
Biomarkers and Mechanisms of Oxidative Stress—Last 20 Years of Research with an Emphasis on Kidney Damage and Renal Transplantation


Abstract
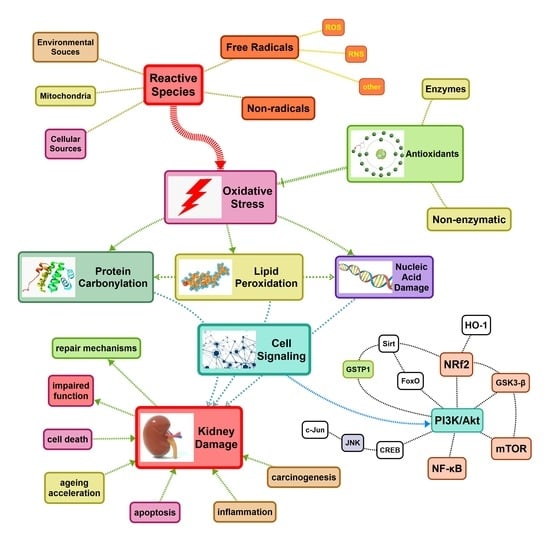
:
1. Introduction
2. Oxidative Stress
3. Oxidative Stress in Kidney Transplantation
3.1. Oxidative Stress in Ischemia-Reperfusion Injury
3.2. Oxidative Stress and Inflammation
3.3. Oxidative Stress and Kidney Damage
4. Biomarkers of Oxidative Stress
4.1. Endogenous Antioxidants
4.2. Lipid Peroxidation (LPO)
4.3. Protein Oxidation
4.4. Nucleic Acid Oxidation
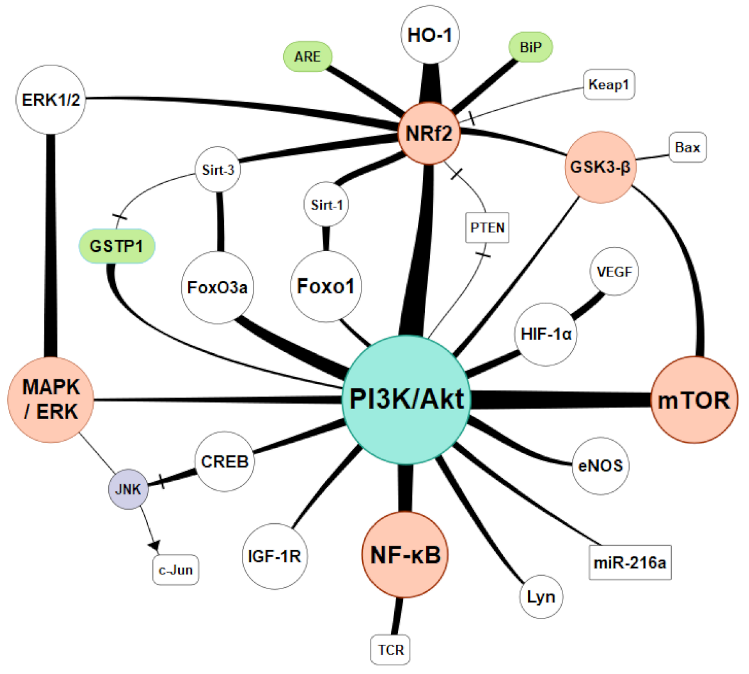
5. Regulatory Pathways and Antioxidative Therapies
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Forcados, G.E.; James, D.B.; Sallau, A.B.; Muhammad, A.; Mabeta, P. Oxidative Stress and Carcinogenesis: Potential of Phytochemicals in Breast Cancer Therapy. Nutr. Cancer 2017, 69, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [Green Version]
- Hentia, C.; Rizzato, A.; Camporesi, E.; Yang, Z.; Muntean, D.M.; Săndesc, D.; Bosco, G. An overview of protective strategies against ischemia/reperfusion injury: The role of hyperbaric oxygen preconditioning. Brain Behav. 2018, 8, e00959. [Google Scholar] [CrossRef]
- Nastos, C.; Kalimeris, K.; Papoutsidakis, N.; Tasoulis, M.-K.; Lykoudis, P.M.; Theodoraki, K.; Nastou, D.; Smyrniotis, V.; Arkadopoulos, N. Global consequences of liver ischemia/reperfusion injury. Oxid. Med. Cell Longev. 2014, 2014, 906965. [Google Scholar] [CrossRef] [Green Version]
- Pantazi, E.; Zaouali, M.A.; Bejaoui, M.; Folch-Puy, E.; Ben Abdennebi, H.; Roselló-Catafau, J. Role of sirtuins in ischemia-reperfusion injury. World J. Gastroenterol. 2013, 19, 7594–7602. [Google Scholar] [CrossRef] [Green Version]
- Panisello-Roselló, A.; Roselló-Catafau, J. Molecular Mechanisms and Pathophysiology of Ischemia-Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 4093. [Google Scholar] [CrossRef] [Green Version]
- Panisello-Roselló, A.; Roselló-Catafau, J.; Adam, R. New Insights in Molecular Mechanisms and Pathophysiology of Ischemia-Reperfusion Injury 2.0, An Updated Overview. Int. J. Mol. Sci. 2020, 22, 28. [Google Scholar] [CrossRef]
- Jones, D.P. Redefining oxidative stress. Antioxid Redox Signal. 2006, 8, 1865–1879. [Google Scholar] [CrossRef]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obs. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Ogura, S.; Shimosawa, T. Oxidative stress and organ damages. Curr. Hypertens. Rep. 2014, 16, 452. [Google Scholar] [CrossRef]
- Efe, U.; Dede, S.; Yüksek, V.; Çetin, S. Apoptotic and Oxidative Mechanisms in Liver and Kidney Tissues of Sheep with Fluorosis. Biol. Trace Elem. Res. 2021, 199, 136–141. [Google Scholar] [CrossRef]
- La Russa, D.; Brunelli, E.; Pellegrino, D. Oxidative imbalance and kidney damage in spontaneously hypertensive rats: Activation of extrinsic apoptotic pathways. Clin. Sci. 2017, 131, 1419–1428. [Google Scholar] [CrossRef]
- Sangeetha Lakshmi, B.; Harini Devi, N.; Suchitra, M.M.; Srinivasa Rao, P.V.L.N.; Siva Kumar, V. Changes in the inflammatory and oxidative stress markers during a single hemodialysis session in patients with chronic kidney disease. Ren. Fail. 2018, 40, 534–540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, K.; Shoji, T.; Kabata, D.; Shintani, A.; Okute, Y.; Tsuchikura, S.; Shimomura, N.; Tsujimoto, Y.; Nakatani, S.; Mori, K.; et al. Oxidative Stress and Inflammation as Predictors of Mortality and Cardiovascular Events in Hemodialysis Patients: The DREAM Cohort. J. Atheroscler. Thromb. 2021, 28, 249–260. [Google Scholar] [CrossRef]
- Yari, Z.; Tabibi, H.; Najafi, I.; Hedayati, M.; Movahedian, M. Effects of soy isoflavones on serum systemic and vascular inflammation markers and oxidative stress in peritoneal dialysis patients: A randomized controlled trial. Phytother. Res. 2020, 34, 3011–3018. [Google Scholar] [CrossRef]
- Russa DLa Pellegrino, D.; Montesanto, A.; Gigliotti, P.; Perri, A.; Russa, A.L.a.; Bonofiglio, R. Oxidative Balance and Inflammation in Hemodialysis Patients: Biomarkers of Cardiovascular Risk? Oxid. Med. Cell Longev. 2019, 2019, 8567275. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Bisht, S.; Faiq, M.; Tolahunase, M.; Dada, R. Oxidative stress and male infertility. Nat. Rev. Urol. 2017, 14, 470–485. [Google Scholar] [CrossRef]
- Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273. [Google Scholar] [CrossRef]
- Kimura, A.; Namekata, K.; Guo, X.; Noro, T.; Harada, C.; Harada, T. Targeting Oxidative Stress for Treatment of Glaucoma and Optic Neuritis. Oxid. Med. Cell Longev. 2017, 2017, 2817252. [Google Scholar] [CrossRef]
- Orzechowski, A.; Cywińska, A.; Rostagno, A.A.; Rizzi, F.M. Oxidative Stress, Chronic Inflammation, and Amyloidoses. Oxid. Med. Cell Longev. 2019, 2019, 6024975. [Google Scholar] [CrossRef]
- Crotty, G.F.; Ascherio, A.; Schwarzschild, M.A. Targeting urate to reduce oxidative stress in Parkinson disease. Exp. Neurol. 2017, 298, 210–224. [Google Scholar] [CrossRef]
- Chielle, E.O.; Trott, A.; da Silva, R.B.; Casarin, J.N.; Fortuna, P.C.; da Cruz, I.B.M.; Moretto, M.B.; Moresco, R.N. Impact of the Ile105Val Polymorphism of the Glutathione S-transferase P1 (GSTP1) Gene on Obesity and Markers of Cardiometabolic Risk in Young Adult Population. Exp. Clin. Endocrinol. Diabetes 2017, 125, 335–341. [Google Scholar] [CrossRef]
