
The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis

1
Département de Pharmacologie et Physiologie, Université de Montréal, Montréal, QC H3C 3J7, Canada
2
Laboratory of Molecular and Cellular Biology, Montréal Heart Institute, Montréal, QC H1T 1C8, Canada
3
Faculty of Pharmacy, Université de Montréal, Montréal, QC H3C 3J7, Canada
4
Centre de Biomédecine, CIUSSS-NÎM/Hôpital du Sacré-Cœur, Montréal, QC H4J 1C5, Canada
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(15), 8074; https://doi.org/10.3390/ijms22158074
Submission received: 26 June 2021
/
Revised: 13 July 2021
/
Accepted: 15 July 2021
/
Published: 28 July 2021
(This article belongs to the Special Issue Cardiovascular Diseases–a Focus on Atherosclerosis, Its Prophylaxis, Complications and Recent Advancements in Therapies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Hypercholesterolemia plays a causal role in the development of atherosclerosis and is one of the main risk factors for cardiovascular disease (CVD), the leading cause of death worldwide especially in developed countries. Current data show that the role of microbiota extends beyond digestion by being implicated in several metabolic and inflammatory processes linked to several diseases including CVD. Studies have reported associations between bacterial metabolites and hypercholesterolemia. However, such associations remain poorly investigated and characterized. In this review, the mechanisms of microbial derived metabolites such as primary and secondary bile acids (BAs), trimethylamine N-oxide (TMAO), and short-chain fatty acids (SCFAs) will be explored in the context of cholesterol metabolism. These metabolites play critical roles in maintaining cardiovascular health and if dysregulated can potentially contribute to CVD. They can be modulated via nutritional and pharmacological interventions such as statins, prebiotics, and probiotics. However, the mechanisms behind these interactions also remain unclear, and mechanistic insights into their impact will be provided. Therefore, the objectives of this paper are to present current knowledge on potential mechanisms whereby microbial metabolites regulate cholesterol homeostasis and to discuss the feasibility of modulating intestinal microbes and metabolites as a novel therapeutic for hypercholesterolemia.
1. Introduction
The gut is home to trillions of microorganisms that include fungi, parasites, viruses, and bacteria. The microbiota is a hidden organ of the human system establishing homeostasis or disease in an individual. In a healthy state, these interactions are largely symbiotic and influence a host’s nutrition, metabolism, energy, and immunity [1]. Modern methodologies have given insights into the role of the gut in many processes such as the absorption, distribution and extraction of nutrients, synthesis of vitamins, immunomodulation, and protection against pathogens [2]. Composition of the microbiota can vary between people of the same ethnicity, age, or lifestyle because there is a complex interaction between classic genetic and environmental factors. There are over 50 bacterial phyla in the gut, yet there are two predominant phyla governing the gut microbiome: the Gram-positive Firmicutes (e.g., Enterococcus and Lactobacillus) and the Gram-negative Bacteroidetes (e.g., Bacteroides). The other phyla are present in variable numbers and include Actinobacteria (Bifidobacterium), Proteobacteria, and Verrucomicrobia [3]. Overall, numerous factors such as the physiological conditions of the host (e.g., age and stress), dietary habits, environmental factors (e.g., use of medications especially antibiotic therapy), and intestinal infections can alter the diversity and composition in intestinal microorganisms creating a state of dysbiosis. This state has been associated with the pathogenesis of both intestinal and extraintestinal disorders such as irritable bowel syndrome, metabolic syndrome, obesity, diabetes, and cardiovascular system disorders [4]. Lower abundance of Bacteroides; higher abundance of Lactobacillus, Enterobacteriaceae, and Streptococcus spp.; and increased Firmicutes to Bacteroidetes ratio have been associated with cardiovascular disease (CVD) [5]. Most of these disorders may increase the permeability of the intestine, leading to increased systemic levels of bacterial products which in turn cause low-grade chronic inflammation [6]. This inflammation may promote the development of atherosclerosis and has also been hypothesized to alter plasma lipid and lipoprotein levels [7]. Despite extensive research in human and in animal models, the specific protective microorganisms depleted in dysbiotic cases and the precise molecular mechanisms causing disease remain unknown due to the complex interplay between genetic and nongenetic factors [8]. Evidence suggests that the gut microbiome is involved in the development of CVD as studies show its association with variations in body mass index and blood lipids levels, independent of age, sex, and host genetics [9].
Many findings suggest that the gut microbiota has the capacity to alter blood lipid composition in particular cholesterol, through their role in bile acid metabolism and the generation of microbial products [4,10,11,12]. The metabolism of cholesterol by gut microorganisms has been known for decades, although the specifics of these interactions on human health have only started to be considered. Given that cholesterol is an important signaling molecule in the body regulating multiple physiological functions such as BA production and hormone regulation, a disruption in its formation has many health repercussions [4]. High levels of blood cholesterol, also known as hypercholesterolemia, is a major risk factor for the development of atherosclerosis, a chronic lipid-driven inflammatory disease of the arteries, promoting myocardial infarctions and strokes [13,14]. The reduction of total cholesterol (TC) and low-density lipoprotein (LDL) cholesterol (LDLc) concentrations in the blood significantly decreases the risk of CVD [15]. Reduction of cholesterol levels can be achieved with changes to the diet and increase in physical activity, while lipid-lowering drugs are indicated only in selected situations where lifestyle modification is not possible or sufficient to reach desired target blood cholesterol concentration. It is important to note that nonpharmacological management should, as much as possible, accompany lipid-lowering therapy as it maximizes a patient’s outcome [16]. Lifestyle improvement can reduce LDLc levels by 5–15% and may lead to a meaningful CVD risk reduction [17]. Interestingly, factors influencing plasma lipids and atherosclerosis such as the consumption of dietary fatty acids, carbohydrates, and dietary fibers as well to tobacco use, obesity and level of physical activity, are also associated to changes in the gut microbial ecosystem [18].
Gut microbes are involved in the biosynthesis of an array of compounds contributing to normal human physiological functions or eliciting disease. Research over the past decade has uncovered several key microbial metabolites such as secondary bile acids, trimethylamine-N-oxide (TMAO) and short chain fatty acids (SCFAs) that may contribute to the progression and pathogenesis of atherosclerosis [19,20,21]. Although these metabolites are well studied, the processes by which they contribute to CVD remain unclear. The ability to modulate the production of these metabolites by altering the microbial composition through the diet could represent a non-pharmacological treatment for CVD [22]. The use of probiotics, prebiotics, and synbiotics has gained considerable interest as supplements for food to protect the host from enteral problems [23]. Probiotics introduce additional external microorganisms in the intestinal tract while prebiotics stimulate the growth rate of one or more of these microorganisms in the host. Both these applications can be combined (i.e., synbiotics) to improve the viability of probiotic microorganisms. Thus, the possibility to modulate lipid profiles by targeting microbial communities to control SCFA and TMAO production via dietary interventions could be used as a therapeutic tool to improve human health and decrease hypercholesterolemia. However, it is still unknown which gut microbes contribute the most to CVD and the detailed mechanisms involved require further investigation [24].
1.1. Cholesterol Metabolism
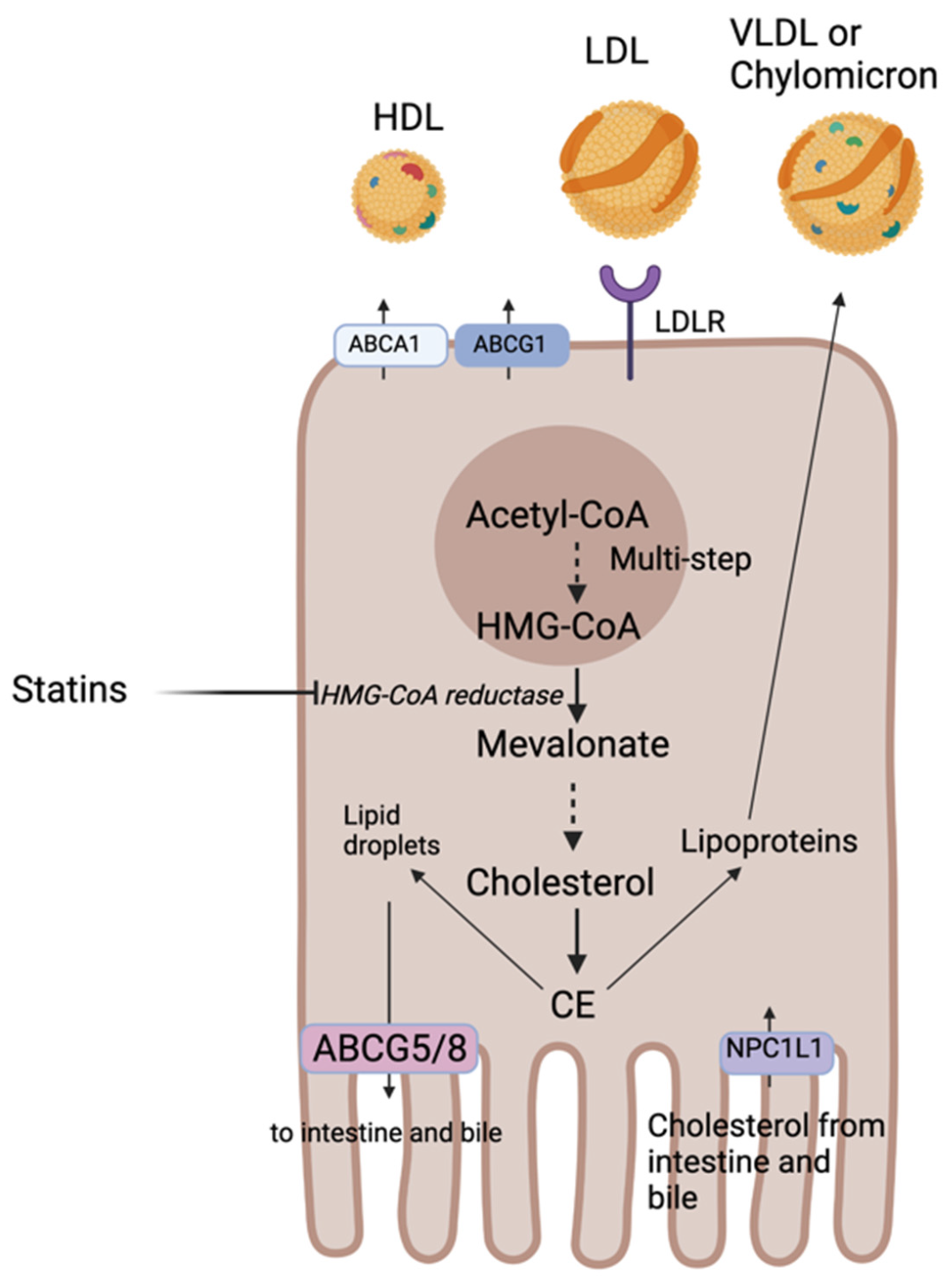
Cholesterol is an essential component of eukaryotic cell membranes and the precursor of corticosteroids, sex hormones, vitamin D, and BAs. Only about one-quarter of the pooled body cholesterol is obtained from the diet mainly from animal and dairy food products, with the remaining ~75% being synthesized endogenously in the liver via the mevalonate pathway [25,26]. Cholesterol metabolism and export in the liver and intestine involve a series of enzymes and transporters (Figure 1). Dietary cholesterol can be transported into the intestine via the Niemann-Pick C1-like 1 (NPC1L1) transporter located on the apical surface of enterocytes. This transporter is also expressed on hepatocytes mediating transport of cholesterol from bile [27,28]. Circulating blood cholesterol, transported with LDL, is taken up by LDL receptors (LDLR) located on the basal surface of polarized cells. The bulk of endogenous cholesterol is synthesized by the liver and starts from acetyl coenzyme A (acetyl-CoA) and concerted actions of more than 20 enzymes. The rate-limiting enzyme of this reaction is 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) and is the target of statins used for primary and secondary prevention of CVD (covered in Section 3.1) [29]. Cholesterol biosynthesis and uptake is tightly regulated by the transcription factor sterol-regulatory element-binding protein 2 (SREBP-2). SREBP-2 is embedded in the endoplasmic reticulum (ER) membrane and forms a complex with SREBP cleavage-activating protein (SCAP), also integral to the ER membrane. SCAP contains a sterol-sensing domain and when ER cholesterol exceeds 5% of total ER lipids it retains SREBP-2 in the ER in its inactive form [30]. When ER cholesterol falls below the 5% threshold, SCAP undergoes a conformational change that allows transport of SREBP-2 in COPII coated vesicles from the ER to the Golgi where two proteases sequentially cleave SREBP-2. Cleavage of SREBP-2 releases its cytosolic basic-helix-loop-helix leucine zipper domain so that it can enter the nucleus to activate transcription of all genes encoding enzymes involved in cholesterol synthesis and of the LDLR gene [31,32,33].
