
Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease


1
Department of Chemistry, Kongju National University, Gongju 32588, Chungcheongnam-do, Korea
2
Department of Chemistry Education, Kongju National University, Gongju 32588, Chungcheongnam-do, Korea
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2021, 22(14), 7697; https://doi.org/10.3390/ijms22147697
Submission received: 28 June 2021
/
Revised: 11 July 2021
/
Accepted: 16 July 2021
/
Published: 19 July 2021
(This article belongs to the Special Issue Redox Active Metals in Neurodegenerative Diseases: Therapeutic Implications)
Abstract
:Redox-active metal ions, Cu(I/II) and Fe(II/III), are essential biological molecules for the normal functioning of the brain, including oxidative metabolism, synaptic plasticity, myelination, and generation of neurotransmitters. Dyshomeostasis of these redox-active metal ions in the brain could cause Alzheimer’s disease (AD). Thus, regulating the levels of Cu(I/II) and Fe(II/III) is necessary for normal brain function. To control the amounts of metal ions in the brain and understand the involvement of Cu(I/II) and Fe(II/III) in the pathogenesis of AD, many chemical agents have been developed. In addition, since toxic aggregates of amyloid-β (Aβ) have been proposed as one of the major causes of the disease, the mechanism of clearing Aβ is also required to be investigated to reveal the etiology of AD clearly. Multiple metalloenzymes (e.g., neprilysin, insulin-degrading enzyme, and ADAM10) have been reported to have an important role in the degradation of Aβ in the brain. These amyloid degrading enzymes (ADE) could interact with redox-active metal ions and affect the pathogenesis of AD. In this review, we introduce and summarize the roles, distributions, and transportations of Cu(I/II) and Fe(II/III), along with previously invented chelators, and the structures and functions of ADE in the brain, as well as their interrelationships.
1. Introduction
In the human brain, various metal ions are essential as cofactors for numerous enzymes for catalytic activities and neurotransmission including synaptic plasticity, myelination, and synthesis of neurotransmitters [1,2]. Moreover, redox-active metal ions [i.e., Cu(I/II) and Fe(II/III)] have critical roles in oxidative metabolism. Therefore, the homeostasis of metal ions is tightly regulated [1,2,3,4,5,6]. Dyshomeostasis of Cu(I/II) and Fe(II/III) could overproduce reactive oxygen species (ROS) via Fenton-like reactions to elevate oxidative stress and induce the malfunctioning of mitochondria. Additionally, defects in energy metabolism, aberrant axonal transport, and inflammation have been observed which potentially lead to neurodegenerative disorders [7,8,9,10]. Once the redox-active metal ions bind to amyloid-β (Aβ), a major risk factor of Alzheimer’s (AD), rapid peptide aggregation and formation of toxic oligomeric species are observed, along with overproduction of ROS [11,12,13,14]. Therefore, the redox-active metal ions, Cu(I/II) and Fe(II/III), could be related to neuronal impairments, subsequently leading to cognitive defects. In order to reduce the risk of neurodegeneration by Cu(I/II) and/or Fe(II/III), the metal chelation strategy has been suggested as the treatment of AD; however, only targeting metal ions could not cure the disease completely (vide infra).
In addition to redox-active metal ions, clearance of Aβ in the brain is critical for ameliorating neurotoxicity. Amyloid degrading enzymes (ADE), including neprilysin (NEP), insulin-degrading enzyme (IDE), and ADAM10, are involved in the Aβ removal process to regulate the protein levels in the brain [15,16,17]. Since the levels and activity of ADE have been reported to be decreased with aging, the risk of AD occurring increases [15,18]. Thus, enhancing the levels and/or actions of ADE could be a potent therapeutic strategy to AD [18,19]. Moreover, although the redox-active metal ions could affect the activity and levels of ADE, the influence of Cu(I/II) and Fe(II/III) on the enzymatic activity of ADE still needs to be investigated in many aspects.
In this review, we summarize the distributions and roles of redox-active metal ions [i.e., Cu(I/II) and Fe(II/III)] in both healthy and diseased brains, as well as previously developed chemicals to regulate the levels of those metal ions. Moreover, the effect of Cu(I/II) and Fe(II/III) on the activity and/or levels of ADE is also introduced.
2. Cu(I/II)
2.1. Cu(I/II) Distributions in the Nervous System
The Cu ion is the third most abundant transition metal ion (ca. 100 mg) in the human body [20]. It works as a cofactor that binds to various metalloenzymes and assists their activation [4,21]. Since it usually exists as cuprous ions [Cu(I)] and cupric ions [Cu(II)], Cu(I/II) can serve as an electron transporter. Cu(I) has an electron configuration of [Ar]3d10, and Cu(II) has [Ar]3d9 [20,22]. In addition, Cu(I/II) plays a key role in energy metabolism, signal transduction, reproduction, and development that are very important for physiological functions. For example, cytochrome c oxidase in mitochondria needs Cu ions for its activation, and dopamine-β-hydroxylase utilizes it for the cellular secretory pathway [23,24,25,26]. Cu ions can be detected in various points of the brain such as the soma of cortical pyramidal and cerebellar granular neurons, neuropil within the cerebral cortex, hippocampus, cerebellum, and spinal cord [27]. On average, ca. 100 μM of Cu ions has been detected in the brain; however, some parts of the brain have a 2- to 3-fold higher concentration than the other regions [28]. In particular, the ceruleus has a 1.3 mM concentration, which is a part of the brain related to stress and panic. The substania nigra, the dopamine-producing region of the brain, also has a high Cu ion concentration (ca. 400 μM) [28].
The extracellular level of Cu ions depends on the cellular environment. In cerebrospinal fluid (CSF), only 0.5–2.5 μM of Cu ions exists, while the synaptic cleft contains 30 μM [29,30,31]. Usually, the Cu ion concentration is 2–3-fold higher in neurons [32]. In the brain, Cu ions exist in two types: (i) those tightly bound to the proteins, or (ii) those in labile pools [33]. Several regions of the brain such as the soma of cortical pyramidal and cerebellar granular neurons, the hippocampus, the cerebellum, and the spinal cord have labile copper stores [27]. There are also labile pools with a low concentration in the extracellular regions [34].
2.2. Regulations of Cu by Metallochaperones
2.2.1. Copper Chaperone for Superoxide Dismutase (CCS)
A 54 kDa metalloprotein, CCS, is found in the cytosol, mitochondria, and nucleus and transports Cu(I) to superoxide dismutase (SOD1) which is the major antioxidant [35,36,37]. For its activation, SOD1 needs catalytic metal ions such as Cu or Mn ions. CCS has three domains for its function: (i) domain I at the N-terminus with Cu binding motif MXCXXC, (ii) domain II with a similar structure to SOD1 which can bind to SOD1, and (iii) domain III containing Cu(I) with the CXC Cu binding motif [35,36,38]. Once docked to SOD1, a disulfide bond is formed between Cys244 of CCS and Cys57 of SOD1, leading to activation of SOD1. On the other hand, SOD1 activity was greatly decreased (70–90%) upon the CCS gene [35,36].
2.2.2. Antioxidant Protein 1 (Atox1)
Atox1 is a cytosolic metallochaperone protein that delivers cytosolic Cu to ATP7A and ATP7B via a ligand exchange [39,40,41]. The role of Atox1 is to protect cells from attacking ROS. Sequestrated Cu ions by Atox1 are transferred to the trans-Golgi network (TGN) of secretory vesicles [42,43]. Atox1 consists of four β-sheets and two α-helices forming a βαββαβ structure. First βα has a Cu binding motif, MXCXXC, and binding of Cu(I) induces a conformational change to form a bent S–Cu(I)–S bond. Recently, it has been found that tumors need a high level of Cu ions. Breast cancer cells need Atox1 for migration and identification of partner proteins [44].
2.2.3. Cytochrome C Oxidase Assembly: Cox11, Cox17, Cox19, Sco1, and Sco2
Cox11, a 28 kDa metallochaperone, is located in the mitochondrial inner membrane. A single transmembrane helix allows it to be tethered to the membrane [45]. The Cu(I) binding domain is located at the C-terminus of the protein, which forms a dimer. Each monomer can coordinate one Cu(I) via three thiolate ligands [46]. It has been reported that Cox11 helps the formation of the CuB site of cytochrome c oxidase [47]. On the other hand, Cox17, an 8 kDa metallochaperone, delivers Cu(I) to form both CuA and CuB sites [48,49]. Cox17 is found in the cytosol and mitochondrial intermembrane space. Among the seven Cys residues of the protein, three Cys residues are important for the function of Cox17 (CCXC motif), and one Cox17 can bind to three Cu(I) [50]. This protein delivers Cu(I) to Sco1, an intermediate protein on the way to cytochrome oxidase [51]. Another Cu(I)-binding protein, Cox19, has a similar structure to Cox17, and it is located in the mitochondrial intermembrane space as well. Cox19 is expected to contribute to the transportation of Cu ions into cytochrome c oxidase [52,53].
Both Sco1 and Sco2 are cytochrome c oxidase assembly proteins which are Cu metallochaperones [54,55,56]. Sco1 is a 33.8 kDa protein with three Cu binding amino acid residues at Cys169, Cys173, and His260. It has 7 α-helices, 10 β-strands, and 2 turns [57,58]. Its location is in the inner mitochondrial membrane, where it transports Cu ions to the CuA site on Cox2. The other important role is controlling the localization and abundance of Ctr1 for Cu homeostasis [59,60]. Cytochrome c oxidase assembly becomes defective when there are mutations in Sco1 and Sco2, inducing Cu ion deficiency [61]. Sco2 is a 15.1 kDa protein consisting of 136 amino acids and is important for transferring Cu ions to the CuA site of cytochrome c oxidase subunit II. The redox state of the Cys residue in Sco1 is also regulated by Sco2, which is a thiol-disulfide oxidoreductase [61,62].
2.3. Uptake of Cu(I/II) through Blood–Brain Barrier
Important regions in the brain for regulating the uptake and release of Cu(I/II) are the blood–brain barrier (BBB) between the blood and brain interstitial fluid, and the blood–CSF barrier (BCB) between the blood and CSF [63,64,65]. The intracellular concentration of Cu ions should be tightly controlled since they can produce harmful chemical species (e.g., ROS) during the oxidation/reduction process [64,66,67]. Copper transporter-1 (Ctr1), ATP7A, ATP7B, glutathione (GSH), metallothioneins (MTs), and Cu chaperone regulate Cu ion transportation [68]. Reduction of Cu(II) to Cu(I) should be required before entering the brain [69,70]. In yeast, Fre1p and Fre2p, ferric reductases, are responsible for this process [69,70,71]. Reduced Cu(I) can be transported into the brain via multiple pathways driven by numerous proteins [72,73,74]. A more detailed description of this process, along with the related proteins, is summarized in the following sections.
2.3.1. Copper Transporter-1 (Ctr1), Ctr2, and Ctr6
Ctr1 was first discovered in Saccharomyces cerevisiae as a high-affinity Cu uptake protein. It is a membrane protein composed of 190 amino acids and that is involved in the transport of Cu ions from the blood to cells [72,73]. The N-terminus is an extracellular part which is less conserved, while the C-terminus is a cytoplasmic part and highly conserved. Between them, there are three transmembrane domains. Met- and His-rich motifs (7MGMSYM12, 40MMMMPM45, 3HSHH6, and 22HHH24) at the N-terminus are involved in Cu binding [74,75,76]. Fluorescence resonance energy transfer (FRET) experiments revealed that Cu binding to the extracellular part of Ctr1 induces a conformational change in the cytosolic part, and this conformational change is a driving force for releasing Cu ions into the cytosol [77,78,79]. Ctr1 has a Km of 2–5 μM and does not need ATP consumption for Cu ion transport. Ctr1 only uptakes Cu(I), not Cu(II), and Ctr1 transports Cu ions in both the liver and intestine of mice, allowing various proteins to function in the cytosol [80]. Ctr1 is mainly present in the plasma membrane and the intracellular vesicle. Under a high Cu concentration, Ctr1 moves from the plasma membrane to the cytosol and is decomposed [74]. The Ctr2 gene found in the human genome has homology with Ctr1, but it differs from Ctr1 in that there is no extended portion of the N-terminus, but the sequences of the transmembrane domain, which are considered essential for Cu transport, are almost the same [81,82]. Unlike Ctr1, the proportion of Ctr2 distributed on the plasma membrane is less than 5%, and the rest is intracellular [83,84]. Therefore, it was expected to have a different function from Ctr1, suggesting that the absence of the N-terminus of Ctr2 would function as Cu uptake in the plasma membrane with a low affinity. Recent studies revealed that the vacuole can store and mobilize Cu ions [85,86]. Ctr6, a newly discovered Cu transporter in Schizosaccharomyces pombe, is translated under Cu-deficient conditions. Both Ctr2 and Ctr6 are localized to the vacuole, and their functions transfer Cu ions stored in vacuoles to the cytosol [87,88].
2.3.2. ATP7A and ATP7B
ATP7A and ATP7B are homologous ATP-driven transporters, types of ATPase. Their size is 160–170 kDa, containing eight transmembrane domains and several cytosolic domains. The N-terminus is located in the cytosol and has six Cu binding motifs consisting of GMXCXXC [89]. ATP hydrolysis occurs at the ATP binding domain located between transmembrane domain 6 (TM6) and TM7. The A-domain between TM4 and TM5 plays a role in inducing a conformational change during ATP hydrolysis [90,91,92]. Normally, they are located in the TGN and receive Cu ions from the Cu chaperone Atox1 into the Cu binding domain. This causes a conformational change, and Atox1 can deliver different Cu ions to different binding sites [93,94]. Cu ions are transferred to the TM domain by ATP hydrolysis and phosphorylation of Asp residues, which cause Cu ions to be exposed to the lumen of TGN. ATPase is dephosphorylated while releasing Cu ions and returns to its original state. The low pH of TGN plays a key role during this action [95]. There is about 60% sequence homology between ATP7A and ATP7B, but the functional aspects are not identical. ATP7A is faster, whereas ATP7B has a higher affinity for Cu [96,97]. Unlike Ctr, they are responsible for the transport of Cu ions out of the cytosol using the energy from ATP hydrolysis. The released Cu ions move to the other tissues by ATP7A and to the bile by ATP7B. Since neither ATPase exists in the plasma membrane, Cu ions are not directly transported across the plasma membrane but are transported to intracellular vesicles, allowing the vesicles to be fused with the plasma membrane for release [98,99,100]. Additionally, these Cu-APTases deliver Cu ions to many Cu-binding proteins. Cu-binding proteins in the plasma membrane receive Cu ions while ATP7A and APT7B export Cu ions through the vesicle [101].
2.3.3. Glutathione (GSH)
GSH is a tripeptide (Glu-Cys-Gly) existing in high concentrations (0.5–10 mM) which is deeply involved in the oxidation/reduction reactions occurring in the body. As an antioxidant, it prevents cell damage caused by ROS and heavy metals [102,103]. There are a reduced form of GSH and an oxidized form of GSSG, and the ratio between these two forms is a measure of the oxidative stress of cells [104]. In normal cells and tissues, the GSH form is more than 90% [105]. Cu(II) is reduced by GSH to become Cu(I), and then it forms a Cu(I)–GSH complex [106]. GSH plays a role in regulating the function of Cu ion uptake by Ctr1 [107,108]. It is known that GSH also regulates the functions of ATP7A and ATP7B, which are involved in the transport of Cu ions, by regulating the binding of Cu ions to ATPase [109,110].
2.3.4. Metallothioneins (MTs)
MTs are small molecule proteins present in almost all living organisms and have very high affinity with essential or toxic mono- and divalent transition metal cations. Due to their Cys-rich characteristic, they form metal–thiolate clusters selectively with metal ions having an electron configuration of d10 [111]. In particular, the binding to Zn(II) or Cu(I) plays a biologically important role [112,113,114]. They protect the body by releasing highly toxic transition metal ions and reduce oxidative stress induced by ROS and/or reactive nitrogen species [112,115]. There are four types of mammalian copper MT, from 1 to 4, and they consist of 61 to 68 amino acids. A total of 20 Cys are arranged in the form of Cys-Cys, Cys-X-Cys, and Cys-X-X-Cys [116,117]. They have a shape similar to a dumbbell, and two metal-thiolate clusters at both ends are connected by flexible sequences. MTs also exist in the liver, kidneys, and intestines and are widely distributed in the brain and neurons [118,119].
2.4. Cu in Normal and Diseased Conditions
2.4.1. Cu in Nervous Systems under Normal Conditions
In normal conditions, Cu ions have two important physiological roles. First, Cu(I/II) provides a driving force for several oxidation/reduction reactions performed by enzymes. As a cofactor for various enzymes, Cu(I/II) becomes a redox-active metal center in those enzymes [120]. In an electron transport chain occurring in mitochondria, Cu(I/II) can regulate the levels and activity of enzymes for energy production. In addition, Cu(I/II) regulates neurotransmitters, neuropeptides, and dietary amines. Second, Cu(I/II) acts as a signal and excitotoxicity modulator in neurotransmission [120]. In the brain, specific enzymes such as dopamine β-monooxygenase (DβM), peptidylglycine α-hydroxylating monooxygenase (PHM), tyrosinase, and Cu amine oxidase use Cu ions for their cofactor [121]. Labile Cu ions are also utilized for neuronal activity in the brain. Upon depolarization, micromolar range of Cu ions is excreted to the extracellular space using hypothalamus tissues or synaptosomes [34,122]. The intracellular origin of these Cu ions is ATP7A activated by NMDA receptors [123]. The basic roles of Cu(I/II) in the normal brain are summarized in Figure 1.
2.4.2. Cu under Diseased Conditions
It is important to maintain the homeostasis and precise compartmentalization of metal ions in the neuronal signaling process to prevent toxic effects and dysfunctional activity [1]. Failure in homeostasis and compartmentalization can induce neuronal toxicity, eventually leading to AD. Moreover, redox-active Cu(I/II) binds to Aβ, which can generate ROS by Fenton chemistry and/or the Haber-Weiss reaction through the redox cycle, leading to oxidative stress, and form toxic Cu–Aβ aggregates which could lead to neurotoxicity and neurodegeneration [4,20,22]. About 0.4 mM of Cu ions was present in the senile plaques composed of Aβ aggregates in the brain of AD patients [4]. The dissociation constant for Cu(II)–Aβ is in the nanomolar range, which is found under a physiological environment [124,125]. There are two components of Cu(II) binding to Aβ depending on the pH; at a physiological environment, Aβ favors Cu(II) binding via component I, while under a basic environment, Aβ prefers component II [126,127]. In the synaptic cleft, Cu ions play an important role as a secondary messenger, and the concentration of Cu ions is estimated as ca. 100 to 250 μM, depending on the size of the cleft [128]. In addition, it was observed that the degree of Cu ion excretion into the extracellular space increased through depolarization by 50 mM of K(I) [34]. In a study about the binding of Cu ions to Aβ at the synaptic cleft using reaction-diffusion simulation, the binding of Zn(II) was very low, about 0.1% of the entire Aβ, while the binding of Cu ions was high enough to reach 30%. Therefore, Zn(II) does not significantly affect the formation of Aβ dimers in the neurotransmission process at the synaptic cleft, but it is believed that Cu ions play an important role in the early stages of Aβ oligomerization [129].
Cu–Aβ acts as a catalyst in oxidizing monoamine-based neurotransmitters such as dopamine, epinephrine, and serotonin. In particular, dopamine has an 85-fold higher tendency than autoxidation in Cu–Aβ40 [130]. Among the various Cu–Aβs, Cu–Aβ16 showed the greatest activity in the oxidation of several monoamine-based neurotransmitters. In the presence of hydrogen peroxide, ROS were generated in pathological conditions, and the catalytic capacity of Cu–Aβ was observed to increase, and it is believed that this may affect AD by promoting the oxidation of neurotransmitters. Conversely, it was known that direct interaction between Cu–Aβ and dopamine/dopamine derivatives can inhibit the aggregation of Cu–Aβ [131]. According to Nam et al., dopamine and its derivatives promote the oxidation of Aβ40 and Aβ42 in the presence of Cu(II), leading to modifying the aggregate form of Aβ. Instead of a general fibril form, amorphous and more compact forms were generated, or they decomposed existing Aβ aggregates [131].
