
Hepatic LKB1 Reduces the Progression of Non-Alcoholic Fatty Liver Disease via Genomic Androgen Receptor Signaling


Abstract
:1. Introduction
2. Results
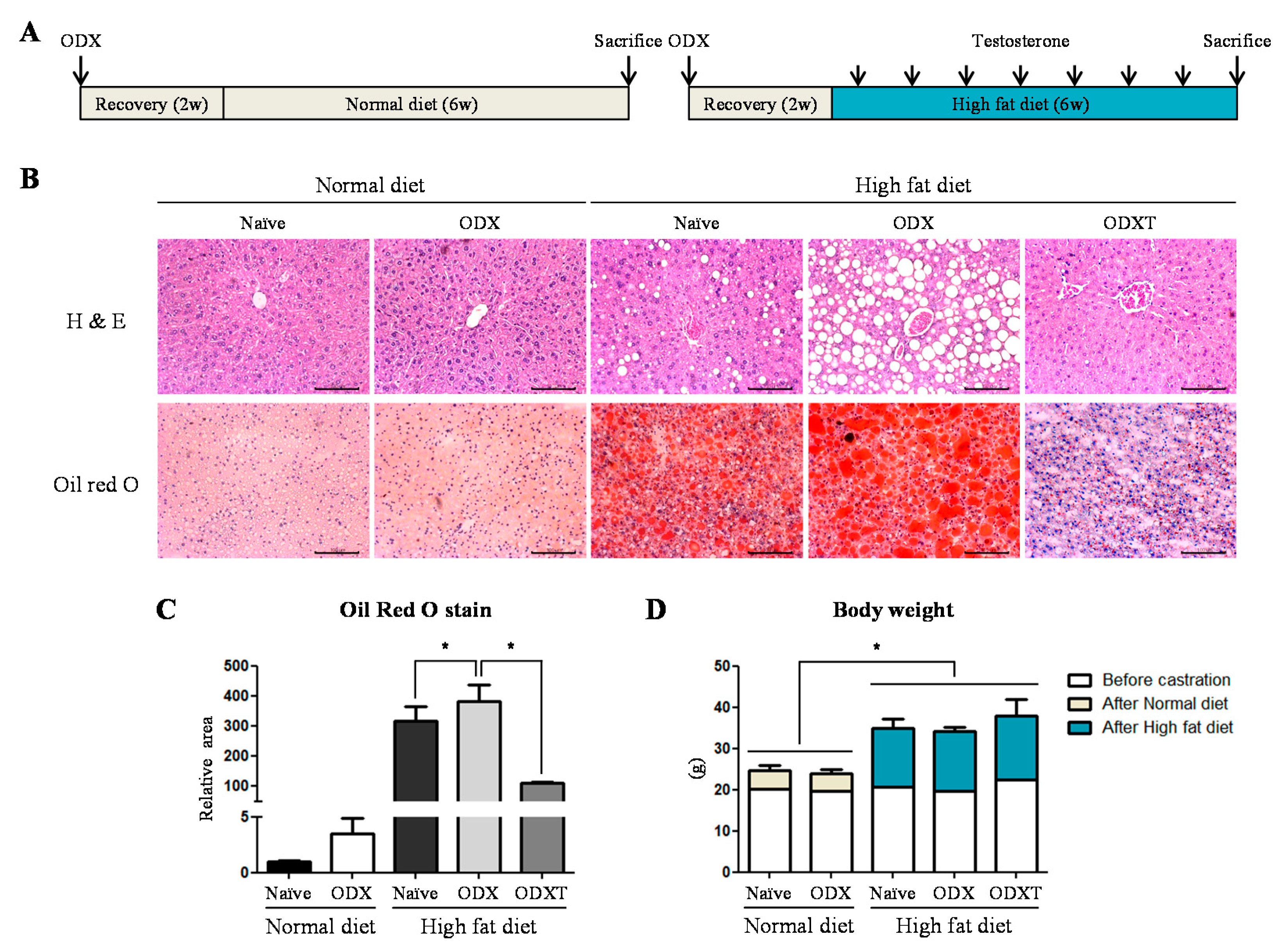
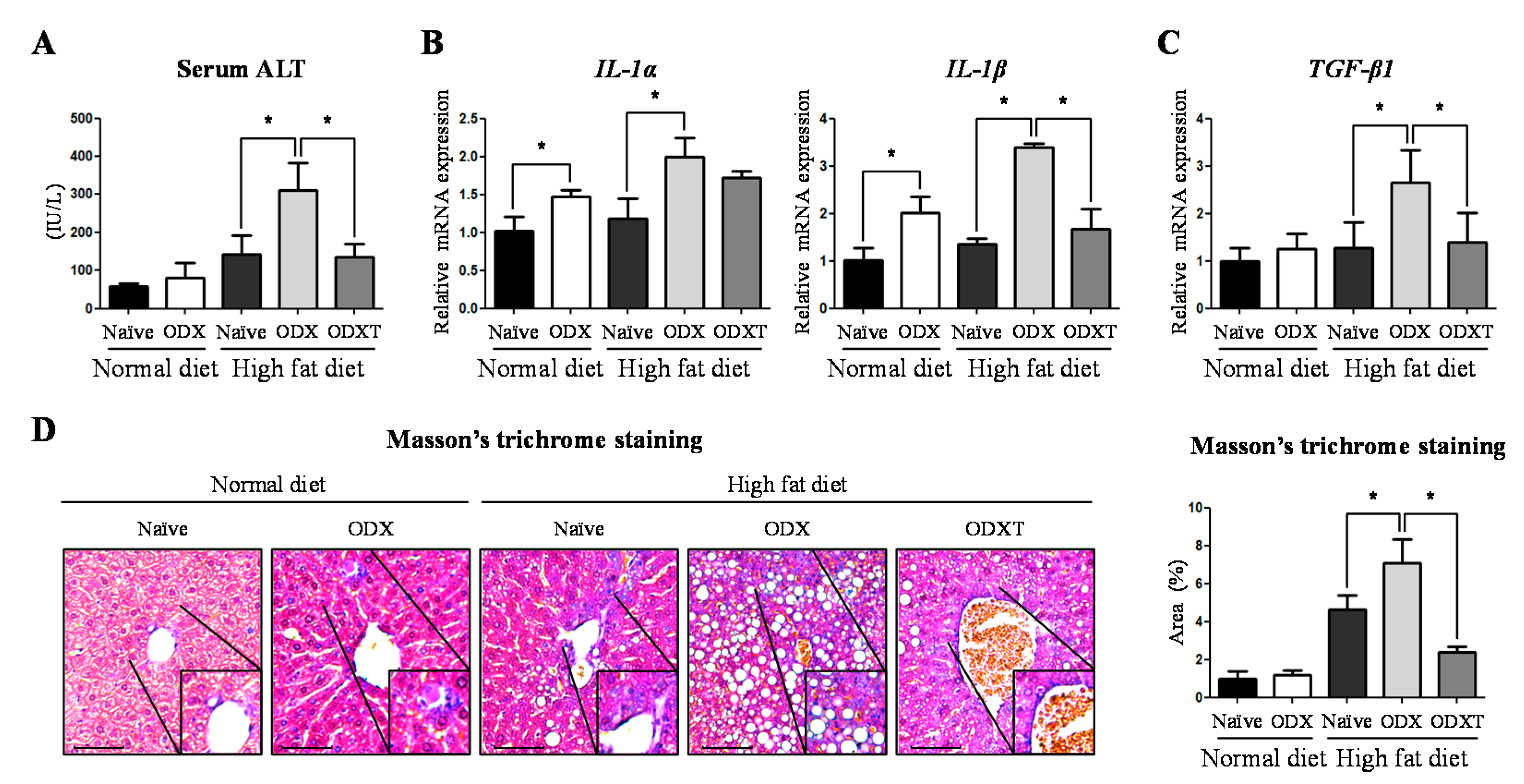
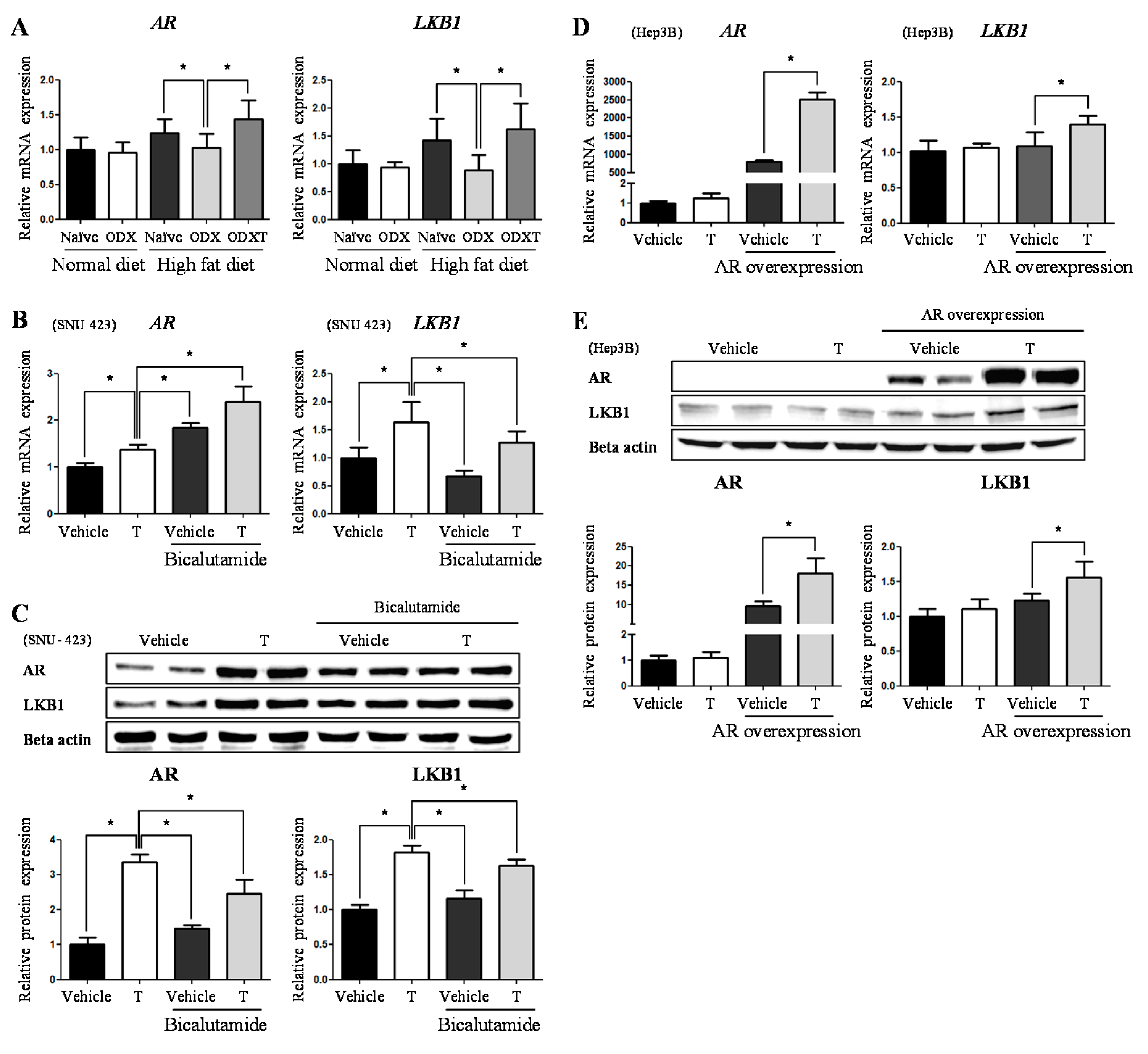
2.1. Androgens Suppressed Hepatic TG Accumulation Involving in Inflammation and Fibrosis
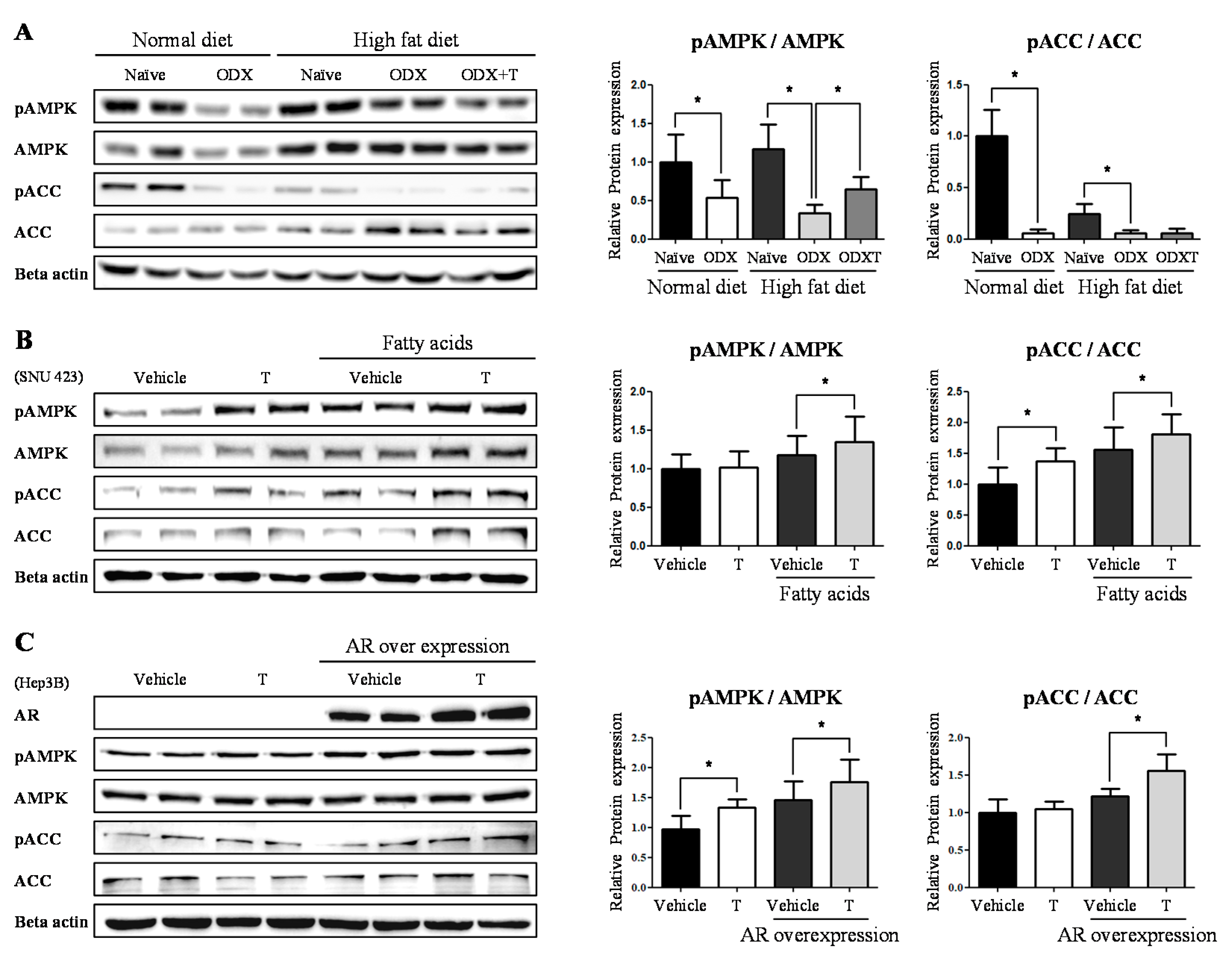
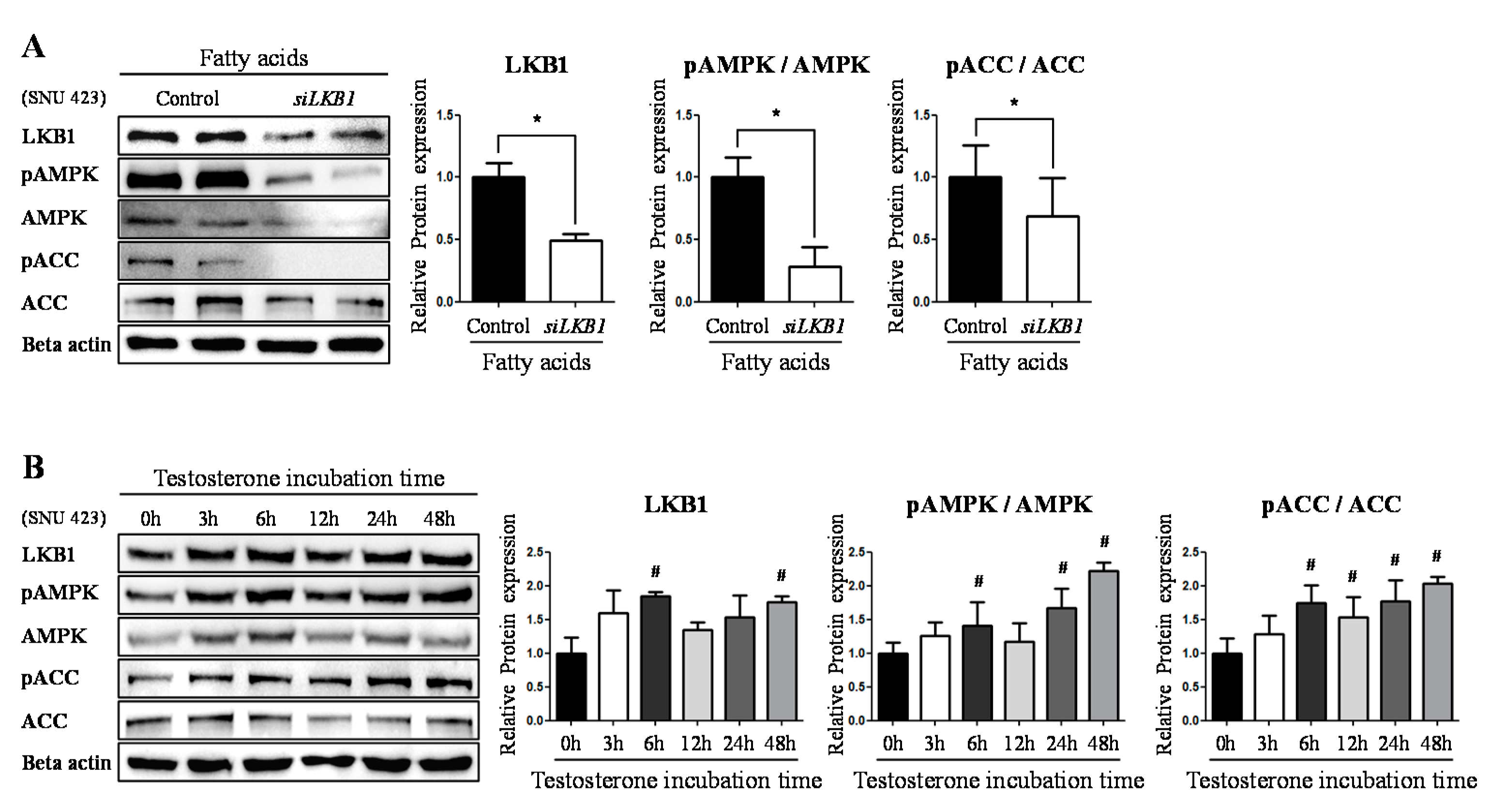
2.2. Testosteronereduces Lipid Synthesis by Modulating AMPK-ACC Signaling via LKB1
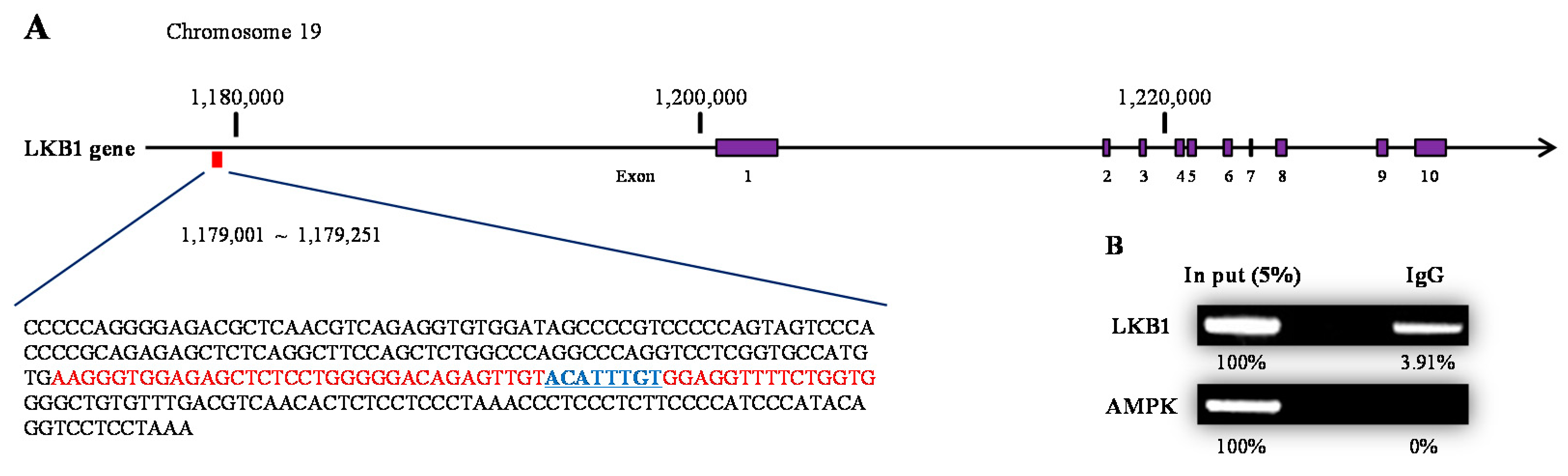
2.3. Testosterone Modulatethe Expression Level of LKB1 by Binding to AR
3. Discussion
4. Materials and Methods
4.1. Antibody
4.2. Animals
4.3. Blood Alanine Transferase Level
4.4. Hematoxylin & Eosin Staining and Masson’s Trichrome Staining
4.5. Oil-Red-O Staining
4.6. Cell Culture
4.7. Gene Overexpression Assay
4.8. Gene Knock-Down Assay
4.9. Total RNA Extraction and Real-Time Quantitative PCR
4.10. Western Blotting
4.11. Chromatin Immunoprecipitation Assay
4.12. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Finelli, C.; Tarantino, G. Is visceral fat reduction necessary to favour metabolic changes in the liver? J. Gastrointest. Liver Dis. 2012, 21, 205–208. [Google Scholar]