- Arfianti, E.; Larter, C.Z.; Lee, S.; Barn, V.; Haigh, G.; Yeh, M.M.; Ioannou, G.N.; Teoh, N.C.; Farrell, G.C. Obesity and diabetes accelerate hepatocarcinogenesis via hepatocyte proliferation independent of NF-κB or Akt/mTORC1. J. Clin. Transl. Res. 2016, 2, 26–37. [Google Scholar]
- Nteeba, J.; Ganesan, S.; Keating, A.F. Impact of obesity on ovotoxicity induced by 7,12-dimethylbenz[a]anthracene in mice. Biol. Reprod. 2014, 90, 68. [Google Scholar] [CrossRef]
- Nteeba, J.; Ross, J.W.; Perfield, J.W., 2nd; Keating, A.F. High fat diet induced obesity alters ovarian phosphatidylinositol-3 kinase signaling gene expression. Reprod. Toxicol. 2013, 42, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Karam, B.S.; Chavez-Moreno, A.; Koh, W.; Akar, J.G.; Akar, F.G. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc. Diabetol. 2017, 16, 120. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Mills, K.; le Cessie, S.; Noordam, R.; van Heemst, D. Ageing, age-related diseases and oxidative stress: What to do next? Ageing Res. Rev. 2020, 57, 100982. [Google Scholar] [CrossRef]
- Halliwell, B.; Whiteman, M. Measuring reactive species and oxidative damage in vivo and in cell culture: How should you do it and what do the results mean? Br. J. Pharm. 2004, 142, 231–255. [Google Scholar] [CrossRef] [Green Version]
- Mattila, H.; Khorobrykh, S.; Havurinne, V.; Tyystjärvi, E. Reactive oxygen species: Reactions and detection from photosynthetic tissues. J. Photochem. Photobiol. B 2015, 152, 176–214. [Google Scholar] [CrossRef]
- Phaniendra, A.; Jestadi, D.B.; Periyasamy, L. Free radicals: Properties, sources, targets, and their implication in various diseases. Indian J. Clin. Biochem. 2015, 30, 11–26. [Google Scholar] [CrossRef] [Green Version]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [Green Version]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Évora, P.; Castro-E-Silva, O. Ischemia/Reperfusion Injury Revisited: An Overview of the Latest Pharmacological Strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [Green Version]
- Bretón-Romero, R.; Lamas, S. Hydrogen peroxide signaling in vascular endothelial cells. Redox Biol. 2014, 2, 529–534. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.-H.; Huang, C.-C.; Chang, C.-P.; Lin, M.-T.; Niu, K.-C.; Tian, Y.-F. Heat Shock Protein 70 (HSP70) Reduces Hepatic Inflammatory and Oxidative Damage in a Rat Model of Liver Ischemia/Reperfusion Injury with Hyperbaric Oxygen Preconditioning. Med. Sci. Monit Int. Med. J. Exp. Clin. Res. 2018, 24, 8096–8104. [Google Scholar] [CrossRef]
- Wang, L.; Chen, H.; Liu, X.-H.; Chen, Z.-Y.; Weng, X.-D.; Qiu, T.; Liu, L.; Zhu, H.-C. Ozone oxidative preconditioning inhibits renal fibrosis induced by ischemia and reperfusion injury in rats. Exp. Med. 2014, 8, 1764–1768. [Google Scholar] [CrossRef] [Green Version]
- Fernández, A.R.; Sánchez-Tarjuelo, R.; Cravedi, P.; Ochando, J.; López-Hoyos, M. Review: Ischemia Reperfusion Injury-A Translational Perspective in Organ Transplantation. Int. J. Mol. Sci. 2020, 21, 8549. [Google Scholar] [CrossRef]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef]
- Acar, A.; Akil, E.; Alp, H.; Evliyaoglu, O.; Kibrisli, E.; Inal, A.; Unan, F.; Tasdemir, N. Oxidative damage is ameliorated by curcumin treatment in brain and sciatic nerve of diabetic rats. Int. J. Neurosci. 2012, 122, 367–372. [Google Scholar] [CrossRef]
- Prabhakar, O. Cerebroprotective effect of resveratrol through antioxidant and anti-inflammatory effects in diabetic rats. Naunyn. Schmiedebergs Arch. Pharm. 2013, 386, 705–710. [Google Scholar] [CrossRef]
- Lu, L.; Zhou, H.; Ni, M.; Wang, X.; Busuttil, R.; Kupiec-Weglinski, J.; Zhai, Y. Innate Immune Regulations and Liver Ischemia-Reperfusion Injury. Transplantation 2016, 100, 2601–2610. [Google Scholar] [CrossRef] [Green Version]
- Coppolino, G.; Leonardi, G.; Andreucci, M.; Bolignano, D. Oxidative Stress and Kidney Function: A Brief Update. Curr. Pharm. Des. 2018, 24, 4794–4799. [Google Scholar] [CrossRef]
- Podkowińska, A.; Formanowicz, D. Chronic Kidney Disease as Oxidative Stress- and Inflammatory-Mediated Cardiovascular Disease. Antioxid 2020, 9. [Google Scholar] [CrossRef]
- Snoeijs, M.G.J.; van Heurn, L.W.E.; Buurman, W.A. Biological modulation of renal ischemia-reperfusion injury. Curr. Opin Organ. Transplantation 2010, 15, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Duni, A.; Liakopoulos, V.; Roumeliotis, S.; Peschos, D.; Dounousi, E. Oxidative Stress in the Pathogenesis and Evolution of Chronic Kidney Disease: Untangling Ariadne’s Thread. Int. J. Mol. Sci. 2019, 20, 3711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sifuentes-Franco, S.; Padilla-Tejeda, D.E.; Carrillo-Ibarra, S.; Miranda-Díaz, A.G. Oxidative Stress, Apoptosis, and Mitochondrial Function in Diabetic Nephropathy. Int. J. Endocrinol. 2018, 2018, 1875870. [Google Scholar] [CrossRef]
- Nagata, M. Podocyte injury and its consequences. Kidney Int. 2016, 89, 1221–1230. [Google Scholar] [CrossRef]
- Coresh, J.; Heerspink, H.J.L.; Sang, Y.; Matsushita, K.; Arnlov, J.; Astor, B.C.; Black, C.; Brunskill, N.J.; Carrero, J.-J.; Feldman, H.I.; et al. Change in albuminuria and subsequent risk of end-stage kidney disease: An individual participant-level consortium meta-analysis of observational studies. Lancet Diabetes Endocrinol. 2019, 7, 115–127. [Google Scholar] [CrossRef]
- Daehn, I.; Casalena, G.; Zhang, T.; Shi, S.; Fenninger, F.; Barasch, N.; Yu, L.; D’Agati, V.; Schlondorff, D.; Kriz, W.; et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J. Clin. Investig. 2014, 124, 1608–1621. [Google Scholar] [CrossRef] [Green Version]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef] [Green Version]