Once cholesterol is formed, it is either esterified to cholesterol ester (CE) and stored in lipid droplets or secreted into the bloodstream as lipoproteins. Excess cholesterol can be excreted by ABC subfamily G member 5 and member 8 (ABCG5/8) transporters to the intestine or to the bile or packaged with lipoproteins for subsequent secretion into the blood [29]. Lipoproteins carrying endogenous cholesterol are secreted by hepatocytes and are known as very low-density lipoproteins (VLDL) and the ones secreted by the intestine carry cholesterol absorbed from diet and are known as chylomicrons (covered in detail in Section 1.2). In macrophages, excess cholesterol can also be excreted via the adenosine triphosphate-binding cassette subfamily A member 1 (ABCA1) or subfamily G member 1 (ABCG1) to the blood and incorporated into the high-density lipoprotein (HDL). ABCG1 mediates the efflux of cholesterol from macrophages and thereby prevent foam cell formation and protect against atherosclerosis and CVD [34]. ABCA1 mediates the first step of reverse cholesterol transport (RCT) (further explored in Section 1.3). Mutations in ABCA1 cause Tangier disease characterized by a severe accumulation of cholesterol in macrophages responsible of prevalent atherosclerosis and premature CVD [35].
When intracellular cholesterol levels are high, liver X receptors (LXR) are activated and mediate cholesterol efflux via increased expression of the ABCG5/8 transporters [36]. Patients with mutations in genes encoding these transporters have a condition called sitosterolemia which is a disorder of sterol metabolism characterized by increased absorption and reduced biliary excretion of all neutral sterols and may exhibit hypercholesterolemia and premature heart disease [37]. The liver is the only organ capable of getting rid of excess cholesterol via its secretion into bile for conversion into BAs (further explored in Section 2.1). This is a crucial step to maintain cholesterol homeostasis in the body. Excess cholesterol decreases membrane fluidity, lipid raft signaling and generates toxic oxidative molecules such as oxysterols [33].
1.2. Cholesterol Transport
Because cholesterol is insoluble in water, it is packaged with triglycerides (TGs), phospholipids, and apolipoproteins (Apo) into lipoproteins that enable their transport throughout the body. There are four major classes of plasma lipoproteins—chylomicrons, VLDL, LDL, and HDL, each with unique size, density, and characteristic protein and lipid composition. They all act as lipid carriers but exert different functions throughout the body. The Apo found on their surface stabilize the complex can serve as ligands and can be cofactors for enzymes involves in the metabolism of lipoproteins [29].
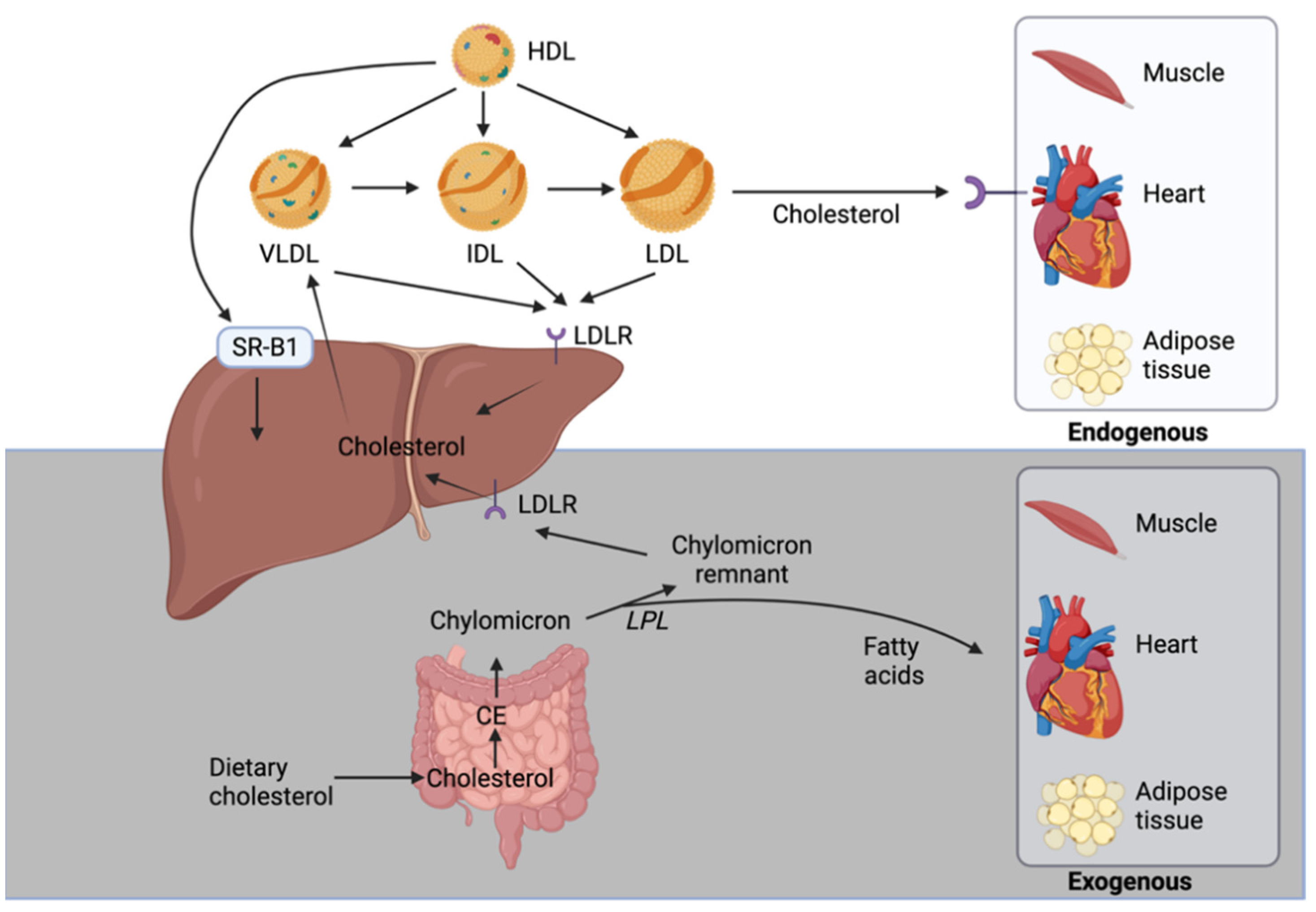
Exogenous and Endogenous Cholesterol Pathways
In the exogenous cholesterol pathway, the dietary cholesterol is absorbed in the duodenum and proximal jejunum via NPC1L1. In enterocytes, it is esterified to CE and packaged along with apolipoproteins and TGs into chylomicrons [38]. In the circulation, the TGs of these particles are hydrolyzed by lipoprotein lipase to release fatty acids that are taken up by muscle cells and adipocytes and results in the conversion of chylomicrons to chylomicron remnants (Figure 2). The liver is responsible for removing these remnants from the circulation but smaller ones can penetrate the endothelial monolayer of an artery and participate in plaque buildup. Clinical data support the concept of chylomicron remnants increasing the risk for atherosclerosis and coronary heart disease [7].
The endogenous cholesterol pathway begins in the liver with de novo synthesis of cholesterol that is then assembled along with ApoB100, TG, and phospholipids into VLDL particles. VLDL’s TGs are broken down by lipoprotein lipase in the blood releasing fatty acids and forming intermediate density lipoproteins (IDL), which are further metabolized to cholesterol-enriched LDL (Figure 2). LDL, which carries about 70% of cholesterol in the blood, supply cholesterol to numerous tissues through LDLR-mediated endocytosis including the liver that is the predominant site of uptake. LDL can also reach the artery wall to promote atherosclerosis. Circulating LDL particles directly bind to endothelial scavenger receptor class B type 1 (SR-B1) that mediates its transcellular transport and delivery into the subendothelial space [39]. LDL particles can be trapped and oxidized in the subendothelial extracellular matrix of arteries and cause a buildup of immune cells and cholesterol forming atherosclerotic plaques [40]. Monocytes are attracted to plaques where they differentiate into macrophages, which internalize oxidized LDL particles causing them to become foam cells. Foam cells are highly inflammatory as they release chemotactic cytokines propagating a monocyte response [40]. High levels of LDL accelerate plaques progression that can ultimately block blood flow and oxygen delivery to organs such as the heart and brain.
1.3. Reverse Cholesterol Transport (RCT)
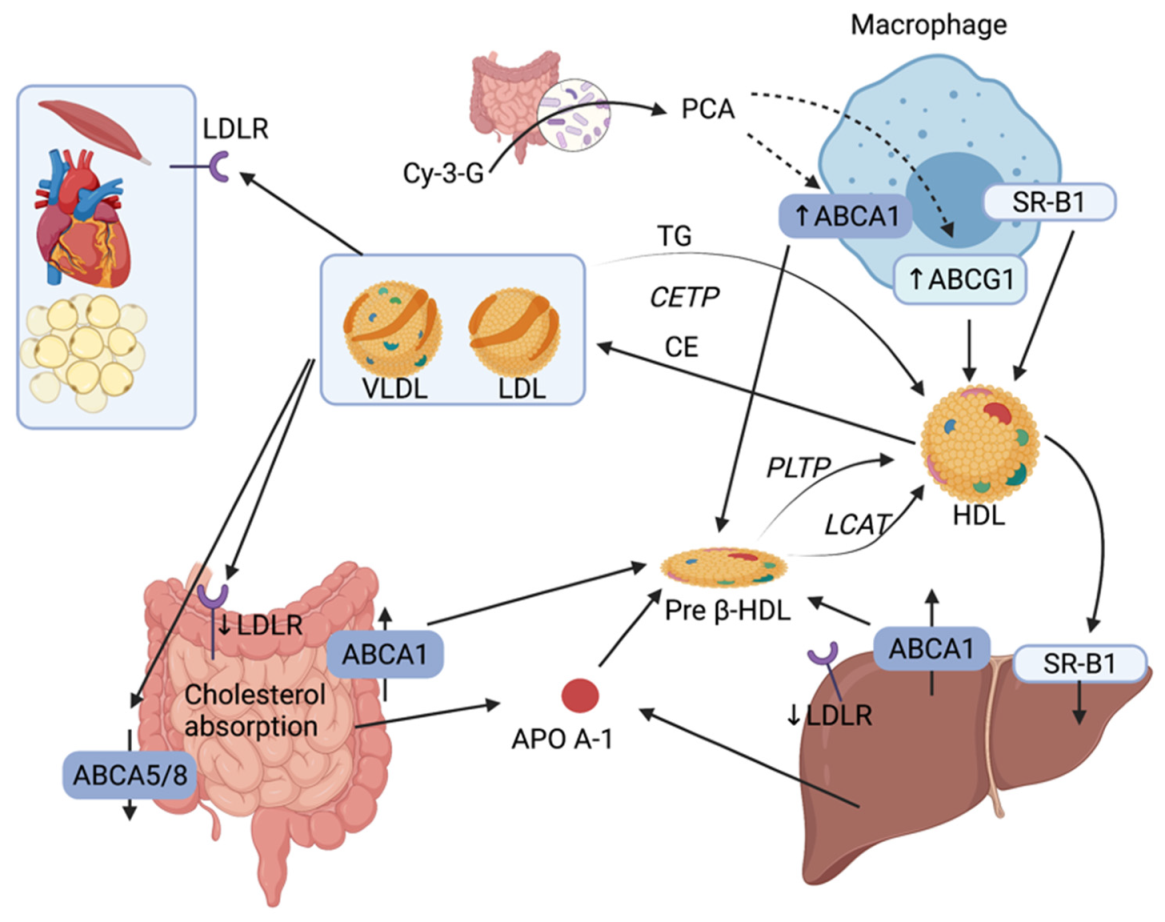
Cholesterol efflux from macrophages and its excretion by the liver is one of the most critical mechanisms for RCT. This transport plays an important role of countering toxic intracellular cholesterol accumulation and progression of atherosclerotic plaque. RCT begins in the liver and the intestine with the synthesis of ApoA-1 (Figure 3). ApoA-1 associates with phospholipids and free cholesterol through an interaction with ABCA1 located on various cell types (enterocytes, hepatocytes, and macrophages) [7]. Free cholesterol is esterified by lecithin-cholesterol acyltransferase (LCAT) and the process results in the formation of pre-β HDL particles. Pre-β HDL particles subsequently interact with scavenger receptor class B type 1 (SR-B1) and ABCG1 to incorporate more cholesterol as well as phospholipids from TG-rich lipoproteins through phospholipid transfer protein (PLTP) to form mature HDL particles. Cholesterol can then be delivered to the liver directly via hepatic SR-B1 or indirectly by transferring CE to LDL or VLDL through a process utilizing cholesteryl ester transfer protein (CETP). In addition to hepatobiliary cholesterol excretion (covered in the Section 2.1), the small intestine can also eliminate endogenous cholesterol through a pathway called the trans-intestinal cholesterol efflux (TICE) which accounts for 45% of fecal cholesterol excretion [41]. In this pathway, cholesterol enters enterocytes via LDLR and is effluxed in the intestinal lumen by ABCG5/8 transporters.
Experimental data suggest that the gut microbiota influences reverse transport by different mechanisms, including the increase in the expression of ABCA1 and ABCG1 in macrophages by a miRNA-10b-dependent mechanism involving PCA, a gut microbiota metabolite of the Cy-3-G, or by TMAO, a microbiota metabolite of L-carnitine, which inhibits the reverse transport and affect the level of circulating cholesterol (further explored in Section 2.2) [42].
2. Gut Bacterial Metabolites and Cholesterol Metabolism
2.1. Bile Acids
Cholesterol can be secreted into the bile in the unesterified form via the ABCG5/G8 heterodimer or converted into BAs in the liver. BA excretion is the major pathway for cholesterol export from the body, apart from neutral sterol excretion, and plays a key role in cholesterol, triglycerides, glucose, and energy homeostasis through receptor mediated processes [4,43]. It is also crucial for absorption of dietary fats and lipid-soluble vitamins [44]. Bacteria in the lower intestine can process cholesterol to coprostanol and the latter, unlike cholesterol, is poorly absorbed by the intestinal mucosa as it is the most abundant sterol in the feces [45]. Notably, a high efficiency of cholesterol to coprostanol metabolism was suggested to reduce CVD risk [4]. The main bacteria taxa carrying this conversion involve Lactobacillus and Eubacterium (Eubacterium coprostanoligenes), however bacterial enzymes responsible for this conversion remain unknown [4].