- Cobbina, E.; Akhlaghi, F. Non-alcoholic fatty liver disease (NAFLD)—Pathogenesis, classification, and effect on drug metabolizing enzymes and transporters. Drug Metab. Rev. 2017, 49, 197–211. [Google Scholar] [CrossRef]
- Ratziu, V.; Bellentani, S.; Cortez-Pinto, H.; Day, C.; Marchesini, G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J. Hepatol. 2010, 53, 372–384. [Google Scholar] [CrossRef] [Green Version]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Lee, S.R.; Kwon, S.W.; Kaya, P.; Lee, Y.H.; Lee, J.G.; Kim, G.; Lee, G.S.; Baek, I.J.; Hong, E.J. Loss of progesterone receptor membrane component 1 promotes hepatic steatosis via the induced de novo lipogenesis. Sci. Rep. 2018, 8, 15711. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Lee, Y.H.; Yang, H.; Lee, H.W.; Lee, G.S.; An, B.S.; Jeung, E.B.; Park, B.K.; Hong, E.J. Sex hormone-binding globulin suppresses NAFLD-triggered hepatocarcinogenesis after menopause. Carcinogenesis 2019, 40, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, J.M.; Vermeulen, A. The decline of androgen levels in elderly men and its clinical and therapeutic implications. Endocr. Rev. 2005, 26, 833–876. [Google Scholar] [CrossRef]