In the case of Aβ found in the brain of AD patients, there are many N-terminally truncated forms, especially Aβ4-x. This form contains an H2N-X-X-His (ATCUN) motif that has a higher Cu(II) affinity than Aβ1-x [132]. Glutamate, one of the neurotransmitters, promotes the transfer of Cu(II) from Cu–Aβ4-16 to Zn7-metallothionein-3 (Zn7-MT-3). This indicates that glutamate, which is instantaneously elevated during neurotransmission, forms an instant ternary complex with Cu–Aβ4-16 and promotes the transmission of Cu(II) [133].
In addition to Aβ, Cu ions can also interact with the tau protein which might be a potential pathogen for AD. Depending on the pH and stoichiometry, the R2 and R3 domains of tau can be Cu ion binding sites [134]. The His residue in this region is sensitive to pH changes, and it is responsible for Cu ion binding. Since R3 has two His residues close to each other, its binding affinity to Cu ions is higher than that of R2. The His residue in the R1 region can also bind to Cu(II), but its affinity is much weaker than the others [135,136]. The simplified effects of Cu(I/II) in the AD-affected brain are presented in Figure 1.
2.5. Regulators of Cu(I/II)
To reduce the risk of onset and/or progression of AD by Cu(I/II), various chemical agents have been developed to regulate the levels of the metal ions (Figure 2). The chemicals were examined by their ability to bind to Cu(I/II) in numerous studies. In this section, we summarize the chemical agents capable of interacting with Cu(I/II).
Clioquinol (CQ), an 8-hydroxyquinolin (8-HQ) derivative, is a candidate for treatment of AD by targeting Cu(II) and Aβ (Figure 2; top row) [137,138,139]. The CQ–Cu(II) complex has a square planar structure where two CQs coordinate Cu(II) in a crystal structure. In the solution phase, its symmetry is broken, and it becomes a tetragonally distorted structure [140]. Regardless of this, the structure in solution is still closely related to that observed in crystallography. It has nanomolar affinity for Cu(II) and can cross the BBB [141]. In 2003, Prana Biotechnology reported the results of a phase II trial on CQ in AD patients due to its abilities of metal chelation and modulation of metal-free and metal-bound Aβ aggregation. However, further processes were stopped during the phase II/III trial since a toxic compound was detected during the manufacturing steps. Although it showed toxicity, CQ has recently been examined for its potential neuroprotective effect in C. elegans [142].
PBT-2 is also an 8-HQ derivative and the next generation of CQ developed for treatment of AD and Huntington’s disease (Figure 2; top row) [143,144]. It has improved BBB penetrance and pharmacokinetics compared to CQ [145]. Interactions between Cu ions and Aβ are disrupted by PBT-2, preventing the accumulation of toxic Aβ species in the brain. The extracellular Cu ion concentration is reduced since PBT-2 transports Cu ions to the intracellular space. This effectively reduces the chance for Cu–Aβ interactions which could trigger Aβ aggregation [146,147]. Prana started the first phase II trial of PBT-2 for AD in 2007, and the second phase II trial in 2011. In 2014, Prana reported that there is not much difference between treated and non-treated groups [148]. An additional phase II trial revealed that 250 mg/day, which the patients could tolerate, of PBT-2 has positive results in terms of cognitive ability [149,150]. The clinical trial information of PBT-2 is summarized in Table 1.
DP-109 and DP-460 are lipophilic chelators that chelate Cu, Fe, and Zn in the membrane [151]. In AD mice, DP-109 administration reduced amyloid plaques and the degree of cerebral amyloid angiopathy in the brain [152]. Furthermore, both molecules slightly extended (10% and 9%, for DP-109 and DP-460, respectively) the life span of G93A-transgenic ALS mice [153]. However, a recent study presented that DP-460 has detrimental effects on learning based on the Morris water maze test [154]. Moreover, bathocuproine (BC) and bathocuproine disulfonate (BCS), presented in the middle row of Figure 2, could chelate Cu(I) and Cu(II) by N as electron donor atoms near methyl groups. BC prefers Cu(I) to Zn(II) due to its size [155]. BCS has two negative charges which provide water solubility to the molecule. Thus, BCS can be used to chelate extracellular Cu(II) as well [156,157,158].
Multiple chelators against Cu have been developed to treat other diseases such as Wilson’s disease. D-penicillamine is one of those Cu ion chelators (Figure 2; bottom row) [159,160,161]. It chelates and reduces Cu(II), which is then excreted in the urine at up to 1.5 mg/day, which is a four-five times increased amount compared to untreated cases in patients [162]. Additionally, upon treatment of D-penicillamine in a hydrogel form, it could improve the cognitive ability of APP/PS1 mice through the activation of ADAM10 [163]. Trientine (triethylenetetramine) is a selective Cu(II) chelator that suppresses oxidative stress. Once Cu(II) binds to trientine, a Cu(II)–trientine complex is excreted in the urine, but not as much as D-penicillamine (Figure 2; bottom row) [164,165].
Tetrathiomolybdate, shown in the bottom row of Figure 2, consists of a central molybdenum surrounded by four sulfhydryl groups and has been suggested as a therapeutic agent for Wilson’s disease to control the levels of Cu(I/II) as well. Once it binds to Cu ions in foods, it forms a very stable complex and is excreted in the stool [166]. Tetrathiomolybdate also forms a complex with serum albumin and labile Cu ions in the blood. Since Cu–tetrathiomolybdate cannot be reabsorbed, the complex is metabolized in the liver. The resulting metabolized fragments are excreted in bile. Moreover, upon treatment with tetrathiomolybdate, the production of inflammatory cytokines decreased in APP/PS1 transgenic mice [167]. If tetrathiomolybdate is used at more than a specified dose of 120 mg/day, two side effects might appear, namely, anemia, and elevation of the transaminase level [168].
There are several multifunctional molecules where other scaffolds are conjugated to metal-chelating moieties for reducing possible side effects or improving metal chelation for treatment of AD [1]. Since there are many factors that cause AD, multifunctional chemical agents that target multiple risk factors of the disease would be an effective strategy to treat/cure AD. In particular, many chemical agents targeting both Cu(I/II) and Aβ or Cu(I/II) and ROS have been developed. Based on multifunctionality, a few chemical agents, although they have firstly been suggested as Cu(I/II) chelators, are still examined for their potentials as therapeutic agents for AD in various animals (Table 2) [142,152,153,154,163,167].
More than ten multifunctional molecules based on 8-HQ have been developed. They have tetra-O-benzyl-β-D-glucopyranoside, rasagiline, trehalose, glutathione, and β-cyclodextrine conjugated to 8-HQ [169,170,171,172,173,174,175,176]. By applying thioflavin-T (ThT), an imaging agent for aggregated Aβ, several multifunctional molecules were developed by conjugating CQ, DTPA, di-(2-picolyl)amine, and/or N-(2-pyridylmethyl)amine [177,178,179,180,181,182,183,184]. Multifunctional molecules based on p-I stilbene (pISTIB) can chelate Cu(II) to regulate Cu(II)-induced Aβ aggregation [185,186,187,188,189,190,191,192,193,194]. Even though the exact role of triazole has not yet been elucidated, triazole-based chemicals have been developed having a quinoline ring and a phenol [195,196,197]. Multifunctional molecules affected metal-free Aβ aggregation, and others reduced Cu(II)-induced Aβ aggregation when selegiline, aurone, and chromone were conjugated [198,199,200]. An antioxidant molecule, resveratrol, was used to invent multifunctional molecules by incorporation with CQ and deferiprone (DFP) [201,202]. DFP is a well-known Fe(II/III) chelator which is being examined for its potential to treat neurodegeneration. The information of clinical trials (phase II) is summarized in Table 1, and more information of DFP is discussed in Section 3.4 (vide infra).
3. Fe(II/III)
3.1. Fe(II/III) Distributions in the Nervous Systems
Fe is the most abundant metal in the brain [203]. Mostly, the oxidation states of Fe are ferrous [Fe(II)] and ferric [Fe(III)]. Due to the oxidation and reduction process between the ferrous and ferric states of Fe, it is called a redox-active metal ion, as with Cu(I/II). This redox-active property is important for the activity of various enzymes and proteins related to (i) O2 chemistry, (ii) electron transfer, (iii) gene regulation, and (iv) cell growth and differentiation [204,205,206,207]. To maintain the normal functions of the brain, Fe(II/III) are bound to the proteins in common, while labile Fe ions have been observed in intracellular pools with a concentration of up to 100 μM [208]. The concentrations of Fe(II/III) are different depending on the regions and cell types of the brain; 20–30 μM has been found in blood serum, and 0.5 to 1 mM has been found in neurons [209,210].
3.2. Homeostasis of Fe
Since the Fe ion is a redox-active metal ion, regulation of its level is essential to maintaining the normal biological functions [65]. The homeostasis of Fe(II/III) is tightly controlled by multiple proteins: heme proteins, transporters, Fe chaperones, ferrireductases, ferritin, transferrin (Tf), transferrin receptor 1 (TfR1), divalent metal transporter 1 (DMT1), and ferroportin (Fpn) [211]. Generally, in the human body, Fe(II/III) are bound to heme, ferritin, and Tf [212,213]. Two mechanisms of Fe(II/III) transportation have been reported: with and without Tf [214,215,216]. Non-Tf-bound Fe(II/III) would be delivered into the brain by astrocytes, oligodendrocytes, microglia, and/or albumin; however, heme- or ferritin-bound Fe(II/III) are not considered as non-Tf-bound Fe [217,218].
The major role of ferritin is storing Fe(III). It forms a 24 mer complex storing up to 4500 Fe(III)s composed of two subunits (chains): heavy (H) and light (L) chains [219,220]. These two chains present different functions. At a ferroxidase site in the H chain, Fe(II) is oxidized by coupling with O2. The ferroxidase site of human ferritin contains two Fe(II)s [221,222,223]. Glu27, Glu62, and His65, along with a water molecule, coordinate to Fe(II), and another Fe(II) is bound to Glu61, Glu62, and Glu107, with the assistance of Tyr34 and Gln141, in order to stabilize the structure [221,222,223]. The L chain promotes the nucleation of Fe core minerals such as ferrihydrite [224]. In addition to storing Fe ions, ferritin could reduce the generation of ROS to decrease oxidative stress, as well as lowering the toxicity induced by Fe(III) by inhibiting the interactions and reactions between Fe(III) and other biological molecules [219,224].
3.2.1. Fe(II/III) Cross the BBB by Involvement of Tf
Most Fe ions are bound to Tf to be transported into the brain. Tf is a glycoprotein which is generated in the liver and has two Fe(II/III) binding sites at both the N- and C-terminal lobes composed of an aspartate, two Tyr, His residues (Asp63, Tyr95, Tyr188, and His249 on the N-terminal lobe; Asp392, Tyr426, Tyr517, and His585 on the C-terminal lobe), and a carbonate ion showing a high binding affinity towards Fe(III) (ca. Ka = 1022 M−1 at pH 7.4) [220,221,225,226]. About 34% of Tf has only one Fe(III) at the N-terminal lobe or C-terminal lobe, and 27% of Tf contains two Fe(III)s [226,227]. The suggested mechanism for the transportation of the Fe(III)-bound Tf complex through the BBB is receptor-mediated endocytosis [226,228,229]. At the membrane of brain capillary endothelial cells (BCEC), which maintain the integrity of the BBB, up to two Fe(III)–Tf complexes are captured by transferrin receptor-1 (TfR1) [226,228,230]. Then, Fe(III)–Tf tightly interacts with the receptor to be transported into endosomes, and the structural changes in Tf lead to opening the cleft of the Fe binding site to release the reduced form of Fe ions, Fe(II) [229]. Although the exact mechanism remains unclear, ferrireductases (e.g., STEAP3) have been proposed to be involved in this process [231].
Next, the released Fe(II) from Tf is bound to divalent metal transporter 1 (DMT1) to cross the endosomal membranes [232]. Fe(II) transport by DMT1 is an active and proton-dependent process; DMT1 could deliver Fe(II) the most effectively under mild acidic conditions [233,234]. Fe(II) from Tf could bind to Zrt-/Irt-like protein 8/14 (ZIP8/14) as well [235,236]. Although ZIP8 and ZIP14 are mostly involved in regulating Zn(II) levels, they have a significant role in the cellular uptake of Fe(II/III), with a poor understanding of the exact mechanism [237]. Unlike DMT1, ZIP8 and ZIP14 presented their optimal ability of Fe(II/III) transport under physiological pH [238,239].
The Fe ions are then exported to the brain by Fpn located on the cytoplasmic side of the plasma membrane [240]. For this process, extracellular ferroxidases are required [241]. Ceruloplasmin is one of the ferroxidases interacting with Fpn to enhance the incorporation of Fe ions into Tf, and efflux of the Fe ions from astrocytes [230,242].
3.2.2. Fe(II/III) Transport without Tf
In CSF and interstitial fluids, labile Fe(II/III) could bind to other transporters such as albumin, lactoferrin, and p97 [243]. The lactoferrin receptor- and glycosylphosphatidylinositol-anchored p97-secreted pathways have also been reported to transport Fe(II/III) through the BBB [244,245]. Fe ions could bind to citrate and ascorbate as well. Most non-Tf-bound Fe(III) is bound to citrate in CSF, while the Fe(II/III)–ascorbate complex is observed with a low concentration (ca. nanomolar range). When Fe ions are not bound to Tf, known as non-Tf-bound Fe(II/III), the toxicity could be increased due to the high propensity to generate ROS [243].
Although numerous studies tried to reveal the exact mechanisms of Fe(II/III) transport across the BBB, they still remain unclear [246]. In order to understand the implications of Fe(II/III) in the brain, more details about Fe(II/III) miscompartmentalization, along with production of ROS in the pathogenesis of neurodegeneration and transport of Fe(II/III) without Tf, should be investigated.
3.3. Physiological and Pathological Functions of Fe in Nervous Systems
3.3.1. Fe under Normal Conditions
Fe(II/III) are used for various enzymatic activities based on their common redox activity: mitochondrial respiration and O2 transport [247,248,249]. Specifically, Fe(II/III) help to generate energy in the form of ATP by the involvement of the electron transport chain as Fe(II/III)–S complexes and enzymatic cofactors of cytochromes. Since ATP synthesis requires O2, both hemoglobin and myoglobin containing Fe(II/III) are related to the process [250,251]. The generated energy is used for axonal and synaptic signaling in particular. For normally functioning mitochondria, the energy supplied by ATP is necessary [251].
In addition, Fe(II/III) are involved in the synthesis of myelin and neurotransmitters [247,248,249,252,253]. Myelin is a lipid-rich substance and required for fast information transmission (electronic signaling) from neurons to neurons by insulating nerve cell axons. For the myelination, the levels of Fe are important for the composition of myelin at the gestation and early post-natal periods [252,253]. In the process of myelin production, oligodendrocytes play important roles, with high concentrations of Fe(II/III) in the form of ferritin [254]. Cholesterol and lipids are two main components of myelin, and Fe(II/III) have been implicated to synthesize these biomolecules as a cofactor [254].
Additionally, Fe(II/III) are essential for the production of monoamine neurotransmitters, dopamine and serotonin, which regulate cognitive processes including emotion and arousal behaviors [255,256]. Dopaminergic systems, the tryptophan hydroxylases (necessary for serotonin synthesis), and glutamate dehydrogenase, along with γ-aminobutyric acid (GABA) transaminase (responsible for the synthesis and degradation of GABA), are affected by the levels of Fe(II/III) [257]. The basic roles of Fe(II/III) in the normal brain are summarized in Figure 3.
3.3.2. Fe in Diseased Conditions
Under the conditions of AD, an abnormal concentration and distribution of Fe(II/III) have been reported. In particular, once the BBB is compromised due to aging and/or high levels of oxidative stress, the random transportation of Fe(II/III) across the BBB leads to miscompartmentalization of the ions in the brain [258]. Based on post-mortem analyses of AD-affected brains, accumulation of Fe(II/III) (up to 1 mM) was observed in amyloid plaques and neurofibrillary tangles, especially at the parietal cortex, putamen, and hippocampus, while the concentration of Fe(II/III) in the entire brain is similar to a healthy brain [259,260,261,262]. This dyshomeostasis of Fe(II/III) could lead to the malfunction of metalloenzymes and proteins in the brain which require Fe(II/III) as cofactors.
Moreover, both direct and indirect interactions between Fe(II/III) and Aβ/tau could be related to the onset and/or progression of AD, as shown in Figure 3 [263,264,265]. Fe(II/III) binding to Aβ could lead to the aggregation of proteins and toxicity induced by Aβ species. In addition, once Fe(II/III) are bound to Aβ, ROS would be generated by Fenton chemistry [264,265]. As Fe(II/III) themselves, without Aβ, could enhance the amounts of ROS, they could cause oxidative stress [266,267]. Indirectly, a high level of Fe(II/III) could decrease furin which increases the activity of β-secretase involving the amyloidogenic process of APP to generate Aβ [268]. Additionally, Fe(III) could directly bind to tau, causing its phosphorylation and aggregation of hyperphosphorylated tau [263]. Once tau is accumulated, the expression of heme oxygenase-1 would be increased; subsequently, more biliverdin, carbon monoxide, and labile Fe(II) catalyzed from heme would exist. These products from the catalysis of heme could enhance oxidative stress [268,269]. Although a recent study found that ceruloplasmin could reduce ferroptosis [270], these increased Fe(II/III) levels could lead to ferroptosis and damage the cellular antioxidant capacity in general [230,271]. Once the antioxidant ability has been compromised, ROS would accumulate, causing oxidative damage to nucleic acids, lipids, and proteins [230].
3.4. Regulators of Fe(II/III)
In order to reduce the risk of neurodegeneration by Fe(II/III), numerous studies tried to regulate the levels of metal ions, along with the antioxidant ability. Therefore, many chemicals have been invented as Fe(II) or Fe(III) chelators and examined for their ability to bind to Fe(II/III). In this section, we summarize the chemical agents capable of interacting with Fe(II/III).
The oxidation state of Fe should be considered to develop Fe chelators. Since Fe(III) is a hard acid based on the hard-soft acid and base (HSAB) principle, it prefers to bind to a hard base, such as an O donor atom. Fe(II), a borderline hard acid, prefers a borderline base, such as a N donor atom. Thus, various Fe(II/III) chelators have combinations of O and N as electron donor atoms [272,273,274].
Among the chemical agents, deferoxamine (DFO) and DFP are the most well-known Fe(III) chelators (Figure 4) [272,273,275,276]. DFO is applied to Fe(II/III)-poisoning treatment by chelating metal ions (Figure 4). DFO forms the hexadentate Fe(III) coordination mode, forming an octahedral geometry [277]. Although DFO has limitations in cell permeability because of its hydrophilicity, it presented inhibition of Fe accumulation in rats and mice [275,278,279]. Moreover, DFO could improve the cognitive function of APP/PS1 transgenic mice as well as healthy mice [280,281]. It also presented an improvement in memory in an intracerebroventricular streptozotocin rat model assessed through the Morris water maze test (Table 3) [282]. DFP could interact with Fe(III) in a 1:3 metal-to-ligand ratio, forming an octahedral complex, and it has neuroprotective properties in neurons [273,276,283]. In addition, based on the structure of DFP with the benzothiazole moiety of ThT, compound 2d (from reference [284]) was developed, and it showed a strong Fe-chelating ability [284]. Moreover, recent animal studies showed that DFP could regulate the Aβ levels and phosphorylation of tau, leading to improved cognitive function in a transgenic mouse model of tauopathy, rTg4510 (Table 3) [285,286,287]. The results of clinical trials (phase II) of DFP are summarized in Table 1 [288]. Additionally, for chelating Fe(II), bathophenanthroline disulfonate (BPS; Figure 4; right) has been applied [289,290]. It could coordinate to Fe(II), forming an octahedral geometry, with a dissociation constant of 10−17 M [289,290]. Although BPS is water-soluble, due to the negative charges on sulfonate groups, it could not penetrate the cellular membrane [291].