- Nlandu Khodo, S.; Dizin, E.; Sossauer, G.; Szanto, I.; Martin, P.-Y.; Feraille, E.; Krause, K.H.; de Seigneux, S. NADPH-oxidase 4 protects against kidney fibrosis during chronic renal injury. J. Am. Soc. Nephrol. 2012, 23, 1967–1976. [Google Scholar] [CrossRef] [Green Version]
- Zoja, C.; Benigni, A.; Remuzzi, G. The Nrf2 pathway in the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, I19–I24. [Google Scholar] [CrossRef] [Green Version]
- Sureshbabu, A.; Ryter, S.W.; Choi, M.E. Oxidative stress and autophagy: Crucial modulators of kidney injury. Redox Biol. 2015, 4, 208–214. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.I.; Saglam, M.; Caglar, K.; Cakir, E.; Sonmez, A.; Ozgurtas, T.; Aydin, A.; Eyileten, T.; Ozcan, O.; Acikel, C.; et al. The determinants of endothelial dysfunction in CKD: Oxidative stress and asymmetric dimethylarginine. Am. J. Kidney Dis. 2006, 47, 42–50. [Google Scholar] [CrossRef]
- Modlinger, P.S.; Wilcox, C.S.; Aslam, S. Nitric oxide, oxidative stress, and progression of chronic renal failure. Semin. Nephrol. 2004, 24, 354–365. [Google Scholar] [CrossRef]
- Lai, E.Y.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Superoxide modulates myogenic contractions of mouse afferent arterioles. Hypertens 2011, 58, 650–656. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Lai, E.Y.; Wellstein, A.; Welch, W.J.; Wilcox, C.S. Differential effects of superoxide and hydrogen peroxide on myogenic signaling, membrane potential, and contractions of mouse renal afferent arterioles. Am. J. Physiol. Ren. Physiol. 2016, 310, F1197–F1205. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Haridas, B.; Jackson, A.R.; Cortado, H.; Mayne, N.; Kohnken, R.; Bolon, B.; McHugh, K.M.; Schwaderer, A.L.; Spencer, J.D.; et al. Inflammation drives renal scarring in experimental pyelonephritis. Am. J. Physiol. Ren. Physiol. 2017, 312, F43–F53. [Google Scholar] [CrossRef] [Green Version]
- Havasi, A.; Borkan, S.C. Apoptosis and acute kidney injury. Kidney Int. 2011, 80, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Young, I.S.; Woodside, J.V. Antioxidants in health and disease. J. Clin. Pathol. 2001, 54, 176–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef] [PubMed]
- Gathwala, G.; Aggarwal, R. Selenium supplementation for the preterm Indian neonate. Indian J. Public Health 2016, 60, 142–144. [Google Scholar] [CrossRef]
- Campbell, N.K.; Fitzgerald, H.K.; Dunne, A. Regulation of inflammation by the antioxidant haem oxygenase 1. Nat. Rev. Immunol. 2021. [Google Scholar] [CrossRef]
- Stocker, R.; Perrella, M.A. Heme oxygenase-1, A novel drug target for atherosclerotic diseases? Circulation 2006, 114, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Taillé, C.; El-Benna, J.; Lanone, S.; Dang, M.-C.; Ogier-Denis, E.; Aubier, M.; Boczkowski, J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J. Biol. Chem. 2004, 279, 28681–28688. [Google Scholar] [CrossRef] [Green Version]
- Ryter, S.W.; Xi, S.; Hartsfield, C.L.; Choi, A.M.K. Mitogen activated protein kinase (MAPK) pathway regulates heme oxygenase-1 gene expression by hypoxia in vascular cells. Antioxid Redox Signal. 2002, 4, 587–592. [Google Scholar] [CrossRef]
- Morita, T.; Imai, T.; Sugiyama, T.; Katayama, S.; Yoshino, G. Heme oxygenase-1 in vascular smooth muscle cells counteracts cardiovascular damage induced by angiotensin II. Curr. Neurovasc. Res. 2005, 2, 113–120. [Google Scholar] [CrossRef]
- Asher, G.; Tsvetkov, P.; Kahana, C.; Shaul, Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005, 19, 316–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oja, S.S.; Janáky, R.; Varga, V.; Saransaari, P. Modulation of glutamate receptor functions by glutathione. Neurochem. Int. 2000, 37, 299–306. [Google Scholar] [CrossRef]
- Trevisson, E.; DiMauro, S.; Navas, P.; Salviati, L. Coenzyme Q deficiency in muscle. Curr. Opin. Neurol. 2011, 24, 449–456. [Google Scholar] [CrossRef]
- Zhang, J.; Zhou, X.; Wu, W.; Wang, J.; Xie, H.; Wu, Z. Regeneration of glutathione by α-lipoic acid via Nrf2/ARE signaling pathway alleviates cadmium-induced HepG2 cell toxicity. Environ. Toxicol. Pharm. 2017, 51, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Wang, H.; Chen, Z. Alpha-Lipoic Acid Attenuates Cerebral Ischemia and Reperfusion Injury via Insulin Receptor and PI3K/Akt-Dependent Inhibition of NADPH Oxidase. Int. J. Endocrinol. 2015, 2015, 903186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, T.; Bai, J.; Liu, W.; Hu, Y. A systematic review and meta-analysis of α-lipoic acid in the treatment of diabetic peripheral neuropathy. Eur. J. Endocrinol. 2012, 167, 465–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinto, L.; Quinn, J.; Montine, T.; Dodge, H.H.; Woodward, W.; Baldauf-Wagner, S.; Waichunas, D.; Bumgarner, L.; Bourdette, D.; Silbert, L.; et al. A randomized placebo-controlled pilot trial of omega-3 fatty acids and alpha lipoic acid in Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedlak, T.W.; Saleh, M.; Higginson, D.S.; Paul, B.D.; Juluri, K.R.; Snyder, S.H. Bilirubin and glutathione have complementary antioxidant and cytoprotective roles. Proc. Natl. Acad. Sci. USA 2009, 106, 5171–5176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.; Maghzal, G.J.; Ayer, A.; Suarna, C.; Dunn, L.L.; Stocker, R. Absence of the biliverdin reductase-a gene is associated with increased endogenous oxidative stress. Free. Radic. Biol. Med. 2018, 115, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Lee, D.Y.; Kang, S.; Miao, W.; Kim, H.; Lee, Y.; Jon, S. Bilirubin nanoparticle preconditioning protects against hepatic ischemia-reperfusion injury. Biomaterial 2017, 133, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bösch, F.; Thomas, M.; Kogler, P.; Oberhuber, R.; Sucher, R.; Aigner, F.; Semsroth, S.; Wiedemann, D.; Yamashita, K.; Troppmair, J.; et al. Bilirubin rinse of the graft ameliorates ischemia reperfusion injury in heart transplantation. Transpl. Int. Off. J. Eur. Soc. Organ. Transpl. 2014, 27, 504–513. [Google Scholar] [CrossRef] [PubMed]