- Varlamov, O. Western-style diet, sex steroids and metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1147–1155. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.; Wang, L.; Li, Z.; Zhang, H.; Wu, J.; Rahman, N.; Guo, Y.; Li, D.; Li, N.; et al. Differential effects of estrogen/androgen on the prevention of nonalcoholic fatty liver disease in the male rat. J. Lipid Res. 2013, 54, 345–357. [Google Scholar] [CrossRef] [Green Version]
- Ghanim, H.; Dhindsa, S.; Batra, M.; Green, K.; Abuaysheh, S.; Kuhadiya, N.D.; Makdissi, A.; Chaudhuri, A.; Sandhu, S.; Dandona, P. Testosterone increases the expression and phosphorylation of AMP kinase alpha in men with hypogonadism and type 2 diabetes. J. Clin. Endocrinol. Metab. 2020, 105, 1169–1175. [Google Scholar] [CrossRef]
- Imai, K.; Inukai, K.; Ikegami, Y.; Awata, T.; Katayama, S. LKB1, an upstream AMPK kinase, regulates glucose and lipid metabolism in cultured liver and muscle cells. Biochem. Biophys. Res. Commun. 2006, 351, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Mitsuhashi, K.; Senmaru, T.; Fukuda, T.; Yamazaki, M.; Shinomiya, K.; Ueno, M.; Kinoshita, S.; Kitawaki, J.; Katsuyama, M.; Tsujikawa, M.; et al. Testosterone stimulates glucose uptake and GLUT4 translocation through LKB1/AMPK signaling in 3T3-L1 adipocytes. Endocrine 2016, 51, 174–184. [Google Scholar] [CrossRef]