8-HQ and its derivatives [e.g., HLA20, GS(HQ)H, Compound 8g, VK28, M30] have been developed, and they could interact with various metal ions [295,296,297,298]. HLA20 could form a complex with Fe(III) at a 1:3 ratio, and its binding affinity to Fe(II) was 5.4 μM, determined by a fluorescence dequenching experiment of calcein [296]. GS(HQ)H was invented by a combination of glutathione and 8-HQ, which could coordinate with Fe(III) and protect SHSY-5Y human neuroblastoma cells from H2O2- and 6-OHDA-induced damage [299]. Compound 8g (from reference [294]), designed by Knez and colleagues, also has Fe(II) binding ability [297]. VK28 has been examined for its Fe(II/III)-chelating ability across the BBB [298]. M30 containing an N-propargylamine group of rasagiline on the structure of 8-HQ itself could bind Fe(II/III) directly with a 1:3 ratio and induce mRNA expression levels of the major antioxidant defense system composed of catalase, SOD-1, and glutathione peroxidase, in various brain regions [300]. Moreover, M30 and HLA20 significantly improved the cognitive deficits in a rat model [292,293,301]. In particular, M30 could induce a neuroprotective effect and improve the memory of C57BL/J6 mice once they showed memory and behavioral impairment [294]. The information of animal studies performed with the treatment of Fe-targeting chemical agents is summarized in Table 3.
In addition to targeting only Fe(II) and/or Fe(III), multi-target molecules have emerged as a new strategy to investigate the pathology of AD and treat diseases. Multifunctional molecules are designed to simultaneously target multiple neurodegenerative pathological factors, which is anticipated to slow or even reverse the cognitive decline more effectively than single-target agents [1,302]. For targeting the risk factors of AD, the derivatives of resveratrol, compound 3i and compound 4f (from references [201,202]), were invented for interacting with Fe(III) as well as Fe(III)-bound Aβ42 [201,202]. These compounds could bind to Fe(III) with pFe(III) values of 20 and 19, respectively, along with having antioxidant activity in an ABTS assay. Moreover, they could affect the aggregation of Fe(III)–Aβ42 and disassemble preformed Fe(III)–Aβ42 aggregates [202]. Based on multifunctionality, a few chemical agents, although they have firstly been suggested as Fe(II/III) chelators, have recently been examined for their potentials as therapeutic agents for AD in various animals (Table 3) [280,281,282,285,286,287,292,293,294].
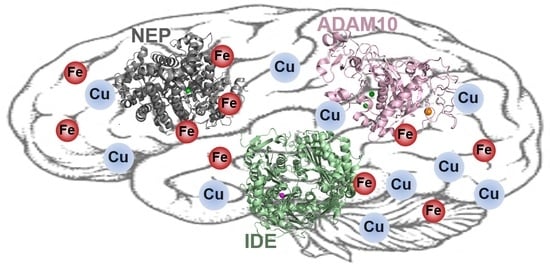
4. Amyloid Degrading Enzymes (ADE)
Although current strategies to treat AD could relieve symptoms temporarily, the disease cannot be cured completely. Therefore, in recent decades, inhibiting the generation of Aβ and modulating its aggregation pathways to form less neurotoxic aggregates have been intensively studied as potent methods to treat AD. In addition to these two aspects, clearance of amyloidogenic proteins from the brain is also important to have less toxic species in the brain. In Section 4 and Section 5, we provide a summary of (i) multiple ADEs, such as NEP, IDE, and ADAM10, along with their roles in the brain, and (ii) interrelations between redox-active metal ions and ADE affecting the activity and/or levels of ADE.
4.1. Neprilysin (NEP)
NEP is a zinc-dependent metalloprotease (type II integral membrane endopeptidase) which has been reported to be involved in the onset and/or progression of multiple diseases such as AD, heart failure, and diabetes [303,304,305,306]. Upon generation in Golgi, NEP exists in neutrophils, the kidney, the lungs, and the cerebral cortex in the brain [306,307,308]. It is mainly composed of α-helical structures with 749 amino acids in 3 domains: an N-terminal intracellular domain (27 amino acids), a transmembrane domain (23 amino acids), and an extracellular catalytic site (699 amino acids) [303,305,309]. For Zn(II) binding, 2N and 2O from His583, His587, Glu584, and Glu646 coordinate to Zn(II) with a tetrahedral geometry [310,311].
The S1, S1′, and S2′ subsites are binding pockets located near the Zn(II) binding site in the extracellular catalytic domain [303,305]. The pocket of the S1′ subsite specifically cleaves large hydrophobic and aromatic side chains at the hydrophobic amino acid residues, while the S2′ subsite could degrade bulky side chains [303,305]. NEP cleaves various vasoactive peptides (e.g., natriuretic peptides, bradykinin, adrenomedullin, angiotensin, substance P, enkephalins, endothelin, and Aβ) between the hydrophobic amino acids. Since Aβ has broad hydrophobic regions, it is an ideal substrate of NEP [15,312,313]. To have a high selectivity of substrates, the enzyme has a sterically hindered active site. The peptide bonds between Glu3 and Phe4, Gly9 and Tyr10, Phe19 and Phe20, and/or Ala30 and Ile31 of monomeric Aβ could be cleaved by NEP [314,315,316]. About 73% of monomeric Aβ40 and 27% of monomeric Aβ42 were degraded by NEP [317]. Oligomeric Aβ species could be degraded by NEP with a lower degree compared to monomeric Aβ, while it cannot cleave the amyloid precursor protein (APP) [312,317,318].
Based on the amyloid hypothesis, the accumulation of Aβ aggregates in the cerebral cortex and gray matter regions in the brain could cause AD. Therefore, the Aβ degrading activity and concentrations of NEP could affect the onset and/or progression of AD [319]. A lower level of NEP in CSF was shown in the early stage of AD which could progress to AD [320]. Its activity is varied based on pH. NEP presents the maximum cleavage activity at neutral pH against small peptides containing less than 50 amino acids [321]. The expression levels of NEP are not different between males and females [313].
In the mouse brain, higher Aβ levels were reported upon disruption of NEP expression [318]. Inactivation of NEP in a hAPP mouse model showed cognitive defects and impaired synaptic plasticity [318,322]. In addition, NEP in CSF has been suggested as a biochemical marker to monitor synaptic impairment since its activity was observed to be decreased by 12% in mild AD patients [323]. Furthermore, it has been reported that the increase in NEP levels could be a potent treatment for AD through Aβ metabolism as well as other mechanisms such as producing neuropeptide Y fragments [324,325,326,327]. Moreover, upon treatment of Tf-bound NEP, Wistar rats presented reduced levels of Aβ in both CSF and the brain [328]. Based on a recent meta-analysis of the expression and function of NEP in AD, it was reported that both the expression and activity of NEP were decreased in the cortex of elderly AD patients, supporting the idea that targeting NEP could be a potent strategy to treat and/or cure AD [329].
4.2. Insulin-Degrading Enzyme (IDE)
Another zinc-dependent metallopeptidase, IDE, which is a 113 kDa with 1019 amino acids, could cleave insulin and Aβ [330,331]. IDE is composed of two similar-sized domains: IDE-N and IDE-C. These two domains are connected by a loop containing 28 amino acids. A number of hydrogen bonds between IDE-N and IDE-C could continue to close the catalytic site located in IDE-N [332,333,334]. Since the active site is located in IDE-N, IDE-N itself could perform the proteolytic activity, while IDE-C could not show enzymatic activity [335]. Another difference between IDE-N and IDE-C is that the inner side of IDE-N is neutral or negatively charged, but IDE-C is positively charged. The positive charge of IDE-C could help the enzyme recognize the substrates [334]. The substrates of IDE contain β-structures such as Aβ [336,337,338]. At the catalytic site, the 108HXXEH112 motif is responsible for binding to Zn(II) [332]. In particular, Glu111 has an important role in the hydrolysis of substrates by acting as a base to the active catalytic water [332].
IDE is mostly located in the cytosol as well as mitochondria, peroxisomes, the plasma membrane, and CSF [339]. Therefore, IDE could control the levels of intracellular Aβ and reduce the damage induced by toxic Aβ species [340,341,342]. IDE could cleave the peptide bonds of Aβ between Val12 and His13, His13 and His14, His14 and Gln15, Val18 and Phe19, Phe19 and Phe20, Phe20 and Ala21, and/or Lys28 and Gly29 [333,343]. Additionally, IDE could act as a chaperone to interfere with the fibrillization of Aβ [344]. Furthermore, IDE has been reported as an important enzyme for the clearance of Aβ in hippocampal lysates, the cytoplasm, and cerebrospinal fluid [345,346]. Similar to NEP, IDE shows optimal cleavage activity at neutral pH as well [347].
In AD-affected brains, the levels of IDE were shown to increase, while the activity of IDE was reported to be decreased with aging, particularly in the early stage of the disease [347,348]. IDE and Aβ plaques were shown to be colocalized in the brain, indicating that IDE could be buried in the plaques and/or oxidized. Consequently, IDE could lose its amyloid-degrading ability, leading to a lower clearance of Aβ and higher aggregation of Aβ, resulting in neuronal damage [349,350]. Moreover, in vivo studies showed that IDE knockout animals have relatively high levels of Aβ, suggesting that the action of IDE to remove Aβ is important to control the amount of Aβ in the brain, and regulating the Aβ cleavage activity of IDE could be a potent strategy to treat AD [349,350].
4.3. ADAM10
The ADAM family, composed of zinc-dependent transmembrane metalloproteases, has been reported to cleave APP as well as cell adhesion and the proteolytic activity of signaling molecules and receptors [351,352,353,354]. Usually, ADAM contains ca. 750 amino acids as a signal peptide, a prodomain, a metalloprotease-like domain, a disintegrin-like domain, a Cys-rich domain, an EGF-like domain, a transmembrane domain, and cytoplamic tail [353]. The disintegrin-like domain acts as a ligand for integrin binding; however, this domain is not necessary for ADAM10 protease activity [355,356]. Among the ADAM family, ADAM9, ADAM10, and ADAM17 are involved in the generation of Aβ, and ADAM10 contributes the most to the proteolytic actions on APP [357,358].
ADAM10 is produced in the ER and matured and activated in Golgi by removing the prodomain. The size of matured ADAM10 is 68 kDa without activation (with the prodomain) [359,360]. While proADAM10 is located in Golgi, most of the activated ADAM10 is located on the plasma membrane [361]. ADAM10 exists as a dimeric form, and 383HEVGHNFGSPHD344 forms a Zn(II) binding site which contains the 383HEXXH387 motif. Three His residues play an important role in Zn(II) binding, and Gly between Phe and Ser constitutes a turn, whilst Glu acts as an acid/base catalyst [355,362]. Recently, the catalytic activity of ADAM10 has been revealed, and the activity of the enzymes could be regulated by a modulatory antibody. The C-terminal Cys-rich domain hinders the active site until there is direct binding of the proteolytic substrates [362,363,364,365].
APP, the target of ADAM10, undergoes a proteolytic cleavage reaction by β- and γ-secretases to generate Aβ [356,366]. ADAM10 acts as an α-secretase cleaving APP at different sites from β- and γ-secretases and produces non-toxic soluble APP (sAPPα), which has a neuroprotective function [355]. Therefore, upregulation of ADAM10 could be a promising strategy to treat AD by reducing toxic Aβ species and increasing neuroprotective sAPPα [355,367].
Furthermore, in neuronal systems, ADAM10 has a role in regulating synaptic proteins [368]. In particular, neuronal surface ADAM10 could be associated with AP2 for endocytosis. In AD patients, the level of ADAM–AP2 in the hippocampus was increased, while the activity of ADAM10 decreased upon interaction with AP2 in hippocampal neurons [368,369]. Moreover, in ADAM10 knockout mice, synaptic impairments, decreased neuromotor abilities, and reduced learning abilities were observed, indicating that the low level of ADAM10 could induce postsynaptic defects [370].
5. Redox-Active Metal Ions with ADE
Since both redox-active metal ions and ADE are related to the etiology of AD, investigation of their interactions along with enzymatic activity is necessary to further understand AD pathology. First, Cu ions could interact with NEP; in particular, the extracellular Cu(II) could reduce the levels of NEP in cells [371]. Similar results were obtained using a transgenic drosophila experiment that has a silent Ctr1 protein, along with a dissociation constant of NEP for Cu(II) determined as 1.04 (±0.07) μM. Even 100 μM of Zn(II) was not able to restore the activity of NEP [372,373]. Downregulation of NEP activity by Cu(II) was also observed in mouse neuroblastoma N2a cells. NEP activity was not inhibited at the transcription level. Rather, NEP turnover was blocked by specific proteasome inhibitors such as MG132 and lactacystin, suggesting a possible mechanism of NEP degradation would be the proteosome pathway [371]. Additionally, Fe could be related to decreased NEP activity. Once a chelator, deferasirox, chelates an Fe out from treated 18-month-old rats for 4 months, the Aβ42-degrading activity of NEP was recovered compared to the same aged rats [374].
Cu ions could downregulate the activity of IDE as well [372,375]. Inhibition of IDE activity by Cu(II) is reversible, and addition of Zn(II) restores IDE activity. However, inhibition of IDE activity by Cu(I) is irreversible [375]. Cu(I) binding to IDE blocks two Cys residues, Cys812 and Cys819, rendering the hydrophobic core destabilized. As a result, the Zn(II) binding site is closed, which is irreversibly inactivated [375]. Additionally, the proteolytic activity of IDE was inhibited by Cu(II); however, ubiquitin-activating (E1-like) activity was not affected up to 20 μM [376].
The interactions between Cu ions and ADAM10 have also been reported. There was no significant change in the ADAM10 level after 3-month Cu exposure of transgenic mice, while the ADAM10 level was dramatically increased after 9-month Cu exposure [377]. Unlike Cu ions, the overload of Fe ions could not significantly affect the expression levels of ADAM10, while the expression of ADAM17 increased. Additionally, Fe ions in the presence of peroxidative stress (treating with Fenton) could induce the expression of ADAM10 [378]. Moreover, Fe(II) could induce the mRNA levels of ADAM10 in cells along with the generation of sAPPα [379]. Recently, application of a Cu chelator, tetrathiomolybdate (shown in Figure 2; bottom row), to transgenic mice increased the levels of ADAM10, indicating that Cu chelators could activate the non-amyloidogenic processing of APP (less generation of Aβ) [380]. Nasal injection of a hydrogel containing another Cu-binding molecule, D-penicillamine (presented in Figure 2; bottom row), showed regulation of the ADAM10 level via the MT1/2 pathway, and enhanced lower production of Aβ in a transgenic mice experiment [163].
6. Conclusions
Redox-active metal ions, such as Cu(I/II) and Fe(II/III), are considered as major contributors to the complex chemical and biological reactions in the brain. In particular, Cu(I/II) and Fe(II/III) are important for oxygen consumption and the oxidative capacity of the neurons and glia in the brain. However, accumulation or miscompartmentalization of metal ions could be involved in the onset and/or progression of AD. Although various metal chelators have been developed for (i) regulating metal ions in the brain and (ii) investigating the involvement of metal ions in neurodegeneration, these have not been clearly revealed yet. Due to the complexity of the interrelationships between redox-active metal ions and other biological molecules (i.e., proteins, enzymes), multiple chemical agents have been examined as multifunctional molecules, reducing cytotoxicity associated with Cu(I/II), Fe(II/III), and Aβ. This strategy is promising; however, it is not the perfect way to treat the disease. Therefore, for treating AD, it is necessary to investigate and reveal the involvement of metalloenzymes, which can regulate the levels of Aβ in the brain such as ADE, in the pathogenesis of the disease, along with redox-active metal ions. In particular, Cu(I/II) and Fe(II/III) have potentials to interact with ADE and affect the enzymatic activity; however, their relationships still remain unclear. Current knowledge of the influence of Cu(I/II) and Fe(II/III) on the expression levels and activity of ADE is summarized in Table 4.
Through this review, we summarized (i) the roles, distributions, homeostasis, and transport of Cu(I/II) and Fe(II/III) in both healthy and AD-affected brains, (ii) chemicals designed for targeting metal ions, (iii) functions of multiple IDEs in the brain, and (iv) interrelationships between redox-active metal ions and ADE. Further understanding of the interrelationships between redox-active metal ions and ADE could lead to revealing the etiology of AD more clearly than before and developing successful treatments of the disease.
