J. Mol. Sci. 2021, 22, 8112. https://doi.org/10.3390/ijms22158112
Dvorácskó S, Lázár L, Fülöp F, Palkó M, Zalán Z, Penke B, Fülöp L, Tömböly C, Bogár F. Novel High Affinity Sigma-1 Receptor Ligands from Minimal Ensemble Docking-Based Virtual Screening. International Journal of Molecular Sciences. 2021; 22(15):8112. https://doi.org/10.3390/ijms22158112
Chicago/Turabian StyleDvorácskó, Szabolcs, László Lázár, Ferenc Fülöp, Márta Palkó, Zita Zalán, Botond Penke, Lívia Fülöp, Csaba Tömböly, and Ferenc Bogár. 2021. "Novel High Affinity Sigma-1 Receptor Ligands from Minimal Ensemble Docking-Based Virtual Screening" International Journal of Molecular Sciences 22, no. 15: 8112. https://doi.org/10.3390/ijms22158112