- McInnes, K.J.; Brown, K.A.; Hunger, N.I.; Simpson, E.R. Regulation of LKB1 expression by sex hormones in adipocytes. Int. J. Obes. 2012, 36, 982–985. [Google Scholar] [CrossRef] [Green Version]
- Pratt, D.S.; Kaplan, M.M. Evaluation of abnormal liver-enzyme results in asymptomatic patients. N. Engl. J. Med. 2000, 342, 1266–1271. [Google Scholar] [CrossRef]
- Dewidar, B.; Meyer, C.; Dooley, S.; Meindl-Beinker, A.N. TGF-beta in hepatic stellate cell activation and liver fibrogenesis-updated 2019. Cells 2019, 8, 1419. [Google Scholar] [CrossRef] [Green Version]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef]
- Smith, L.B.; Walker, W.H. The regulation of spermatogenesis by androgens. Semin Cell Dev Biol. 2014, 30, 2–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, W.L.; Lai, H.C.; Yeh, S.; Cai, X.; Chang, C. Androgen receptor roles in hepatocellular carcinoma, fatty liver, cirrhosis and hepatitis. Endocr. Relat. Cancer 2014, 21, R165–R182. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.; Lee, S.O.; Yeh, S.; Chang, T.M. Androgen receptor (AR) differential roles in hormone-related tumors including prostate, bladder, kidney, lung, breast and liver. Oncogene 2014, 