Author Contributions
N.K. and H.J.L. contributed to the literature search as well as writing and designing the manuscript. Both authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the research grant of the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (2020R1G1A1003993 (to H.J.L.); 2019R1F1A1059679 (to N.K.)).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Savelieff, M.G.; Nam, G.; Kang, J.; Lee, H.J.; Lee, M.; Lim, M.H. Development of Multifunctional Molecules as Potential Therapeutic Candidates for Alzheimer’s Disease, Parkinson’s Disease, and Amyotrophic Lateral Sclerosis in the Last Decade. Chem. Rev. 2019, 119, 1221–1322. [Google Scholar] [CrossRef] [PubMed]
- Gromadzka, G.; Tarnacka, B.; Flaga, A.; Adamczyk, A. Copper Dyshomeostasis in Neurodegenerative Diseases-Therapeutic Implications. Int. J. Mol. Sci. 2020, 21, 9259. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Bush, A.I. Biological metals and metal-targeting compounds in major neurodegenerative diseases. Chem. Soc. Rev. 2014, 43, 6727–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kepp, K.P. Bioinorganic chemistry of Alzheimer’s disease. Chem. Rev. 2012, 112, 5193–5239. [Google Scholar] [CrossRef] [Green Version]
- Bonda, D.J.; Lee, H.G.; Blair, J.A.; Zhu, X.; Perry, G.; Smith, M.A. Role of metal dyshomeostasis in Alzheimer’s disease. Metallomics 2011, 3, 267–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolognin, S.; Messori, L.; Zatta, P. Metal ion physiopathology in neurodegenerative disorders. Neuromol. Med. 2009, 11, 223–238. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E. NAD(+) in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477. [Google Scholar] [CrossRef]
- De Ricco, R.; Valensin, D.; Dell’Acqua, S.; Casella, L.; Hureau, C.; Faller, P. Copper(I/II), a/b-Synuclein and Amyloid-b: Menage a Trois? ChemBioChem 2015, 16, 2319–2328. [Google Scholar] [CrossRef] [PubMed]
- Faller, P.; Hureau, C.; Berthoumieu, O. Role of metal ions in the self-assembly of the Alzheimer’s amyloid-b peptide. Inorg. Chem. 2013, 52, 12193–12206. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper imbalance in Alzheimer’s disease and its link with the amyloid hypothesis: Towards a combined clinical, chemical, and genetic etiology. J. Alzheimers Dis. 2021. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Turner, A.J. Targeting amyloid clearance in Alzheimer’s disease as a therapeutic strategy. Br. J. Pharmacol. 2019, 176, 3447–3463. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Ab clearance and degradation by glial cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Nalivaeva, N.N.; Turner, A.J. Role of ageing and oxidative stress in regulation of amyloid-degrading enzymes and development of neurodegeneration. Curr. Aging Sci. 2017, 10, 32–40. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased clearance of CNS b-amyloid in Alzheimer’s disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Hemming, M.L.; Patterson, M.; Reske-Nielsen, C.; Lin, L.; Isacson, O.; Selkoe, D.J. Reducing amyloid plaque burden via ex vivo gene delivery of an Ab-degrading protease: A novel therapeutic approach to Alzheimer disease. PLoS Med. 2007, 4, e262. [Google Scholar] [CrossRef] [Green Version]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef] [PubMed]
- Tikhonova, T.V.; Sorokin, D.Y.; Hagen, W.R.; Khrenova, M.G.; Muyzer, G.; Rakitina, T.V.; Shabalin, I.G.; Trofimov, A.A.; Tsallagov, S.I.; Popov, V.O. Trinuclear copper biocatalytic center forms an active site of thiocyanate dehydrogenase. Proc. Natl. Acad. Sci. USA 2020, 117, 5280–5290. [Google Scholar] [CrossRef] [PubMed]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.T.; Anderson, C.M. Cardiac cytochrome C oxidase activity and contents of subunits 1 and 4 are altered in offspring by low prenatal copper intake by rat dams. J. Nutr. 2008, 138, 1269–1273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nelson, K.T.; Prohaska, J.R. Copper deficiency in rodents alters dopamine b-mono-oxygenase activity, mRNA and protein level. Br. J. Nutr. 2009, 102, 18–28. [Google Scholar] [CrossRef] [Green Version]
- Bisaglia, M.; Bubacco, L. Copper Ions and Parkinson’s Disease: Why Is Homeostasis So Relevant? Biomolecules 2020, 10, 195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jett, K.A.; Leary, S.C. Building the CuA site of cytochrome c oxidase: A complicated, redox-dependent process driven by a surprisingly large complement of accessory proteins. J. Biol. Chem. 2018, 293, 4644–4652. [Google Scholar] [CrossRef] [Green Version]
- Kozma, M.; Szerdahelyi, P.; Kasa, P. Histochemical detection of zinc and copper in various neurons of the central nervous system. Acta Histochem. 1981, 69, 12–17. [Google Scholar] [CrossRef]
- Stockel, J.; Safar, J.; Wallace, A.C.; Cohen, F.E.; Prusiner, S.B. Prion protein selectively binds copper(II) ions. Biochemistry 1998, 37, 7185–7193. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Heja, L.; Simon, A.; Jablonkai, I.; Kovacs, R.; Jemnitz, K. Copper signalling: Causes and consequences. Cell Commun. Signal. 2018, 16, 71. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.R.; Qin, K.; Herms, J.W.; Madlung, A.; Manson, J.; Strome, R.; Fraser, P.E.; Kruck, T.; von Bohlen, A.; Schulz-Schaeffer, W.; et al. The cellular prion protein binds copper in vivo. Nature 1997, 390, 684–687. [Google Scholar] [CrossRef]
- Bush, A.I. Metals and neuroscience. Curr. Opin. Chem. Biol. 2000, 4, 184–191. [Google Scholar] [CrossRef]
- Atwood, C.S.; Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I. Role of free radicals and metal ions in the pathogenesis of Alzheimer’s disease. Met. Ions Biol. Syst. 1999, 36, 309–364. [Google Scholar] [PubMed]
- Schlief, M.L.; Gitlin, J.D. Copper homeostasis in the CNS: A novel link between the NMDA receptor and copper homeostasis in the hippocampus. Mol. Neurobiol. 2006, 33, 81–90. [Google Scholar] [CrossRef]
- Kardos, J.; Kovacs, I.; Hajos, F.; Kalman, M.; Simonyi, M. Nerve endings from rat brain tissue release copper upon depolarization. A possible role in regulating neuronal excitability. Neurosci. Lett. 1989, 103, 139–144. [Google Scholar] [CrossRef]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevitt, T.; Ohrvik, H.; Thiele, D.J. Charting the travels of copper in eukaryotes from yeast to mammals. Biochim. Biophys. Acta 2012, 1823, 1580–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, Y.; Wang, L.; Li, D.; Zhao, C.; Li, J.; Liu, T. Exploring the extended biological functions of the human copper chaperone of superoxide dismutase 1. Protein J. 2019, 38, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Skopp, A.; Boyd, S.D.; Ullrich, M.S.; Liu, L.; Winkler, D.D. Copper-zinc superoxide dismutase (Sod1) activation terminates interaction between its copper chaperone (Ccs) and the cytosolic metal-binding domain of the copper importer Ctr1. Biometals 2019, 32, 695–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klomp, L.W.; Lin, S.J.; Yuan, D.S.; Klausner, R.D.; Culotta, V.C.; Gitlin, J.D. Identification and functional expression of HAH1, a novel human gene involved in copper homeostasis. J. Biol. Chem. 1997, 272, 9221–9226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, N.J.; Winge, D.R. Copper metallochaperones. Annu. Rev. Biochem. 2010, 79, 537–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolgova, N.V.; Yu, C.; Cvitkovic, J.P.; Hodak, M.; Nienaber, K.H.; Summers, K.L.; Cotelesage, J.J.H.; Bernholc, J.; Kaminski, G.A.; Pickering, I.J.; et al. Binding of copper and cisplatin to Atox1 is mediated by glutathione through the formation of metal-sulfur clusters. Biochemistry 2017, 56, 3129–3141. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Lutsenko, S. The Role of Copper Chaperone Atox1 in Coupling Redox Homeostasis to Intracellular Copper Distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatori, Y.; Inouye, S.; Akagi, R. Thiol-based copper handling by the copper chaperone Atox1. IUBMB Life 2017, 69, 246–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blockhuys, S.; Zhang, X.; Wittung-Stafshede, P. Single-cell tracking demonstrates copper chaperone Atox1 to be required for breast cancer cell migration. Proc. Natl. Acad. Sci. USA 2020, 117, 2014–2019. [Google Scholar] [CrossRef] [Green Version]
- Carr, H.S.; Maxfield, A.B.; Horng, Y.C.; Winge, D.R. Functional analysis of the domains in Cox11. J. Biol. Chem. 2005, 280, 22664–22669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carr, H.S.; George, G.N.; Winge, D.R. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J. Biol. Chem. 2002, 277, 31237–31242. [Google Scholar] [CrossRef] [Green Version]
- Radin, I.; Gey, U.; Kost, L.; Steinebrunner, I.; Rödel, G. The mitochondrial copper chaperone COX11 plays an auxiliary role in the defence against oxidative stress. BioRxiv 2018. [Google Scholar] [CrossRef] [Green Version]
- Maxfield, A.B.; Heaton, D.N.; Winge, D.R. Cox17 is functional when tethered to the mitochondrial inner membrane. J. Biol. Chem. 2004, 279, 5072–5080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inesi, G. Molecular features of copper binding proteins involved in copper homeostasis. IUBMB Life 2017, 69, 211–217. [Google Scholar] [CrossRef]
- Heaton, D.N.; George, G.N.; Garrison, G.; Winge, D.R. The mitochondrial copper metallochaperone Cox17 exists as an oligomeric, polycopper complex. Biochemistry 2001, 40, 743–751. [Google Scholar] [CrossRef]
- Glerum, D.M.; Shtanko, A.; Tzagoloff, A. SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J. Biol. Chem. 1996, 271, 20531–20535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rigby, K.; Zhang, L.; Cobine, P.A.; George, G.N.; Winge, D.R. characterization of the cytochrome c oxidase assembly factor Cox19 of Saccharomyces cerevisiae. J. Biol. Chem. 2007, 282, 10233–10242. [Google Scholar] [PubMed] [Green Version]
- Garcia, L.; Mansilla, N.; Ocampos, N.; Pagani, M.A.; Welchen, E.; Gonzalez, D.H. The mitochondrial copper chaperone COX19 influences copper and iron homeostasis in arabidopsis. Plant Mol. Biol. 2019, 99, 621–638. [Google Scholar] [CrossRef]
- Petruzzella, V.; Tiranti, V.; Fernandez, P.; Ianna, P.; Carrozzo, R.; Zeviani, M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics 1998, 54, 494–504. [Google Scholar] [CrossRef] [PubMed]
- Horng, Y.C.; Leary, S.C.; Cobine, P.A.; Young, F.B.; George, G.N.; Shoubridge, E.A.; Winge, D.R. Human Sco1 and Sco2 function as copper-binding proteins. J. Biol. Chem. 2005, 280, 34113–34122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ekim Kocabey, A.; Kost, L.; Gehlhar, M.; Rodel, G.; Gey, U. Mitochondrial Sco proteins are involved in oxidative stress defense. Redox Biol. 2019, 21, 101079. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Sue, C.; Banting, G.S.; Yang, H.; Glerum, D.M.; Hendrickson, W.A.; Schon, E.A. Crystal structure of human SCO1: Implications for redox signaling by a mitochondrial cytochrome c oxidase “assembly” protein. J. Biol. Chem. 2005, 280, 15202–15211. [Google Scholar] [CrossRef] [Green Version]
- Balatri, E.; Banci, L.; Bertini, I.; Cantini, F.; Ciofi-Baffoni, S. Solution structure of Sco1: A thioredoxin-like protein Involved in cytochrome c oxidase assembly. Structure 2003, 11, 1431–1443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leary, S.C.; Kaufman, B.A.; Pellecchia, G.; Guercin, G.H.; Mattman, A.; Jaksch, M.; Shoubridge, E.A. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum. Mol. Genet. 2004, 13, 1839–1848. [Google Scholar] [CrossRef] [Green Version]
- Leary, S.C.; Cobine, P.A.; Kaufman, B.A.; Guercin, G.H.; Mattman, A.; Palaty, J.; Lockitch, G.; Winge, D.R.; Rustin, P.; Horvath, R.; et al. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007, 5, 9–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baker, Z.N.; Cobine, P.A.; Leary, S.C. The mitochondrion: A central architect of copper homeostasis. Metallomics 2017, 9, 1501–1512. [Google Scholar] [CrossRef]
- Leary, S.C.; Sasarman, F.; Nishimura, T.; Shoubridge, E.A. Human SCO2 is required for the synthesis of CO II and as a thiol-disulphide oxidoreductase for SCO1. Hum. Mol. Genet. 2009, 18, 2230–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, W.; Monnot, A.D. Regulation of brain iron and copper homeostasis by brain barrier systems: Implication in neurodegenerative diseases. Pharmacol. Ther. 2012, 133, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheiber, I.F.; Mercer, J.F.; Dringen, R. Metabolism and functions of copper in brain. Prog. Neurobiol. 2014, 116, 33–57. [Google Scholar] [CrossRef] [PubMed]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simao, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassani, S.; Ghaffari, P.; Chahardouli, B.; Alimoghaddam, K.; Ghavamzadeh, A.; Alizadeh, S.; Ghaffari, S.H. Disulfiram/copper causes ROS levels alteration, cell cycle inhibition, and apoptosis in acute myeloid leukaemia cell lines with modulation in the expression of related genes. Biomed. Pharmacother. 2018, 99, 561–569. [Google Scholar] [CrossRef]
- Nishito, Y.; Kambe, T. Absorption mechanisms of iron, copper, and zinc: An overview. J. Nutr. Sci. Vitaminol. 2018, 64, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georgatsou, E.; Mavrogiannis, L.A.; Fragiadakis, G.S.; Alexandraki, D. The yeast Fre1p/Fre2p cupric reductases facilitate copper uptake and are regulated by the copper-modulated Mac1p activator. J. Biol. Chem. 1997, 272, 13786–13792. [Google Scholar] [CrossRef] [Green Version]
- Hassett, R.; Kosman, D.J. Evidence for Cu(II) reduction as a component of copper uptake by Saccharomyces cerevisiae. J. Biol. Chem. 1995, 270, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Jiang, Y.; Yang, Y.; Peng, Y.; Li, C. Copper metabolism in Saccharomyces cerevisiae: An update. Biometals 2021, 34, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Gitschier, J. hCTR1: A human gene for copper uptake identified by complementation in yeast. Proc. Natl. Acad. Sci. USA 1997, 94, 7481–7486. [Google Scholar] [CrossRef] [Green Version]
- Dancis, A.; Yuan, D.S.; Haile, D.; Askwith, C.; Eide, D.; Moehle, C.; Kaplan, J.; Klausner, R.D. Molecular characterization of a copper transport protein in S. cerevisiae: An unexpected role for copper in iron transport. Cell 1994, 76, 393–402. [Google Scholar] [CrossRef]
- Guo, Y.; Smith, K.; Lee, J.; Thiele, D.J.; Petris, M.J. Identification of methionine-rich clusters that regulate copper-stimulated endocytosis of the human Ctr1 copper transporter. J. Biol. Chem. 2004, 279, 17428–17433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefaniak, E.; Plonka, D.; Drew, S.C.; Bossak-Ahmad, K.; Haas, K.L.; Pushie, M.J.; Faller, P.; Wezynfeld, N.E.; Bal, W. The N-terminal 14-mer model peptide of human Ctr1 can collect Cu(ii) from albumin. Implications for copper uptake by Ctr1. Metallomics 2018, 10, 1723–1727. [Google Scholar] [CrossRef]
- Shenberger, Y.; Marciano, O.; Gottlieb, H.E.; Ruthstein, S. Insights into the N-terminal Cu(II) and Cu(I) binding sites of the human copper transporter CTR1. J. Coord. Chem. 2018, 71, 1985–2002. [Google Scholar] [CrossRef] [Green Version]
- Eisses, J.F.; Kaplan, J.H. Molecular characterization of hCTR1, the human copper uptake protein. J. Biol. Chem. 2002, 277, 29162–29171. [Google Scholar] [CrossRef] [Green Version]
- Klomp, A.E.; Juijn, J.A.; van der Gun, L.T.; van den Berg, I.E.; Berger, R.; Klomp, L.W. The N-terminus of the human copper transporter 1 (hCTR1) is localized extracellularly, and interacts with itself. Biochem. J. 2003, 370, 881–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, A.; Lutsenko, S. Human copper transporters: Mechanism, role in human diseases and therapeutic potential. Future Med. Chem. 2009, 1, 1125–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Pena, M.M.; Nose, Y.; Thiele, D.J. Biochemical characterization of the human copper transporter Ctr1. J. Biol. Chem. 2002, 277, 4380–4387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klomp, A.E.; Tops, B.B.; Van Denberg, I.E.; Berger, R.; Klomp, L.W. Biochemical characterization and subcellular localization of human copper transporter 1 (hCTR1). Biochem. J. 2002, 364, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Peng, J.; Zeng, F.; Zhang, K.; Liu, J.; Li, X.; Ouyang, Q.; Wang, G.; Wang, L.; Liu, Z.; et al. Association between polymorphisms in CTR1, CTR2, ATP7A, and ATP7B and platinum resistance in epithelial ovarian cancer. Int. J. Clin. Pharamacol. Ther. 2017, 55, 774–780. [Google Scholar] [CrossRef]
- van den Berghe, P.V.; Folmer, D.E.; Malingre, H.E.; van Beurden, E.; Klomp, A.E.; van de Sluis, B.; Merkx, M.; Berger, R.; Klomp, L.W. Human copper transporter 2 is localized in late endosomes and lysosomes and facilitates cellular copper uptake. Biochem. J. 2007, 407, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Bertinato, J.; Swist, E.; Plouffe, L.J.; Brooks, S.P.; L’Abbe, M.R. Ctr2 is partially localized to the plasma membrane and stimulates copper uptake in COS-7 cells. Biochem. J. 2008, 409, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Rees, E.M.; Thiele, D.J. Identification of a vacuole-associated metalloreductase and its role in Ctr2-mediated intracellular copper mobilization. J. Biol. Chem. 2007, 282, 21629–21638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Portnoy, M.E.; Schmidt, P.J.; Rogers, R.S.; Culotta, V.C. Metal transporters that contribute copper to metallochaperones in Saccharomyces cerevisiae. Mol. Genet. Genom. Med. 2001, 265, 873–882. [Google Scholar] [CrossRef]