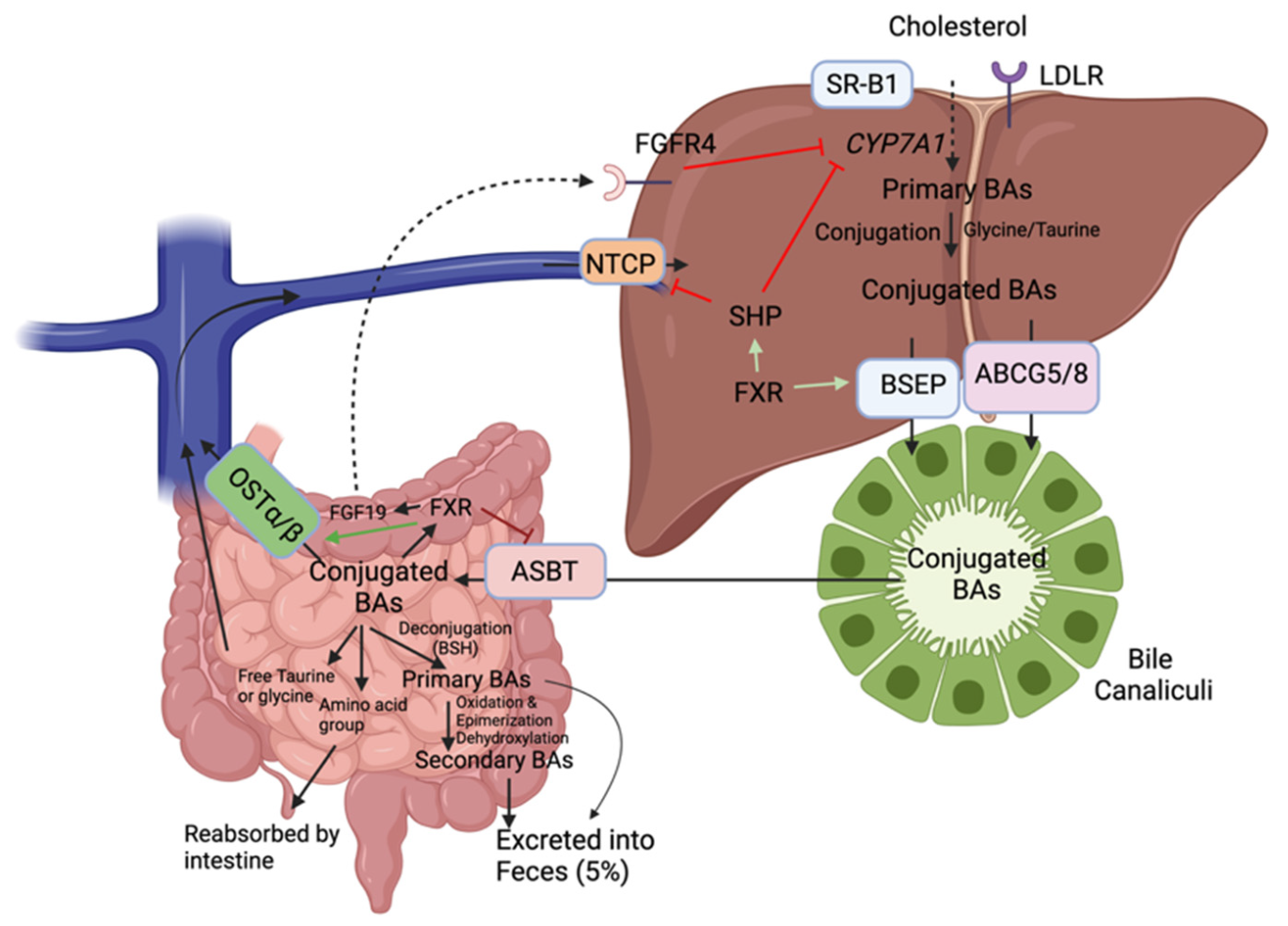
In the liver, cholesterol is metabolized to BAs by hepatic cytochromes, notably 90% of the time by the rate-limiting enzyme cholesterol 7 α-hydroxylase (CYP7A1) (classical pathway) (Figure 4). When the BA pool (i.e., total amount of BAs present in the enterohepatic circulation) increases, nuclear farnesoid X receptor (FXR) induces the expression of small heterodimer partner (SHP), which in turn represses CYP7A1expression, the rate-limiting enzyme of BA synthesis [46]. The remaining 10% of the time, cholesterol is processed by sterol 27-hydrolyase (CYP27A1) in extrahepatic sites including the vascular endothelium and macrophages (alternative pathway). The initial products are cholic acid and chenodeoxycholic acid (primary BAs) which are then conjugated with the amino acids glycine (mainly in humans) or taurine (mainly in rodents) for excretion into bile and further passage in the intestine [47]. These conjugated BAs are actively transported into bile via the bile salt export pump (BSEP) or via ABCG5/8. They can be stored temporarily as mixed micelles along with cholesterol and phospholipids in the gallbladder to be released into the duodenum upon meals to facilitate emulsion.
Approximately 95% of BAs are reabsorbed from the intestine, predominantly as conjugated BAs, mainly via the apical sodium-dependent bile acid transporter (ASBT) and a small amount can also enter the hepatocyte via passive diffusion (Figure 4) [48]. Then, they are secreted into the portal circulation by the organic solute transporters (OST α/β) to be transported back to the liver. In the ileum, BAs can activate FXR, which inhibits ASBT, induces OST α/β and the production and secretion of fibroblast growth factor 19 (FGF19) in the portal circulation. This factor will bind to the fibroblast growth factor receptor 4 (FGFR4) and activate a signaling pathway that will repress CYP7A1 transcription [49]. On the basolateral membrane of hepatocytes, Na+/taurochlorate cotransporting polypeptide (NTCP) mediates the uptake of conjugated BAs and organic anion transporting proteins that of unconjugated BAs. This process is called enterohepatic circulation and it occurs about six times a day in humans [47]. It is tightly negatively regulated by the FXR receptor both in the liver and intestine [46]. Not only can this receptor inhibit hepatic CYP7A1, but it can also regulate basolateral uptake by stimulating BSEP expression and repressing NTCP expression via SHP to prevent intrahepatic BA accumulation [44].
In the small intestine, certain gut microbes contain an enzyme known as bile salt hydrolase (BSH) capable of deconjugating (i.e., remove glycine or taurine conjugates) BAs preventing their reuptake by the ASBT transporter (Figure 4). BSH genes have been detected in Gram-positive gut bacteria including Lactobacillus, Clostridium, Listeria, and Bifidobacterium. Members of the Bacteroides genus are the only Gram-negative bacteria with BSH activity [4]. This deconjugation produces amino acid groups and free taurine or glycine which are reabsorbed into the intestine and return to the liver, respectively. It also produces free cholic acids (unconjugated bile salts) that are less efficiently reabsorbed which results in their excretion into the feces. Primary BAs can be further metabolized through gut microbial 7α-dehydroxylation to secondary BA known as lithocholic acid (from chenodeoxycholic acid) and deoxycholic acid (from cholic acid) [47]. The known bacterial species that possess 7α-dehydroxylation activity are members of the Firmicutes phylum (Clostridium and Eubacterium) [4]. Quantitatively, these members possess the most important enzymatic activity as secondary BAs predominate in the feces [44].
Secondary BAs have often been discussed as having negative impact on human health and their increased concentration in the intestine may promote the development of carcinomas [50]. Primary and secondary BAs compose the total BA pool, in a relatively fixed proportion [44]. If the ratio of bile salts to cholesterol changes, cholesterol can precipitate with calcium salts and bile pigments and may drive diseases such as gallstone disease and non-alcoholic fatty liver diseases [51,52]. Furthermore, serum levels of primary BA were reduced and specific secondary BAs increased in patient with chronic heart failure [53]. Microbial metabolism leads to a more hydrophobic BA pool facilitating excretion into feces of the 5% remaining BAs.
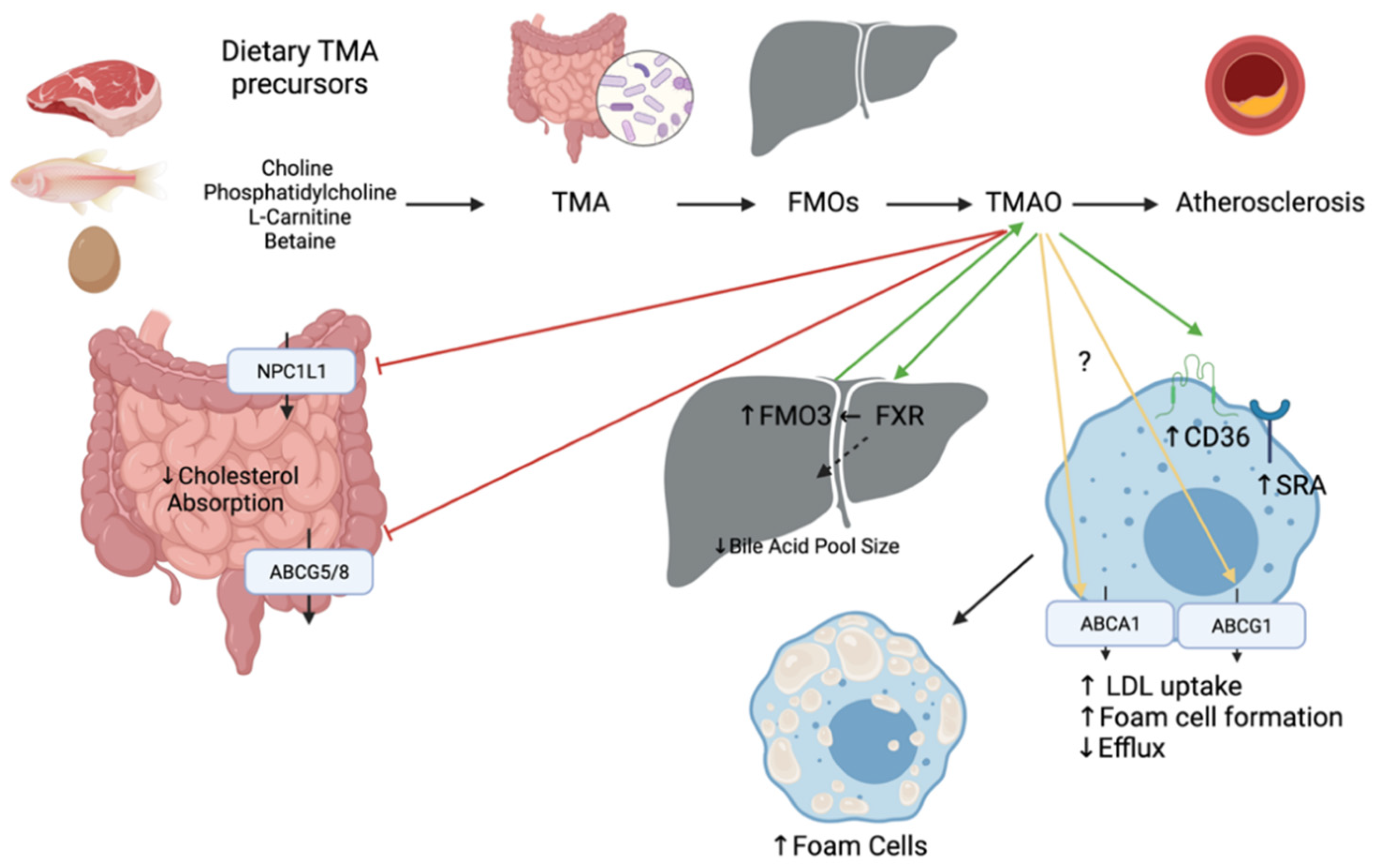
2.2. Trimethylamine (TMA) and Trimethylamine-N-Oxide (TMAO)
The intestinal microbiota metabolizes choline, phosphatidylcholine, L-carnitine, and betaine to trimethylamine (TMA), which is oxidized into TMAO by hepatic flavin monooxygenases (FMO3) (Figure 5). Foods of animal origin such as dairy products, eggs, and red meat are all rich in TMA precursors, thus a potential source of TMAO. TMA and TMAO can also be acquired from fish and other seafood [54]. When fish were compared to other foods rich in carnitine and choline, fish have the highest levels of TMAO and marked metabolites [54]. TMAO levels in serum are dictated by genetic variations, gut microbiota, and the diet [55]. Elevated blood TMAO levels have been linked to poor outcomes in patients with CVDs such as coronary heart disease and chronic heart failure and can be a valuable prognostic factor for adverse cardiac events for patients with chronic heart failure after myocardial infarction [19,56]. Inhibition of gut microbiota-dependent TMAO production has been shown a promising strategy for the treatment of atherosclerosis [57].