33, 3225–3234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J.; American Association for the Study of Liver Diseases; American College of Gastroenterology; et al. The diagnosis and management of non-alcoholic fatty liver disease: Practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology 2012, 142, 1592–1609. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Phan, F.; Bourron, O.; Ferre, P.; Foufelle, F. Steatosis and NASH in type 2 diabetes. Biochimie 2017, 143, 37–41. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, E.; Sullivan, S.; Klein, S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010, 51, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, S.; Storer, T.W.; Berman, N.; Yarasheski, K.E.; Clevenger, B.; Phillips, J.; Lee, W.P.; Bunnell, T.J.; Casaburi, R. Testosterone replacement increases fat-free mass and muscle size in hypogonadal men. J. Clin. Endocrinol. Metab. 1997, 82, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.E.; Ramos-Roman, M.A.; Browning, J.D.; Parks, E.J. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014, 146, 726–735. [Google Scholar] [CrossRef]
- Foretz, M.; Even, P.C.; Viollet, B. AMPK activation reduces hepatic lipid content by increasing fat oxidation in vivo. Int. J. Mol. Sci. 2018, 19, 2826. [Google Scholar] [CrossRef] [Green Version]
- Kokontis, J.; Takakura, K.; Hay, N.; Liao, S. Increased androgen receptor activity and altered c-myc expression in prostate cancer cells after long-term androgen deprivation. Cancer Res. 1994, 54, 1566–1573. [Google Scholar]
- Massie, C.E.; Lynch, A.; Ramos-Montoya, A.; Boren, J.; Stark, R.; Fazli, L.; Warren, A.; Scott, H.; Madhu, B.; Sharma, N.; et al. The androgen receptor fuels prostate cancer by regulating central metabolism and biosynthesis. EMBO J. 2011, 30, 2719–2733. [Google Scholar] [CrossRef] [Green Version]
- Masiello, D.; Cheng, S.; Bubley, G.J.; Lu, M.L.; Balk, S.P. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J. Biol. Chem. 2002, 277, 26321–26326. [Google Scholar] [CrossRef] [Green Version]
- Hong, E.J.; Sahu, B.; Janne, O.A.; Hammond, G.L. Cytoplasmic accumulation of incompletely glycosylated SHBG enhances androgen action in proximal tubule epithelial cells. Mol. Endocrinol. 2011, 25, 269–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, B.G.; Bort, A.; Mateos-Gomez, P.A.; Rodriguez-Henche, N.; Diaz-Laviada, I. Combination of the natural product capsaicin and docetaxel synergistically kills human prostate cancer cells through the metabolic regulator AMP-activated kinase. Cancer Cell. Int. 2019, 19, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Type | Lot. | Inc. |