- Bellemare, D.R.; Shaner, L.; Morano, K.A.; Beaudoin, J.; Langlois, R.; Labbe, S. Ctr6, a vacuolar membrane copper transporter in Schizosaccharomyces pombe. J. Biol. Chem. 2002, 277, 46676–46686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plante, S.; Normant, V.; Ramos-Torres, K.M.; Labbe, S. Cell-surface copper transporters and superoxide dismutase 1 are essential for outgrowth during fungal spore germination. J. Biol. Chem. 2017, 292, 11896–11914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strausak, D.; La Fontaine, S.; Hill, J.; Firth, S.D.; Lockhart, P.J.; Mercer, J.F. The role of GMXCXXC metal binding sites in the copper-induced redistribution of the Menkes protein. J. Biol. Chem. 1999, 274, 11170–11177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephenson, S.E.; Dubach, D.; Lim, C.M.; Mercer, J.F.; La Fontaine, S. A single PDZ domain protein interacts with the Menkes copper ATPase, ATP7A. A new protein implicated in copper homeostasis. J. Biol. Chem. 2005, 280, 33270–33279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, M.J.; Jones, E.E.; Levy, E.R.; Martin, R.L.; Ponnambalam, S.; Monaco, A.P. Identification of a di-leucine motif within the C terminus domain of the Menkes disease protein that mediates endocytosis from the plasma membrane. J. Cell Sci. 1999, 112, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.H.; Dolgova, N.V.; Dmitriev, O.Y. Dynamics of the metal binding domains and regulation of the human copper transporters ATP7B and ATP7A. IUBMB life 2017, 69, 226–235. [Google Scholar] [CrossRef]
- Walker, J.M.; Huster, D.; Ralle, M.; Morgan, C.T.; Blackburn, N.J.; Lutsenko, S. The N-terminal metal-binding site 2 of the Wilson’s Disease Protein plays a key role in the transfer of copper from Atox1. J. Biol. Chem. 2004, 279, 15376–15384. [Google Scholar] [CrossRef] [Green Version]
- Achila, D.; Banci, L.; Bertini, I.; Bunce, J.; Ciofi-Baffoni, S.; Huffman, D.L. Structure of human Wilson protein domains 5 and 6 and their interplay with domain 4 and the copper chaperone HAH1 in copper uptake. Proc. Natl. Acad. Sci. USA 2006, 103, 5729–5734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huster, D.; Lutsenko, S. The distinct roles of the N-terminal copper-binding sites in regulation of catalytic activity of the Wilson’s disease protein. J. Biol. Chem. 2003, 278, 32212–32218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, N.; Tsivkovskii, R.; Tsivkovskaia, N.; Lutsenko, S. The copper-transporting ATPases, menkes and wilson disease proteins, have distinct roles in adult and developing cerebellum. J. Biol. Chem. 2005, 280, 9640–9645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogorek, M.; Lenartowicz, M.; Starzynski, R.; Jonczy, A.; Staron, R.; Doniec, A.; Krzeptowski, W.; Bednarz, A.; Pierzchala, O.; Lipinski, P.; et al. Atp7a and Atp7b regulate copper homeostasis in developing male germ cells in mice. Metallomics 2017, 9, 1288–1303. [Google Scholar] [CrossRef] [PubMed]
- Nyasae, L.; Bustos, R.; Braiterman, L.; Eipper, B.; Hubbard, A. Dynamics of endogenous ATP7A (Menkes protein) in intestinal epithelial cells: Copper-dependent redistribution between two intracellular sites. Am. J. Physiol. Gastrointest Liver Physiol. 2007, 292, G1181–G1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pase, L.; Voskoboinik, I.; Greenough, M.; Camakaris, J. Copper stimulates trafficking of a distinct pool of the Menkes copper ATPase (ATP7A) to the plasma membrane and diverts it into a rapid recycling pool. Biochem. J. 2004, 378, 1031–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadini-Buoninsegni, F.; Smeazzetto, S. Mechanisms of charge transfer in human copper ATPases ATP7A and ATP7B. IUBMB Life 2017, 69, 218–225. [Google Scholar] [CrossRef] [Green Version]
- Hellman, N.E.; Kono, S.; Mancini, G.M.; Hoogeboom, A.J.; De Jong, G.J.; Gitlin, J.D. Mechanisms of copper incorporation into human ceruloplasmin. J. Biol. Chem. 2002, 277, 46632–46638. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [CrossRef] [Green Version]
- Adeoye, O.; Olawumi, J.; Opeyemi, A.; Christiania, O. Review on the role of glutathione on oxidative stress and infertility. JBRA Assist. Reprod. 2018, 22, 61–66. [Google Scholar] [CrossRef]
- Pastore, A.; Piemonte, F.; Locatelli, M.; Lo Russo, A.; Gaeta, L.M.; Tozzi, G.; Federici, G. Determination of blood total, reduced, and oxidized glutathione in pediatric subjects. Clin. Chem. 2001, 47, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- Halprin, K.M.; Ohkawara, A. The measurement of glutathione in human epidermis using glutathione reductase. J. Invest. Dermatol. 1967, 48, 149–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefaniak, E.; Plonka, D.; Szczerba, P.; Wezynfeld, N.E.; Bal, W. Copper Transporters? Glutathione Reactivity of Products of Cu-Abeta Digestion by Neprilysin. Inorg. Chem. 2020, 59, 4186–4190. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol. Cell Physiol. 2013, 304, C768–C779. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharjee, A.; Chakraborty, K.; Shukla, A. Cellular copper homeostasis: Current concepts on its interplay with glutathione homeostasis and its implication in physiology and human diseases. Metallomics 2017, 9, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.M.; Cater, M.A.; Mercer, J.F.; La Fontaine, S. Copper-dependent interaction of glutaredoxin with the N termini of the copper-ATPases (ATP7A and ATP7B) defective in Menkes and Wilson diseases. Biochem. Biophys. Res. Commun. 2006, 348, 428–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singleton, W.C.J.; McInnes, K.T.; Cater, M.A.; Winnall, W.R.; McKirdy, R.; Yu, Y.; Taylor, P.E.; Ke, B.X.; Richardson, D.R.; Mercer, J.F.B.; et al. Role of glutaredoxin1 and glutathione in regulating the activity of the copper-transporting P-type ATPases, ATP7A and ATP7B. J. Biol. Chem. 2010, 285, 27111–27121. [Google Scholar] [CrossRef] [Green Version]
- Ziller, A.; Fraissinet-Tachet, L. Metallothionein diversity and distribution in the tree of life: A multifunctional protein. Metallomics 2018, 10, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Calvo, J.; Jung, H.; Meloni, G. Copper metallothioneins. IUBMB Life 2017, 69, 236–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.Z.; Lin, Y.J.; Zhang, Q. Metallothioneins enhance chromium detoxification through scavenging ROS and stimulating metal chelation in Oryza sativa. Chemosphere 2019, 220, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tan, S.; Wang, Y.; Luo, J.; Long, Y.; Mei, X.; Tang, Y. Role of metallothionein-1 and metallothionein-2 in the neuroprotective mechanism of sevoflurane preconditioning in mice. J. Mol. Neurosci. 2020, 70, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Thornalley, P.J.; Vasak, M. Possible role for metallothionein in protection against radiation-induced oxidative stress. Kinetics and mechanism of its reaction with superoxide and hydroxyl radicals. Biochim. Biophys. Acta 1985, 827, 36–44. [Google Scholar] [CrossRef]
- Binz, P.-A.; Kägi, J.H.R. Metallothionein: Molecular evolution and classification. In Metallothionein IV; Klaassen, C.D., Ed.; Birkhauser: Basel, Switzerland, 1999; pp. 7–13. [Google Scholar]
- Sakatoku, A.; Ishikawa, M.; Yamazaki, K.; Nakamachi, T.; Kamachi, H.; Tanaka, D.; Nakamura, S. Molecular identification, characterization, and expression analysis of a metallothionein gene from septifer virgatus. Mar. Biothenol. 2020, 22, 488–497. [Google Scholar] [CrossRef]
- Cherian, M.G.; Jayasurya, A.; Bay, B.H. Metallothioneins in human tumors and potential roles in carcinogenesis. Mutat. Res. 2003, 533, 201–209. [Google Scholar] [CrossRef]
- Rahman, M.T.; Haque, N.; Abu Kasim, N.H.; De Ley, M. Origin, function, and fate of metallothionein in human blood. Rev. Physiol. Biochem. Pharmacol. 2017, 173, 41–62. [Google Scholar]
- Kim, B.E.; Nevitt, T.; Thiele, D.J. Mechanisms for copper acquisition, distribution and regulation. Nat. Chem. Biol. 2008, 4, 176–185. [Google Scholar] [CrossRef]
- Alwan, K.B.; Welch, E.F.; Blackburn, N.J. Catalytic M center of copper monooxygenases probed by rational design. effects of selenomethionine and histidine substitution on structure and reactivity. Biochemistry 2019, 58, 4436–4446. [Google Scholar] [CrossRef] [PubMed]
- Hartter, D.E.; Barnea, A. Evidence for release of copper in the brain: Depolarization-induced release of newly taken-up 67copper. Synapse 1988, 2, 412–415. [Google Scholar] [CrossRef] [PubMed]
- Schlief, M.L.; Craig, A.M.; Gitlin, J.D. NMDA receptor activation mediates copper homeostasis in hippocampal neurons. J. Neurosci. 2005, 25, 239–246. [Google Scholar] [CrossRef] [Green Version]
- Alies, B.; Renaglia, E.; Rozga, M.; Bal, W.; Faller, P.; Hureau, C. Cu(II) affinity for the Alzheimer’s peptide: Tyrosine fluorescence studies revisited. Anal. Chem. 2013, 85, 1501–1508. [Google Scholar] [CrossRef]
- Weibull, M.G.M.; Simonsen, S.; Oksbjerg, C.R.; Tiwari, M.K.; Hemmingsen, L. Effects of Cu(II) on the aggregation of amyloid-b. J. Biol. Inorg. Chem. 2019, 24, 1197–1215. [Google Scholar] [CrossRef]
- Drew, S.C.; Noble, C.J.; Masters, C.L.; Hanson, G.R.; Barnham, K.J. Pleomorphic copper coordination by Alzheimer’s disease amyloid-beta peptide. J. Am. Chem. Soc. 2009, 131, 1195–1207. [Google Scholar] [CrossRef]
- Summers, K.L.; Schilling, K.M.; Roseman, G.; Markham, K.A.; Dolgova, N.V.; Kroll, T.; Sokaras, D.; Millhauser, G.L.; Pickering, I.J.; George, G.N. X-ray Absorption spectroscopy investigations of copper(II) coordination in the human amyloid b peptide. Inorg. Chem. 2019, 58, 6294–6311. [Google Scholar] [CrossRef] [PubMed]
- Duce, J.A.; Bush, A.I. Biological metals and Alzheimer’s disease: Implications for therapeutics and diagnostics. Prog. Neurobiol. 2010, 92, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Branch, T.; Barahona, M.; Dodson, C.A.; Ying, L. Kinetic Analysis Reveals the Identity of Abeta-Metal Complex Responsible for the Initial Aggregation of Abeta in the Synapse. ACS Chem. Neurosci. 2017, 8, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- da Silva, G.F.; Ming, L.J. Metallo-ROS in Alzheimer’s disease: Oxidation of neurotransmitters by CuII-beta-amyloid and neuropathology of the disease. Angew. Chem. Int. Ed. 2007, 46, 3337–3341. [Google Scholar] [CrossRef]
- Nam, E.; Derrick, J.S.; Lee, S.; Kang, J.; Han, J.; Lee, S.J.C.; Chung, S.W.; Lim, M.H. Regulatory Activities of Dopamine and Its Derivatives toward Metal-Free and Metal-Induced Amyloid-beta Aggregation, Oxidative Stress, and Inflammation in Alzheimer’s Disease. ACS Chem. Neurosci. 2018, 9, 2655–2666. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Wezynfeld, N.E.; Stefaniak, E.; Pomorski, A.; Plonka, D.; Krezel, A.; Bal, W.; Faller, P. Cu transfer from amyloid-beta4-16 to metallothionein-3: The role of the neurotransmitter glutamate and metallothionein-3 Zn(ii)-load states. Chem. Commun. 2018, 54, 12634–12637. [Google Scholar] [CrossRef]
- Margerum, D.W.; Dukes, G.R. Metal Ions in Biological Systems. In Metal Ions in Biological Systems; Sigel, H., Ed.; Marcel Dekker: New York, NY, USA, 1974; Volume 1, pp. 157–212. [Google Scholar]
- Ma, Q.F.; Li, Y.M.; Du, J.T.; Kanazawa, K.; Nemoto, T.; Nakanishi, H.; Zhao, Y.F. Binding of copper (II) ion to an Alzheimer’s tau peptide as revealed by MALDI-TOF MS, CD, and NMR. Biopolymers 2005, 79, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lukacs, M.; Szunyog, G.; Grenacs, A.; Lihi, N.; Kallay, C.; Di Natale, G.; Campagna, T.; Lanza, V.; Tabbi, G.; Pappalardo, G.; et al. Copper(II) Coordination Abilities of the Tau Protein’s N-Terminus Peptide Fragments: A Combined Potentiometric, Spectroscopic and Mass Spectrometric Study. ChemPlusChem 2019, 84, 1697–1708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacchella, C.; Gentili, S.; Bellotti, D.; Quartieri, E.; Draghi, S.; Baratto, M.C.; Remelli, M.; Valensin, D.; Monzani, E.; Nicolis, S.; et al. Binding and Reactivity of Copper to R1 and R3 Fragments of tau Protein. Inorg. Chem. 2020, 59, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Di Vaira, M.; Bazzicalupi, C.; Orioli, P.; Messori, L.; Bruni, B.; Zatta, P. Clioquinol, a drug for Alzheimer’s disease specifically interfering with brain metal metabolism: Structural characterization of its zinc(II) and copper(II) complexes. Inorg. Chem. 2004, 43, 3795–3797. [Google Scholar] [CrossRef]
- Chen, D.; Cui, Q.C.; Yang, H.; Barrea, R.A.; Sarkar, F.H.; Sheng, S.; Yan, B.; Reddy, G.P.; Dou, Q.P. Clioquinol, a therapeutic agent for Alzheimer’s disease, has proteasome-inhibitory, androgen receptor-suppressing, apoptosis-inducing, and antitumor activities in human prostate cancer cells and xenografts. Cancer Res. 2007, 67, 1636–1644. [Google Scholar] [CrossRef] [Green Version]
- Tahmasebinia, F.; Emadi, S. Effect of metal chelators on the aggregation of b-amyloid peptides in the presence of copper and iron. Biometals 2017, 30, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Pushie, M.J.; Nienaber, K.H.; Summers, K.L.; Cotelesage, J.J.; Ponomarenko, O.; Nichol, H.K.; Pickering, I.J.; George, G.N. The solution structure of the copper clioquinol complex. J. Inorg. Biochem. 2014, 133, 50–56. [Google Scholar] [CrossRef]
- Opazo, C.; Luza, S.; Villemagne, V.L.; Volitakis, I.; Rowe, C.; Barnham, K.J.; Strozyk, D.; Masters, C.L.; Cherny, R.A.; Bush, A.I. Radioiodinated clioquinol as a biomarker for b-amyloid: Zn complexes in Alzheimer’s disease. Aging Cell 2006, 5, 69–79. [Google Scholar] [CrossRef]
- Pretsch, D.; Rollinger, J.M.; Schmid, A.; Genov, M.; Wohrer, T.; Krenn, L.; Moloney, M.; Kasture, A.; Hummel, T.; Pretsch, A. Prolongation of metallothionein induction combats ass and a-synuclein toxicity in aged transgenic Caenorhabditis elegans. Sci. Rep. 2020, 10, 11707. [Google Scholar] [CrossRef]
- Barnham, K.J.; Ciccotosto, G.D.; Tickler, A.K.; Ali, F.E.; Smith, D.G.; Williamson, N.A.; Lam, Y.H.; Carrington, D.; Tew, D.; Kocak, G.; et al. Neurotoxic, redox-competent Alzheimer’s b-amyloid is released from lipid membrane by methionine oxidation. J. Biol. Chem. 2003, 278, 42959–42965. [Google Scholar] [CrossRef] [Green Version]
- Cherny, R.A.; Ayton, S.; Finkelstein, D.I.; Bush, A.I.; McColl, G.; Massa, S.M. PBT2 Reduces Toxicity in a C. elegans model of polyQ aggregation and extends lifespan, reduces striatal atrophy and improves motor Performance in the R6/2 mouse model of Huntington’s disease. J. Huntingt. Dis. 2012, 1, 211–219. [Google Scholar] [CrossRef] [Green Version]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Ab. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [Green Version]
- Bush, A.I.; Tanzi, R.E. Therapeutics for Alzheimer’s disease based on the metal hypothesis. Neurotherapeutics 2008, 5, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, M.; Vendier, L.; Stigliani, J.L.; Meunier, B.; Robert, A. Structures of the copper and zinc complexes of PBT2, a chelating agent evaluated as potential drug for neurodegenerative diseases. Eur. J. Inorg. Chem. 2017, 2017, 600–608. [Google Scholar] [CrossRef]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Ab as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet. Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimers Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villemagne, V.L.; Rowe, C.C.; Barnham, K.J.; Cherny, R.; Woodward, M.; Bozinosvski, S.; Salvado, O.; Bourgeat, P.; Perez, K.; Fowler, C.; et al. A randomized, exploratory molecular imaging study targeting amyloid beta with a novel 8-OH quinoline in Alzheimer’s disease: The PBT2-204 IMAGINE study. Alzheimers Dement. 2017, 3, 622–635. [Google Scholar] [CrossRef]
- Kozak, A.; Shapiro, I. Lipophilic Diesters of Chelating Agents. U.S. Patent WO 99/16741, 1 July 1999. [Google Scholar]
- Lee, J.Y.; Friedman, J.E.; Angel, I.; Kozak, A.; Koh, J.Y. The lipophilic metal chelator DP-109 reduces amyloid pathology in brains of human beta-amyloid precursor protein transgenic mice. Neurobiol. Aging 2004, 25, 1315–1321. [Google Scholar] [CrossRef]
- Petri, S.; Calingasan, N.Y.; Alsaied, O.A.; Wille, E.; Kiaei, M.; Friedman, J.E.; Baranova, O.; Chavez, J.C.; Beal, M.F. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J. Neurochem. 2007, 102, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Raz, L.; Yang, Y.; Thompson, J.; Hobson, S.; Pesko, J.; Mobashery, S.; Chang, M.; Rosenberg, G. MMP-9 inhibitors impair learning in spontaneously hypertensive rats. PLoS ONE 2018, 13, e0208357. [Google Scholar] [CrossRef]
- Chen, D.; Darabedian, N.; Li, Z.; Kai, T.; Jiang, D.; Zhou, F. An improved Bathocuproine assay for accurate valence identification and quantification of copper bound by biomolecules. Anal. Biochem. 2016, 497, 27–35. [Google Scholar] [CrossRef]
- Haas, K.L.; Franz, K.J. Application of metal coordination chemistry to explore and manipulate cell biology. Chem. Rev. 2009, 109, 4921–4960. [Google Scholar] [CrossRef] [Green Version]
- La Mendola, D. Nerve growth factor catches copper in neuronal inning. Neural Regen. Res. 2020, 15, 665–666. [Google Scholar] [CrossRef]
- Hwang, S.; Kim, J.K. Fluoxetine Induces Apoptotic and Oxidative Neuronal Death Associated with The Influx of Copper Ions in Cultured Neuronal Cells. Chonnam Med. J. 2020, 56, 20–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walshe, J.M. Penicillamine, a new oral therapy for Wilson’s disease. Am. J. Med. 1956, 21, 487–495. [Google Scholar] [CrossRef]
- Walshe, J.M. Copper chelation in patients with Wilson’s disease. A comparison of penicillamine and triethylene tetramine dihydrochloride. Q. J. Med. 1973, 42, 441–452. [Google Scholar] [PubMed]
- Brewer, G.J. Zinc and tetrathiomolybdate for the treatment of Wilson’s disease and the potential efficacy of anticopper therapy in a wide variety of diseases. Metallomics 2009, 1, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, C.; Bronstein, J.M. Wilson Disease: An Overview and Approach to Management. Neurol. Clin. 2020, 38, 417–432. [Google Scholar] [CrossRef] [PubMed]
- Zhong, M.; Kou, H.; Zhao, P.; Zheng, W.; Xu, H.; Zhang, X.; Lan, W.; Guo, C.; Wang, T.; Guo, F.; et al. Nasal delivery of D-penicillamine hydrogel upregulates a disintegrin and metalloprotease 10 expression via melatonin receptor 1 in Alzheimer’s disease models. Front. Aging Neurosci. 2021, 13, 660249. [Google Scholar] [CrossRef]
- Cooper, G.J. Therapeutic potential of copper chelation with triethylenetetramine in managing diabetes mellitus and Alzheimer’s disease. Drugs 2011, 71, 128–1320. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Castoldi, F.; Zischka, H.; Kroemer, G. Extending the mode of action of triethylenetetramine (trientine): Autophagy besides copper chelation. J. Hepatol. 2020, 73, 970–972. [Google Scholar] [CrossRef]