The underlying mechanisms whereby TMAO contributes to CVD are not fully elucidated, yet mechanisms related to cholesterol metabolism have been proposed (Figure 5). A study performed with ApoE−/− mice fed TMAO for 8 weeks demonstrated accelerated aortic lesion formation. Mechanistically, it downregulates CYP7A1 and CYP27A1 enzymes involved in the BA synthesis, which might be mediated by the activation of SHP and FXR [58]. The activation of FXR inhibits cholesterol absorption by modulating BA pool size and composition, thus leading to decreased RCT [46]. Injection of FXR has been shown to induce the expression of FMO3 and the production of TMAO in mice. Dietary supplementation of TMAO was also shown to reduce expression of both intestinal transporters NPC1L1 and ABCG5/8 in vivo [59]. However, another study has shown that TMAO caused an upregulation of ABCG5/8 in the small intestine in rats fed a high fat high cholesterol diet [60]. Expression of ABCA1 and ABCG1 in macrophages also seems contradictory as some studies show no differences and others show a downregulation upon TMAO treatment [59,60,61]. Last, TMAO is thought to upregulate cluster of differentiation 36 (CD36) and scavenger receptor A (SRA) located on macrophages and involved in the uptake of oxidized LDL [62]. This enhances the formation of foam cells which are the most critical component directly related to atherosclerosis.
There is strong evidence demonstrating that TMAO is not only an independent risk factor for CVD, but also a therapeutic target for CVD on the basis of a large amount of experimental and clinical data [63]. TMA and FMO3 are also important targets to consider as they participate in the formation of TMAO, but they have also shown clinical evidence of disrupting cholesterol homeostasis. Inhibition of TMA lyase has been shown to increase loss of fecal neutral sterol in the form of coprostanol and alter the total amount of composition of circulating bile acids [64]. This inhibition was achieved with small molecule drugs directly targeting TMA lyase which have previously shown potent anti-atherothrombotic activity and hold therapeutic potential for diverse cardiometabolic disorders [4]. In contrast to the microbiota’s cholesterol-lowering effects by coprostanol generation and the formation of secondary bile acids, the causative role of TMAO in atherogenesis is under debate [65]. FMO3 knockdown has been shown to alter biliary excretion, intestinal absorption and limit the production of hepatic oxysterols and cholesteryl esters in cholesterol-fed mice [66]. Thus, these dietary metabolites represent markers mediating disease and are an interesting avenue to further explore treatments for CVD risk reduction.
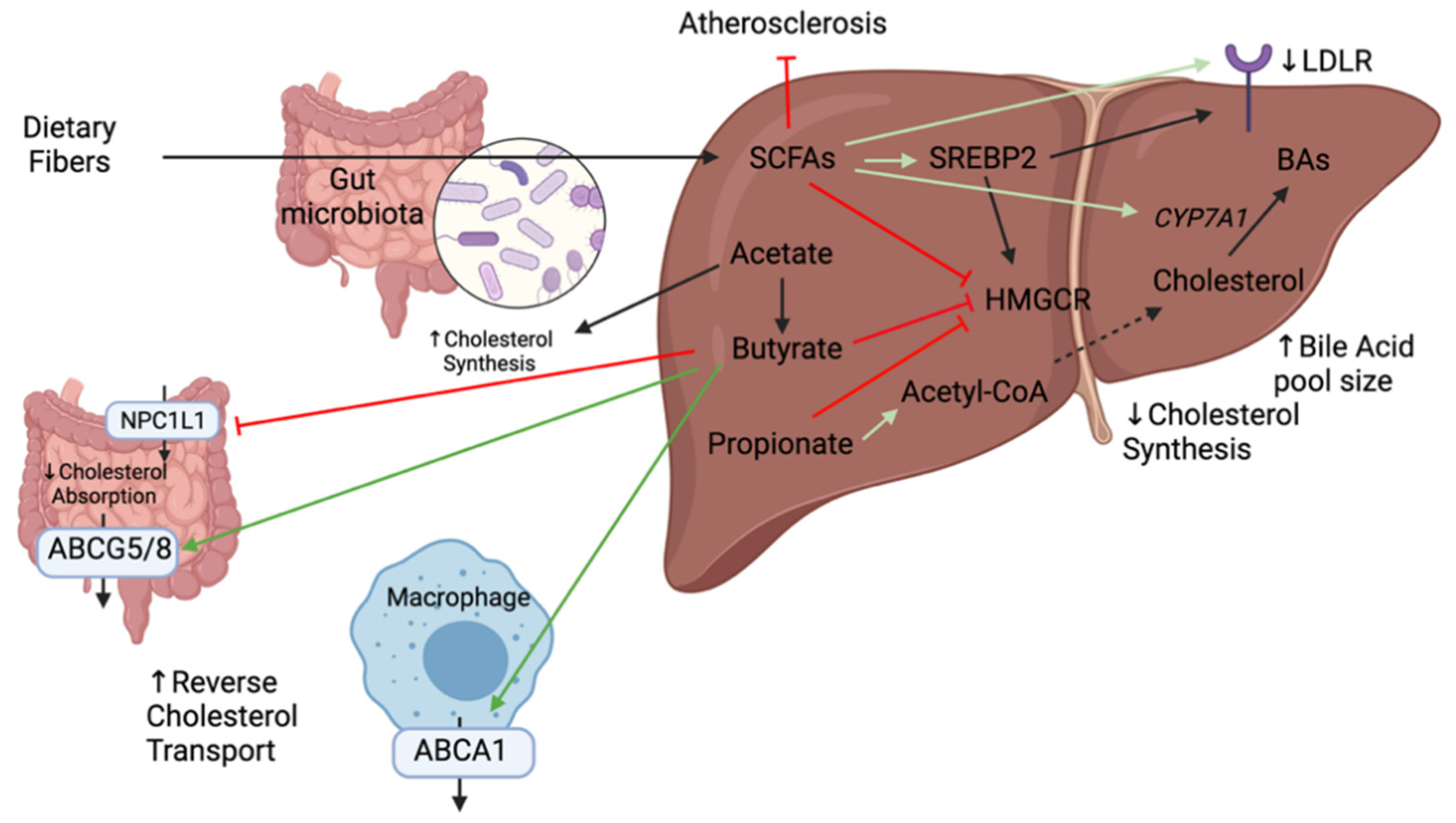
2.3. Short-Chain Fatty Acids
SCFAs are crucial for intestinal health as they mediate the interaction between the diet, the gut, and the host. In doing so, they are involved in multiple metabolic processes such as the synthesis of lipids, fat storage, glucose uptake, and inflammation [20]. Once dietary fibers and resistant starches enter the colon, they are fermented and transformed to SCFAs such as butyrate, acetate, and propionate which account for ~90% of total SCFAs in the human colon [20]. Their production depends on the microflora present in the colon as well as the substrate source (diet), environment condition including pH and the gut transit time [67]. The main bacteria producing SCFAs are the clostridial clusters IV and XIVa of Firmicutes including species of Eubacterium, Roseburia, Faecalibacterium, and Coprococcus [44]. Hundreds of gut bacterial species across many taxa share the genes for fermenting carbohydrates into SCFAs [68]. Acetate and propionate are mainly produced by Bacteroidetes, whereas Firmicutes are the primary contributors of butyrate [69]. The concentrations of SCFAs changes along the GI tract with the highest concentrations in the proximal colon and cecum where they can be used locally by the enterocytes or transported across the epithelium into the bloodstream [20]. Between 90% to 99% of SCFAs are absorbed in the gut or used by the microbiota [20]. High SCFA concentration in the gut lumen can lower colonic pH and impact microbial communities inhibiting the growth of potential pathogens including Salmonella spp. and Escherichia coli and promote the growth of beneficial bacteria such as Lactobacilli and Bifidobacteria, which are known to reduce the risk of CVDs [48,70,71].
Numerous mechanisms that link dietary fibers to the reduction of cholesterol levels have been elucidated notably through the production of SCFAs (Figure 6). SCFAs have been shown to lower cholesterol synthesis rate thereby reducing cholesterol in the serum [70]. The addition of SCFAs in the diet of hamsters on a high-cholesterol diet showed an upregulation of SREBP2, LDLR, and CYP7A1 genes in the liver thereby promoting hepatic uptake of serum cholesterol and fecal excretion of BAs [72]. The cholesterol lowering properties of SCFAs can be further extended to individual SCFAs. Propionate has been shown to inhibit the incorporation of acetate into fatty acids and cholesterol in rat hepatocytes thereby decreasing cholesterol serum levels [71]. It can also stimulate the hepatic synthesis of bile salts by upregulating CYP7A1 [73]. Acetate, the most abundant SCFA in peripheral circulation, is a substrate for cholesterol and, therefore, promotes cholesterol synthesis [71]. Therefore, a decrease in the acetate to propionate ratio may result in a decrease in serum lipids which is an interesting avenue to decrease cardiovascular risk. It can also be transformed into butyrate and participate in long-chain fatty acid synthesis [20]. Butyrate is the most beneficial in terms of colonic health as it is the key energy source for colonocytes and enterocytes and promotes normal cell differentiation and proliferation [74]. It also has numerous beneficial health effects as it protects from pathogen invasion, modulates the immune system and reduces cancer progression. It is locally consumed, where other absorbed SCFAs drain into the portal vein [74]. It has been shown to inhibit the HMGCR activity, thus preventing cholesterol synthesis [70]. As SCFAs levels are usually low in the circulation, local effects of butyrate were evaluated on Caco-2 cells (intestinal permeability model). Butyrate was shown to suppress cholesterol uptake in a dose-dependent fashion by inhibiting NPC1L1 and increasing expression of ABCG5/8 [75]. It was also shown to protect against high-fat induced atherosclerosis by upregulating ABCA1 in ApoE−/− mice [76]. At present, oral butyrate is commonly used as a supplement but has low bioavailability and most butyrate producing bacteria are anaerobic which limits the production capacity of this metabolite. Studies are being done to evaluate butyrate concentration in the small intestine in order to develop oral formulations that maintain pharmacologically active butyrate concentrations [77]. Collectively, several studies point towards an important role of SCFAs in cholesterol levels. However, there are contradicting results on their mechanisms of action and further studies demonstrating solid evidence are crucial to move into human studies.
3. Cholesterol-Lowering Agents
Recently, evidence indicated that gut microbiota can be modified by drugs and by nutritional interventions. Interactions between statins and gut microbiota will be explored to determine the involvement of the gut microbiota in their beneficial effects. After that, probiotics will be further investigated as a potential therapy to reduce the levels of cholesterol.
3.1. Statins
Statin therapy represents the privileged method to achieve therapeutic circulating LDLc targets in patients for both primary and secondary prevention of CVD. It is one of the most potent treatments to decrease LDLc levels and, as per current guidelines, first-line therapy for atherosclerotic CVD. Statins inhibit HMGCR which is the rate-limiting enzyme of the mevalonate pathway that forms cholesterol [33]. As a result, cholesterol synthesis is inhibited in the liver leading to the activation of SREBP2 and upregulation of LDLR on the membrane of hepatocytes. This ultimately leads to an increased uptake of LDLc by the liver and lower circulating cholesterol levels [78]. Together with age, sex, blood pressure, diabetes and smoking, and total cholesterol, LDLc and HDLc levels are important risk factors used to calculate the Framingham Risk Score (FRS; Canadian guidelines) or the clinician–patient risk discussion (ACC/AHA guidelines) that will determine if a statin will be recommended to a patient for primary prevention of CVD. The higher the ratio of LDLc (or total cholesterol)/HDLc, the higher the probability that a statin will be recommended as LDLc is directly associated with CVD while HDLc is inversely associated with CVD [79,80]. The effects of statins are pleiotropic meaning they go beyond cholesterol reduction. They exhibit anti-inflammatory, immunomodulatory and antithrombotic actions [81]. They have also been reported to alter the gut microbiome as their use has been associated with decreased TMAO [82]. However, statins present many limitations. They present increased risk of diabetes, myopathy and hepatic damage [78]. Furthermore, treatment response is individual and varied. A meta-analysis evaluated statin interventional trials encompassing 32,258 patients from 37 trials and found that 5.3% to 53.3% percent of patients experience a suboptimal response (less than 30% reduction in LDLc levels). This study concluded that there is high interindividual variation in LDLc reduction at all doses of simvastatin, atorvastatin, and rosuvastatin [83]. Other factors such as smoking, diet, exercise, and genetic polymorphisms can affect LDLc levels and, thus, statin doses must be adjusted. Even when doses are adjusted, suboptimal adherence to the therapy represents another challenge [84]. As statin therapy exhibits potential antibacterial activities, the gut microbiota may modulate the therapeutic actions of these drugs [85].
Hyperlipidemic patients often display intestinal disorders that may further impair lipid metabolism [86]. An in vivo study was conducted on 64 hyperlipidemic patients treated with rosuvastatin for 4 to 8 weeks and investigated the role of the microflora in mediating the lipid-reduction efficacy of rosuvastatin. They found variations in the community composition, taxa, and diversity of the gut microbiota in association to the LDLc-lowering response of the statin [87]. These results indicate that it is possible to modulate the gut microflora for a better statin response. A study conducted by Sun et al. assessed bacterial composition and diversity of 202 hyperlipidemic patients with either statin-sensitive or statin-resistant response. The results indicated that statin-sensitive patients exhibited a higher gut biodiversity, and this group showed an increased genera of Lactobacillus, Eubacterium, Faecalibacterium, and Bifidobacterium and decreased genus of Clostridium when compared to the statin-resistant group [88].