- Sundararaghavan, V.L.; Binepal, S.; Stec, D.E.; Sindhwani, P.; Hinds, T.D.J. Bilirubin, a new therapeutic for kidney transplant? Transpl. Rev. 2018, 32, 234–240. [Google Scholar] [CrossRef]
- Blankenhaus, B.; Braza, F.; Martins, R.; Bastos-Amador, P.; González-García, I.; Carlos, A.R.; Mahu, I.; Faisca, P.; Nunes, J.M.; Ventura, P.; et al. Ferritin regulates organismal energy balance and thermogenesis. Mol. Metab. 2019, 24, 64–79. [Google Scholar] [CrossRef]
- Kell, D.B.; Pretorius, E. Serum ferritin is an important inflammatory disease marker, as it is mainly a leakage product from damaged cells. Metallomics 2014, 6, 748–773. [Google Scholar] [CrossRef] [Green Version]
- Ong, D.S.T.; Wang, L.; Zhu, Y.; Ho, B.; Ding, J.L. The response of ferritin to LPS and acute phase of Pseudomonas infection. J. Endotoxin Res. 2005, 11, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Dąbrowska, N.; Wiczkowski, A. Analytics of oxidative stress markers in the early diagnosis of oxygen DNA damage. Adv. Clin. Exp. Med. 2017, 26, 155–166. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, England, 2015. [Google Scholar] [CrossRef]
- Marnett, L.J. Lipid peroxidation-DNA damage by malondialdehyde. Mutat. Res. 1999, 424, 83–95. [Google Scholar] [CrossRef]
- Collodel, G.; Moretti, E.; Micheli, L.; Menchiari, A.; Moltoni, L.; Cerretani, D. Semen characteristics and malondialdehyde levels in men with different reproductive problems. Andrology 2015, 3, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Morrow, J.D. F2-isoprostanes as markers of oxidant stress: An overview. Curr. Protoc. Toxicol. 2005, 17, 175. [Google Scholar] [CrossRef]
- Ahmed, O.S.; Galano, J.-M.; Pavlickova, T.; Revol-Cavalier, J.; Vigor, C.; Lee, J.C.-Y.; Oger, C.; Durand, T. Moving forward with isoprostanes, neuroprostanes and phytoprostanes: Where are we now? Essays Biochem. 2020, 64, 463–484. [Google Scholar] [CrossRef]
- Biringer, R.G. The Role of Eicosanoids in Alzheimer’s Disease. Int. J. Environ. Res. Public Health 2019, 16, 2560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, E.A.; Barry, O.P.; Lawson, J.A.; Rokach, J.; FitzGerald, G.A. Effects of vitamin E on lipid peroxidation in healthy persons. JAMA 2001, 285, 1178–1182. [Google Scholar] [CrossRef]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [Green Version]
- Halliwell, B.; Long, L.H.; Yee, T.P.; Lim, S.; Kelly, R. Establishing biomarkers of oxidative stress: The measurement of hydrogen peroxide in human urine. Curr. Med. Chem. 2004, 11, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Marnett, L.J.; Riggins, J.N.; West, J.D. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J. Clin. Investig. 2003, 111, 583–593. [Google Scholar] [CrossRef] [PubMed]
- LoPachin, R.M.; Gavin, T.; Petersen, D.R.; Barber, D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009, 22, 1499–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.S.; Taniguchi, N. Acrolein induces inflammatory response underlying endothelial dysfunction: A risk factor for atherosclerosis. Ann. N. Y. Acad. Sci. 2008, 1126, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Kehrer, J.P.; Biswal, S.S. The molecular effects of acrolein. Toxicol. Sci. 2000, 57, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Hamann, K.; Durkes, A.; Ouyang, H.; Uchida, K.; Pond, A.; Shi, R. Critical role of acrolein in secondary injury following ex vivo spinal cord trauma. J. Neurochem. 2008, 107, 712–721. [Google Scholar] [CrossRef] [Green Version]
- Park, Y.S.; Kim, J.; Misonou, Y.; Takamiya, R.; Takahashi, M.; Freeman, M.R.; Taniguchi, N. Acrolein induces cyclooxygenase-2 and prostaglandin production in human umbilical vein endothelial cells: Roles of p38 MAP kinase. Arter. Thromb. Vasc. Biol. 2007, 27, 1319–1325. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.E.; Lee, S.H.; Ryu, D.S.; Park, C.-S.; Park, K.-S.; Park, Y.S. Differentially-expressed genes related to atherosclerosis in acrolein-stimulated human umbilical vein endothelial cells. BioChip. J. 2010, 4, 264–271. [Google Scholar] [CrossRef]
- Conklin, D.J.; Bhatnagar, A.; Cowley, H.R.; Johnson, G.H.; Wiechmann, R.J.; Sayre, L.M.; Trent, M.B.; Boor, P.J. Acrolein generation stimulates hypercontraction in isolated human blood vessels. Toxicol. Appl. Pharm. 2006, 217, 277–288. [Google Scholar] [CrossRef] [Green Version]
- McCall, M.R.; Tang, J.Y.; Bielicki, J.K.; Forte, T.M. Inhibition of lecithin-cholesterol acyltransferase and modification of HDL apolipoproteins by aldehydes. Arter. Thromb. Vasc. Biol. 1995, 15, 1599–1606. [Google Scholar] [CrossRef]
- Alfredsson, L.; Hammar, N.; Hogstedt, C. Incidence of myocardial infarction and mortality from specific causes among bus drivers in Sweden. Int. J. Epidemiol. 1993, 22, 57–61. [Google Scholar] [CrossRef]