- Brewer, G.J.; Dick, R.D.; Yuzbasiyan-Gurkin, V.; Tankanow, R.; Young, A.B.; Kluin, K.J. Initial therapy of patients with Wilson’s disease with tetrathiomolybdate. Arch. Neurol. 1991, 48, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.H.; Guo, C.; Gao, H.L.; Zhong, M.L.; Huang, T.T.; Liu, N.N.; Guo, R.F.; Lan, T.; Zhang, W.; et al. Tetrathiomolybdate Treatment Leads to the Suppression of Inflammatory Responses through the TRAF6/NFkappaB Pathway in LPS-Stimulated BV-2 Microglia. Front. Againg Neurosci. 2018, 10, 9. [Google Scholar] [CrossRef] [Green Version]
- Brewer, G.J.; Askari, F.; Lorincz, M.T.; Carlson, M.; Schilsky, M.; Kluin, K.J.; Hedera, P.; Moretti, P.; Fink, J.K.; Tankanow, R.; et al. Treatment of Wilson disease with ammonium tetrathiomolybdate: IV. Comparison of tetrathiomolybdate and trientine in a double-blind study of treatment of the neurologic presentation of Wilson disease. Arch. Neurol. 2006, 63, 521–527. [Google Scholar] [CrossRef]
- Dickens, M.G.; Franz, K.J. A prochelator activated by hydrogen peroxide prevents metal-induced amyloid Beta aggregation. Chembiochem Eur. J. Chem. Biol. 2010, 11, 59–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avramovich-Tirosh, Y.; Amit, T.; Bar-Am, O.; Zheng, H.; Fridkin, M.; Youdim, M.B. Therapeutic targets and potential of the novel brain- permeable multifunctional iron chelator-monoamine oxidase inhibitor drug, M-30, for the treatment of Alzheimer’s disease. J. Neurochem. 2007, 100, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Mechlovich, D.; Amit, T.; Mandel, S.A.; Bar-Am, O.; Bloch, K.; Vardi, P.; Youdim, M.B. The novel multifunctional, iron-chelating drugs M30 and HLA20 protect pancreatic b-cell lines from oxidative stress damage. J. Pharmacol. Exp. Ther. 2010, 333, 874–882. [Google Scholar] [CrossRef]
- Zheng, H.; Youdim, M.B.; Fridkin, M. Site-activated chelators targeting acetylcholinesterase and monoamine oxidase for Alzheimer’s therapy. ACS Chem. Biol. 2010, 5, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Youdim, M.B.; Fridkin, M. Site-activated multifunctional chelator with acetylcholinesterase and neuroprotective-neurorestorative moieties for Alzheimer’s therapy. J. Med. Chem. 2009, 52, 4095–4098. [Google Scholar] [CrossRef] [PubMed]
- Gomes, L.M.; Vieira, R.P.; Jones, M.R.; Wang, M.C.; Dyrager, C.; Souza-Fagundes, E.M.; Da Silva, J.G.; Storr, T.; Beraldo, H. 8-Hydroxyquinoline Schiff-base compounds as antioxidants and modulators of copper-mediated Ab peptide aggregation. J. Inorg. Biochem. 2014, 139, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Hu, J.; Yang, X.; Feng, X.; Li, X.; Huang, L.; Chan, A.S.C. Design, synthesis, and evaluation of orally bioavailable quinoline-indole derivatives as innovative multitarget-directed ligands: Promotion of cell proliferation in the adult murine hippocampus for the treatment of Alzheimer’s disease. J. Med. Chem. 2018, 61, 1871–1894. [Google Scholar] [CrossRef] [PubMed]
- Oliveri, V.; Puglisi, A.; Viale, M.; Aiello, C.; Sgarlata, C.; Vecchio, G.; Clarke, J.; Milton, J.; Spencer, J. New cyclodextrin-bearing 8-hydroxyquinoline ligands as multifunctional molecules. Chem. Eur. J. 2013, 19, 13946–13955. [Google Scholar] [CrossRef] [PubMed]
- Groenning, M. Binding mode of Thioflavin T and other molecular probes in the context of amyloid fibrils-current status. J. Chem. Biol. 2010, 3, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedeoglu, A.; Cormier, K.; Payton, S.; Tseitlin, K.A.; Kremsky, J.N.; Lai, L.; Li, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; et al. Preliminary studies of a novel bifunctional metal chelator targeting Alzheimer’s amyloidogenesis. Exp. Gerontol. 2004, 39, 1641–1649. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, C.; Sanchez de Groot, N.; Rimola, A.; Alvarez-Larena, A.; Lloveras, V.; Vidal-Gancedo, J.; Ventura, S.; Vendrell, J.; Sodupe, M.; Gonzalez-Duarte, P. Design, selection, and characterization of thioflavin-based intercalation compounds with metal chelating properties for application in Alzheimer’s disease. J. Am. Chem. Soc. 2009, 131, 1436–1451. [Google Scholar] [CrossRef]
- Rodriguez-Rodriguez, C.; Telpoukhovskaia, M.A.; Ali-Torres, J.; Rodriguez-Santiago, L.; Manso, Y.; Bailey, G.A.; Hidalgo, J.; Sodupe, M.; Orvig, C. Thioflavin-based molecular probes for application in Alzheimer’s disease: From in silico to in vitro models. Metallomics 2015, 7, 83–92. [Google Scholar] [CrossRef]
- Barabash, R.D.; Skobelkin, O.K.; Petukhov, M.I.; Normanskii, V.E.; Riazhskii, G.G.; Mironova, M.I.; Andreeva, K.P.; Kolobanov, A.S. The pharmacokinetics of hematoporphyrin, its derivative and fluorescein during the development of carcinosarcoma. Farmakol. I Toksikol. 1990, 53, 24–26. [Google Scholar]
- Sharma, A.K.; Pavlova, S.T.; Kim, J.; Finkelstein, D.; Hawco, N.J.; Rath, N.P.; Kim, J.; Mirica, L.M. Bifunctional compounds for controlling metal-mediated aggregation of the Ab42 peptide. J. Am.Chem.Soc. 2012, 134, 6625–6636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, J.; Li, M.; Wu, L.; Ren, J.; Qu, X. Liberation of copper from amyloid plaques: Making a risk factor useful for Alzheimer’s disease treatment. J. Med. Chem. 2012, 55, 9146–9155. [Google Scholar] [CrossRef]
- Jones, M.R.; Mu, C.; Wang, M.C.; Webb, M.I.; Walsby, C.J.; Storr, T. Modulation of the Ab peptide aggregation pathway by KP1019 limits Ab-associated neurotoxicity. Metallomics 2015, 7, 129–135. [Google Scholar] [CrossRef]
- Hindo, S.S.; Mancino, A.M.; Braymer, J.J.; Liu, Y.; Vivekanandan, S.; Ramamoorthy, A.; Lim, M.H. Small molecule modulators of copper-induced Ab aggregation. J. Am. Chem. Soc. 2009, 131, 16663–16665. [Google Scholar] [CrossRef] [Green Version]
- Choi, J.S.; Braymer, J.J.; Nanga, R.P.; Ramamoorthy, A.; Lim, M.H. Design of small molecules that target metal-Ab species and regulate metal-induced Ab aggregation and neurotoxicity. Proc. Natl. Acad. Sci. USA 2010, 107, 21990–21995. [Google Scholar] [CrossRef] [Green Version]
- Geldenhuys, W.J.; Ko, K.S.; Stinnett, H.; Schyf, C.J.V.d.; Lim, M.H. Identification of multifunctional small molecule-based reversible monoamine oxidase inhibitors. MedChemComm 2011, 2, 1099–1103. [Google Scholar] [CrossRef]
- Savelieff, M.G.; Liu, Y.; Senthamarai, R.R.; Korshavn, K.J.; Lee, H.J.; Ramamoorthy, A.; Lim, M.H. A small molecule that displays marked reactivity toward copper- versus zinc-amyloid-b implicated in Alzheimer’s disease. Chem. Commun. 2014, 50, 5301–5303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Zheng, X.; Krishnamoorthy, J.; Savelieff, M.G.; Park, H.M.; Brender, J.R.; Kim, J.H.; Derrick, J.S.; Kochi, A.; Lee, H.J.; et al. Rational design of a structural framework with potential use to develop chemical reagents that target and modulate multiple facets of Alzheimer’s disease. J. Am. Chem. Soc. 2014, 136, 299–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beck, M.W.; Derrick, J.S.; Kerr, R.A.; Oh, S.B.; Cho, W.J.; Lee, S.J.; Ji, Y.; Han, J.; Tehrani, Z.A.; Suh, N.; et al. Structure-mechanism-based engineering of chemical regulators targeting distinct pathological factors in Alzheimer’s disease. Nat. Commun. 2016, 7, 13115. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.W.; Derrick, J.S.; Suh, J.M.; Kim, M.; Korshavn, K.J.; Kerr, R.A.; Cho, W.J.; Larsen, S.D.; Ruotolo, B.T.; Ramamoorthy, A.; et al. Minor Structural Variations of Small Molecules Tune Regulatory Activities toward Pathological Factors in Alzheimer’s Disease. ChemMedChem 2017, 12, 1828–1838. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.J.; Korshavn, K.J.; Nam, Y.; Kang, J.; Paul, T.J.; Kerr, R.A.; Youn, I.S.; Ozbil, M.; Kim, K.S.; Ruotolo, B.T.; et al. Structural and mechanistic insights into development of chemical tools to control individual and inter-related pathological features in Alzheimer’s disease. Chem. Eur. J. 2017, 23, 2706–2715. [Google Scholar] [CrossRef] [Green Version]
- Guan, L.; Hao, Y.; Chen, L.; Wei, M.-L.; Jiang, Q.; Liu, W.-Y.; Zhang, Y.-B.; Zhang, J.; Feng, F.; Qu, W. Synthesis and evaluation of neuroprotective 4-O-substituted chrysotoxine derivatives as potential multifunctional agents for the treatment of Alzheimer’s disease. RSC Adv. 2016, 6, 22827–22838. [Google Scholar] [CrossRef]
- Jiang, N.; Wang, X.-B.; Li, Z.-R.; Li, S.-Y.; Xie, S.-S.; Huanga, M.; Kong, L.-Y. Design of a structural framework with potential use to develop balanced multifunctional agents against Alzheimer’s disease. RSC Adv. 2015, 5, 14242–14255. [Google Scholar] [CrossRef]
- Jones, M.R.; Dyrager, C.; Hoarau, M.; Korshavn, K.J.; Lim, M.H.; Ramamoorthy, A.; Storr, T. Multifunctional quinoline-triazole derivatives as potential modulators of amyloid-b peptide aggregation. J. Inorg. Biochem. 2016, 158, 131–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.R.; Mathieu, E.; Dyrager, C.; Faissner, S.; Vaillancourt, Z.; Korshavn, K.J.; Lim, M.H.; Ramamoorthy, A.; Wee Yong, V.; Tsutsui, S.; et al. Multi-target-directed phenol-triazole ligands as therapeutic agents for Alzheimer’s disease. Chem. Sci. 2017, 8, 5636–5643. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Gomes, L.M.F.; Zhang, T.; Storr, T. A small bifunctional chelator that modulates Ab42 aggregation. Can. J. Chem. 2017, 96, 78–82. [Google Scholar] [CrossRef] [Green Version]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef]
- Li, Y.; Qiang, X.; Luo, L.; Li, Y.; Xiao, G.; Tan, Z.; Deng, Y. Synthesis and evaluation of 4-hydroxyl aurone derivatives as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2016, 24, 2342–2351. [Google Scholar] [CrossRef]
- Li, F.; Wu, J.J.; Wang, J.; Yang, X.L.; Cai, P.; Liu, Q.H.; Kong, L.Y.; Wang, X.B. Synthesis and pharmacological evaluation of novel chromone derivatives as balanced multifunctional agents against Alzheimer’s disease. Bioorg. Med. Chem. 2017, 25, 3815–3826. [Google Scholar] [CrossRef]
- Li, S.Y.; Wang, X.B.; Kong, L.Y. Design, synthesis and biological evaluation of imine resveratrol derivatives as multi-targeted agents against Alzheimer’s disease. Eur. J. Med. Chem. 2014, 71, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Zhang, M.; Sheng, R.; Ma, Y. Synthesis and biological evaluation of deferiprone-resveratrol hybrids as antioxidants, Ab1-42 aggregation inhibitors and metal-chelating agents for Alzheimer’s disease. Eur. J. Med. Chem. 2017, 127, 174–186. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimers Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Mason, H.S. Mechanisms of oxygen metabolism. Science 1957, 125, 1185–1188. [Google Scholar] [CrossRef] [PubMed]
- Lieu, P.T.; Heiskala, M.; Peterson, P.A.; Yang, Y. The roles of iron in health and disease. Mol. Asp. Med. 2001, 22, 1–87. [Google Scholar] [CrossRef]
- Nunez, M.T.; Chana-Cuevas, P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 2018, 11, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiban, J.; So, M.; Kaguni, L.S. Iron-sulfur clusters in mitochondrial metabolism: Multifaceted roles of a simple cofactor. Biochemistry 2016, 81, 1066–1080. [Google Scholar] [CrossRef]
- Que, E.L.; Domaille, D.W.; Chang, C.J. Metals in neurobiology: Probing their chemistry and biology with molecular imaging. Chem. Rev. 2008, 108, 1517–1549. [Google Scholar] [CrossRef]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2016, 139, 179–197. [Google Scholar] [CrossRef] [Green Version]
- Nnah, I.C.; Wessling-Resnick, M. Brain Iron Homeostasis: A Focus on Microglial Iron. Pharmaceuticals 2018, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, D.J.; Merlot, A.M.; Huang, M.L.; Bae, D.H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, S.; Paris, J.; Ryvar, R.; Botteron, C. Brain copper elevation and neurological changes in north ronaldsay sheep: A model for neurodegenerative disease? J. Comp. Pathol. 2008, 139, 252–255. [Google Scholar] [CrossRef]
- Gerlach, M.; Ben-Shachar, D.; Riederer, P.; Youdim, M.B. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J. Neurochem. 1994, 63, 793–807. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef] [Green Version]
- Whitnall, M.; Suryo Rahmanto, Y.; Huang, M.L.; Saletta, F.; Lok, H.C.; Gutierrez, L.; Lazaro, F.J.; Fleming, A.J.; St Pierre, T.G.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishop, G.M.; Dang, T.N.; Dringen, R.; Robinson, S.R. Accumulation of non-transferrin-bound iron by neurons, astrocytes, and microglia. Neurotox. Res. 2011, 19, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Bishop, G.M.; Koeppe, M.; Dang, T.N.; Robinson, S.R. The pivotal role of astrocytes in the metabolism of iron in the brain. Neurochem. Res. 2007, 32, 1884–1890. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Rosengren Nielsen, T.; Skjorringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef]
- Ponka, P.; Beaumont, C.; Richardson, D.R. Function and regulation of transferrin and ferritin. Semin. Hematol. 1998, 35, 35–54. [Google Scholar]
- Salvador, G.A. Iron in neuronal function and dysfunction. BioFactors 2010, 36, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, C.; Di Pisa, F.; Bernacchioni, C.; Ciambellotti, S.; Turano, P.; Mangani, S. Iron binding to human heavy-chain ferritin. Acta Crystallogr. D 2015, 71, 1909–1920. [Google Scholar] [CrossRef]
- Pfaffen, S.; Abdulqadir, R.; Le Brun, N.E.; Murphy, M.E. Mechanism of ferrous iron binding and oxidation by ferritin from a pennate diatom. J. Biol. Chem. 2013, 288, 14917–14925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfaffen, S.; Bradley, J.M.; Abdulqadir, R.; Firme, M.R.; Moore, G.R.; Le Brun, N.E.; Murphy, M.E.P. A diatom ferritin optimized for iron oxidation but not iron storage. J. Biol. Chem. 2015, 290, 28416–28427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pozzi, C.; Ciambellotti, S.; Bernacchioni, C.; Di Pisa, F.; Mangani, S.; Turano, P. Chemistry at the protein-mineral interface in L-ferritin assists the assembly of a functional (m3-oxo)Tris[(m2-peroxo)] triiron(III) cluster. Proc. Natl. Acad. Sci. USA 2017, 114, 2580–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, T.E.; Mason, A.B.; He, Q.Y.; Halbrooks, P.J.; Briggs, S.K.; Smith, V.C.; MacGillivray, R.T.; Everse, S.J. The position of arginine 124 controls the rate of iron release from the N-lobe of human serum transferrin. A structural study. J. Biol. Chem. 2003, 278, 6027–6033. [Google Scholar]
- Mason, A.B.; Halbrooks, P.J.; James, N.G.; Connolly, S.A.; Larouche, J.R.; Smith, V.C.; MacGillivray, R.T.; Chasteen, N.D. Mutational analysis of C-lobe ligands of human serum transferrin: Insights into the mechanism of iron release. Biochemistry 2005, 44, 8013–8021. [Google Scholar] [CrossRef]
- McCarthy, R.C.; Kosman, D.J. Iron transport across the blood-brain barrier: Development, neurovascular regulation and cerebral amyloid angiopathy. Cell. Mol. Life Sci. 2015, 72, 709–727. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.; Moreton, K. The distribution of iron between the metal-binding sites of transferrin human serum. Biochem. J. 1980, 185, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Connor, J.R.; Fine, R.E. The distribution of transferrin immunoreactivity in the rat central nervous system. Brain Res. 1986, 368, 319–328. [Google Scholar] [CrossRef]
- Masaldan, S.; Bush, A.I.; Devos, D.; Rolland, A.S.; Moreau, C. Striking while the iron is hot: Iron metabolism and ferroptosis in neurodegeneration. Free Radic. Biol. Med. 2019, 133, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Burkhart, A.; Skjorringe, T.; Johnsen, K.B.; Siupka, P.; Thomsen, L.B.; Nielsen, M.S.; Thomsen, L.L.; Moos, T. Expression of iron-related proteins at the neurovascular unit supports reduction and reoxidation of iron for transport through the blood-brain barrier. Mol. Neurobiol. 2016, 53, 7237–7253. [Google Scholar] [CrossRef]
- Pinero, D.J.; Connor, J.R. Iron in the brain: An important contributor in normal and diseased states. Neuroscientist 2000, 6, 435–453. [Google Scholar] [CrossRef]
- Skjorringe, T.; Burkhart, A.; Johnsen, K.B.; Moos, T. Divalent metal transporter 1 (DMT1) in the brain: Implications for a role in iron transport at the blood-brain barrier, and neuronal and glial pathology. Front. Mol. Neurosci. 2015, 8, 19. [Google Scholar] [PubMed] [Green Version]
- Garrick, M.D.; Singleton, S.T.; Vargas, F.; Kuo, H.C.; Zhao, L.; Knopfel, M.; Davidson, T.; Costa, M.; Paradkar, P.; Roth, J.A.; et al. DMT1: Which metals does it transport? Biol. Res. 2006, 39, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natal. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkitkasemwong, S.; Wang, C.Y.; Mackenzie, B.; Knutson, M.D. Physiologic implications of metal-ion transport by ZIP14 and ZIP8. Biometals 2012, 25, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Andrews, N.C. The iron transporter DMT1. Int. J. Biochem. Cell Biol. 1999, 31, 991–994. [Google Scholar] [CrossRef]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, N.; Gao, J.; Enns, C.A.; Knutson, M.D. ZRT/IRT-like protein 14 (ZIP14) promotes the cellular assimilation of iron from transferrin. J. Biol. Chem. 2010, 285, 32141–32150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, A.E.; Mendez, M.J.; Hokanson, C.A.; Rees, D.C.; Bjorkman, P.J. Investigation of the biophysical and cell biological properties of ferroportin, a multipass integral membrane protein iron exporter. J. Mol. Biol. 2009, 386, 717–732. [Google Scholar] [CrossRef] [Green Version]
- Ward, D.M.; Kaplan, J. Ferroportin-mediated iron transport: Expression and regulation. Biochim. Biophys. Acta 2012, 1823, 1426–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qian, Z.M.; Shen, X. Brain iron transport and neurodegeneration. Trends Mol. Med. 2001, 7, 103–108. [Google Scholar] [CrossRef]
- Qian, Z.M.; Wang, Q. Expression of iron transport proteins and excessive iron accumulation in the brain in neurodegenerative disorders. Brain Res. Rev. 1998, 27, 257–267. [Google Scholar] [PubMed]
- Malecki, E.A.; Devenyi, A.G.; Beard, J.L.; Connor, J.R. Existing and emerging mechanisms for transport of iron and manganese to the brain. J. Neurosci. Res. 1999, 56, 113–122. [Google Scholar] [CrossRef]