Participants with a poor treatment response to rosuvastatin also showed a significant increase in TMAO values compared to other participants treated with placebo [89]. Another study evaluated gut microbial communities in hypercholesterolemic patients treated with atorvastatin. When compared with healthy and untreated subjects, statin-treated patients exhibited an increased abundance of anti-inflammation-associated bacteria (Faecalibacterium prausnitzii and Akkermansia muciniphila) and a decreased abundance of bile-associated species (Bifidobacterium bifidum) [90]. Thus, defining the gut microbiota diversity and metabolites could be used as a valuable tool to predict a patient’s statin response and optimize statin dosages in personalized CVD treatment regimens. Further research is needed to determine whether these changes in the gut are directly caused by statins or if alterations in the microbiota are a consequence of host response to these statins.
3.2. Probiotics
Because of the high cost and deleterious side effects of many pharmacological agents, recent interest has sparked for probiotics therapies as a convenient approach to lower serum cholesterol levels. Probiotics are currently defined by the World Health Organization as « live microorganisms that, when administered in adequate quantities, confer health benefits to the host » [91]. They present a cheap, noninvasive approach with minimal to no side effects to reduce coexisting CVD risk factors such as TC, LDLc, weight, waist, and inflammation markers [92]. Probiotics strains must meet safety and functionality criteria and maintain their properties through distribution and storage. Some required properties include human or animal origin, non-pathogenic, isolated from a healthy GI tract, resist bile salts and enzymes and be able to antagonize pathogens such as Clostridium difficile and Helicobacter pylori [23]. Probiotics can be found in fermented foods of vegetable origin such as sauerkraut and pickles, and may also be added to unfermented foods like milk, juices, and cereals [93]. Their benefits are specific to species and strains and thus cannot be extrapolated to other strains without confirmation [94].
A meta-analysis of 30 randomized controlled trials found that 1624 participants treated with probiotics showed a reduction of TC and LDLc compared to control subjects by 7.8 mg/dL and 7.3 mg/dL, respectively. No change was observed for levels of HDL or TGs [95]. A larger meta-analysis incorporating 32 randomized controlled trials, showed that the probiotic group had lower serum TC levels than the control group with a mean difference (MD) of −13.27 mg/dL [96]. Another meta-analysis by Shimizu et al. including 11 randomized controlled trials, found that probiotic supplementation could be useful in the treatment of hypercholesterolemia as they observed a reduction of TC (MD −0.17 mmol/L) and LDLc (MD −0.22 mmol/L) [97]. As these meta-analyses had many limitations, the authors concluded that the existing clinical evidence was not strong enough to endorse probiotics as an alternative therapy to improve blood lipid profiles [95,96,97]. A review assessed probiotic supplementation in healthy adults and found 14 studies on lipid profiles and cardiovascular risk. Authors determined that there was insufficient evidence to support the role of probiotics in improving blood lipid profiles [98]. The most well-studied human probiotic microorganisms for their cholesterol-lowering effects in humans and animals belong mostly to Lactobacillus and Bifidobacterium genera [99]. Based on meta-analysis studies, specific probiotic strains that reduced TC in patients are B. lactis and L. acidophilus (MD of −8.30 mg/dL), VSL #3 (MD −11.04 mg/dL), and L. plantarum (MD −1.56 mg/dL) [96]. Consuming Lactobacillus significantly reduced TC by 0.26 mmol/L and LDLc by 0.23 mmol/L. Among probiotic Lactobacillus genus, sub-analyses showed the greatest reduction of TC was observed with L. plantarum and the greatest reduction of LDLc was with L. plantarum and L. reuteri [100]. Another study demonstrated that L. acidophilus strain has been shown to cause the greatest TC and LDLc reductions (MD −0.35 mmol/L) in normal to mildly hypercholesterolemic individuals [97,101].
3.2.1. Combination of Probiotics
Recently, studies have tried combining multiple strains of probiotics to increase their hypocholesterolemic action. In a study conducted by Kim et al. low, medium and high doses of a probiotic mixture containing two Lactobacilli (L. reuteri and L. plantarum) and three Bifidobacteria (B. longum, B. lactis, and B. breve) strains were given to hypercholesterolemic rats [102]. These treated groups, compared to the control group not given probiotics, experienced a TC reduction by 1.2-fold, 1.5-fold, and 1.3-fold, respectively. TG levels were also decreased by 1.32-fold, 1.4-fold, and 1.4-fold in low, medium, and high doses of probiotic-treated groups when compared to the control. A similar reduction (1.3-fold, 1.4-fold, and 1.5-fold in low, medium, and high doses of probiotic-treated groups, respectively) was observed for LDLc [102]. A multi-strain probiotic mixture VSL #3 containing 4 strains of Lactobacilli (including L. plantarum), 3 strains of Bifidobacteria and Streptococcus thermophilus showed reduced inflammation and atherosclerotic plaque development in ApoE−/− mice [103]. In humans, a combination of two most popular probiotics (L. acidophilus and B. bifidum) showed decreased serum TC (MD −40.1 mg/dL) and LDLc (MD −28.2 mg/dL) levels in hypercholesterolemic patients treated over a six-week period [101,104].
3.2.2. Probiotics and TMAO
Research concerning the impact of probiotic supplementation on decreasing serum TMAO levels is limited. Probiotics could modulate the microbiome in a way to inhibit pathogenic strains responsible of TMAO synthesis. In female BALBc mice, diet supplementation with Enterobacter aerogenes (ZDY01) showed a 53% reduction in serum TMAO, which could be explained by the reduction in cecal TMA [105]. Lactobacillus plantarum (ZDY04) was able to reduce serum TMAO levels via the modulation of gut microbiota, increasing abundance of Lachnospiraceae by 78.8% and Bacteroidaceae from 1.84% ± 2.55% to 3.56% ± 3.47% and decreasing the relative abundance of Mucispirillum genus by 67%. Furthermore, it mediated an atheroprotective effect in ApoE−/− mice by decreasing atherosclerotic lesion size observed in aortic sinus sections and whole aortas [106]. Enterococcus faecium (WEFA23) was also shown to improve the diversity of the gut microbiota in rats fed a high-fat diet and decreased TMAO and cholesterol levels [107]. This probiotic showed enhanced synthesis of BAs via CYP7A1 upregulation, increased the expression of LDLR involved in cholesterol transport and downregulated the expression of HMGCR involved in cholesterol synthesis.
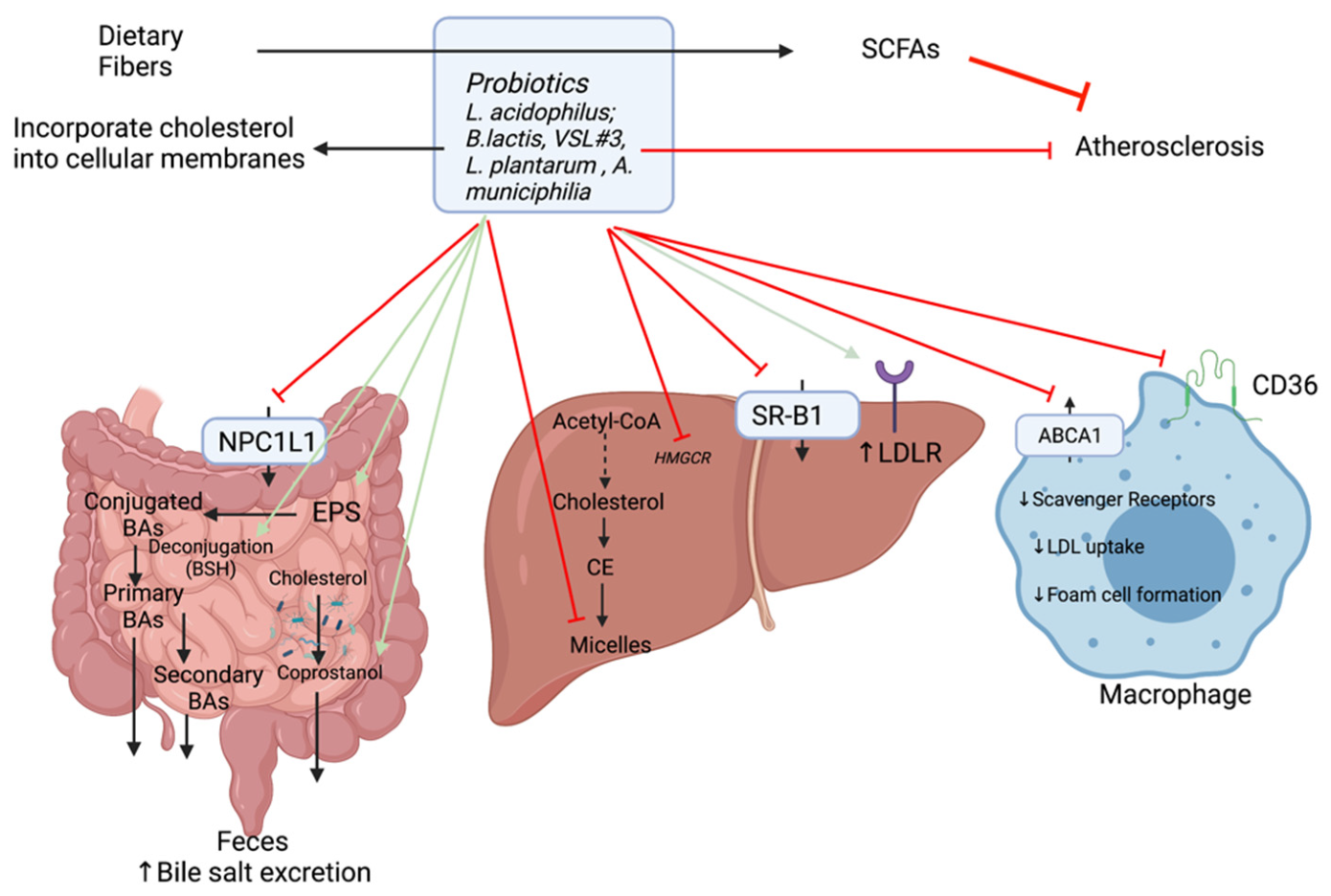
3.2.3. Suggested Hypocholesterolemic Mechanisms of Probiotics
Based on in vitro and animal experiments, there are several possible mechanisms for the removal of cholesterol (Figure 7):
- (1)
- Some probiotics are thought to reduce the enterohepatic circulation of bile salts through the regulation of BSH activity (Section 2.1). As the excretion of deconjugated BAs increases, there is a greater need to mobilize systemic cholesterol to the liver for de novo BA production. This provokes an increase in LDLR expression and hepatic uptake of LDLc from the circulation. On this account, serum LDLc (1.28 mM vs. 2.46 mM) and TC concentrations (3.49 mM vs. 4.47mM) are reduced in mice given probiotics [73,108]. Moreover, with increased BSH activity, there are fewer BAs to participate in micelle formation. Micelles play a role in the absorption of cholesterol in the intestine and are composed of cholesterol, bile salts, and phospholipids. The cholesterol assimilation abilities of selected strains of Lactobacillus were shown to vary from one strain to another. For example, in in vitro experiments L. acidophilus KU41 and M23 were found to reduce cholesterol levels by 50% while L. casei MB3 decreased cholesterol slightly below 30%. Thus, these strains were able to remove cholesterol by inhibiting micelle formation [109].
- (2)
- Another mechanism whereby probiotics reduce cholesterolemia is the incorporation of cholesterol into bacterial cellular membranes [110,111]. Cholesterol assimilation was significantly higher in growing cells than resting and dead cells. Both live and heat-killed Lactococcus lactis subsp. lactis N7 cells were able to remove cholesterol from growth media [111].
- (3)
- Subsequently, the gut microbiota plays a role on cholesterol conversion into coprostanol making it easily eliminable with defection and reducing its absorption [4]. The efficiency of this conversion depends on the abundance of cholesterol-reducing bacteria (possessing reductase enzyme). This enzyme is found in some strains of probiotic bacteria such as L. acidophilus ATCC 314, L. acidophilus FTCC 0291, L. bulgaricus FTCC 0411, and B. bifidum PRL2010 [73].
- (4)
- (5)
- They can also modulate cholesterol synthesis by inhibiting HMGCR notably through the production of SCFAs allowing a redistribution of cholesterol from the plasma to the liver [20].
- (6)
- In addition to the production of SCFAs, gut microorganisms notably lactic acid bacteria (LAB) (including Lactobacillus and Bifidobacterium species) can synthesize different polysaccharides such as exopolysaccharides (EPS). EPS are extracellular polysaccharides secreted or attached to the bacterial cell wall which can affect adhesion and provide protection from environmental stresses. In addition, they have the potential to act as prebiotics as they can affect the GI microbiome and lower serum cholesterol levels [70]. There is a correlation between the amount of EPS produced and the quantity of cholesterol assimilated by the strain. The EPS produced by Lb. plantarum BR2 and L. paracasei M7 exhibited cholesterol-lowering properties [114]. The proposed mechanism for this action is the binding to free BAs, which thereby increases their excretion via the feces.
These mechanisms are hypothetical and further studies are needed to elucidate which ones play dominant roles.