- van der Toorn, M.; Smit-de Vries, M.P.; Slebos, D.-J.; de Bruin, H.G.; Abello, N.; van Oosterhout, A.J.M.; Bischoff, R.; Kauffman, H.F. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2007, 293, L1156–L1162. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yang, Z.; Pan, X.; Zhu, M.; Xie, J. Crotonaldehyde induces oxidative stress and caspase-dependent apoptosis in human bronchial epithelial cells. Toxicol. Lett. 2010, 195, 90–98. [Google Scholar] [CrossRef]
- Araya, J.; Tsubouchi, K.; Sato, N.; Ito, S.; Minagawa, S.; Hara, H.; Hosaka, Y.; Ichikawa, A.; Saito, N.; Kadota, T.; et al. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 2019, 15, 510–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lingappan, K.; Jiang, W.; Couroucli, X.I.; Welty, S.E.; Shivanna, B.; Barrios, R.; Wang, G.; Khan, M.F.; Gonzalez, F.J.; et al. Disruption of cytochrome P4501A2 in mice leads to increased susceptibility to hyperoxic lung injury. Free. Radic. Biol. Med. 2015, 82, 147–159. [Google Scholar] [CrossRef] [Green Version]
- Nègre-Salvayre, A.; Garoby-Salom, S.; Swiader, A.; Rouahi, M.; Pucelle, M.; Salvayre, R. Proatherogenic effects of 4-hydroxynonenal. Free. Radic. Biol. Med. 2017, 111, 127–139. [Google Scholar] [CrossRef]
- Helmschrodt, C.; Becker, S.; Schröter, J.; Hecht, M.; Aust, G.; Thiery, J.; Ceglarek, U. Fast LC-MS/MS analysis of free oxysterols derived from reactive oxygen species in human plasma and carotid plaque. Clin. Chim. Acta 2013, 425, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Shi, M.; Guo, Z.; Brisbon, W.; Hoover, R.; Yang, H. Different cytotoxic injuries induced by lysophosphatidylcholine and 7-ketocholesterol in mouse endothelial cells. Endothelium 2006, 13, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Pirinen, E.; Mirsaidi, A.; Kunze, F.A.; Richards, P.J.; Auwerx, J.; Hottiger, M.O. ARTD1-induced poly-ADP-ribose formation enhances PPARγ ligand binding and co-factor exchange. Nucleic Acids Res. 2015, 43, 129–142. [Google Scholar] [CrossRef] [Green Version]
- Müller, K.H.; Hayward, R.; Rajan, R.; Whitehead, M.; Cobb, A.M.; Ahmad, S.; Sun, M.; Goldberga, I.; Li, R.; Bashtanova, U.; et al. Poly(ADP-Ribose) Links the DNA Damage Response and Biomineralization. Cell Rep. 2019, 27, 3124–3138.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esterbauer, H.; Schaur, R.J.; Zollner, H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free. Radic. Biol. Med. 1991, 11, 81–128. [Google Scholar] [CrossRef]
- Coskun, C.; Kural, A.; Döventas, Y.; Koldas, M.; Ozturk, H.; Inal, B.B.; Gümüs, A. Hemodialysis and protein oxidation products. Ann. N. Y. Acad. Sci. 2007, 1100, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Pennathur, S.; Byun, J.; Crowley, J.; Mueller, D.; Gischler, J.; Heinecke, J.W. NADPH oxidase of neutrophils elevates o,o’-dityrosine cross-links in proteins and urine during inflammation. Arch. Biochem. Biophys. 2001, 395, 69–77. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Rasmussen, J.E.; Hsu, F.F.; Mueller, D.M.; Pennathur, S.; Heinecke, J.W. Mass spectrometric quantification of markers for protein oxidation by tyrosyl radical, copper, and hydroxyl radical in low density lipoprotein isolated from human atherosclerotic plaques. J. Biol. Chem. 1997, 272, 3520–3526. [Google Scholar] [CrossRef] [Green Version]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Milzani, A.; Colombo, R. Protein carbonyl groups as biomarkers of oxidative stress. Clin. Chim. Acta 2003, 329, 23–38. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Jaruga, P.; Birincioglu, M.; Rodriguez, H. Free radical-induced damage to DNA: Mechanisms and measurement. Free. Radic. Biol. Med. 2002, 32, 1102–1115. [Google Scholar] [CrossRef]
- Chiorcea-Paquim, A.-M.; Oliveira-Brett, A.M. Nanostructured material-based electrochemical sensing of oxidative DNA damage biomarkers 8-oxoguanine and 8-oxodeoxyguanosine: A comprehensive review. Mikrochim. Acta 2021, 188, 58. [Google Scholar] [CrossRef]
- Karahan, M.; Yildirim, M.; Kucuk, H.F.; Turunc, V.; Demir, H.; Salturk, C.; Yavuz, A.; Demir, T.; Ari, E. Oxidative DNA Damage Is Increased in Living Kidney Donors. Transpl. Proc. 2019, 51, 1049–1053. [Google Scholar] [CrossRef]
- Loft, S.; Poulsen, H.E. Markers of oxidative damage to DNA: Antioxidants and molecular damage. Methods Enzym. 1999, 300, 166–184. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, P.; Unni, S.; Krishnappa, G.; Padmanabhan, B. The Keap1-Nrf2 pathway: Promising therapeutic target to counteract ROS-mediated damage in cancers and neurodegenerative diseases. Biophys. Rev. 2017, 9, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Hou, W.; Wu, W.; Zhao, Y.; Dong, X.; Bai, X.; Peng, L.; Song, L. 6′-O-Galloylpaeoniflorin Attenuates Cerebral Ischemia Reperfusion-Induced Neuroinflammation and Oxidative Stress via PI3K/Akt/Nrf2 Activation. Oxid. Med. Cell Longev. 2018, 2018, 8678267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Zhang, L.; Ding, M.; Li, M. Protective Effect of Vasicine Against Myocardial Infarction in Rats via Modulation of Oxidative Stress, Inflammation, and the PI3K/Akt Pathway. Drug Des. Devel. 2019, 13, 3773–3784. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Tang, Z.; Chu, P.; Song, Y.; Yang, Y.; Sun, B.; Niu, M.; Qaed, E.; Shopit, A.; Han, G.; et al. Neuroprotective effect of phosphocreatine on oxidative stress and mitochondrial dysfunction induced apoptosis in vitro and in vivo: Involvement of dual PI3K/Akt and Nrf2/HO-1 pathways. Free. Radic. Biol. Med. 2018, 120, 228–238. [Google Scholar] [CrossRef]