- Nam, G.; Yi, Y.; Lee, H.J.; Lee, J.; Kang, J.; Lim, M.H. Metalloneurochemistry. In Comprehensive Coordination Chemistry, 3rd ed.; Constable, E., Parkin, G., Que, L., Eds.; Elsevier: Amsterdam, The Netherlands, 2021. [Google Scholar] [CrossRef]
- Beard, J.L. Iron biology in immune function, muscle metabolism and neuronal functioning. J. Nutr. 2001, 131, 568S–579S. [Google Scholar] [CrossRef] [PubMed]
- Singh, N. The role of iron in prion disease and other neurodegenerative diseases. PLoS Pathog. 2014, 10, e1004335. [Google Scholar] [CrossRef]
- Agarwal, K.N. Iron and the brain: Neurotransmitter receptors and magnetic resonance spectroscopy. Br. J. Nutr. 2001, 85, S147–S150. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R.; Hoffmann, B.; Molik, S.; Pierik, A.J.; Rietzschel, N.; Stehling, O.; Uzarska, M.A.; Webert, H.; Wilbrecht, C.; Muhlenhoff, U. The role of mitochondria in cellular iron-sulfur protein biogenesis and iron metabolism. Biochim. Biophys. Acta 2012, 1823, 1491–1508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ortiz, E.; Pasquini, J.M.; Thompson, K.; Felt, B.; Butkus, G.; Beard, J.; Connor, J.R. Effect of manipulation of iron storage, transport, or availability on myelin composition and brain iron content in three different animal models. J. Neurosci. Res. 2004, 77, 681–689. [Google Scholar] [CrossRef]
- Yu, G.S.; Steinkirchner, T.M.; Rao, G.A.; Larkin, E.C. Effect of prenatal iron deficiency on myelination in rat pups. Am. J. Pathol. 1986, 125, 620–624. [Google Scholar] [PubMed]
- Connor, J.R.; Menzies, S.L. Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17, 83–93. [Google Scholar] [CrossRef]
- Kim, J.; Wessling-Resnick, M. Iron and mechanisms of emotional behavior. J. Nutr. Biochem. 2014, 25, 1101–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwik-Uribe, C.L.; Gietzen, D.; German, J.B.; Golub, M.S.; Keen, C.L. Chronic marginal iron intakes during early development in mice result in persistent changes in dopamine metabolism and myelin composition. J. Nutr. 2000, 130, 2821–2830. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, D.M.; Ruskin, B.; Lovenberg, W. Tryptophan hydroxylase. The role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J. Biol. Chem. 1980, 255, 4137–4143. [Google Scholar] [CrossRef]
- Bradbury, M.W. Transport of iron in the blood-brain-cerebrospinal fluid system. J. Neurochem. 1997, 69, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Perry, G.; Harris, P.L.; Liu, Y.; Schubert, K.A.; Smith, M.A. In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer’s disease: A central role for bound transition metals. J. Neurochem. 2000, 74, 270–279. [Google Scholar] [CrossRef]
- Suh, S.W.; Jensen, K.B.; Jensen, M.S.; Silva, D.S.; Kesslak, P.J.; Danscher, G.; Frederickson, C.J. Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res. 2000, 852, 274–278. [Google Scholar] [CrossRef]
- Bartzokis, G.; Sultzer, D.; Cummings, J.; Holt, L.E.; Hance, D.B.; Henderson, V.W.; Mintz, J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch. Gen. Psychiatry 2000, 57, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovell, M.A.; Robertson, J.D.; Teesdale, W.J.; Campbell, J.L.; Markesbery, W.R. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998, 158, 47–52. [Google Scholar] [CrossRef]
- Yamamoto, A.; Shin, R.W.; Hasegawa, K.; Naiki, H.; Sato, H.; Yoshimasu, F.; Kitamoto, T. Iron (III) induces aggregation of hyperphosphorylated tau and its reduction to iron (II) reverses the aggregation: Implications in the formation of neurofibrillary tangles of Alzheimer’s disease. J. Neurochem. 2002, 82, 1137–1147. [Google Scholar] [CrossRef]
- Huang, X.; Moir, R.D.; Tanzi, R.E.; Bush, A.I.; Rogers, J.T. Redox active metals, oxidative stress, and Alzheimer’s disease pathology. Ann. N. Y. Acad. Sci. 2004, 1012, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Schubert, D.; Chevion, M. The role of iron in beta amyloid toxicity. Biochem. Biophys. Res. Commun. 1995, 216, 702–707. [Google Scholar] [CrossRef] [PubMed]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative stress and neurodegeneration: The involvement of iron. Biometals 2018, 31, 715–735. [Google Scholar] [CrossRef]
- Uranga, R.M.; Salvador, G.A. Unraveling the burden of iron in neurodegeneration: Intersections with amyloid b peptide pathology. Oxid. Med. Cell Longev. 2018, 2018, 2850341. [Google Scholar] [CrossRef]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef] [Green Version]
- Bansal, S.; Biswas, G.; Avadhani, N.G. Mitochondria-targeted heme oxygenase-1 induces oxidative stress and mitochondrial dysfunction in macrophages, kidney fibroblasts and in chronic alcohol hepatotoxicity. Redox Biol. 2014, 2, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shang, Y.; Luo, M.; Yao, F.; Wang, S.; Yuan, Z.; Yang, Y. Ceruloplasmin suppresses ferroptosis by regulating iron homeostasis in hepatocellular carcinoma cells. Cell. Signal. 2020, 72, 109633. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keberle, H. The Biochemistry of Desferrioxamine and Its Relation to Iron Metabolism. Ann. N.Y. Acad. Sci. 1964, 119, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ma, Y.; Kong, X.; Hider, R.C. Design of iron chelators with therapeutic application. Dalton Trans. 2012, 41, 6371–6389. [Google Scholar] [CrossRef] [PubMed]
- Fredenburg, A.M.; Sethi, R.K.; Allen, D.D.; Yokel, R.A. The pharmacokinetics and blood-brain barrier permeation of the chelators 1,2 dimethly-, 1,2 diethyl-, and 1-[ethan-1’ol]-2-methyl-3-hydroxypyridin-4-one in the rat. Toxicology 1996, 108, 191–199. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Eshel, G.; Finberg, J.P.; Youdim, M.B. The iron chelator desferrioxamine (Desferal) retards 6-hydroxydopamine-induced degeneration of nigrostriatal dopamine neurons. J. Neurochem. 1991, 56, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Kalinowski, D.S.; Richardson, D.R. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol. Rev. 2005, 57, 547–583. [Google Scholar] [CrossRef]
- Evers, A.; Hancock, R.D.; Martell, A.E.; Motekaitis, R.J. Metal ion recognition in ligands with negatively charged oxygen donor groups. Complexation of iron(III), gallium(III), indium(III), aluminum(III), and other highly charged metal ions. Inorg. Chem. 1989, 28, 2189–2195. [Google Scholar] [CrossRef]
- Lan, J.; Jiang, D.H. Desferrioxamine and vitamin E protect against iron and MPTP-induced neurodegeneration in mice. J. Neural Transm. 1997, 104, 469–481. [Google Scholar] [CrossRef] [PubMed]
- Cable, H.; Lloyd, J.B. Cellular uptake and release of two contrasting iron chelators. J. Pharm. Pharmacol. 1999, 51, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, M.L. Deferoxamine enhances alternative activation of microglia and inhibits amyloid beta deposits in APP/PS1 mice. Brain Res. 2017, 1677, 8692. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.M.; Kosyakovsky, J.; Baillargeon, A.M.; Tokarev, J.V.; Cooner, J.M.; Svitak, A.L.; Faltesek, K.A.; Frey, W.H., II; Hanson, L.R. Intranasal deferoxamine can improve memory in healthy C57 mice, suggesting a partially non-disease-specific pathway of functional neurologic improvement. Brain Behav. 2020, 10, e01536. [Google Scholar] [CrossRef] [PubMed]
- Fine, J.M.; Forsberg, A.C.; Stroebel, B.M.; Faltesek, K.A.; Verden, D.R.; Hamel, K.A.; Raney, E.B.; Crow, J.M.; Haase, L.R.; Knutzen, K.E.; et al. Intranasal deferoxamine affects memory loss, oxidation, and the insulin pathway in the streptozotocin rat model of Alzheimer’s disease. J. Neurol. Sci. 2017, 380, 164–171. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Gaeta, A.; Francis, P.T.; Williams, R.J.; Hider, R.C. Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY-5Y cells. J. Neurochem. 2008, 105, 2466–2476. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Marques, S.M.; Quintanova, C.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Multifunctional iron-chelators with protective roles against neurodegenerative diseases. Dalton Trans. 2013, 42, 6058–6073. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Portbury, S.D.; Lago, L.; McColl, G.; Finkelstein, D.I.; Bush, A.I.; Adlard, P.A. The Iron Chelator Deferiprone Improves the Phenotype in a Mouse Model of Tauopathy. J. Alzheimers Dis. 2020, 77, 753–771. [Google Scholar] [CrossRef] [PubMed]
- Prasanthi, J.R.; Schrag, M.; Dasari, B.; Marwarha, G.; Dickson, A.; Kirsch, W.M.; Ghribi, O. Deferiprone reduces amyloid-beta and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J. Alzheimers Dis. 2012, 30, 167–182. [Google Scholar] [CrossRef] [Green Version]
- Gleason, A.; Bush, A.I. Iron and ferroptosis as therapeutic targets in Alzheimer’s disease. Neurotherapeutics 2021, 18, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Mudasir, X.; Arai, M.; Yoshioka, N.; Inoue, H. Reversed-phase high-performance liquid chromatography of iron(II) and copper(II) chelates with 4,7-diphenyl-1,10-phenanthroline disulfonate. J. Chromatogr. A 1998, 799, 171–176. [Google Scholar] [CrossRef]
- Yokoyama, T.; Nakano, K.; Zenki, M. Specific separation of nickel ion with bathophenanthroline disulfonic acid by capillary zone electrophoresis. Anal. Chim. Acta 1999, 396, 117–123. [Google Scholar] [CrossRef]
- Randell, E.W.; Parkes, J.G.; Olivieri, N.F.; Templeton, D.M. Uptake of non-transferrin-bound iron by both reductive and nonreductive processes is modulated by intracellular iron. J. Biol. Chem. 1994, 269, 16046–16053. [Google Scholar] [CrossRef]
- Amit, T.; Bar-Am, O.; Mechlovich, D.; Kupershmidt, L.; Youdim, M.B.H.; Weinreb, O. The novel multitarget iron chelating and propargylamine drug M30 affects APP regulation and processing activities in Alzheimer’s disease models. Neuropharmacology 2017, 123, 359–367. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, P.; Chen, B.; Lun, P.; Dou, Y.; Cao, W.W. A study on the effect of ion chelating agent on Alzheimer disease. Biomed. Res. 2017, 28, 8022–8026. [Google Scholar]
- Peters, C.; Bascunan, D.; Burgos, C.F.; Bobadilla, C.; Gonzalez-Sanmiguel, J.; Boopathi, S.; Riffo, N.; Fernandez-Perez, E.J.; Tarnok, M.E.; Aguilar, L.F.; et al. Characterization of a new molecule capable of inhibiting several steps of the amyloid cascade in Alzheimer’s disease. Neurobiol. Dis. 2020, 141, 104938. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Ho, T.-L. A New Glycosylation Method Based on 8-Quinolyl Glycosides. J. Chin. Chem. Soc. 2006, 53, 1567–1570. [Google Scholar] [CrossRef]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef]
- Knez, D.; Brus, B.; Coquelle, N.; Sosic, I.; Sink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorg. Med. Chem. 2015, 23, 4442–4452. [Google Scholar] [CrossRef]
- Shachar, D.B.; Kahana, N.; Kampel, V.; Warshawsky, A.; Youdim, M.B. Neuroprotection by a novel brain permeable iron chelator, VK-28, against 6-hydroxydopamine lession in rats. Neuropharmacology 2004, 46, 254–263. [Google Scholar] [CrossRef]
- Cacciatore, I.; Cornacchia, C.; Fornasari, E.; Baldassarre, L.; Pinnen, F.; Sozio, P.; Di Stefano, A.; Marinelli, L.; Dean, A.; Fulle, S.; et al. A glutathione derivative with chelating and in vitro neuroprotective activities: Synthesis, physicochemical properties, and biological evaluation. ChemMedChem 2013, 8, 1818–1829. [Google Scholar] [CrossRef]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 2011, 189, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Salkovic-Petrisic, M.; Knezovic, A.; Osmanovic-Barilar, J.; Smailovic, U.; Trkulja, V.; Riederer, P.; Amit, T.; Mandel, S.; Youdim, M.B. Multi-target iron-chelators improve memory loss in a rat model of sporadic Alzheimer’s disease. Life Sci. 2015, 136, 108–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braymer, J.J.; Detoma, A.S.; Choi, J.S.; Ko, K.S.; Lim, M.H. Recent Development of Bifunctional Small Molecules to Study Metal-Amyloid-beta Species in Alzheimer’s Disease. Int. J. Alzheiers Dis. 2010, 2011, 623051. [Google Scholar]
- Moss, S.; Subramanian, V.; Acharya, K.R. High resolution crystal structure of substrate-free human neprilysin. J. Struct. Biol. 2018, 204, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Oefner, C.; Pierau, S.; Schulz, H.; Dale, G.E. Structural studies of a bifunctional inhibitor of neprilysin and DPP-IV. Acta Crystallogr. D 2007, 63, 975–981. [Google Scholar] [CrossRef]
- Oefner, C.; D’Arcy, A.; Hennig, M.; Winkler, F.K.; Dale, G.E. Structure of human neutral endopeptidase (Neprilysin) complexed with phosphoramidon. J. Mol. Biol. 2000, 296, 341–349. [Google Scholar] [CrossRef]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-beta protein clearance and degradation (ABCD) pathways and their role in Alzheimer’s disease. Curr. Alzheimers Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [Green Version]
- Hellstrom-Lindahl, E.; Ravid, R.; Nordberg, A. Age-dependent decline of neprilysin in Alzheimer’s disease and normal brain: Inverse correlation with Ab levels. Neurobiol. Aging 2008, 29, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Erdos, E.G.; Skidgel, R.A. Neutral endopeptidase 24.11 (enkephalinase) and related regulators of peptide hormones. FASEB J. 1989, 3, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Moss, S.; Subramanian, V.; Acharya, K.R. Crystal structure of peptide-bound neprilysin reveals key binding interactions. FEBS Lett. 2020, 594, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Feygina, E.E.; Katrukha, A.G.; Semenov, A.G. Neutral Endopeptidase (Neprilysin) in Therapy and Diagnostics: Yin and Yang. Biochemistry 2019, 84, 1346–1358. [Google Scholar] [CrossRef] [PubMed]
- Bayes-Genis, A.; Barallat, J.; Richards, A.M. A Test in Context: Neprilysin: Function, Inhibition, and Biomarker. J. Am. Coll. Cardiol. 2016, 68, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Howell, S.; Nalbantoglu, J.; Crine, P. Neutral endopeptidase can hydrolyze b-amyloid(1-40) but shows no effect on b-amyloid precursor protein metabolism. Peptides 1995, 16, 647–652. [Google Scholar] [CrossRef]
- Bayes-Genis, A.; Barallat, J.; Galan, A.; de Antonio, M.; Domingo, M.; Zamora, E.; Urrutia, A.; Lupon, J. Soluble neprilysin is predictive of cardiovascular death and heart failure hospitalization in heart failure patients. J. Am. Coll. Cardiol. 2015, 65, 657–665. [Google Scholar] [CrossRef]
- Carson, J.A.; Turner, A.J. b-amyloid catabolism: Roles for neprilysin (NEP) and other metallopeptidases? J. Neurochem. 2002, 81, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Hinton, D.J.; Choi, D.S. Protein kinase C-regulated abeta production and clearance. Int. J. Alzheimers Dis. 2011, 2011, 857368. [Google Scholar] [PubMed] [Green Version]
- Stock, A.J.; Kasus-Jacobi, A.; Pereira, H.A. The role of neutrophil granule proteins in neuroinflammation and Alzheimer’s disease. J. Neuroinflamm. 2018, 15, 240. [Google Scholar] [CrossRef]
- Kanemitsu, H.; Tomiyama, T.; Mori, H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 2003, 350, 113–116. [Google Scholar] [CrossRef]
- Huang, S.M.; Mouri, A.; Kokubo, H.; Nakajima, R.; Suemoto, T.; Higuchi, M.; Staufenbiel, M.; Noda, Y.; Yamaguchi, H.; Nabeshima, T.; et al. Neprilysin-sensitive synapse-associated amyloid-beta peptide oligomers impair neuronal plasticity and cognitive function. J. Biol. Chem. 2006, 281, 17941–17951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bush, A.I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. [Google Scholar] [CrossRef]
- Huang, J.Y.; Hafez, D.M.; James, B.D.; Bennett, D.A.; Marr, R.A. Altered NEP2 expression and activity in mild cognitive impairment and Alzheimer’s disease. J. Alzheimers. Dis. 2012, 28, 433–441. [Google Scholar] [CrossRef]
- Wang, S.; Wang, R.; Chen, L.; Bennett, D.A.; Dickson, D.W.; Wang, D.S. Expression and functional profiling of neprilysin, insulin-degrading enzyme, and endothelin-converting enzyme in prospectively studied elderly and Alzheimer’s brain. J. Neurochem. 2010, 115, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, W.; Schutz, S.G.; Cirrito, J.R.; Shankar, G.M.; Sun, X.; George, A.; Leissring, M.A.; Walsh, D.M.; Qiu, W.Q.; Holtzman, D.M.; et al. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol. 2007, 171, 241–251. [Google Scholar] [CrossRef] [Green Version]
- Maruyama, M.; Higuchi, M.; Takaki, Y.; Matsuba, Y.; Tanji, H.; Nemoto, M.; Tomita, N.; Matsui, T.; Iwata, N.; Mizukami, H.; et al. Cerebrospinal fluid neprilysin is reduced in prodromal Alzheimer’s disease. Ann. Neurol. 2005, 57, 832–842. [Google Scholar] [CrossRef]
- Rose, J.B.; Crews, L.; Rockenstein, E.; Adame, A.; Mante, M.; Hersh, L.B.; Gage, F.H.; Spencer, B.; Potkar, R.; Marr, R.A.; et al. Neuropeptide Y fragments derived from neprilysin processing are neuroprotective in a transgenic model of Alzheimer’s disease. J. Neurosci. 2009, 29, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, T.; Iwata, N.; Tsubuki, S.; Takaki, Y.; Takano, J.; Huang, S.M.; Suemoto, T.; Higuchi, M.; Saido, T.C. Somatostatin regulates brain amyloid b peptide Ab42 through modulation of proteolytic degradation. Nature Med. 2005, 11, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Mizukami, H.; Shirotani, K.; Takaki, Y.; Muramatsu, S.; Lu, B.; Gerard, N.P.; Gerard, C.; Ozawa, K.; Saido, T.C. Presynaptic localization of neprilysin contributes to efficient clearance of amyloid-beta peptide in mouse brain. J. Neurosci. 2004, 24, 991–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwata, N.; Sekiguchi, M.; Hattori, Y.; Takahashi, A.; Asai, M.; Ji, B.; Higuchi, M.; Staufenbiel, M.; Muramatsu, S.; Saido, T.C. Global brain delivery of neprilysin gene by intravascular administration of AAV vector in mice. Sci. Rep. 2013, 3, 1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campos, C.R.; Kemble, A.M.; Niewoehner, J.; Freskgard, P.O.; Urich, E. Brain shuttle neprilysin reduces central amyloid-b levels. PLoS ONE 2020, 15, e0229850. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Wang, Y.; Huang, H.; Zhao, Y.; Zhou, H. Meta-analysis of expression and function of neprilysin in Alzheimer’s disease. Neurosci. Lett. 2017, 657, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Cakir, B.; Dagliyan, O.; Dagyildiz, E.; Baris, I.; Kavakli, I.H.; Kizilel, S.; Turkay, M. Structure based discovery of small molecules to regulate the activity of human insulin degrading enzyme. PLoS ONE 2012, 7, e31787. [Google Scholar] [CrossRef] [PubMed]