4. Conclusions
This study provides a general understanding of cholesterol transport and metabolism in the body and deregulated mechanisms when cholesterol homeostasis is disrupted. Lowering TC and LDLc plasma levels during hypercholesterolemia is extremely useful for diminishing the risk of CVD. It also highlights potential nutritional interventions that may restore microbial populations and can be used as an adjuvant to traditional treatments for hypercholesteremia. According to the literature at this time, we believe the first of line treatment to restoring a healthy gut microbiota should be lifestyle interventions such as improving dietary habits and achieving exercise recommendations. A greater understanding of the host–microbiota–metabolite interactions may promote a shift towards the development of customized nutritional plans and probiotics that are specifically designed according to bacterial deficiencies. Statin therapy has prescribed and evidence-based lipid-lowering drug for lowering LDLc but in higher doses, is associated with an increased risk of hepatic and muscular side effects (as discussed in Section 3.1). A subset of patients on statins cannot achieve target plasma cholesterol levels even at the highest doses and decide to stop treatment altogether than deal with the side effects [115]. Co-therapy involving a statin in combination with a probiotic is another interesting avenue to explore that can exert a combine cholesterol-lowering effect thereby decreasing statin doses [116]. However, it is presently very difficult to utilize knowledge of microbiota composition and function in clinical practice. There are no outcome studies proving that nutraceuticals can prevent CVD morbidity or mortality. Many of the proposed mechanisms and experimental evidence targeting cholesterol-lowering effects remain controversial and have never been directly observed in humans. Furthermore, there exists a gap in the knowledge about the impact of gut microbiota on CVD. Attempts to establish a common “human core microbiome” are challenging due to strong influence of genetic and environmental factors. As research on the gut increases, the correlation of some bacterial populations and different physiological states strengthens and molecular mechanisms contributing to disease are slowly being discerned. With the current advances in technology, metabolic profiling and others “omic” analytic platforms can be useful tools to discover candidate microbial communities and their derived metabolites that impact human health [117]. Such advances will help discern the gut microbial mechanisms that explain the relationship between intestinal microbiota with cholesterol metabolism. This will lead to deeper understanding of the microbiota and encourage the development of microbiota-based therapies and microbiome-informed precision medicine in order to reduce the global prevalence of CVD.
Author Contributions
Writing, review and editing: M.V., G.M., and G.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The figures were made using Biorender.com (accessed on 26 June 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Malard, F.; Dore, J.; Gaugler, B.; Mohty, M. Introduction to host microbiome symbiosis in health and disease. Mucosal Immunol. 2021, 14, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.K.; Dubey, V.; Ghosh, A.R. Obesity: An overview of possible role(s) of gut hormones, lipid sensing and gut microbiota. Metabolism 2016, 65, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Kriaa, A.; Bourgin, M.; Potiron, A.; Mkaouar, H.; Jablaoui, A.; Gerard, P.; Maguin, E.; Rhimi, M. Microbial impact on cholesterol and bile acid metabolism: Current status and future prospects. J. Lipid Res. 2019, 60, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, N.; Yamashita, T.; Hirata, K.-I. Gut Microbiome and Cardiovascular Diseases. Diseases 2018, 6, 56. [Google Scholar] [CrossRef] [Green Version]
- Cani, P.D. Human gut microbiome: Hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. The Effect of Inflammation and Infection on Lipids and Lipoproteins. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., Dungan, K., Grossman, A., Hershman, J.M., Kaltsas, G., Koch, C., Kopp, P., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Kazemian, N.; Mahmoudi, M.; Halperin, F.; Wu, J.C.; Pakpour, S. Gut microbiota and cardiovascular disease: Opportunities and challenges. Microbiome 2020, 8, 36. [Google Scholar] [CrossRef] [Green Version]
- Jie, Z.; Xia, H.; Zhong, S.-L.; Feng, Q.; Li, S.; Liang, S.; Zhong, H.; Liu, Z.; Gao, Y.; Zhao, H.; et al. The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 2017, 8, 845. [Google Scholar] [CrossRef] [Green Version]
- Rabot, S.; Membrez, M.; Bruneau, A.; Gérard, P.; Harach, T.; Moser, M.; Raymond, F.; Mansourian, R.; Chou, C.J. Germ-free C57BL/6J mice are resistant to high-fat-diet-induced insulin resistance and have altered cholesterol metabolism. J. Fed. Am. Soc. Exp. Biol. 2010, 24, 4948–4959. [Google Scholar]
- Le Roy, T.; Lécuyer, E.; Chassaing, B.; Rhimi, M.; Lhomme, M.; Boudebbouze, S.; Ichou, F.; Haro Barceló, J.; Huby, T.; Guerin, M.; et al. The intestinal microbiota regulates host cholesterol homeostasis. BMC Biol. 2019, 17, 94. [Google Scholar] [CrossRef] [Green Version]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [Green Version]
- Mortensen, M.B.; Nordestgaard, B.G. Elevated LDL cholesterol and increased risk of myocardial infarction and atherosclerotic cardiovascular disease in individuals aged 70–100 years: A contemporary primary prevention cohort. Lancet 2020, 396, 1644–1652. [Google Scholar] [CrossRef]
- Amarenco, P.; Kim, J.S.; Labreuche, J.; Charles, H.; Abtan, J.; Béjot, Y.; Cabrejo, L.; Cha, J.K.; Ducrocq, G.; Giroud, M.; et al. A Comparison of Two LDL Cholesterol Targets after Ischemic Stroke. N. Engl. J. Med. 2020, 382, 9. [Google Scholar] [CrossRef]
- Ference, B.A.; Graham, I.; Tokgozoglu, L.; Catapano, A.L. Impact of Lipids on Cardiovascular Health: JACC Health Promotion Series. J. Am. Coll. Cardiol. 2018, 72, 1141–1156. [Google Scholar] [CrossRef]
- Banach, M.; Jankowski, P.; Jóźwiak, J.; Cybulska, B.; Windak, A.; Guzik, T.; Mamcarz, A.; Broncel, M.; Tomasik, T.; Rysz, J.; et al. PoLA/CFPiP/PCS Guidelines for the Management of Dyslipidaemias for Family Physicians 2016. Arch. Med. Sci. 2017, 13, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Cicero, A.F.G.; Colletti, A.; Bajraktari, G.; Descamps, O.; Djuric, D.M.; Ezhov, M.; Fras, Z.; Katsiki, N.; Langlois, M.; Latkovskis, G.; et al. Lipid-lowering nutraceuticals in clinical practice: Position paper from an International Lipid Expert Panel. Nutr. Rev. 2017, 75, 731–767. [Google Scholar] [CrossRef] [PubMed]
- Villette, R.; Kc, P.; Beliard, S.; Salas Tapia, M.F.; Rainteau, D.; Guerin, M.; Lesnik, P. Unraveling Host-Gut Microbiota Dialogue and Its Impact on Cholesterol Levels. Front. Pharm. 2020, 11, 278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Jin, M.; Liu, L.; Yu, Z.; Lu, X.; Zhang, H. Trimethylamine N-oxide and cardiovascular outcomes in patients with chronic heart failure after myocardial infarction. Eur. Soc. Cardiol. Heart Fail. Assoc. 2020, 7, 188–193. [Google Scholar] [CrossRef] [Green Version]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charach, G.; Argov, O.; Geiger, K.; Charach, L.; Rogowski, O.; Grosskopf, I. Diminished bile acids excretion is a risk factor for coronary artery disease: 20-year follow up and long-term outcome. Ther. Adv. Gastroenterol. 2017, 11, 1756283X17743420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markowiak, P.; Śliżewska, K. Effects of Probiotics, Prebiotics, and Synbiotics on Human Health. Nutrients 2017, 9, 1021. [Google Scholar] [CrossRef] [PubMed]
- Molinero, N.; Ruiz, L.; Sánchez, B.; Margolles, A.; Delgado, S. Intestinal Bacteria Interplay With Bile and Cholesterol Metabolism: Implications on Host Physiology. Front. Physiol. 2019, 10, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, J.L.; Brown, M.S. Regulation of the mevalonate pathway. Nature 1990, 343, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Kapourchali, F.R.; Surendiran, G.; Goulet, A.; Moghadasian, M.H. The Role of Dietary Cholesterol in Lipoprotein Metabolism and Related Metabolic Abnormalities: A Mini-review. Crit. Rev. Food Sci. Nutr. 2016, 56, 2408–2415. [Google Scholar] [CrossRef]
- Yamamoto, H.; Yamanashi, Y.; Takada, T.; Mu, S.; Tanaka, Y.; Komine, T.; Suzuki, H. Hepatic Expression of Niemann-Pick C1-Like 1, a Cholesterol Reabsorber from Bile, Exacerbates Western Diet–Induced Atherosclerosis in LDL Receptor Mutant Mice. Mol. Pharmacol. 2019, 96, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Yu, L. The structure and function of Niemann-Pick C1-like 1 protein. Curr. Opin. Lipidol. 2008, 19, 263–269. [Google Scholar] [CrossRef]
- Luo, J.; Yang, H.; Song, B.-L. Mechanisms and regulation of cholesterol homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 225–245. [Google Scholar] [CrossRef]
- Radhakrishnan, A.; Goldstein, J.L.; McDonald, J.G.; Brown, M.S. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: A delicate balance. Cell Metab. 2008, 8, 512–521. [Google Scholar] [CrossRef] [Green Version]
- Goldstein, J.L.; Brown, M.S. A century of cholesterol and coronaries: From plaques to genes to statins. Cell 2015, 161, 161–172. [Google Scholar] [CrossRef] [Green Version]
- Moon, Y.A. The SCAP/SREBP Pathway: A Mediator of Hepatic Steatosis. Endocrinol. Metab. 2017, 32, 6–10. [Google Scholar] [CrossRef]
- Riscal, R.; Skuli, N.; Simon, M.C. Even Cancer Cells Watch Their Cholesterol! Mol. Cell 2019, 76, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Hardy, L.M.; Frisdal, E.; Le Goff, W. Critical Role of the Human ATP-Binding Cassette G1 Transporter in Cardiometabolic Diseases. Int. J. Mol. Sci. 2017, 18, 1892. [Google Scholar] [CrossRef] [Green Version]
- Ceccanti, M.; Cambieri, C.; Frasca, V.; Onesti, E.; Biasiotta, A.; Giordano, C.; Bruno, S.M.; Testino, G.; Lucarelli, M.; Arca, M.; et al. A Novel Mutation in ABCA1 Gene Causing Tangier Disease in an Italian Family with Uncommon Neurological Presentation. Front. Neurol. 2016, 7, 185. [Google Scholar] [CrossRef]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters ABCG5 and ABCG8 by the liver X receptors alpha and beta. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.-W.; Qu, J.; Black, D.D.; Tso, P. Regulation of intestinal lipid metabolism: Current concepts and relevance to disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Xiao, C.; Morgantini, C.; Lewis, G.F. New Insights into the Regulation of Chylomicron Production. Annu. Rev. Nutr. 2015, 35, 265–294. [Google Scholar] [CrossRef]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef]
- Jakulj, L.; Van Dijk, T.H.; De Boer, J.F.; Kootte, R.S.; Schonewille, M.; Paalvast, Y.; Boer, T.; Bloks, V.W.; Boverhof, R.; Nieuwdorp, M.; et al. Transintestinal Cholesterol Transport Is Active in Mice and Humans and Controls Ezetimibe-Induced Fecal Neutral Sterol Excretion. Cell Metab. 2016, 24, 783–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Xia, M.; Yan, X.; Li, D.; Wang, L.; Xu, Y.; Jin, T.; Ling, W. Gut microbiota metabolism of anthocyanin promotes reverse cholesterol transport in mice via repressing miRNA-10b. Circ. Res. 2012, 111, 967–981. [Google Scholar] [CrossRef] [Green Version]
- Busnelli, M.; Manzini, S.; Chiesa, G. The Gut Microbiota Affects Host Pathophysiology as an Endocrine Organ: A Focus on Cardiovascular Disease. Nutrients 2019, 12, 79. [Google Scholar] [CrossRef] [Green Version]
- Fiorucci, S.; Distrutti, E. Bile Acid-Activated Receptors, Intestinal Microbiota, and the Treatment of Metabolic Disorders. Trends Mol. Med. 2015, 21, 702–714. [Google Scholar] [CrossRef]
- Botham, K.M.; Mayes, P.A. Cholesterol Synthesis, Transport & Excretion. In Harper’s Illustrated Biochemistry, 30th ed.; Rodwell, V.W., Bender, D.A., Botham, K.M., Kennelly, P.J., Weil, P.A., Eds.; McGraw-Hill Education: New York, NY, USA, 2016. [Google Scholar]