- Zhuang, Y.; Wu, H.; Wang, X.; He, J.; He, S.; Yin, Y. Resveratrol Attenuates Oxidative Stress-Induced Intestinal Barrier Injury through PI3K/Akt-Mediated Nrf2 Signaling Pathway. Oxid. Med. Cell Longev. 2019, 2019, 7591840. [Google Scholar] [CrossRef] [Green Version]
- Zarogoulidis, P.; Lampaki, S.; Turner, J.F.; Huang, H.; Kakolyris, S.; Syrigos, K.; Zarogoulidis, K. mTOR pathway: A current, up-to-date mini-review (Review). Oncol. Lett. 2014, 8, 2367–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Wei, F.; Li, C.; Lv, G.; Wang, G.; Liu, T.; Bellail, A.C.; Hao, C. Combination of mTOR and EGFR kinase inhibitors blocks mTORC1 and mTORC2 kinase activity and suppresses the progression of colorectal carcinoma. PLoS ONE 2013, 8, e73175. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Zheng, L.; Wang, F.; Su, Y.; Kong, H.; Xin, H. Mangiferin ameliorates placental oxidative stress and activates PI3K/Akt/mTOR pathway in mouse model of preeclampsia. Arch. Pharm. Res. 2020, 43, 233–241. [Google Scholar] [CrossRef]
- Wang, M.; Hu, R.; Wang, Y.; Liu, L.; You, H.; Zhang, J.; Wu, X.; Pei, T.; Wang, F.; Lu, L.; et al. Atractylenolide III Attenuates Muscle Wasting in Chronic Kidney Disease via the Oxidative Stress-Mediated PI3K/AKT/mTOR Pathway. Oxid. Med. Cell Longev. 2019, 2019, 1875471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, J.; Chen, Y.; Wang, J.; Qiu, J.; Chang, C.; Bi, F.; Wu, X.; Liu, W. EGCG protects vascular endothelial cells from oxidative stress-induced damage by targeting the autophagy-dependent PI3K-AKT-mTOR pathway. Ann. Transl. Med. 2020, 8, 200. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Salama, Y.; Takahashi, S.; Osada, T.; Hattori, K. The multifaceted role of plasminogen in inflammation. Cell Signal. 2020, 75, 109761. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Rahmani, F.; Asgharzadeh, F.; Avan, A.; Barneh, F.; Parizadeh, M.R.; Ferns, G.A.; Ryzhikov, M.; Ahmadian, M.R.; Elisa, G.J.M.; Khazaei, M.; et al. Rigosertib potently protects against colitis-associated intestinal fibrosis and inflammation by regulating PI3K/AKT and NF-κB signaling pathways. Life Sci. 2020, 249, 117470. [Google Scholar] [CrossRef]
- Wang, F.; Wang, F.; Li, F.; Wang, D.; Li, H.; He, X.; Zhang, J. Methane attenuates lung ischemia-reperfusion injury via regulating PI3K-AKT-NFκB signaling pathway. J. Recept. Signal. Transduct Res. 2020, 40, 209–217. [Google Scholar] [CrossRef]
- Li, Z.; Chen, C.; Zhu, X.; Li, Y.; Yu, R.; Xu, W. Glycyrrhizin Suppresses RANKL-Induced Osteoclastogenesis and Oxidative Stress Through Inhibiting NF-κB and MAPK and Activating AMPK/Nrf2. Calcif. Tissue Int. 2018, 103, 324–337. [Google Scholar] [CrossRef]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef]
- Chuang, P.Y.; Dai, Y.; Liu, R.; He, H.; Kretzler, M.; Jim, B.; Cohen, C.D.; He, J.C. Alteration of forkhead box O (foxo4) acetylation mediates apoptosis of podocytes in diabetes mellitus. PLoS ONE 2011, 6, e23566. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, L.; Liu, X.; Jiang, Y.; Zhang, L.; Zhang, X.; Li, X.; Zhang, Y. α-Melanocyte-stimulating hormone protects retinal vascular endothelial cells from oxidative stress and apoptosis in a rat model of diabetes. PLoS ONE 2014, 9, e93433. [Google Scholar] [CrossRef]
- Nakayoshi, T.; Sasaki, K.-I.; Kajimoto, H.; Koiwaya, H.; Ohtsuka, M.; Ueno, T.; Chibana, H.; Itaya, N.; Sasaki, M.; Yokoyama, S.; et al. FOXO4-knockdown suppresses oxidative stress-induced apoptosis of early pro-angiogenic cells and augments their neovascularization capacities in ischemic limbs. PLoS ONE 2014, 9, e92626. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO transcription factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Dinda, B.; Dinda, M.; Kulsi, G.; Chakraborty, A.; Dinda, S. Therapeutic potentials of plant iridoids in Alzheimer’s and Parkinson’s diseases: A review. Eur. J. Med. Chem. 2019, 169, 185–199. [Google Scholar] [CrossRef]
- Duangjan, C.; Rangsinth, P.; Gu, X.; Zhang, S.; Wink, M.; Tencomnao, T. Glochidion zeylanicum leaf extracts exhibit lifespan extending and oxidative stress resistance properties in Caenorhabditis elegans via DAF-16/FoxO and SKN-1/Nrf-2 signaling pathways. Phytomedicine 2019, 64, 153061. [Google Scholar] [CrossRef]
- Mahmoud, A.R.; Ali, F.E.M.; Abd-Elhamid, T.H.; Hassanein, E.H.M. Coenzyme Q(10) protects hepatocytes from ischemia reperfusion-induced apoptosis and oxidative stress via regulation of Bax/Bcl-2/PUMA and Nrf-2/FOXO-3/Sirt-1 signaling pathways. Tissue Cell 2019, 60, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, J.; Singh, R.R.; Riggle, C.; Haugrud, B.; Abdalla, M.Y.; Reindl, K.M. JNK inhibition blocks piperlongumine-induced cell death and transcriptional activation of heme oxygenase-1 in pancreatic cancer cells. Apoptosis 2019, 24, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Okamura, T.; Antoun, G.; Keir, S.T.; Friedman, H.; Bigner, D.D.; Ali-Osman, F. Phosphorylation of Glutathione S-Transferase P1 (GSTP1) by Epidermal Growth Factor Receptor (EGFR) Promotes Formation of the GSTP1-c-Jun N-terminal kinase (JNK) Complex and Suppresses JNK Downstream Signaling and Apoptosis in Brain Tumor Cells. J. Biol. Chem. 2015, 290, 30866–30878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pljesa-Ercegovac, M.; Savic-Radojevic, A.; Kravic-Stevovic, T.; Bumbasirevic, V.; Mimic-Oka, J.; Simic, T. Co-localization of GSTP1 and JNK in transitional cell carcinoma of urinary bladder. Genet. Mol. Biol. 2010, 33, 460–462. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wolf, C.R.; Henderson, C.J.; Cai, Y.; Board, P.G.; Foster, P.S.; Webb, D.C. Glutathione transferase P1, An endogenous inhibitor of allergic responses in a mouse model of asthma. Am. J. Respir. Crit. Care Med. 2008, 178, 1202–1210. [Google Scholar] [CrossRef]