| Beta-actin | Mouse monoclonal | sc-47778 | Santa Cruz biotechnology |
| Beta-actin | Rabbit polyclonal | sc-130656 | Santa Cruz biotechnology |
| ACC | Rabbit monoclonal | #3676T | Cell signaling technology |
| pACC | Rabbit monoclonal | #11818T | Cell signaling technology |
| AMPK-alpha | Rabbit monoclonal | #5831T | Cell signaling technology |
| pAMPK-alpha | Rabbit monoclonal | #2535T | Cell signaling technology |
| AR | Rabbit monoclonal | #5153 | Cell signaling technology |
| LKB1 | Rabbit polyclonal | #A2122 | Company ABclonal, Inc. |
| Secondary Antibody | Type | Lot. | Inc. |
| Anti-Mouse IgG | Goat | 121507 | Jackonimmuno |
| Anti-Rabbit IgG | Mouse | 123213 | Jackonimmuno |
| Gene Name | Upper Primer (5′-3′) | Lower Primer (5′-3′) | Species |
|---|---|---|---|
| 18 s | GGA CAC GGA CAG GAT TGA CA | AGA CTG TGT CCC TGT GGA GA | Human |
| AR | GAC GAC CAG ATG GCT GTC ATT | GGG CGA AGT AGA GCA TCC T | Human |
| LKB1 | GGT TCC GGA AGA AAC ATC CT | TGT GAC TGG CCT CCT CTT CT | Human |
| RPLP0 | GCA GCA GAT CCG CAT GTC GCT CCG | GAG CTG GCA CAG TGA CCTCAC ACG G | Mouse |