- Tundo, G.R.; Di Muzio, E.; Ciaccio, C.; Sbardella, D.; Di Pierro, D.; Polticelli, F.; Coletta, M.; Marini, S. Multiple allosteric sites are involved in the modulation of insulin-degrading-enzyme activity by somatostatin. FEBS J. 2016, 283, 3755–3770. [Google Scholar] [CrossRef] [Green Version]
- Im, H.; Manolopoulou, M.; Malito, E.; Shen, Y.; Zhao, J.; Neant-Fery, M.; Sun, C.Y.; Meredith, S.C.; Sisodia, S.S.; Leissring, M.A.; et al. Structure of substrate-free human insulin-degrading enzyme (IDE) and biophysical analysis of ATP-induced conformational switch of IDE. J. Biol. Chem. 2007, 282, 25453–25463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malito, E.; Hulse, R.E.; Tang, W.J. Amyloid beta-degrading cryptidases: Insulin degrading enzyme, presequence peptidase, and neprilysin. Cell. Mol. Life Sci. 2008, 65, 2574–2585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.; Joachimiak, A.; Rosner, M.R.; Tang, W.J. Structures of human insulin-degrading enzyme reveal a new substrate recognition mechanism. Nature 2006, 443, 870–874. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Kuo, W.L.; Yousef, M.; Rosner, M.R.; Tang, W.J. The C-terminal domain of human insulin degrading enzyme is required for dimerization and substrate recognition. Biochem. Biophys. Res. Commun. 2006, 343, 1032–1037. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Guarnera, E.; Berezovsky, I.N. Insulin-degrading enzyme in the fight against Alzheimer’s disease. Trends Pharmacol. Sci. 2018, 39, 49–58. [Google Scholar] [CrossRef]
- Kurochkin, I.V.; Goto, S. Alzheimer’s b-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Amata, O.; Marino, T.; Russo, N.; Toscano, M. Human insulin-degrading enzyme working mechanism. J. Am. Chem. Soc. 2009, 131, 14804–14811. [Google Scholar] [CrossRef]
- Tundo, G.R.; Sbardella, D.; Ciaccio, C.; Grasso, G.; Gioia, M.; Coletta, A.; Polticelli, F.; Di Pierro, D.; Milardi, D.; Van Endert, P.; et al. Multiple functions of insulin-degrading enzyme: A metabolic crosslight? Crit. Rev. Biochem. Bol. Biol. 2017, 52, 554–582. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.Q.; Walsh, D.M.; Ye, Z.; Vekrellis, K.; Zhang, J.; Podlisny, M.B.; Rosner, M.R.; Safavi, A.; Hersh, L.B.; Selkoe, D.J. Insulin-degrading enzyme regulates extracellular levels of amyloid b-protein by degradation. J. Biol. Chem. 1998, 273, 32730–32738. [Google Scholar] [CrossRef] [Green Version]
- Perez, A.; Morelli, L.; Cresto, J.C.; Castano, E.M. Degradation of soluble amyloid b-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 2000, 25, 247–255. [Google Scholar] [CrossRef]
- Leal, M.C.; Magnani, N.; Villordo, S.; Buslje, C.M.; Evelson, P.; Castano, E.M.; Morelli, L. Transcriptional regulation of insulin-degrading enzyme modulates mitochondrial amyloid b (Ab) peptide catabolism and functionality. J. Biol. Chem. 2013, 288, 12920–12931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sikanyika, N.L.; Parkington, H.C.; Smith, A.I.; Kuruppu, S. Powering amyloid b degrading enzymes: A possible therapy for Alzheimer’s disease. Neurochem. Res. 2019, 44, 1289–1296. [Google Scholar] [CrossRef] [PubMed]
- de Tullio, M.B.; Castelletto, V.; Hamley, I.W.; Martino Adami, P.V.; Morelli, L.; Castano, E.M. Proteolytically inactive insulin-degrading enzyme inhibits amyloid formation yielding non-neurotoxic Ab peptide aggregates. PLoS ONE 2013, 8, e59113. [Google Scholar] [CrossRef] [Green Version]
- Kurochkin, I.V. Insulin-degrading enzyme: Embarking on amyloid destruction. Trends Biochem. Sci. 2001, 26, 421–425. [Google Scholar] [CrossRef]
- Portelius, E.; Mattsson, N.; Pannee, J.; Zetterberg, H.; Gisslen, M.; Vanderstichele, H.; Gkanatsiou, E.; Crespi, G.A.; Parker, M.W.; Miles, L.A.; et al. Ex vivo 18O-labeling mass spectrometry identifies a peripheral amyloid b clearance pathway. Mol. Neurodegener. 2017, 12, 18. [Google Scholar] [CrossRef] [Green Version]
- Stargardt, A.; Gillis, J.; Kamphuis, W.; Wiemhoefer, A.; Kooijman, L.; Raspe, M.; Benckhuijsen, W.; Drijfhout, J.W.; Hol, E.M.; Reits, E. Reduced amyloid-b degradation in early Alzheimer’s disease but not in the APPswePS1dE9 and 3xTg-AD mouse models. Aging Cell 2013, 12, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xiang, Z.; Haroutunian, V.; Buxbaum, J.D.; Stetka, B.; Pasinetti, G.M. Insulin degrading enzyme activity selectively decreases in the hippocampal formation of cases at high risk to develop Alzheimer’s disease. Neurobiol. Aging 2007, 28, 824–830. [Google Scholar] [CrossRef]
- Miller, B.C.; Eckman, E.A.; Sambamurti, K.; Dobbs, N.; Chow, K.M.; Eckman, C.B.; Hersh, L.B.; Thiele, D.L. Amyloid-b peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 6221–6226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid b-protein, and the b-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcello, E.; Borroni, B.; Pelucchi, S.; Gardoni, F.; Di Luca, M. ADAM10 as a therapeutic target for brain diseases: From developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. [Google Scholar] [CrossRef]
- Huovila, A.P.; Turner, A.J.; Pelto-Huikko, M.; Karkkainen, I.; Ortiz, R.M. Shedding light on ADAM metalloproteinases. Trends Biochem. Sci. 2005, 30, 413–422. [Google Scholar] [CrossRef]
- Yuan, X.Z.; Sun, S.; Tan, C.C.; Yu, J.T.; Tan, L. The Role of ADAM10 in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 303–322. [Google Scholar] [CrossRef]
- Wolfsberg, T.G.; Primakoff, P.; Myles, D.G.; White, J.M. ADAM, a novel family of membrane proteins containing A Disintegrin And Metalloprotease domain: Multipotential functions in cell-cell and cell-matrix interactions. J. Cell. Biol. 1995, 131, 275–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kamp, F.; Wagner, J.; Nuscher, B.; Zilow, S.; Ludwig, A.K.; Willem, M.; Haass, C. Translational repression of the disintegrin and metalloprotease ADAM10 by a stable G-quadruplex secondary structure in its 5′-untranslated region. J. Biol. Chem. 2011, 286, 45063–45072. [Google Scholar] [CrossRef] [Green Version]
- Hartl, D.; May, P.; Gu, W.; Mayhaus, M.; Pichler, S.; Spaniol, C.; Glaab, E.; Bobbili, D.R.; Antony, P.; Koegelsberger, S.; et al. A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol. Psychiatry 2020, 25, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Endres, K.; Deller, T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front. Mol. Neurosci. 2017, 10, 56. [Google Scholar] [CrossRef]
- Seipold, L.; Altmeppen, H.; Koudelka, T.; Tholey, A.; Kasparek, P.; Sedlacek, R.; Schweizer, M.; Bar, J.; Mikhaylova, M.; Glatzel, M.; et al. In vivo regulation of the A disintegrin and metalloproteinase 10 (ADAM10) by the tetraspanin 15. Cell Mol. Life Sci. 2018, 75, 3251–3267. [Google Scholar] [CrossRef]
- Lammich, S.; Kojro, E.; Postina, R.; Gilbert, S.; Pfeiffer, R.; Jasionowski, M.; Haass, C.; Fahrenholz, F. Constitutive and regulated a-secretase cleavage of Alzheimer’s amyloid precursor protein by a disintegrin metalloprotease. Proc. Natl. Acad. Sci. USA 1999, 96, 3922–3927. [Google Scholar] [CrossRef] [Green Version]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural basis for regulated proteolysis by the a-Secretase ADAM10. Cell 2017, 171, 1638–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stawikowska, R.; Cudic, M.; Giulianotti, M.; Houghten, R.A.; Fields, G.B.; Minond, D. Activity of ADAM17 (a disintegrin and metalloprotease 17) is regulated by its noncatalytic domains and secondary structure of its substrates. J. Biol. Chem. 2013, 288, 22871–22879. [Google Scholar] [CrossRef] [Green Version]
- Tape, C.J.; Willems, S.H.; Dombernowsky, S.L.; Stanley, P.L.; Fogarasi, M.; Ouwehand, W.; McCafferty, J.; Murphy, G. Cross-domain inhibition of TACE ectodomain. Proc. Natl. Acad. Sci. USA 2011, 108, 5578–5583. [Google Scholar] [CrossRef] [Green Version]
- Janes, P.W.; Saha, N.; Barton, W.A.; Kolev, M.V.; Wimmer-Kleikamp, S.H.; Nievergall, E.; Blobel, C.P.; Himanen, J.P.; Lackmann, M.; Nikolov, D.B. Adam meets Eph: An ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell 2005, 123, 291–304. [Google Scholar] [CrossRef] [Green Version]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.Q.; Qu, D.H.; Wang, K. Therapeutic approaches to Alzheimer’s disease through stimulating of non-amyloidogenic processing of amyloid precursor protein. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 2389–2403. [Google Scholar]
- Marcello, E.; Saraceno, C.; Musardo, S.; Vara, H.; de la Fuente, A.G.; Pelucchi, S.; Di Marino, D.; Borroni, B.; Tramontano, A.; Perez-Otano, I.; et al. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. J. Clin. Investig. 2013, 123, 2523–2538. [Google Scholar] [CrossRef] [PubMed]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]
- Prox, J.; Bernreuther, C.; Altmeppen, H.; Grendel, J.; Glatzel, M.; D’Hooge, R.; Stroobants, S.; Ahmed, T.; Balschun, D.; Willem, M.; et al. Postnatal disruption of the disintegrin/metalloproteinase ADAM10 in brain causes epileptic seizures, learning deficits, altered spine morphology, and defective synaptic functions. J. Neurosci. 2013, 33, 12915–12928. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, M.; Liu, Y.; Yu, J.; Yang, H.; Fan, D.; Chui, D. Copper downregulates neprilysin activity through modulation of neprilysin degradation. J. Alzheimers Dis. 2010, 19, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Lang, M.; Fan, Q.; Wang, L.; Zheng, Y.; Xiao, G.; Wang, X.; Wang, W.; Zhong, Y.; Zhou, B. Inhibition of human high-affinity copper importer Ctr1 orthologous in the nervous system of drosophila ameliorates Ab42-induced Alzheimer’s disease-like symptoms. Neurobiol. Aging 2013, 34, 2604–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mital, M.; Bal, W.; Fraczyk, T.; Drew, S.C. Interplay between copper, neprilysin, and N-truncation of b-amyloid. Inorg. Chem. 2018, 57, 6193–6197. [Google Scholar] [CrossRef]
- Banerjee, P.; Sahoo, A.; Anand, S.; Bir, A.; Chakrabarti, S. The oral iron chelator, deferasirox, reverses the age-dependent alterations in iron and amyloid-b homeostasis in rat brain: Implications in the therapy of Alzheimer’s disease. J. Alzheimers Dis. 2016, 49, 681–693. [Google Scholar] [CrossRef]
- Grasso, G.; Pietropaolo, A.; Spoto, G.; Pappalardo, G.; Tundo, G.R.; Ciaccio, C.; Coletta, M.; Rizzarelli, E. Copper(I) and copper(II) inhibit Ab peptides proteolysis by insulin-degrading enzyme differently: Implications for metallostasis alteration in Alzheimer’s disease. Chem. Eur. J. 2011, 17, 2752–2762. [Google Scholar] [CrossRef] [Green Version]
- Bellia, F.; Lanza, V.; Ahmed, I.M.M.; Garcia-Vinuales, S.; Veiss, E.; Arizzi, M.; Calcagno, D.; Milardi, D.; Grasso, G. Site directed mutagenesis of insulin-degrading enzyme allows singling out the molecular basis of peptidase versus E1-like activity: The role of metal ions. Metallomics 2019, 11, 278–281. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Laferla, F.M. Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD. J. Neurochem. 2009, 108, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Maras, J.S.; Das, S.; Sharma, S.; Sukriti, S.; Kumar, J.; Vyas, A.K.; Kumar, D.; Bhat, A.; Yadav, G.; Choudhary, M.C.; et al. Iron-Overload triggers ADAM-17 mediated inflammation in Severe Alcoholic Hepatitis. Sci. Rep. 2018, 8, 10264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H.; Yoo, Y.M. Altered APP Carboxyl-Terminal Processing Under Ferrous Iron Treatment in PC12 Cells. Korean J. Physiol. Pharmacol. 2013, 17, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, Y.H.; Zhang, W.; Gao, H.L.; Zhong, M.L.; Huang, T.T.; Guo, R.F.; Liu, N.N.; Li, D.D.; Li, Y.; et al. Copper chelators promote nonamyloidogenic processing of AbPP via MT1/2 /CREB-dependent signaling pathways in AbPP/PS1 transgenic mice. J. Pineal Res. 2018, 65, e12502. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The roles and influence of Cu(I/II) on normal (left) and AD-affected (right) conditions.

Figure 2.
Selected Cu(I/II)-targeting molecules developed to maintain Cu(I/II) homeostasis. Top row: 8-hydroxyquinoline (8-HQ) and its derivatives, clioquinol (CQ), 5-chloro-7-iodoquinolin-8-ol, and BPT-2, 5,7-dichloro-2-[(dimethylamino)methyl]quinolin-8-ol; middle row: bathocuproine (BC), 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline, and bathocuproine disulfonate (BCS), 4,40-(2,9-dimethyl-1,10-phenanthroline-4,7-diyl)dibenzenesulfonate; bottom row: D-penicillamine, (2S)-2-amino-3-methyl-3-sulfanylbutanoic acid, trientine, triethylenetetramine, and tetramolybdate.
Figure 2.
Selected Cu(I/II)-targeting molecules developed to maintain Cu(I/II) homeostasis. Top row: 8-hydroxyquinoline (8-HQ) and its derivatives, clioquinol (CQ), 5-chloro-7-iodoquinolin-8-ol, and BPT-2, 5,7-dichloro-2-[(dimethylamino)methyl]quinolin-8-ol; middle row: bathocuproine (BC), 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline, and bathocuproine disulfonate (BCS), 4,40-(2,9-dimethyl-1,10-phenanthroline-4,7-diyl)dibenzenesulfonate; bottom row: D-penicillamine, (2S)-2-amino-3-methyl-3-sulfanylbutanoic acid, trientine, triethylenetetramine, and tetramolybdate.

Figure 3.
The roles and effects of Fe(II/III) in normal (left) and AD-affected (right) brains.

Figure 4.
Representatives of Fe(II/III)-targeting molecules. Left: deferoxamine (DFO), N1-(5-aminopentyl)-N1-hydroxy-N4-(5-(N-hydroxy-4-((5-(N-hydroxyacetamido)pentyl)amino)-4-oxobutanamido)pentyl)succinamide; middle: deferiprone (DFP), 3-hydroxy-1,2-dimethylpyridin-4(1H)-one; right: bathophenanthroline disulfonate (BPS), 4,7-diphenyl-1,10-phenanthroline-3,8-disulfonate.
Figure 4.
Representatives of Fe(II/III)-targeting molecules. Left: deferoxamine (DFO), N1-(5-aminopentyl)-N1-hydroxy-N4-(5-(N-hydroxy-4-((5-(N-hydroxyacetamido)pentyl)amino)-4-oxobutanamido)pentyl)succinamide; middle: deferiprone (DFP), 3-hydroxy-1,2-dimethylpyridin-4(1H)-one; right: bathophenanthroline disulfonate (BPS), 4,7-diphenyl-1,10-phenanthroline-3,8-disulfonate.


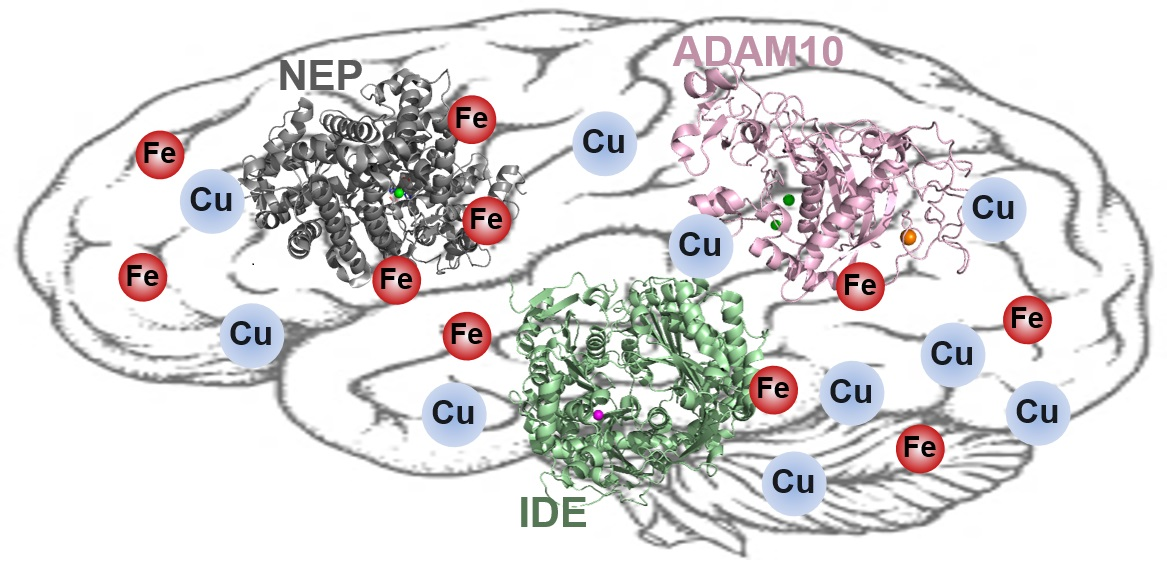{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Clinical trials of chemical agents. PBT-2 and deferiprone (DFP) have been examined their potential as treatments for neurodegeneration.
Table 1.
Clinical trials of chemical agents. PBT-2 and deferiprone (DFP) have been examined their potential as treatments for neurodegeneration.
| Chemical Agents | Clinical Trials (Periods) | Status |
|---|---|---|
| PBT-2 | Phase 2 (December, 2006-December, 2007) | Completed (NCT00471211) |
| Phase 2 (September, 2011-January, 2014) | Completed (ACTRN12611001008910) | |
| Phase 2 (July, 2013-January, 2015) | Completed (ACTRN12613000777796) | |
| Deferiprone (DFP) | Phase 2 (October, 2016-September, 2019) | Completed (NCT02728843) |
| Phase 2 (November, 2017-October, 2019) | Completed (ACTRN12617001578392) | |
| Phase 2 (January, 2018-December, 2021 (Estimated)] | Recruiting (NCT03234686) |
Table 2.
Recent animal studies conducted with Cu-targeting chemical agents. Results of in vivo experiments performed with clioquinol (CQ), DP-109, DP-460, D-penicillamine, and tetrathiomolybdate are summarized.
Table 2.
Recent animal studies conducted with Cu-targeting chemical agents. Results of in vivo experiments performed with clioquinol (CQ), DP-109, DP-460, D-penicillamine, and tetrathiomolybdate are summarized.
| Chemical Agents | Animal Models | Outcomes | References |
|---|---|---|---|
| Clioquinol (CQ) | C. elegans | Enhancement of neuroprotective effect | [142] |
| DP-109 | Transgenic mouse | Extension of life span and Reduction of amyloid plaques | [152,153] |
| DP-460 | Transgenic mouse | Extension of life span and Reduction of amyloid plaques | [152,153] |
| Rat | Detrimental effect on learning | [154] | |
| D-Penicillamine | Transgenic mouse | Improvement of cognitive ability | [163] |
| Tetrathiomolybdate | Transgenic mouse | Decrease of the inflammatory cytokines | [167] |
Table 3.
Recent animal studies conducted with Fe-targeting chemical agents. Results of in vivo experiments performed with deferoxamine (DFO), deferiprone (DFP), M30, and HLA20 are summarized.
Table 3.
Recent animal studies conducted with Fe-targeting chemical agents. Results of in vivo experiments performed with deferoxamine (DFO), deferiprone (DFP), M30, and HLA20 are summarized.
| Chemical Agents | Animal Model | Outcomes | References |
|---|---|---|---|
| Deferoxamine (DFO) | Transgenic mouse | Improvement of cognitive function | [280] |
| Mouse | Improvement of memory | [281] | |
| Rat | Improvement of memory | [282] | |
| Deferiprone (DFP) | Transgenic mouse | Improvement of cognitive function | [285,286,287] |
| M30 | Rat | Recovery of memory impairment | [292,293] |
| Mouse | Increase of neuroprotective effect Improvement of memory | [294] | |
| HLA20 | Rat | Recovery of memory impairment | [292,293] |
Table 4.
The reported relations between redox-active metal ions and ADE. The changes in expression levels and/or activity of NEP, IDE, and ADAM10 associated with Cu(I/II) and Fe(II/III).
Table 4.
The reported relations between redox-active metal ions and ADE. The changes in expression levels and/or activity of NEP, IDE, and ADAM10 associated with Cu(I/II) and Fe(II/III).
| NEP | IDE | ADAM10 | ||
|---|---|---|---|---|
| Cu(I/II) | Expression Levels | Decreased | Need to study | Increased |
| Activity | Decreased | Decreased | Need to study | |
| Fe(II/III) | Expression Levels | Need to study | Need to study | Increased |
| Activity | Decreased | Need to study | Need to study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kim, N.; Lee, H.J. Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7697. https://doi.org/10.3390/ijms22147697
AMA Style
Kim N, Lee HJ. Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(14):7697. https://doi.org/10.3390/ijms22147697
Chicago/Turabian StyleKim, Namdoo, and Hyuck Jin Lee. 2021. "Redox-Active Metal Ions and Amyloid-Degrading Enzymes in Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 14: 7697. https://doi.org/10.3390/ijms22147697
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.