- Chauhan, A.K.; Mittra, N.; Singh, B.K.; Singh, C. Inhibition of glutathione S-transferase-pi triggers c-jun N-terminal kinase-dependent neuronal death in Zn-induced Parkinsonism. Mol. Cell Biochem. 2019, 452, 95–104. [Google Scholar] [CrossRef]




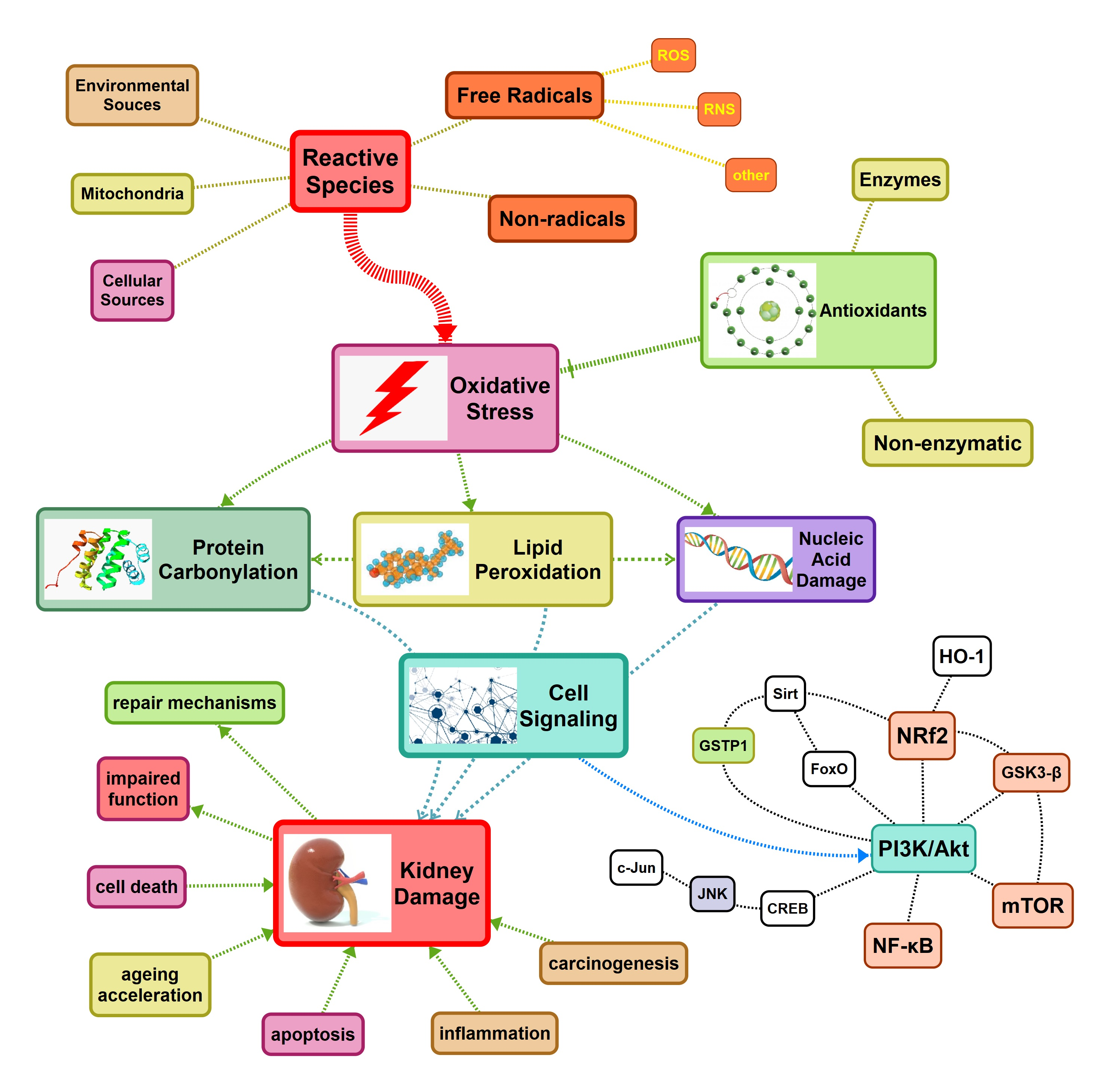{kind=link}
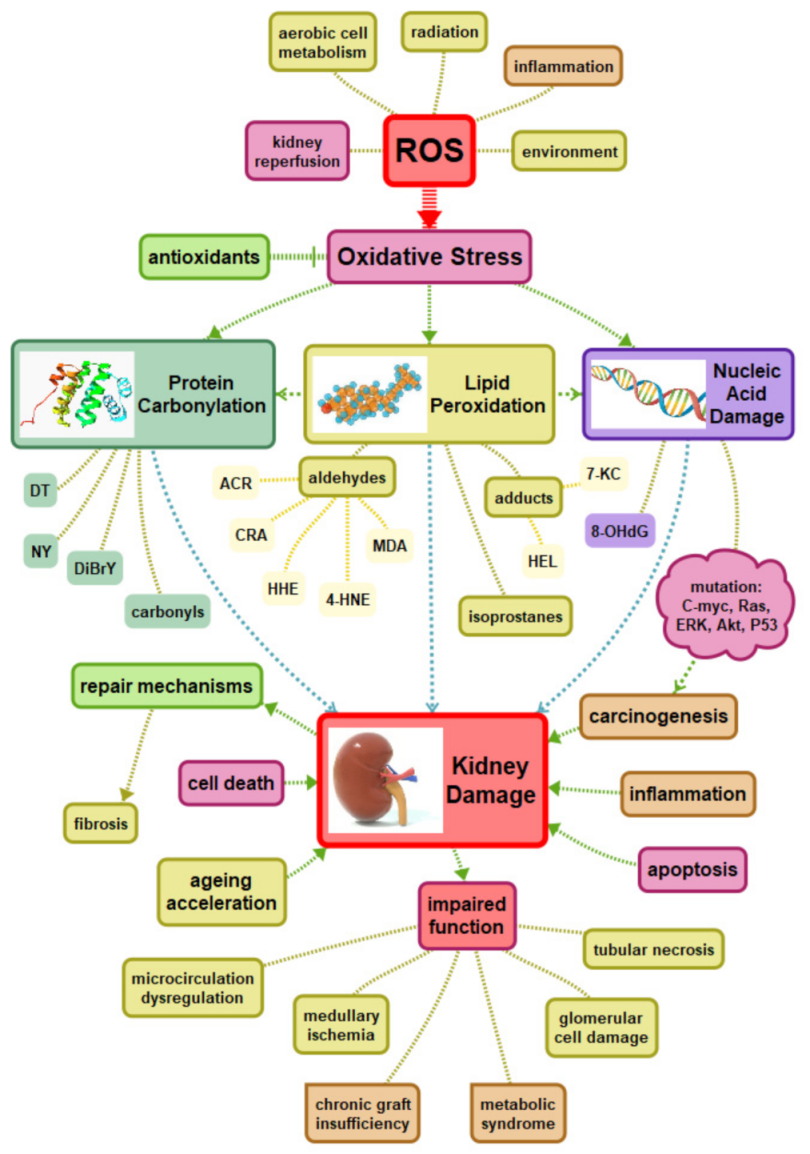{kind=link}
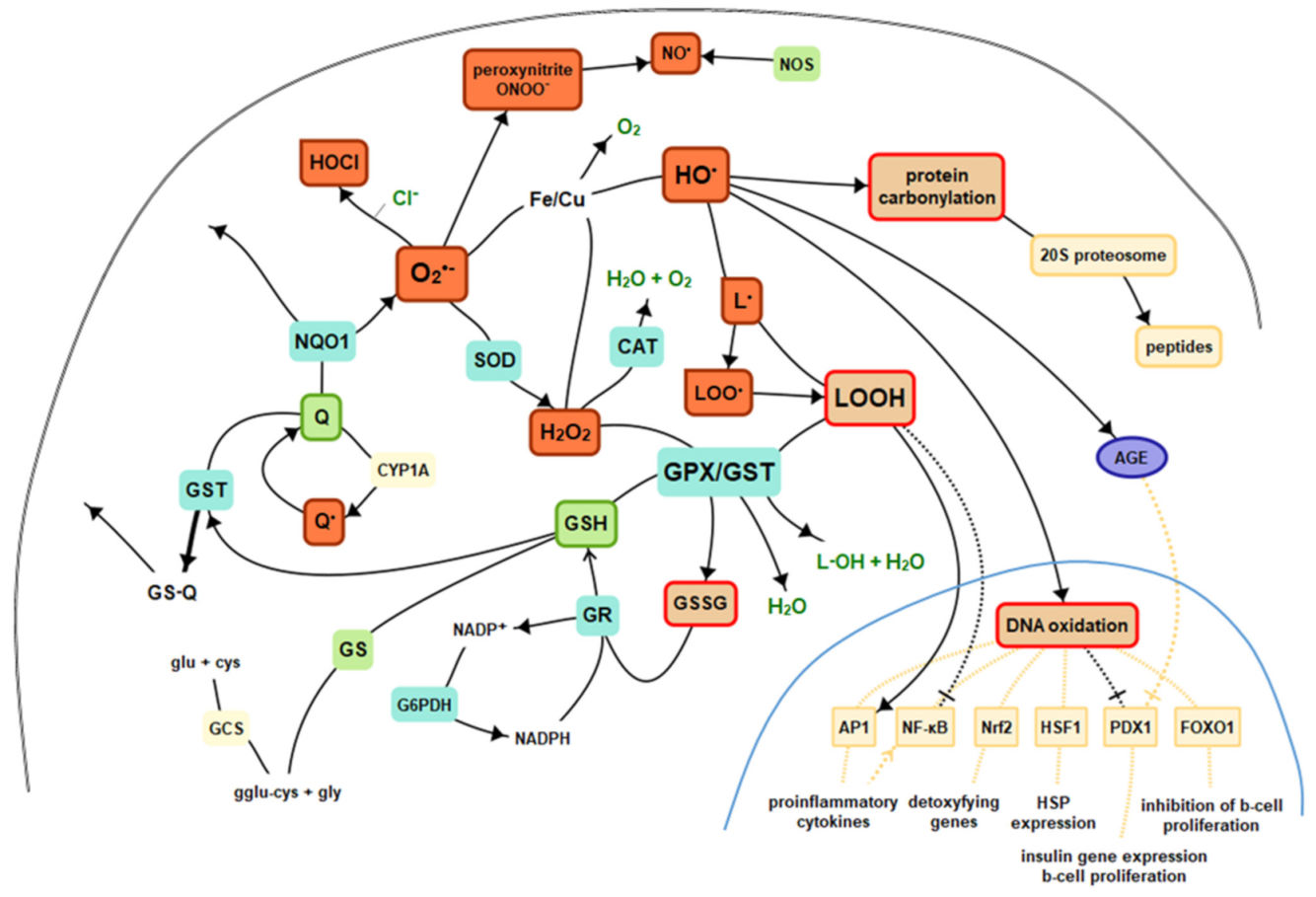{kind=link}
{kind=link}
| ROS | Description | General | Transplantation |
|---|---|---|---|
| O2•− superoxide anion | one-electron reduction state of O2, formed in autoxidation reactions and by the electron transport chain, can release Fe2+ from iron-sulfur proteins and ferritin, undergoes dismutation to H2O2 spontaneously or by enzymatic catalysis, the precursor for metal-catalyzed OH• formation, detectable as a biomarker |  1843 |  102 |
| OH• hydroxyl radical | three-electron reduction state, formed by Fenton reaction and decomposition of peroxynitrite; extremely reactive, attack most of the cellular components |  2154 |  30 |
| RO•, alkoxyl ROO•, peroxyl radicals | oxygen centered organic radicals, lipid forms participate in lipid peroxidation reactions, produced in the presence of oxygen by radical addition to double bonds or hydrogen abstraction |  203 |  4 |
| H2O2 hydrogen peroxide | two-electron reduction state, formed by dismutation of O2•− or by direct reduction of O2; stable, lipid-soluble, able to diffuse across membranes |  2073 |  155 |
| 1O2 singlet oxygen | systematically dioxidene, highly reactive toward organic compounds (more than O2) and unstable, responsible for the photodegradation of many materials, pollutes urban atmospheres |  2226 |  5 |
| O3 ozone | allotrope of oxygen, colorless or pale blue gas, a powerful oxidant, damage mucous and respiratory tissues, harmful to people at levels currently found in urban areas, affects the respiratory, cardiovascular, and central nervous system |  582 |  19 |
| ROOH organic hydroperoxide | stable, formed by radical reactions with cellular components such as lipids and nucleobases |  64 |  2 |
| HOCl hypochlorous acid | formed from H2O2 by myeloperoxidase, lipid-soluble and highly reactive, oxidizes protein constituents, including thiol groups, amino groups, and methionine |  744 |  14 |
| HOBr hypobromous acid | weak, unstable acid, “reactive bromine species”; generated biologically as a disinfectant, especially by eosinophils; an oxidizer; especially effective when used in combination with its congener, hypochlorous acid |  233 |  2 |
| NO• nitric oxide | colorless gas; free radical; signaling molecule; “Molecule of the Year” in 1992; 1998 Nobel Prize in Physiology or Medicine; endothelium-derived relaxing factor (EDRF); converted to nitrates and nitrites by oxygen and water |  3962 |  81 |
| NO2• nitrogen dioxide | reddish-brown gas; good oxidizer; exposed and are at risk for occupational lung diseases; chemically reacts with antioxidant and lipid molecules; classified as an extremely hazardous substance |  1544 |  24 |
| ONOO– peroxynitrite | formed in a rapid reaction between O2•− and NO•, lipid-soluble and similar in reactivity to HOCl, protonation forms peroxynitrous acid, which can undergo homolytic cleavage to form hydroxyl radical and nitrogen dioxide |  2722 |  62 |
| Biomarker | Description | General | Transplantation |
|---|---|---|---|
| CAT catalase | tetramer, enzyme, biomarker, catalyzes the decomposition of hydrogen peroxide to water and oxygen, has one of the highest turnover numbers of all enzymes, first noticed in 1818 |  1068 |  356 |
| SOD superoxide dismutase | enzyme, biomarker, metalloprotein connected in humans with Zn/Cu or Mn, discovered in 1968 |  2117 |  715 |
| GPx glutathione peroxidase | selenium-containing enzyme family, several isozymes are encoded by different genes, biomarker, the protective system depends heavily on the presence of selenium, discovered in 1957 |  763 |  296 |
| GST glutathione S-transferase | family of metabolic isoenzymes, biomarker, catalyze the conjugation of the reduced form of glutathione (GSH) to xenobiotic substrates, nomenclature first proposed in 1992 |  104 |  95 |
| GR glutathione reductase | homodimer disulfide oxidoreductase enzyme encoded by the GSR gene, biomarker, catalyzes GSSH and regenerates it to GSH, involves NADPH and FAD binding domains, first purified in 1955 |  159 |  63 |
| HO-1 heme oxygenase 1 | an enzyme, mediates the first step of heme catabolism, cleaves heme to biliverdin, carbon monoxide, and ferrous iron, encoded by HMOX1 gene induced in oxidative stress and inflammation, first characterized in 1962 |  1457 |  404 |
| NQO1 NADPH-quinone oxidoreductase-1 | protein homodimer, detoxifying enzyme, present in cytosol, performs a two-electron reduction of quinones to hydroquinones and of other redox dyes without the formation of ROS, |  1837 |  86 |
| GSH glutathione | L-γ-glutamyl-L-cysteinyl-glycine, non-enzymatic, biomarker, reduced form (GSH) and glutathione disulfide (GSSG)—primary redox couple in animal cells |  5118 |  798 |