- Xu, Y.; Li, F.; Zalzala, M.; Xu, J.; Gonzalez, F.J.; Adorini, L.; Lee, Y.-K.; Yin, L.; Zhang, Y. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology 2016, 64, 1072–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlström, A.; Sayin, S.I.; Marschall, H.-U.; Bäckhed, F. Intestinal Crosstalk between Bile Acids and Microbiota and Its Impact on Host Metabolism. Cell Metab. 2016, 24, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Richards, L.B.; Li, M.; Van Esch, B.C.A.M.; Garssen, J.; Folkerts, G. The effects of short-chain fatty acids on the cardiovascular system. PharmaNutrition 2016, 4, 68–111. [Google Scholar] [CrossRef]
- Li, T.; Chiang, J.Y.L. Bile acid-based therapies for non-alcoholic steatohepatitis and alcoholic liver disease. Hepatobiliary Surg. Nutr. 2020, 9, 152–169. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.-J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.-Y.; Kim, S.J.; Ko, E.K.; Ahn, S.-H.; Seo, H.; Sung, M.-K. Gut microbiota-associated bile acid deconjugation accelerates hepatic steatosis in ob/ob mice. J. Appl. Microbiol. 2016, 121, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hao, C.; Yao, W.; Zhu, D.; Lu, H.; Li, L.; Ma, B.; Sun, B.; Xue, D.; Zhang, W. Intestinal flora imbalance affects bile acid metabolism and is associated with gallstone formation. BioMed. Cent. Gastroenterol. 2020, 20, 59. [Google Scholar] [CrossRef]
- Mayerhofer, C.C.K.; Ueland, T.; Broch, K.; Vincent, R.P.; Cross, G.F.; Dahl, C.P.; Aukrust, P.; Gullestad, L.; Hov, J.R.; Trøseid, M. Increased Secondary/Primary Bile Acid Ratio in Chronic Heart Failure. J. Card. Fail. 2017, 23, 666–671. [Google Scholar] [CrossRef] [Green Version]
- Cho, C.E.; Taesuwan, S.; Malysheva, O.V.; Bender, E.; Tulchinsky, N.F.; Yan, J.; Sutter, J.L.; Caudill, M.A. Trimethylamine-N-oxide (TMAO) response to animal source foods varies among healthy young men and is influenced by their gut microbiota composition: A randomized controlled trial. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Canyelles, M.; Tondo, M.; Cedo, L.; Farras, M.; Escola-Gil, J.C.; Blanco-Vaca, F. Trimethylamine N-Oxide: A Link among Diet, Gut Microbiota, Gene Regulation of Liver and Intestine Cholesterol Homeostasis and HDL Function. Int. J. Mol. Sci. 2018, 19, 3228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Heaney, L.M.; Jones, D.J.L.; Ng, L.L. Trimethylamine N-oxide and Risk Stratification after Acute Myocardial Infarction. Clin. Chem. 2017, 63, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Chang, M.; Guo, Y.; Zhang, L.; Xue, C.; Yanagita, T.; Zhang, T.; Wang, Y. Trimethylamine-N-oxide (TMAO)-induced atherosclerosis is associated with bile acid metabolism. Lipids Health Dis. 2018, 17, 286. [Google Scholar] [CrossRef] [Green Version]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Z.-H.; Xin, F.-Z.; Zhou, D.; Xue, Y.-Q.; Liu, X.-L.; Yang, R.-X.; Pan, Q.; Fan, J.-G. Trimethylamine N-oxide attenuates high-fat high-cholesterol diet-induced steatohepatitis by reducing hepatic cholesterol overload in rats. World J. Gastroenterol. 2019, 25, 2450–2462. [Google Scholar] [CrossRef] [PubMed]
- Collins, H.L.; Drazul-Schrader, D.; Sulpizio, A.C.; Koster, P.D.; Williamson, Y.; Adelman, S.J.; Owen, K.; Sanli, T.; Bellamine, A. L-Carnitine intake and high trimethylamine N-oxide plasma levels correlate with low aortic lesions in ApoE−/− transgenic mice expressing CETP. Atherosclerosis 2016, 244, 29–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef]
- Ma, J.; Li, H. The Role of Gut Microbiota in Atherosclerosis and Hypertension. Front. Pharm. 2018, 9, 1082. [Google Scholar] [CrossRef]
- Pathak, P.; Helsley, R.N.; Brown, A.L.; Buffa, J.A.; Choucair, I.; Nemet, I.; Gogonea, C.B.; Gogonea, V.; Wang, Z.; Garcia-Garcia, J.C.; et al. Small molecule inhibition of gut microbial choline trimethylamine lyase activity alters host cholesterol and bile acid metabolism. Am. J. Physiol. 2020, 318, H1474–H1486. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.L.; Caesar, R.; Akrami, R.; Reinhardt, C.; Hållenius, F.F.; Borén, J.; Bäckhed, F. Impact of Gut Microbiota and Diet on the Development of Atherosclerosis in Apoe−/− Mice. Arter. Thromb. Vasc. Biol. 2018, 38, 2318–2326. [Google Scholar] [CrossRef] [PubMed]
- Warrier, M.; Shih, D.M.; Burrows, A.C.; Ferguson, D.; Gromovsky, A.D.; Brown, A.L.; Marshall, S.; McDaniel, A.; Schugar, R.C.; Wang, Z.; et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015, 10, 326–338. [Google Scholar] [CrossRef] [Green Version]
- Hartley, L.; May, M.D.; Loveman, E.; Colquitt, J.L.; Rees, K. Dietary fibre for the primary prevention of cardiovascular disease. Cochrane Database Syst. Rev. 2016, 2016, CD011472. [Google Scholar] [CrossRef] [PubMed]
- Flint, H.J.; Duncan, S.H.; Scott, K.P.; Louis, P. Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 2015, 74, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Feng, W.; Ao, H.; Peng, C. Gut Microbiota, Short-Chain Fatty Acids, and Herbal Medicines. Front. Pharm. 2018, 9. [Google Scholar] [CrossRef]
- Korcz, E.; Kerenyi, Z.; Varga, L. Dietary fibers, prebiotics, and exopolysaccharides produced by lactic acid bacteria: Potential health benefits with special regard to cholesterol-lowering effects. Food Funct. 2018, 9, 3057–3068. [Google Scholar] [CrossRef]
- Prasad, P.D.; Gurav, A.; Zhu, H.; Martin, P.M.; Vijay-Kumar, M.; Singh, N. Chapter 5—The Relationship Between Probiotics and Dietary Fiber Consumption and Cardiovascular Health. In Dietary Fiber for the Prevention of Cardiovascular Disease; Samaan, R.A., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 73–90. [Google Scholar]
- Zhao, Y.; Liu, J.; Hao, W.; Zhu, H.; Liang, N.; He, Z.; Ma, K.Y.; Chen, Z.-Y. Structure-Specific Effects of Short-Chain Fatty Acids on Plasma Cholesterol Concentration in Male Syrian Hamsters. J. Agric. Food Chem. 2017, 65, 10984–10992. [Google Scholar] [CrossRef] [PubMed]
- Reis, S.A.; Conceição, L.L.; Rosa, D.D.; Siqueira, N.P.; Peluzio, M.C.G. Mechanisms responsible for the hypocholesterolaemic effect of regular consumption of probiotics. Nutr. Res. Rev. 2017, 30, 36–49. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Xu, C.; Huang, R.; Song, J.; Li, D.; Xia, M. Butyrate from pectin fermentation inhibits intestinal cholesterol absorption and attenuates atherosclerosis in apolipoprotein E-deficient mice. J. Nutr. Biochem. 2018, 56, 175–182. [Google Scholar] [CrossRef]
- Du, Y.; Li, X.; Su, C.; Xi, M.; Zhang, X.; Jiang, Z.; Wang, L.; Hong, B. Butyrate protects against high-fat diet-induced atherosclerosis via up-regulating ABCA1 expression in apolipoprotein E-deficiency mice. Br. J. Pharmacol. 2020, 177, 1754–1772. [Google Scholar] [CrossRef]
- Korsten, S.G.P.J.; Smits, E.A.W.; Garssen, J.; Vromans, H. Modeling of the luminal butyrate concentration to design an oral formulation capable of achieving a pharmaceutical response. PharmaNutrition 2019, 10, 100166. [Google Scholar] [CrossRef]
- Young, S.G.; Fong, L.G. Lowering plasma cholesterol by raising LDL receptors-revisited. N. Engl. J. Med. 2012, 366, 1154–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnett, D.K.; Blumenthal, R.S.; Albert, M.A.; Buroker, A.B.; Goldberger, Z.D.; Hahn, E.J.; Himmelfarb, C.D.; Khera, A.; Lloyd-Jones, D.; McEvoy, J.W.; et al. 2019 ACC/AHA Guideline on the Primary Prevention of Cardiovascular Disease: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation 2019, 140, e596–e646. [Google Scholar] [CrossRef]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Francis, G.A.; Genest, J.; Grégoire, J.; Grover, S.A.; et al. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021. [Google Scholar] [CrossRef]
- Kavalipati, N.; Shah, J.; Ramakrishan, A.; Vasnawala, H. Pleiotropic effects of statins. Indian J. Endocrinol. Metab. 2015, 19, 554–562. [Google Scholar]
- Li, D.Y.; Wang, Z.; Li, X.S.; Hazen, S.L.; Tang, W.H.W. Relationship Between Statin Use and Trimethylamine N-Oxide in Cardiovascular Risk Assessment. J. Am. Coll. Cardiol. 2018, 71 (Suppl. S11), A115. [Google Scholar] [CrossRef]
- Karlson, B.W.; Wiklund, O.; Palmer, M.K.; Nicholls, S.J.; Lundman, P.; Barter, P.J. Variability of low-density lipoprotein cholesterol response with different doses of atorvastatin, rosuvastatin, and simvastatin: Results from VOYAGER. Eur. Heart J.-Cardiovasc. Pharmacother. 2016, 2, 212–217. [Google Scholar] [CrossRef] [Green Version]
- Guglielmi, V.; Bellia, A.; Pecchioli, S.; Della-Morte, D.; Parretti, D.; Cricelli, I.; Medea, G.; Sbraccia, P.; Lauro, D.; Cricelli, C.; et al. Effectiveness of adherence to lipid lowering therapy on LDL-cholesterol in patients with very high cardiovascular risk: A real-world evidence study in primary care. Atherosclerosis 2017, 263, 36–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, H.; An, J.; Song, Y.; Lee, C.; Kim, K.; Kong, H. Alterations in Gut Microbiota by Statin Therapy and Possible Intermediate Effects on Hyperglycemia and Hyperlipidemia. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Jiao, L.; He, C.; Sun, H.; Cai, Q.; Han, T.; Hu, H. Alteration of gut microbiota in association with cholesterol gallstone formation in mice. BioMed. Cent. Gastroenterol. 2017, 17, 74. [Google Scholar] [CrossRef]
- Liu, Y.; Song, X.; Zhou, H.; Zhou, X.; Xia, Y.; Dong, X.; Zhong, W.; Tang, S.; Wang, L.; Wen, S.; et al. Gut Microbiome Associates With Lipid-Lowering Effect of Rosuvastatin in Vivo. Front. MicroBiol. 2018, 9, 530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Li, L.; Zhou, X. Comparative analysis of the gut microbiota in distinct statin response patients in East China. J. Microbiol. 2018, 56, 886–892. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Solberg, O.G.; Storm-Larsen, C.; Holm, K.; Ragnarsson, A.; Trøseid, M.; Vestad, B.; Skårdal, R.; Yndestad, A.; Ueland, T.; et al. Rosuvastatin alters the genetic composition of the human gut microbiome. Sci. Rep. 2020, 10, 5397. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.J.; Ahmed, Y.M.; Zamzami, M.A.; Siddiqui, A.M.; Khan, I.; Baothman, O.A.S.; Mehanna, M.G.; Kuerban, A.; Kaleemuddin, M.; Yasir, M. Atorvastatin Treatment Modulates the Gut Microbiota of the Hypercholesterolemic Patients. J. Integr. Biol. 2018, 22, 154–163. [Google Scholar] [CrossRef]
- Liaskovskii, T.M.; Podgorskii, V.S. Assessment of probiotics according to the international organizations (FAO/WHO). Mikrobiolohichnyi Zhurnal 2005, 67, 104–112. [Google Scholar]
- Sun, J.; Buys, N. Effects of probiotics consumption on lowering lipids and CVD risk factors: A systematic review and meta-analysis of randomized controlled trials. Ann. Med. 2015, 47, 430–440. [Google Scholar] [CrossRef]
- Zhao, W.; Liu, Y.; Latta, M.; Ma, W.; Wu, Z.; Chen, P. Probiotics database: A potential source of fermented foods. Int. J. Food Prop. 2019, 22, 198–217. [Google Scholar] [CrossRef] [Green Version]
- Horackova, S.; Plockova, M.; Demnerova, K. Importance of microbial defence systems to bile salts and mechanisms of serum cholesterol reduction. Biotechnol. Adv. 2018, 36, 682–690. [Google Scholar] [CrossRef]
- Cho, Y.A.; Kim, J. Effect of Probiotics on Blood Lipid Concentrations: A Meta-Analysis of Randomized Controlled Trials. Medicine 2015, 94, e1714. [Google Scholar] [CrossRef]
- Wang, L.; Guo, M.-J.; Gao, Q.; Yang, J.-F.; Yang, L.; Pang, X.-L.; Jiang, X.-J. The effects of probiotics on total cholesterol: A meta-analysis of randomized controlled trials. Medicine 2018, 97, e9679. [Google Scholar] [CrossRef]
- Shimizu, M.; Hashiguchi, M.; Shiga, T.; Tamura, H.-o.; Mochizuki, M. Meta-Analysis: Effects of Probiotic Supplementation on Lipid Profiles in Normal to Mildly Hypercholesterolemic Individuals. PLoS ONE 2015, 10, e0139795. [Google Scholar] [CrossRef]
- Khalesi, S.; Bellissimo, N.; Vandelanotte, C.; Williams, S.; Stanley, D.; Irwin, C. A review of probiotic supplementation in healthy adults: Helpful or hype? Eur. J. Clin. Nutr. 2019, 73, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Miremadi, F.; Sherkat, F.; Stojanovska, L. Hypocholesterolaemic effect and anti-hypertensive properties of probiotics and prebiotics: A review. J. Funct. Foods 2016, 25, 497–510. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, Q.; Ren, Y.; Ruan, Z. Effect of probiotic Lactobacillus on lipid profile: A systematic review and meta-analysis of randomized, controlled trials. PLoS ONE 2017, 12, e0178868. [Google Scholar] [CrossRef] [PubMed]