| AR | CTG GGA AGG GTC TAC CCAC | GGT GCT ATG TTA GCG GCC TC | Mouse |
| LKB1 | TAT GTG GCA TGC AGG AGA TG | TGG TGG TGA GTA GCA GGT TG | Mouse |
| IL-1α- | AGT ATC AGC AAC GTC AAG CAA | TCC AGA TCA TGG GTT ATG GAC TG | Mouse |
| IL-1β | GAA ATG CCA CCT TTT GAC AG | CTG GAT GCT CTC TCA TCA GGA CA | Mouse |
| TGF-β1 | GAC GTC ACT GGA GTT GTA CG | GGT TCA TGTCAT GGA TGG TG | Mouse |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, J.H.; Lee, S.R.; Jo, S.L.; Ko, J.-W.; Kwon, H.-J.; Hong, E.-J. Hepatic LKB1 Reduces the Progression of Non-Alcoholic Fatty Liver Disease via Genomic Androgen Receptor Signaling. Int. J. Mol. Sci. 2021, 22, 7904. https://doi.org/10.3390/ijms22157904
Heo JH, Lee SR, Jo SL, Ko J-W, Kwon H-J, Hong E-J. Hepatic LKB1 Reduces the Progression of Non-Alcoholic Fatty Liver Disease via Genomic Androgen Receptor Signaling. International Journal of Molecular Sciences. 2021; 22(15):7904. https://doi.org/10.3390/ijms22157904
Chicago/Turabian StyleHeo, Jun H., Sang R. Lee, Seong Lae Jo, Je-Won Ko, Hyo-Jung Kwon, and Eui-Ju Hong. 2021. "Hepatic LKB1 Reduces the Progression of Non-Alcoholic Fatty Liver Disease via Genomic Androgen Receptor Signaling" International Journal of Molecular Sciences 22, no. 15: 7904. https://doi.org/10.3390/ijms22157904