| coenzyme Q ubiquinone | benzoquinone derivate, localized in the mitochondrial respiratory chain and other internal membranes, coenzyme, first isolate was in 1950 in the lining of a horse’s gut |  185 |  22 |
| ALA α-lipoic acid | delivers antioxidant activity in nonpolar and polar mediums, effective in recharging enzymes in the mitochondria, might counteract NF-κB activation |  1430 |  62 |
| BR bilirubin | product of heme degradation works as an antioxidant in cycle BR-biliverdin, relevantly documented in 1827 |  1941 |  64 |
| ferritin | intracellular globular iron-binding protein, small amounts are secreted into the serum, prevents ROS generation via the Fenton reaction by binding iron |  1678 |  60 |
| Biomarker | Description | General | Transplantation |
|---|---|---|---|
| TBARS TBA-reactive substances | the oldest and one of the most widely used nonspecific by-products of lipid peroxidation, reacts with thiobarbituric acid (TBA), forming a pink chromogen (TBARS) measured at 532–535 nm |  7431 |  200 |
| MDA malondialdehyde | CH2(CHO)2, colorless liquid, highly reactive, a product of LPO of polyunsaturated fatty acids, form covalent protein adducts referred to as advanced lipoxidation end-products (ALE), in analogy to advanced glycation end-products (AGE) |  30,269 |  2808 |
| 4-HNE 4-hydroxynonenal | α,β-unsaturated hydroxyalkenal, produced by lipid peroxidation (arachidonic or linoleic groups) in cells in higher quantities during oxidative stress, possible role in cell signal transduction, first reported in 1991 [114], they can also come from omega-3 fatty acids |  2418 |  73 |
| ACR acrolein (propenal) | the simplest unsaturated aldehyde, named and characterized in 1839, electrophilic, reactive and toxic, contact herbicide to weeds, present in tobacco smoke increases the risk of cancer, produced during cyclophosphamide treatment |  547 |  29 |
| F2-isoprostanes | prostaglandin-like compounds formed in vivo from the free radical-catalyzed peroxidation of arachidonic acid, discovered in 1990, |  623 |  22 |
| F4-isoprostanes | prostaglandin-like compounds formed in vivo from the free radical-catalyzed peroxidation of docosahexaenoic acid, potent biological activity as anti-inflammatory mediators |  3 | 0 |
| CRA crotonaldehyde | CH3CH, representative carcinogenic aldehyde formed endogenously through lipid peroxidation, CRA is a highly reactive aldehyde and reacts with a lysine residue in the protein, reaction with CRA and lysine residue leads to the formation of numerous numbers of adducts |  74 |  53 |
| HHE 4-hydroxy-trans-2-hexenal | oxygenated α,β-unsaturated aldehyde, other coming from omega-3 fatty acids: 4-oxo-trans-2-nonenal, 4-hydroperoxy-trans-2-nonenal, and 4,5-epoxy-trans-2-decenal |  64 |  11 |
| 7KC 7-ketocholesterol (7-oxocholesterol) | toxic oxysterol, produced from oxidized cholesterol, induces: NOX, pro-inflammatory cytokines and TNF-α |  162 |  6 |
| Biomarker | Description | General | Transplantation |
|---|---|---|---|
| DiBrY dibromotyrosine | product of the reaction of hypobromous acid (HOBr) from hydrogen peroxide (H2O2) and bromide ion (Br–), detected by anti-dibromo-tyrosine [DiBrY], mAb (3A5) JAI-MBY-020P |  52 | 5 1951, 1989, 1995, 1999, 2018 |
| DiY/DT dityrosine (bityrosine) | biphenyl compound comprising two tyrosine residues linked at carbon-3 of their benzene rings |  1014 | 6 2001, 2005, 2010, 2012, 2020(2) |
| m-Tyrosine o-Tyrosine | abnormal tyrosine isomers, derive from oxidation of the benzyl ring of the phenylalanine by hydroxyl radical, adversely affect cells and tissues |  62 | 4 2003, 2007, 2008, 2012 |
| NY 3-nitrotyrosine | specific marker of attack of peroxynitrite (ONOO–) upon proteins, measured by immunostaining, HPLC, and MS in human tissues |  3554 |  201 |
| protein carbonyls | measurement of protein CO groups after their derivatization with DNPH is the most widely utilized measure of protein oxidation |  9122 |  325 |
| Biomarker | Description | General | Transplantation |
|---|---|---|---|
| 8OHdG 8-hydroxy-2′ -deoxyguanosine | oxidized derivative of deoxyguanosine, major products of DNA oxidation, increased levels are found during carcinogenesis, increases with age, linked to the enzyme OGG1 and transcription factor NFκB |  5189 |  212 |
| 8-oxo-Gua 8-hydroxyguanine | one of the most common DNA lesions resulting from reactive oxygen species, modifying guanine, and can result in a mismatched pairing with adenine resulting in G to T and C to A → mutation |  199 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tejchman, K.; Kotfis, K.; Sieńko, J. Biomarkers and Mechanisms of Oxidative Stress—Last 20 Years of Research with an Emphasis on Kidney Damage and Renal Transplantation. Int. J. Mol. Sci. 2021, 22, 8010. https://doi.org/10.3390/ijms22158010
Tejchman K, Kotfis K, Sieńko J. Biomarkers and Mechanisms of Oxidative Stress—Last 20 Years of Research with an Emphasis on Kidney Damage and Renal Transplantation. International Journal of Molecular Sciences. 2021; 22(15):8010. https://doi.org/10.3390/ijms22158010
Chicago/Turabian StyleTejchman, Karol, Katarzyna Kotfis, and Jerzy Sieńko. 2021. "Biomarkers and Mechanisms of Oxidative Stress—Last 20 Years of Research with an Emphasis on Kidney Damage and Renal Transplantation" International Journal of Molecular Sciences 22, no. 15: 8010. https://doi.org/10.3390/ijms22158010