- Tarrah, A.; Dos Santos Cruz, B.C.; Sousa Dias, R.; Da Silva Duarte, V.; Pakroo, S.; Licursi de Oliveira, L.; Gouveia Peluzio, M.C.; Corich, V.; Giacomini, A.; Oliveira de Paula, S. Lactobacillus paracasei DTA81, a cholesterol-lowering strain having immunomodulatory activity, reveals gut microbiota regulation capability in BALB/c mice receiving high-fat diet. J. Appl. MicroBiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Park, S.H.; Sin, H.-S.; Jang, S.-H.; Lee, S.-W.; Kim, S.-Y.; Kwon, B.; Yu, K.-Y.; Kim, S.Y.; Yang, D.K. Hypocholesterolemic Effects of Probiotic Mixture on Diet-Induced Hypercholesterolemic Rats. Nutrients 2017, 9, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, Y.K.; El-Nezami, H.; Chen, Y.; Kinnunen, K.; Kirjavainen, P.V. Probiotic mixture VSL#3 reduce high fat diet induced vascular inflammation and atherosclerosis in ApoE−/− mice. AMB Express 2016, 6, 61. [Google Scholar]
- Rerksuppaphol, S.; Rerksuppaphol, L. A Randomized Double-blind Controlled Trial of Lactobacillus acidophilus Plus Bifidobacterium bifidum versus Placebo in Patients with Hypercholesterolemia. J. Clin. Diagn. Res. 2015, 9, KC01–KC04. [Google Scholar] [CrossRef]
- Qiu, L.; Yang, D.; Tao, X.; Yu, J.; Xiong, H.; Wei, H. Enterobacter aerogenes ZDY01 Attenuates Choline-Induced Trimethylamine N-Oxide Levels by Remodeling Gut Microbiota in Mice. J. Microbiol. Biotechnol. 2017, 27, 1491–1499. [Google Scholar] [CrossRef]
- Qiu, L.; Tao, X.; Xiong, H.; Yu, J.; Wei, H. Lactobacillus plantarum ZDY04 exhibits a strain-specific property of lowering TMAO via the modulation of gut microbiota in mice. Food Funct. 2018, 9, 4299–4309. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, F.; Xu, D.; Zhang, Z.; Xu, F.; Tao, X.; Qiu, L.; Wei, H. Enterococcus faecium WEFA23 from infants lessens high-fat-diet-induced hyperlipidemia via cholesterol 7-alpha-hydroxlase gene by altering the composition of gut microbiota in rats. J. Dairy Sci. 2018, 101, 7757–7767. [Google Scholar] [CrossRef]
- Wang, G.; Huang, W.; Xia, Y.; Xiong, Z.; Ai, L. Cholesterol-lowering potentials of Lactobacillus strain overexpression of bile salt hydrolase on high cholesterol diet-induced hypercholesterolemic mice. Food Funct. 2019, 10, 1684–1695. [Google Scholar] [CrossRef]
- Song, M.; Yun, B.; Moon, J.-H.; Park, D.-J.; Lim, K.; Oh, S. Characterization of Selected Lactobacillus Strains for Use as Probiotics. Korean J. Food Sci. Anim. Resour. 2015, 35, 551–556. [Google Scholar] [CrossRef] [Green Version]
- Kumar, R.; Grover, S.; Batish, V.K. Bile Salt Hydrolase (Bsh) Activity Screening of Lactobacilli: In Vitro Selection of Indigenous Lactobacillus Strains with Potential Bile Salt Hydrolysing and Cholesterol-Lowering Ability. Probiotics Antimicrob. Proteins 2012, 4, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Shehata, M.G.; El-Sahn, M.A.; El Sohaimy, S.A.; Youssef, M.M. In vitro assessment of hypocholesterolemic activity of Lactococcus lactis subsp. lactis. Bull. Natl. Res. Cent. 2019, 43, 60. [Google Scholar] [CrossRef]
- Le, B.; Yang, S.-H. Identification of a Novel Potential Probiotic Lactobacillus plantarum FB003 Isolated from Salted-Fermented Shrimp and its Effect on Cholesterol Absorption by Regulation of NPC1L1 and PPARα. Probiotics Antimicrob. Proteins 2019, 11, 785–793. [Google Scholar] [CrossRef]
- Lim, F.T.; Lim, S.M.; Ramasamy, K. Pediococcus acidilactici LAB4 and Lactobacillus plantarum LAB12 assimilate cholesterol and modulate ABCA1, CD36, NPC1L1 and SCARB1 in vitro. Benef. Microbes 2017, 8, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Sasikumar, K.; Kozhummal Vaikkath, D.; Devendra, L.; Nampoothiri, K.M. An exopolysaccharide (EPS) from a Lactobacillus plantarum BR2 with potential benefits for making functional foods. Bioresour. Technol. 2017, 241, 1152–1156. [Google Scholar] [CrossRef]
- Korneva, V.; Kuznetsova, T.; Julius, U. Efficiency and problems of statin therapy in patients with heterozygous familial hypercholesterolemia. Atheroscler. Suppl. 2019, 40, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhou, B.; Zhou, X.; Wang, Y.; Wang, H.; Jia, S.; Zhang, Z.; Chu, C.; Mu, J. Combined Lowering Effects of Rosuvastatin and L. acidophilus on Cholesterol Levels in Rat. J. MicroBiol. Biotechnol. 2019, 29, 473–481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, M.; Qian, Z.; Yin, J.; Xu, W.; Zhou, X. The Role of Intestinal Microbiota in Cardiovascular Disease. J. Cell. Mol. Med. 2019, 23, 2343–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Metabolism, import, and export of cholesterol in polarized cell. The bulk of endogenous cholesterol is synthesized by the liver and starts from acetyl coenzyme A (acetyl-CoA) and concerted actions of more than 20 enzymes. Once cholesterol is formed, it is either esterified to cholesterol ester (CE) and stored in lipid droplets or secreted into the bloodstream as lipoproteins. Excess cholesterol can be excreted by ABC subfamily G member 5 and member 8 (ABCG5/8) transporters to the intestine or to the bile or packaged with lipoproteins for subsequent secretion into the blood.
Figure 1.
Metabolism, import, and export of cholesterol in polarized cell. The bulk of endogenous cholesterol is synthesized by the liver and starts from acetyl coenzyme A (acetyl-CoA) and concerted actions of more than 20 enzymes. Once cholesterol is formed, it is either esterified to cholesterol ester (CE) and stored in lipid droplets or secreted into the bloodstream as lipoproteins. Excess cholesterol can be excreted by ABC subfamily G member 5 and member 8 (ABCG5/8) transporters to the intestine or to the bile or packaged with lipoproteins for subsequent secretion into the blood.

Figure 2.
The endogenous (white) and exogenous (gray) pathways of cholesterol transport. The exogenous pathway starts with the dietary cholesterol whereas the liver is the starting point of the endogenous pathway.
Figure 2.
The endogenous (white) and exogenous (gray) pathways of cholesterol transport. The exogenous pathway starts with the dietary cholesterol whereas the liver is the starting point of the endogenous pathway.

Figure 3.
Reverse cholesterol transport pathways between intestine, liver and macrophage. RCT begins in the liver and the intestine with the synthesis of ApoA-1. ApoA-1 associates with phospholipids and free cholesterol through an interaction with ABCA1 located on various cell types (enterocytes, hepatocytes and macrophages). Gut microbiota modulates the RCT by increasing the level of ABCA1 and ABCG1 via the transformation of Cy-3-G into PCA. LDLR: LDL receptor; TG: Triglycerides; CETP: cholesteryl ester transfer protein; PLTP: phospholipid transfer protein; LCAT: lecithin-cholesterol acyltransferase; Cy-3-G: cyanidin-3-O-ß-glucoside; PCA: protocatechuic acid.
Figure 3.
Reverse cholesterol transport pathways between intestine, liver and macrophage. RCT begins in the liver and the intestine with the synthesis of ApoA-1. ApoA-1 associates with phospholipids and free cholesterol through an interaction with ABCA1 located on various cell types (enterocytes, hepatocytes and macrophages). Gut microbiota modulates the RCT by increasing the level of ABCA1 and ABCG1 via the transformation of Cy-3-G into PCA. LDLR: LDL receptor; TG: Triglycerides; CETP: cholesteryl ester transfer protein; PLTP: phospholipid transfer protein; LCAT: lecithin-cholesterol acyltransferase; Cy-3-G: cyanidin-3-O-ß-glucoside; PCA: protocatechuic acid.

Figure 4.
Enterohepatic recirculation of bile acids via reverse cholesterol transport. In the liver, cholesterol is metabolized to BAs by hepatic cytochromes, notably 90% of the time by the rate-limiting enzyme cholesterol 7 α-hydroxylase (CYP7A1). When BAs are conjugated, they are actively transported into bile via the bile salt export pump (BSEP) or via ABCG5/8. In the small intestine, certain gut microbes contain an enzyme known as bile salt hydrolase (BSH) capable of deconjugating (i.e., remove glycine or taurine conjugates) BAs preventing their reuptake by the ASBT transporter BAs: Bile acids; apical sodium-dependent bile acid transporter: ASBT; organic solute transporters: OST α/β; nuclear farnesoid X receptor: FXR; bile salt export pump: BSEP; Na+/taurochlorate cotransporting polypeptide: NTCP; fibroblast growth factor receptor 4 (FGFR4).
Figure 4.
Enterohepatic recirculation of bile acids via reverse cholesterol transport. In the liver, cholesterol is metabolized to BAs by hepatic cytochromes, notably 90% of the time by the rate-limiting enzyme cholesterol 7 α-hydroxylase (CYP7A1). When BAs are conjugated, they are actively transported into bile via the bile salt export pump (BSEP) or via ABCG5/8. In the small intestine, certain gut microbes contain an enzyme known as bile salt hydrolase (BSH) capable of deconjugating (i.e., remove glycine or taurine conjugates) BAs preventing their reuptake by the ASBT transporter BAs: Bile acids; apical sodium-dependent bile acid transporter: ASBT; organic solute transporters: OST α/β; nuclear farnesoid X receptor: FXR; bile salt export pump: BSEP; Na+/taurochlorate cotransporting polypeptide: NTCP; fibroblast growth factor receptor 4 (FGFR4).

Figure 5.
Mechanisms linking the pro-atherosclerotic activity of gut microbial metabolite TMAO and cholesterol transport and synthesis in the intestine, liver, and macrophage. The intestinal microbiota metabolizes choline, phosphatidylcholine, L-carnitine, and betaine to trimethylamine (TMA), which is oxidized into TMAO by hepatic flavin monooxygenases (FMO). Green arrows indicate an increase of expression or activation whereas red arrows indicate a reduction of the expression. Yellow arrows indicate that the results in the literature are controversial.
Figure 5.
Mechanisms linking the pro-atherosclerotic activity of gut microbial metabolite TMAO and cholesterol transport and synthesis in the intestine, liver, and macrophage. The intestinal microbiota metabolizes choline, phosphatidylcholine, L-carnitine, and betaine to trimethylamine (TMA), which is oxidized into TMAO by hepatic flavin monooxygenases (FMO). Green arrows indicate an increase of expression or activation whereas red arrows indicate a reduction of the expression. Yellow arrows indicate that the results in the literature are controversial.

Figure 6.
Mechanisms linking the anti-atherosclerotic activity of short-chain fatty acids (SCFAs) and cholesterol transport and synthesis in the intestine and liver. Dietary fibers could be fermented and transformed into short chain fatty acids (SCFA) by some species present in the gut. Green arrows indicate an increase of activity or expression whereas the red arrows indicate an inhibition or reduction of expression.
Figure 6.
Mechanisms linking the anti-atherosclerotic activity of short-chain fatty acids (SCFAs) and cholesterol transport and synthesis in the intestine and liver. Dietary fibers could be fermented and transformed into short chain fatty acids (SCFA) by some species present in the gut. Green arrows indicate an increase of activity or expression whereas the red arrows indicate an inhibition or reduction of expression.

Figure 7.
Hypocholesterolemic mechanisms of action of probiotics. It has been observed that some probiotics could reduce cholesterol and ultimately atherosclerosis. Some of the major strains used as probiotics are indicated. The mechanisms are still speculative and could be associated with an increase (green arrows) or a decrease (red arrows) of the expression or activities.
Figure 7.
Hypocholesterolemic mechanisms of action of probiotics. It has been observed that some probiotics could reduce cholesterol and ultimately atherosclerosis. Some of the major strains used as probiotics are indicated. The mechanisms are still speculative and could be associated with an increase (green arrows) or a decrease (red arrows) of the expression or activities.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Vourakis, M.; Mayer, G.; Rousseau, G. The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis. Int. J. Mol. Sci. 2021, 22, 8074. https://doi.org/10.3390/ijms22158074
AMA Style
Vourakis M, Mayer G, Rousseau G. The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis. International Journal of Molecular Sciences. 2021; 22(15):8074. https://doi.org/10.3390/ijms22158074
Chicago/Turabian StyleVourakis, Margaret, Gaétan Mayer, and Guy Rousseau. 2021. "The Role of Gut Microbiota on Cholesterol Metabolism in Atherosclerosis" International Journal of Molecular Sciences 22, no. 15: 8074. https://doi.org/10.3390/ijms22158074
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.